Melanoma is the most dangerous form of skin cancer.
These invasive growths develop when unrepaired DNA damage causes
mutations in skin cells resulting in prompt proliferation and the
formation of malignant tumors. If melanoma is detected early, it
can be easily cured with appropriate treatment such as surgical
removal. However, metastatic melanoma often proves fatal and
certain patients possess high-risk features for developing
metastases. Melanoma can metastasize almost anywhere, from nearby
tissues to distant major organs. The most typical metastatic sites
are the lymph nodes, lungs, liver, brain and bones. Many academic
reports have been published since 2010 on treatments for metastatic
melanoma, from chemotherapies to molecular-targeted therapies.
Research concerning the application of stem
cell-based therapies for cancer has recently emerged due to their
potential function as a drug delivery vehicle for therapeutic genes
directly to tumor sites. Stem cells, such as mesenchymal stem cells
(MSCs) and neural stem cells (NSCs), are attractive delivery
systems, as they are able to target tumor sites specifically due to
the secretion of chemoattractant factors from tumors. Their ability
to migrate and aggregate around the tumor at a high concentration
gives them the potential as a vector of enzyme/prodrug gene in
gene-directed enzyme prodrug therapy (GDEPT) of human cancers
(1). This review discusses the
current treatment options for metastatic melanoma patients and
elucidates the possibility of applying NSC therapy for melanoma and
justifying it with prior research on other stem cell-based
therapies for melanoma.
Identifying melanoma in its early stages is
extremely important since patients with early stage melanoma can be
surgically healed with relatively limited associated morbidity
(2). Increasing patient survival
can be accomplished by accompanying effective palliative management
of local disease with removal of systemic, especially solitary
lung, melanoma metastases (3). Of
144 patients who underwent surgical resection of non-regional
melanoma metastases, 20% had a 5-year survival rate (4), and in a phase II trial conducted by
the Southwest Oncology Group the overall 3- and 4-year survival
rates of stage IV melanoma patients were 36 and 31%, respectively
(5). Surgical tumor removal can
prevent metastasis; however, surgical removal cannot be applied on
microscopic metastases. Therefore, it must be used with other
therapies such as surgical resection concomitant with systemic
targeted therapies.
A total of 1–6% of patients with melanoma undergo
radiation therapy in the USA. In particular, radiation therapy is
used in patients with brain metastases as adjunct palliative
therapy. Radiation therapy, in contrast to surgical management, has
the benefit of potentially inducing an abscopal effect in which
both the treated tumor and the non-irradiated site respond to the
therapy (2). This abscopal effect
is believed to be generated through immune system mediation, as
radiation therapy can induce cross-priming in which released tumor
antigens are expressed in MHC class I molecules by dendritic cells.
Activated CD8+ T cells can then migrate to far-off
tumors and promote lysis (6).
Dacarbazine (DTIC) is well known as a primary
chemotherapeutic treatment for metastatic melanoma. It is the first
and only alkylating agent approved by the FDA with intravenous
administration every 3–4 weeks at a dose of 800–1,000
mg/m2 (7). DTIC
functions by adding an alkyl group to the bases in DNA, which then
prevents cells from replicating. As the sole agent of treatment,
DTIC creates a partial response in up to 25% of melanomas and a
complete response in ~5% (8,9). Oral
delivery of the DTIC derivative, temozolamide, which showed a
similar response rate in metastatic melanoma, was developed more
recently. Temozolamide has the added ability to cross the
blood-brain barrier, which led to it becoming a first-line therapy
for brain metastases (10).
Analysis of combinations of chemotherapies such as cisplatin,
vinblastine, and DTIC has shown encouraging response rates, but
they have failed to prolong overall survival (OS) when compared
with the single agent DTIC (11,12).
Activated T cells express CTLA-4. This acts as a
negative regulator of T cells and helps preserve immunologic
homeostasis. Ipilimumab is an antibody that blocks CTLA-4 from
mediating T-cell downregulation and reinforces the antitumor
effects of T cells (13). Response
rates for ipilimumab alone range from 5 to 15% due to changes in
the dosage and patient selection in clinical trials (14–17). A
total of 1,861 patients were analyzed in 12 separate studies, and
the median OS was 11.4 months (95% CI, 10.7–12.1 months) with a
plateau at 22% in the survival curve around year 3 (18). Unfortunately, diarrhea, dermatitis,
hepatitis, endocrinopathies, and immune-related adverse events
accompanied the treatment (19).
Clinical trials which focused on a combination of immunotherapy and
chemotherapy such as ipilimumab and DTIC exhibited greater efficacy
than monotherapy. Patients treated with both ipilimumab and DTIC
showed higher OS than those who were treated with only DTIC. The
estimated survival rates for patients treated with a combination
therapy of ipilimumab and DTIC were 47.3% for 1 year, 28.5% for 2
years, and 20.8% for 3 years compared to survival rates of 36.3,
17.9 and 12.2% for patients treated with DTIC monotherapy (14). Adverse effects of ipilimumab such as
gastrointestinal perforations, diarrhea and colitis were less
common in groups treated with a combination of ipilimumab and DTIC
rather than ipilimumab alone at the same dose, but there were
reports of elevated liver function values (14).
T cells upregulate a surface receptor called PD-1 at
later stages of T-cell activation in contrast to CTLA-4, which is
upregulated in the early stages of T-cell activation. PD-1
regulates the immune system by binding to T cells and attenuating
their activity. Tumors are thought to avoid an immune response by
upregulating PD-LI, a ligand of PD-1 (20,21).
Therefore, preventing the PD-1 ligand from binding to the PD-1
receptor on tumor cells can recover the tumor-fighting function of
immune cells. Nivolumab and pembrolizumab are antagonists of the
PD-1 receptor that can disrupt the interaction of PD-1 and PD-L1.
This disruption can allow T cells to proliferate, infiltrate the
tumor and increase effector function (22). Pembrolizumab showed a 38% response
rate with median survival of >7 months in initial clinical
trials. In comparison with other melanoma treatments, the
side-effects were significantly diminished (14,23).
Nivolumab had parallel results for the treatment of
ipilumumab-resistant or BRAF inhibitor and ipilimumab-resistant
advanced melanoma. With nivolumab, 31.7% of patients had an
objective response compared to 10.6% of patients who were treated
with the investigator's choice of chemotherapy (ICC) (24). Adopting a combination therapy of
nivolumab and ipilimumab has shown the highest response rates.
Using these therapies simultaneously in phase I and II trials
demonstrated a 53–61% response rate with >80% tumor reduction in
all responding patients (25,26).
This result shows a synergistic effect between CTLA-4 and PD-1
inhibition and a recent report [Larkin et al (2015)] showed
that the median progression-free survival (PFS) of the combination
therapy was 11.5 months (95% CI, 8.9–16.7) compared with 2.9 months
(95% CI, 2.8–3.4) with ipilimumab alone and 6.9 months (95% CI,
4.3–9.5) with nivolumab alone (27). Vitiligo, colitis, hepatitis,
hypophysitis, and thyroiditis are adverse events of nivolumab, but
they appear less often compared to the treatment with a CTLA-4
antagonist. In addition, inflammatory pneumonitis along with a dry
cough, dyspnea, and ground opacities are unique to PD-1 blockade
and are potentially lethal (28).
The BRAF gene encodes a serine/threonine kinase that
is engaged in the mitogen-activated protein kinase (MAPK)/ERK
signaling pathway (29). The
MAPK/ERK signaling pathway is associated in transferring signals
for cellular proliferation and survival from the cell surface to
the nucleus, and ~50% of cutaneous melanomas are caused by a
mutation in the BRAF oncogene, which leads to fundamental
activation of the MAPK signaling pathway and uncontrolled cellular
proliferation (30,31). Vemurafenib and dabrafenib are potent
BRAF inhibitors with distinct antitumor effects specific to
melanoma cell lines with the BRAF V600E and V600E/K mutations
(32–35). In its initials trials, treatment
with vemurafenib induced complete or partial tumor regression in
81% of patients with melanoma containing the V600E BRAF mutation
(32). Vemurafenib received
approval for BRAF inhibitor monotherapy in 2011. Dabrafenib is
another BRAF-targeted therapy for melanoma which functions as a
reversible ATP-competitive inhibitor for BRAF and was approved in
March 2013 (31). Median PFS for
vemurafenib is 6.8 months compared to 5.1 months for dabrafenib,
which signifies that dabrafenib is not more effective than
vemurfaneib monotherapy (34,36).
However, a study showed that dabrafenib demonstrated efficacy for
patients with brain metastases and remains an effective therapeutic
option for this particular population (37). Vemurafenib showed favorable in
vitro and in vivo results and a 69% objective response
rate in phase I clinical trials (38,39).
As clinical trials proceeded to later phases, however, 90% of
patients gained resistance and showed disease progression within 9
months.
Arthralgia, fatigue, aminotransferase elevation,
nausea, vomiting and decreased kidney function were reported as
general side-effects of vemurafenib, and ~11% of patients
administered dabrafenib reported pyrexia as a side-effect (13,34).
Repeated exposure to mutant BRAF inhibitors can
alter not only the RAS-RAF-MEK-ERK signaling pathway but also
several other kinase pathways (36). As a result, expression levels of
RAS, CRAF and MEK were increased due to ERK pathway reactivation
(40,41). ERK signaling reactivation is driven
by the amplification or alternative splicing of BRAF causing BRAF
dimerization that prevents inhibitors from binding to BRAF V600E
monomers (42,43). For example, activation of the
PI3K/AKT signaling pathway promotes BRAF inhibitor resistance in
melanoma and is therefore a form of adaptive resistance (44). Changes in the tumor microenvironment
caused by increased levels of growth factors such as hepatocyte
growth factor (HGF) can be another mechanism for BRAF inhibitor
resistance and were found to be linked to poor clinical outcomes
(45,46). However, changes in tumor
microenvironment are not hypothesized to be the primary cause of
drug resistance, but they are considered to be a secondary
contributor which could be a targetable option for preventing
adaptive resistance in melanoma tumors (47).
While direct targeting of mutated oncogenic BRAF has
been successful for those with mutated BRAF metastatic melanoma,
blocking MEK, a protein located downstream of BRAF in the MAPK
signaling pathway, showed remarkable success as well. Compared to
oncogenic BRAF mutations, oncogenic MEK mutations are less common
in melanoma. However, because of BRAF inhibitor resistance,
targeting downstream of BRAF for therapeutic efficacy has become a
research topic of interest (13,31).
The common MEK mutation C121S accelerates melanoma growth and
confers resistance to BRAF V600E mutant melanoma cells to
vemurafenib. C121S creates an active kinase that allows for
activation of downstream ERK without upstream activation by BRAF
(48). A MEK inhibitor called
trametinib has been FDA approved as a single agent for melanoma
patients with BRAF V600E or V600K mutations as of June 2013
(13). Trametinib impedes the
progression of advanced melanoma, especially in BRAF-mutant
patients (17,49). Trametinib showed a 33% response rate
for BRAF mutants with 5.6 months of median PFS in recent clinical
trials compared to a 10% response rate for BRAF wild-type tumors
(50,51). Although trametinib showed more
improvement in PFS and OS compared with chemotherapy, the objective
response rate was still lower than that of BRAF inhibitors
(13). Furthermore, trametinib
produced side-effects including diarrhea, peripheral edema,
hypertension and fatigue, which are typical of other MEK inhibitors
as well (52). Many resistance
pathways found in other treatments, especially BRAF inhibitors,
depend upon MEK signaling. Thus, MEK inhibition by trametinib in
combination with other treatments was able to increase their
potential as therapeutic agents and attenuate resistance in
clinical trials (50,51).
BRAF resistance from BRAF kinase inhibitors is
generated by reactivation of the MAPK pathway. In order to solve
this problem, Flaherty et al performed a combined treatment
with a selective BRAF inhibitor, dabrafenib, and a selective MEK
inhibitor, trametinib, in phase I and II trials (53). Vemurafenib was found to inhibit MAPK
signaling in melanoma patients with the BRAF V600E mutation and
produce prolonged survival and PFS in randomized phase III trials
in patients who had not previously received melanoma treatments.
Trametinib restricts MEK, a protein downstream of BRAF in the MAPK
pathway, and it showed an improvement in progression-free survival
and OS in BRAF V600E and V600K mutant melanomas. Rapid reactivation
of the MAPK signaling pathway has been related to BRAF inhibitor
resistance in preclinical models, but stimulation of cell death in
BRAF V600 mutant melanoma requires complete inhibition of the MAPK
pathway. This can be attained by combining a BRAF inhibitor with an
MEK inhibitor (53). The median OS
for combined treatment with trametinib and dabrafenib in a
multicenter, double-blind, phase III randomized controlled trial on
BRAF-mutant melanoma patients was 25.1 months (95% CI, 19.2-not
reached) and 18.7 months (15.2–23.7) for the dabrafenib
only-treated BRAF-mutant melanoma patients [hazard ratio (HR),
0.71; 95% CI, 0.55–0.92; p=0.0107]. Median PFS for the dabrafenib
and trametinib-combined therapy was 11.0 months (95% CI, 8.0–13.9)
and for the dabrafenib only-treated group this value was 8.8 months
(5.5–9.3) (HR, 0.67; 95% CI, 0.53–0.84; p=0.0004) (54). Flaherty et al examined the
adverse side-effects of combination therapy with dabrafenib and
trametinib. Patients who received both dabrafenib and trametinib
treatment had more constant and severe pyrexia and chills compared
to those who had only dabrafenib treatment. They also had more
persistent gastrointestinal toxic effects, such as nausea and
vomiting, but the majority were grade 1 or 2 events (53).
NSCs are self-renewing created by the
differentiation of embryonic tissue and generate the neurons and
glia of the developing brain. NSCs can be isolated, genetically
engineered and differentiated in vitro and reinstated into
the central nervous system (CNS). NSCs have potential for use in
cell replacement therapies in various neurologic disorders as has
been shown in several academic reports (55,56).
NSCs can be defined as cells that self-renew constantly and have
the potential to form intermediate and mature cells of neuronal and
glial lineages (57). From the year
2000 onward, there have been many reports concerning the adoption
of NSCs as a drug delivery vehicle specific to brain sites instead
of solely cell replacement. NSCs were found to appear near
metastatic tumor cells far from where they were transplanted into
animal models of brain neoplasia in these reports. This opens the
possibility to track down and destroy malignant cells by
manufacturing NSCs with chemotherapeutic qualities (58–60).
NSCs have the unique ability to integrate into the host's brain
without interfering with normal functions and can proliferate for
long periods (61). This uniqueness
could allow NSCs to be suitable as therapeutic delivery vehicles
for CNS disorders. In addition, their tropic migration towards
neoplasms is another favorable characteristic for their use as
vehicles for targeted delivery (55). Benedetti et al and Aboody
et al demonstrated that the progression of cancer xenografts
was suppressed by the cytotoxic effects of NSCs that were
manufactured with antitumor gene products (58,60).
These studies opened the doors to the potential of drug-equipped
NSCs as a tumor-homing therapy. NSCs migrate not only to injured
areas but also towards tumor foci. The tumor-tropic homing of NSCs
is directed by chemoattractants produced by cells in the normal
brain wounded by tumor growth or directly released from
glioblastoma multiforme (GBM) cells (62,63).
In hypoxic conditions, GBM cells upregulate the expression of
numerous pro-angiogenic factors and chemoattractants. The relevance
of hypoxia in the tumor-tropic migration of NSCs towards GBM was
demonstrated through several siRNA-mediated knockdowns. The
expression of the chemoattractant factor stromal cell-derived
factor-1 (SDF-1), uPA and vascular endothelial growth factor (VEGF)
was reduced with knockdown of HIG-α in GBM cells, which led to no
tumor-tropic migration of NSCs (62). More cytokines, growth factors, and
receptors have been addressed such as (SCF)/c-Kit (64), monocyte chemoattractant protein-1
(MCP-1)/CCL2 (65), Annexin A2
(66), HGF/c-Met (67) and HMGB1/RAGE (68) for arbitrating the tumor-tropic
migration of NSCs. Engineered NSCs could be designed to express a
plurality of receptors, so they can be deployed wherever
chemotactic signals are emitted from brain pathologies. Various
groups have revealed the potential of migrating towards not only
tumors of glial origin but also metastatic breast cancer and
melanoma foci in the brain (69–71).
Due to their intrinsic migratory and tumor-tropic properties, NSCs
epitomize a novel and potentially efficacious approach for the
treatment of invasive tumors.
Conventional treatments of cancer are impeded by
their inadequacy in being selective and specific to de novo
tumors. They harm normal and healthy tissues by their toxicity.
HB1.F3 cells, a parental cell line of the HB1.F3.CD/CE cell line,
show migration to subcutaneous xenografts of various solid tumors
such as prostate cancer, breast cancer, melanoma, glioma and
neuroblastoma. This suggests that these cell lines do not show
tissue-specific characteristics for therapeutic use (70).
GDEPT is a promising approach for advancing the
selectivity of conventional chemotherapeutics. GDEPT improves
selectivity by delivering 'suicide' genes such as cytosine
deaminase (CD), carboxylesterase (CE), and herpes simplex virus
type 1 thymidine kinase (HSV1-tk), to cancer cells, which lets them
convert non- or low-cytotoxic prodrugs to cytotoxic drugs (58,72,73).
Using GDEPT allows human tumors to be selectively targeted and
specifically treated to increase efficacy and diminish the
side-effects of biological drugs (74). For example, CD converts
5-fluorocytosine (5-FC), a non-toxic drug, to 5-fluorouracil
(5-FU), a toxic agent, CE converts CPT-11 to SN-38, and HSV1-tk
converts GCV to an active metabolite. An essential aspect of GDEPT
is a foreign enzyme expressed only at the tumor site where it is
able to shift a prodrug into its cytotoxic metabolite in
vivo (58). The therapeutic
efficacy of a polymerase chain reaction (PCR) vector which conveyed
a suicide gene, yeast CD, that converts the prodrug 5-FC to the
cytotoxic 5-FU was exhibited after delivery by infusion into the
regional circulation in a multifocal hepatic metastasis model of
colon cancer (75). A noticeable
boost in apoptotic cells and a decrease in proliferated cells in
human breast cancer cell lines was detected when combined treatment
was used with the CD/5-FC suicide system and hTNFα expression
(76).
Selective penetration to tumor sites is the primary
handicap that current gene therapy strategies are confronting, but
this can be overcome by using NSCs. NSCs are able to serve as a
delivery vehicle to target and propagate therapeutic gene products
over tumor sites. The human NSC line HB1.F3.CD was implanted
intracranially at distant sites from the tumor, and the NSCs
selectively migrated to the GBM tumor mass while bypassing normal
tissue which resulted in 80–85% reduction in tumor volume after
injection of the prodrug 5-FC (69,77,78).
NSCs are assumed to have a bystander effect through their
selectively eliminating behavior against dividing tumor cells
wherein toxic prodrugs and their metabolites circulate across gap
junctions and interstitial space to surrounding cells (74). Although the selective migration
towards tumor sites of HB1.F3 parental cells, the HB1.F3.CD/CE cell
line and other stem/progenitor cells has not been fully explained,
biological factors such as SDF-1, scatter factor (SCF; HGF), VEGF
and MCP-1 expressed in tumor cells seem to participate in
chemotaxis to human tumors (55,64,68,79–83).
Adopting the tumor-tropic behavior of NSCs could lead to
significant utility for the treatment of a variety of metastatic
tumors.
Among many types of stem cells, MSCs have emerged as
a potential transporter for not only regenerative medicine but also
cancer therapy. There have been several studies suggesting that
MSCs are able to migrate to both primary and metastatic tumor sites
through associations with various chemokines and cytokines
(1). Similar to NSCs, MSCs can
track specifically to tumor sites via chemokines and cytokines
emitted from tumors (84–87). There is a large body of research
concerning the application of MSCs as carriers of anticancer agents
for melanoma treatments. For example, bone marrow-derived MSCs
engineered to carry the P450 gene showed the ability to inhibit the
growth of malignant melanoma in vitro and in vivo by
reinforcing the expression of CYP2E1 (88). A study by Jing et al used
adipose tissue-derived mesenchymal stromal cells (AT-MSCs) as a
carrier to deliver enhanced expression of TRAIL protein for
impeding melanoma growth. TRAIL protein induced apoptosis by
readjusting the expression of members of the PI3K-AKT signaling
pathway (89). Seo et al
demonstrated the antitumor effect of engineered canine AT-MSC
(cAT-MSC)-producing interferon-β with cisplatin in mouse melanoma
models. The combination of cAT-MSC-IFN-β and cisplatin had more
compelling results than the cisplatin-alone group in inhibiting the
growth of melanoma and increasing the survival rate (90). Tyciakova et al used
engineered AT-MSC-secreting TNFα protein to assess its therapeutic
effect on melanoma. AT-MSC-TNFα restrained melanoma cells from
growth in vitro by inducing apoptosis via activating
caspase-3/7 and inhibited the tumor mass up to 97.5% (91). All these studies suggest that stem
cells are satisfactory as a carrier of both anticancer drugs and
genes for targeting cancers. Overall, the data obtained from
alternative stem cell-based therapies on melanoma propose the
feasibility of NSCs as a delivery system for targeted agents in the
treatment of melanoma.
As we learn more about the mechanisms of melanoma,
treatment has been revolutionized. The advancement of
immunotherapies and targeted therapies has significantly improved
clinical results. However, although there are currently more
wide-ranging treatment options than in the past, it has become
apparent that monotherapy will likely be unsuccessful due to the
aggressiveness and hypermutable nature of melanoma tumors. Thus
far, combination therapy has produced the most convincing clinical
results. Although both immunotherapy and targeted therapies have
conspicuous advantages and disadvantages, preclinical results show
that the combination of these treatments could enhance patient
outcomes. However, related data are inadequate to make a concrete
determination, as the data of patients treated with combination
therapy are limited. The toxicity and resistance issues plaguing
many existing treatments must also be carefully considered with
combination therapy. Therefore, the ultimate efficacy of
combination therapies remains unclear until further data are
gathered.
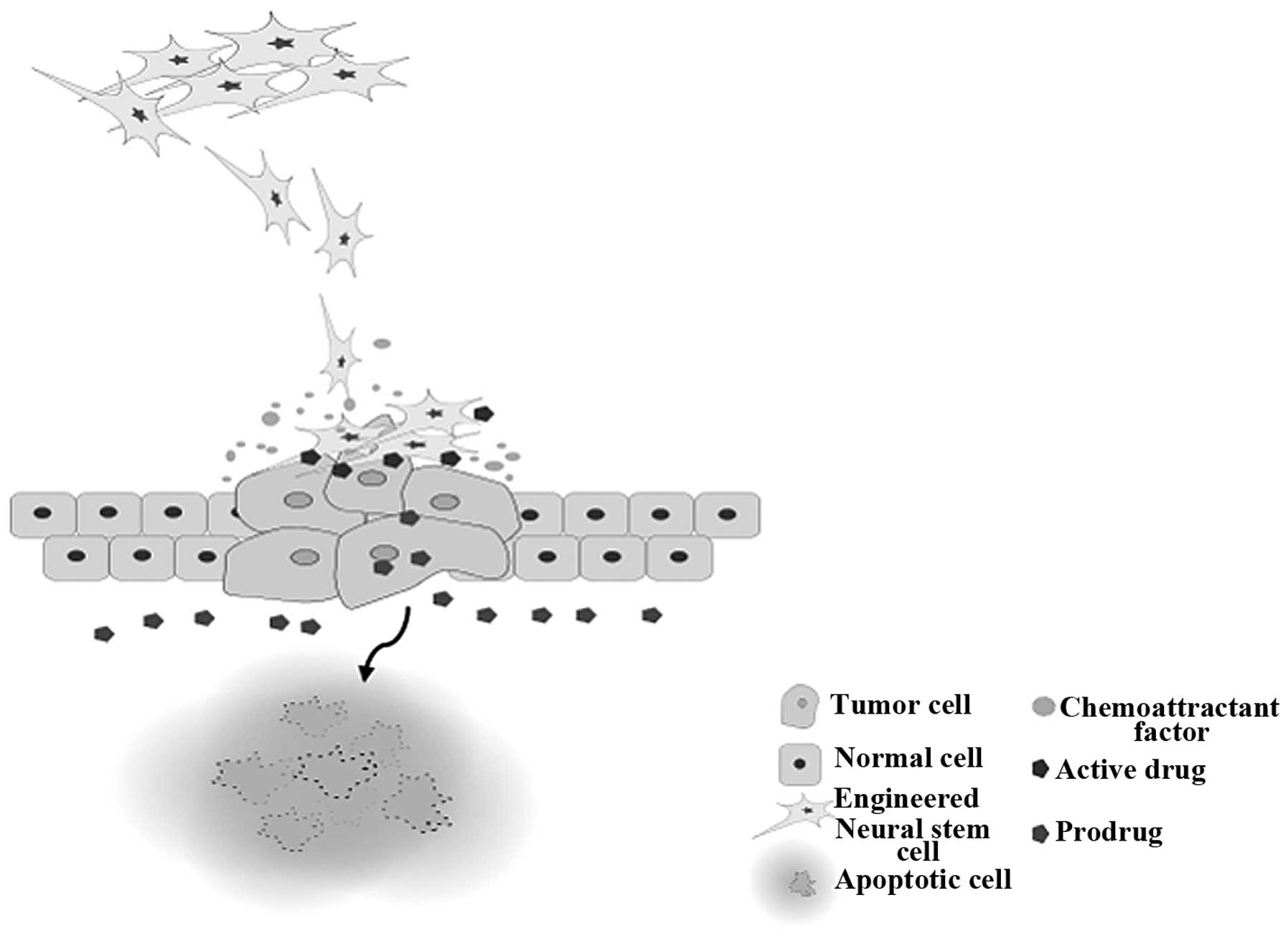
The common obstacle that current melanoma treatment
options confront is damage to other tissues. This issue has placed
patients in situations where whether to continue their treatment or
not has been a serious consideration for maximizing their chances
of survival. However, a parental cell line of HB1.F3.CD/CE has been
demonstrated to exhibit migratory behavior to subcutaneous
xenografts of various solid tumors in the prostate and breast as
well as melanoma, glioma, and neuroblastoma (Fig. 1). We can interpret that engineered
NSCs with suicide genes can be used to selectively target not only
melanoma but also tumors that have already metastasized to other
sites without damaging normal tissues for therapeutic use, as
demonstrated in Table I. Although,
to date, research is lacking regarding the use of engineered NSCs
for melanoma, data from other stem cell-based therapies on melanoma
and the features of NSCs indicate that NSC therapy could be the
next paradigm in gene therapy for melanoma and other cancers in
preclinical and clinical cases. Thus, their potential as a
specialized delivery vehicle should be explored in future
studies.
This study was supported by the Basic Science
Research Program through the National Research Foundation (NRF) of
Korea funded by the Ministry of Education, Science and Technology
(MEST) (2013R1A1A2059092). In addition, this study was supported by
a grant from the Next-Generation BioGreen 21 Program (no.
PJ011355), Rural Development Administration, Republic of Korea.
|
1
|
Mirzaei H, Sahebkar A, Avan A, Jaafari MR,
Salehi R, Salehi H, Baharvand H, Rezaei A, Hadjati J, Pawelek JM,
et al: Application of mesenchymal stem cells in melanoma: A
potential therapeutic strategy for delivery of targeted agents.
Curr Med Chem. 23:455–463. 2016. View Article : Google Scholar
|
|
2
|
Maverakis E, Cornelius LA, Bowen GM, Phan
T, Patel FB, Fitzmaurice S, He Y, Burrall B, Duong C, Kloxin AM, et
al: Metastatic melanoma - a review of current and future treatment
options. Acta Derm Venereol. 95:516–524. 2015. View Article : Google Scholar
|
|
3
|
Younes R, Abrao FC and Gross J: Pulmonary
metastasectomy for malignant melanoma: Prognostic factors for
long-term survival. Melanoma Res. 23:307–311. 2013.PubMed/NCBI
|
|
4
|
Wong JH, Skinner KA, Kim KA, Foshag LJ and
Morton DL: The role of surgery in the treatment of nonregionally
recurrent melanoma. Surgery. 113:389–394. 1993.PubMed/NCBI
|
|
5
|
Sosman JA, Moon J, Tuthill RJ, Warneke JA,
Vetto JT, Redman BG, Liu PY, Unger JM, Flaherty LE and Sondak VK: A
phase 2 trial of complete resection for stage IV melanoma: Results
of Southwest Oncology Group Clinical Trial S9430. Cancer.
117:4740–4706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
National Cancer Institute of Canada
Melanoma Group: Vinblastine, bleomycin, and cis-platinum for the
treatment of metastatic malignant melanoma. J Clin Oncol.
2:131–134. 1984.
|
|
7
|
Kim T, Amaria RN, Spencer C, Reuben A,
Cooper ZA and Wargo JA: Combining targeted therapy and immune
checkpoint inhibitors in the treatment of metastatic melanoma.
Cancer Biol Med. 11:237–246. 2014.
|
|
8
|
Eggermont AM and Kirkwood JM:
Re-evaluating the role of dacarbazine in metastatic melanoma: What
have we learned in 30 years? Eur J Cancer. 40:1825–1836. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huncharek M, Caubet JF and McGarry R:
Single-agent DTIC versus combination chemotherapy with or without
immunotherapy in metastatic melanoma: A meta-analysis of 3273
patients from 20 randomized trials. Melanoma Res. 11:75–81. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Middleton MR, Grob JJ, Aaronson N,
Fierlbeck G, Tilgen W, Seiter S, Gore M, Aamdal S, Cebon J, Coates
A, et al: Randomized phase III study of temozolomide versus
dacarbazine in the treatment of patients with advanced metastatic
malignant melanoma. J Clin Oncol. 18:158–166. 2000.PubMed/NCBI
|
|
11
|
Chapman PB, Einhorn LH, Meyers ML, Saxman
S, Destro AN, Panageas KS, Begg CB, Agarwala SS, Schuchter LM,
Ernstoff MS, et al: Phase III multicenter randomized trial of the
Dartmouth regimen versus dacarbazine in patients with metastatic
melanoma. J Clin Oncol. 17:2745–2751. 1999.PubMed/NCBI
|
|
12
|
Legha SS, Ring S, Papadopoulos N, Plager
C, Chawla S and Benjamin R: A prospective evaluation of a
triple-drug regimen containing cisplatin, vinblastine, and
dacarbazine (CVD) for metastatic melanoma. Cancer. 64:2024–2029.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
A Schindler K and Postow MA: Current
options and future directions in the systemic treatment of
metastatic melanoma. J Community Support Oncol. 12:20–26. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Robert C, Thomas L, Bondarenko I, O'Day S,
Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al:
Ipilimumab plus dacarbazine for previously untreated metastatic
melanoma. N Engl J Med. 364:2517–2526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hersh EM, O'Day SJ, Powderly J, Khan KD,
Pavlick AC, Cranmer LD, Samlowski WE, Nichol GM, Yellin MJ and
Weber JS: A phase II multicenter study of ipilimumab with or
without dacarbazine in chemotherapy-naïve patients with advanced
melanoma. Invest New Drugs. 29:489–498. 2011. View Article : Google Scholar
|
|
16
|
Kaplan MG: Ipilimumab plus dacarbazine in
melanoma. N Engl J Med. 365:1256–1257; author reply 1257–1258.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luke JJ, Callahan MK, Postow MA, Romano E,
Ramaiya N, Bluth M, Giobbie-Hurder A, Lawrence DP, Ibrahim N, Ott
PA, et al: Clinical activity of ipilimumab for metastatic uveal
melanoma: A retrospective review of the Dana-Farber Cancer
Institute, Massachusetts General Hospital, Memorial Sloan-Kettering
Cancer Center, and University Hospital of Lausanne experience.
Cancer. 119:3687–3695. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schadendorf D, Hodi FS, Robert C, Weber
JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM and Wolchok JD:
Pooled analysis of long-term survival data from phase II and phase
III trials of ipilimumab in unresectable or metastatic melanoma. J
Clin Oncol. 33:1889–1894. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weber JS, Kähler KC and Hauschild A:
Management of immune-related adverse events and kinetics of
response with ipilimumab. J Clin Oncol. 30:2691–2697. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zou W and Chen L: Inhibitory B7-family
molecules in the tumour microenvironment. Nat Rev Immunol.
8:467–477. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Keir ME, Liang SC, Guleria I, Latchman YE,
Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH and Sharpe
AH: Tissue expression of PD-L1 mediates peripheral T cell
tolerance. J Exp Med. 203:883–895. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tumeh PC, Harview CL, Yearley JH, Shintaku
IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu
V, et al: PD-1 blockade induces responses by inhibiting adaptive
immune resistance. Nature. 515:568–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hamid O, Robert C, Daud A, Hodi FS, Hwu
WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al:
Safety and tumor responses with lambrolizumab (anti-PD-1) in
melanoma. N Engl J Med. 369:134–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weber JS, D'Angelo SP, Minor D, Hodi FS,
Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD,
et al: Nivolumab versus chemotherapy in patients with advanced
melanoma who progressed after anti-CTLA-4 treatment (CheckMate
037): A randomised, controlled, open-label, phase 3 trial. Lancet
Oncol. 16:375–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wolchok JD, Kluger H, Callahan MK, Postow
MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K,
et al: Nivolumab plus ipilimumab in advanced melanoma. N Engl J
Med. 369:122–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Postow MA, Chesney J, Pavlick AC, Robert
C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK,
Agarwala SS, et al: Nivolumab and ipilimumab versus ipilimumab in
untreated melanoma. N Engl J Med. 372:2006–2017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Larkin J, Hodi FS and Wolchok JD: Combined
nivolumab and ipilimumab or monotherapy in untreated melanoma. N
Engl J Med. 373:1270–1271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Topalian SL, Hodi FS, Brahmer JR,
Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD,
Sosman JA, Atkins MB, et al: Safety, activity, and immune
correlates of anti-PD-1 antibody in cancer. N Engl J Med.
366:2443–2454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davies MA, Stemke-Hale K, Lin E, Tellez C,
Deng W, Gopal YN, Woodman SE, Calderone TC, Ju Z, Lazar AJ, et al:
Integrated molecular and clinical analysis of AKT activation in
metastatic melanoma. Clin Cancer Res. 15:7538–7546. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Davey RJ, van der Westhuizen A and Bowden
NA: Metastatic melanoma treatment: Combining old and new therapies.
Crit Rev Oncol Hematol. 98:242–253. 2016. View Article : Google Scholar
|
|
32
|
Flaherty KT, Puzanov I, Kim KB, Ribas A,
McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et
al: Inhibition of mutated, activated BRAF in metastatic melanoma. N
Engl J Med. 363:809–819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ribas A and Flaherty KT: BRAF targeted
therapy changes the treatment paradigm in melanoma. Nat Rev Clin
Oncol. 8:426–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bollag G, Hirth P, Tsai J, Zhang J,
Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al:
Clinical efficacy of a RAF inhibitor needs broad target blockade in
BRAF-mutant melanoma. Nature. 467:596–599. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sosman JA, Kim KB, Schuchter L, Gonzalez
R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ,
Flaherty KT, et al: Survival in BRAF V600-mutant advanced melanoma
treated with vemurafenib. N Engl J Med. 366:707–714. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Long GV, Trefzer U, Davies MA, Kefford RF,
Ascierto PA, Chapman PB, Puzanov I, Hauschild A, Robert C, Algazi
A, et al: Dabrafenib in patients with Val600Glu or Val600Lys
BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A
multicentre, open-label, phase 2 trial. Lancet Oncol. 13:1087–1095.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Heakal Y, Kester M and Savage S:
Vemurafenib (PLX4032): An orally available inhibitor of mutated
BRAF for the treatment of metastatic melanoma. Ann Pharmacother.
45:1399–1405. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Luke JJ and Hodi FS: Vemurafenib and BRAF
inhibition: A new class of treatment for metastatic melanoma. Clin
Cancer Res. 18:9–14. 2012. View Article : Google Scholar
|
|
40
|
Lo RS and Shi H: Detecting mechanisms of
acquired BRAF inhibitor resistance in melanoma. Methods Mol Biol.
1102:163–174. 2014. View Article : Google Scholar
|
|
41
|
Sullivan RJ and Flaherty KT: Resistance to
BRAF-targeted therapy in melanoma. Eur J Cancer. 49:1297–1304.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Poulikakos PI, Persaud Y, Janakiraman M,
Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al:
RAF inhibitor resistance is mediated by dimerization of aberrantly
spliced BRAF(V600E). Nature. 480:387–390. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shi H, Hugo W, Kong X, Hong A, Koya RC,
Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al:
Acquired resistance and clonal evolution in melanoma during BRAF
inhibitor therapy. Cancer Discov. 4:80–93. 2014. View Article : Google Scholar :
|
|
44
|
Villanueva J, Vultur A, Lee JT,
Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu
X, Gimotty PA, Kee D, et al: Acquired resistance to BRAF inhibitors
mediated by a RAF kinase switch in melanoma can be overcome by
cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 18:683–695. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Koefinger P, Wels C, Joshi S, Damm S,
Steinbauer E, Beham-Schmid C, Frank S, Bergler H and Schaider H:
The cadherin switch in melanoma instigated by HGF is mediated
through epithelial-mesenchymal transition regulators. Pigment Cell
Melanoma Res. 24:382–385. 2011. View Article : Google Scholar :
|
|
46
|
Topcu-Yilmaz P, Kiratli H, Saglam A,
Söylemezoglu F and Hascelik G: Correlation of clinicopathological
parameters with HGF, c-Met, EGFR, and IGF-1R expression in uveal
melanoma. Melanoma Res. 20:126–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gajewski TF: Identifying and overcoming
immune resistance mechanisms in the melanoma tumor
microenvironment. Clin Cancer Res. 12:2326s–2330s. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wagle N, Emery C, Berger MF, Davis MJ,
Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE,
Hahn WC, et al: Dissecting therapeutic resistance to RAF inhibition
in melanoma by tumor genomic profiling. J Clin Oncol. 29:3085–3096.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim KB, Kefford R, Pavlick AC, Infante JR,
Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, et
al: Phase II study of the MEK1/MEK2 inhibitor Trametinib in
patients with metastatic BRAF-mutant cutaneous melanoma previously
treated with or without a BRAF inhibitor. J Clin Oncol. 31:482–489.
2013. View Article : Google Scholar
|
|
50
|
Menzies AM and Long GV: Dabrafenib and
trametinib, alone and in combination for BRAF-mutant metastatic
melanoma. Clin Cancer Res. 20:2035–2043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim MO, Kim SH, Oi N, Lee MH, Yu DH, Kim
DJ, Cho EJ, Bode AM, Cho YY, Bowden TG, et al: Embryonic
stem-cell-preconditioned microenvironment induces loss of cancer
cell properties in human melanoma cells. Pigment Cell Melanoma Res.
24:922–931. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kirkwood JM, Bastholt L, Robert C, Sosman
J, Larkin J, Hersey P, Middleton M, Cantarini M, Zazulina V,
Kemsley K, et al: Phase II, open-label, randomized trial of the
MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in
patients with advanced melanoma. Clin Cancer Res. 18:555–567. 2012.
View Article : Google Scholar
|
|
53
|
Flaherty KT, Infante JR, Daud A, Gonzalez
R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N,
et al: Combined BRAF and MEK inhibition in melanoma with BRAF V600
mutations. N Engl J Med. 367:1694–1703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Dabrafenib and trametinib versus dabrafenib and
placebo for Val600 BRAF-mutant melanoma: A multicentre,
double-blind, phase 3 randomised controlled trial. Lancet.
386:444–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Müller FJ, Snyder EY and Loring JF: Gene
therapy: Can neural stem cells deliver? Nat Rev Neurosci. 7:75–84.
2006. View Article : Google Scholar
|
|
56
|
Bago JR, Sheets KT and Hingtgen SD: Neural
stem cell therapy for cancer. Methods. 99:37–43. 2016. View Article : Google Scholar
|
|
57
|
Gage FH: Mammalian neural stem cells.
Science. 287:1433–1438. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Benedetti S, Pirola B, Pollo B, Magrassi
L, Bruzzone MG, Rigamonti D, Galli R, Selleri S, Di Meco F, De
Fraja C, et al: Gene therapy of experimental brain tumors using
neural progenitor cells. Nat Med. 6:447–450. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Herrlinger U, Woiciechowski C,
Sena-Esteves M, Aboody KS, Jacobs AH, Rainov NG, Snyder EY and
Breakefield XO: Neural precursor cells for delivery of
replication-conditional HSV-1 vectors to intracerebral gliomas. Mol
Ther. 1:347–357. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Aboody KS, Brown A, Rainov NG, Bower KA,
Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM, et al:
Neural stem cells display extensive tropism for pathology in adult
brain: Evidence from intracranial gliomas. Proc Natl Acad Sci USA.
97:12846–12851. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Consiglio A, Gritti A, Dolcetta D,
Follenzi A, Bordignon C, Gage FH, Vescovi AL and Naldini L: Robust
in vivo gene transfer into adult mammalian neural stem cells by
lentiviral vectors. Proc Natl Acad Sci USA. 101:14835–14840. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhao D, Najbauer J, Garcia E, Metz MZ,
Gutova M, Glackin CA, Kim SU and Aboody KS: Neural stem cell
tropism to glioma: Critical role of tumor hypoxia. Mol Cancer Res.
6:1819–1829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang S, Luo X, Wan F and Lei T: The roles
of hypoxia-inducible factors in regulating neural stem cells
migration to glioma stem cells and determinating their fates.
Neurochem Res. 37:2659–2666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sun L, Lee J and Fine HA: Neuronally
expressed stem cell factor induces neural stem cell migration to
areas of brain injury. J Clin Invest. 113:1364–1374. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Magge SN, Malik SZ, Royo NC, Chen HI, Yu
L, Snyder EY, O'Rourke DM and Watson DJ: Role of monocyte
chemoattractant protein-1 (MCP-1/CCL2) in migration of neural
progenitor cells toward glial tumors. J Neurosci Res. 87:1547–1555.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
An JH, Lee SY, Jeon JY, Cho KG, Kim SU and
Lee MA: Identification of gliotropic factors that induce human stem
cell migration to malignant tumor. J Proteome Res. 8:2873–2881.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Heese O, Disko A, Zirkel D, Westphal M and
Lamszus K: Neural stem cell migration toward gliomas in vitro.
Neuro Oncol. 7:476–484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schmidt NO, Przylecki W, Yang W, Ziu M,
Teng Y, Kim SU, Black PM, Aboody KS and Carroll RS: Brain tumor
tropism of transplanted human neural stem cells is induced by
vascular endothelial growth factor. Neoplasia. 7:623–629. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kim SK, Kim SU, Park IH, Bang JH, Aboody
KS, Wang KC, Cho BK, Kim M, Menon LG, Black PM, et al: Human neural
stem cells target experimental intracranial medulloblastoma and
deliver a therapeutic gene leading to tumor regression. Clin Cancer
Res. 12:5550–5556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Joo KM, Park IH, Shin JY, Jin J, Kang BG,
Kim MH, Lee SJ, Jo MY, Kim SU and Nam DH: Human neural stem cells
can target and deliver therapeutic genes to breast cancer brain
metastases. Mol Ther. 17:570–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Aboody KS, Bush RA, Garcia E, Metz MZ,
Najbauer J, Justus KA, Phelps DA, Remack JS, Yoon KJ, Gillespie S,
et al: Development of a tumor-selective approach to treat
metastatic cancer. PLoS One. 1:e232006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Schepelmann S, Ogilvie LM, Hedley D,
Friedlos F, Martin J, Scanlon I, Chen P, Marais R and Springer CJ:
Suicide gene therapy of human colon carcinoma xenografts using an
armed oncolytic adenovirus expressing carboxypeptidase G2. Cancer
Res. 67:4949–4955. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Schepelmann S and Springer CJ: Viral
vectors for gene-directed enzyme prodrug therapy. Curr Gene Ther.
6:647–670. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kim SU, Jeung EB, Kim YB, Cho MH and Choi
KC: Potential tumor-tropic effect of genetically engineered stem
cells expressing suicide enzymes to selectively target invasive
cancer in animal models. Anticancer Res. 31:1249–1258.
2011.PubMed/NCBI
|
|
75
|
Hiraoka K, Kimura T, Logg CR, Tai CK, Haga
K, Lawson GW and Kasahara N: Therapeutic efficacy of
replication-competent retrovirus vector-mediated suicide gene
therapy in a multifocal colorectal cancer metastasis model. Cancer
Res. 67:5345–5353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Pastorakova A, Hlubinova K, Jakubikova J
and Altaner C: Combine cancer gene therapy harnessing plasmids
expressing human tumor necrosis factor alpha and Herpes simplex
thymidine kinase suicide gene. Neoplasma. 53:353–362.
2006.PubMed/NCBI
|
|
77
|
Brown AB, Yang W, Schmidt NO, Carroll R,
Leishear KK, Rainov NG, Black PM, Breakefield XO and Aboody KS:
Intravascular delivery of neural stem cell lines to target
intracranial and extracranial tumors of neural and non-neural
origin. Hum Gene Ther. 14:1777–1785. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yip S, Aboody KS, Burns M, Imitola J,
Boockvar JA, Allport J, Park KI, Teng YD, Lachyankar M, McIntosh T,
et al: Neural stem cell biology may be well suited for improving
brain tumor therapies. Cancer J. 9:189–204. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ehtesham M, Yuan X, Kabos P, Chung NH, Liu
G, Akasaki Y, Black KL and Yu JS: Glioma tropic neural stem cells
consist of astrocytic precursors and their migratory capacity is
mediated by CXCR4. Neoplasia. 6:287–293. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sun L, Hui AM, Su Q, Vortmeyer A,
Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey
R, et al: Neuronal and glioma-derived stem cell factor induces
angiogenesis within the brain. Cancer Cell. 9:287–300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Beppu K, Jaboine J, Merchant MS, Mackall
CL and Thiele CJ: Effect of imatinib mesylate on neuroblastoma
tumorigenesis and vascular endothelial growth factor expression. J
Natl Cancer Inst. 96:46–55. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Geminder H, Sagi-Assif O, Goldberg L,
Meshel T, Rechavi G, Witz IP and Ben-Baruch A: A possible role for
CXCR4 and its ligand, the CXC chemokine stromal cell-derived
factor-1, in the development of bone marrow metastases in
neuroblastoma. J Immunol. 167:4747–4757. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kucia M, Reca R, Miekus K, Wanzeck J,
Wojakowski W, Janowska-Wieczorek A, Ratajczak J and Ratajczak MZ:
Trafficking of normal stem cells and metastasis of cancer stem
cells involve similar mechanisms: Pivotal role of the SDF-1-CXCR4
axis. Stem Cells. 23:879–894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Vicari AP and Caux C: Chemokines in
cancer. Cytokine Growth Factor Rev. 13:143–154. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lazennec G and Richmond A: Chemokines and
chemokine receptors: New insights into cancer-related inflammation.
Trends Mol Med. 16:133–144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Loebinger MR and Janes SM: Stem cells as
vectors for antitumour therapy. Thorax. 65:362–369. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Spaeth E, Klopp A, Dembinski J, Andreeff M
and Marini F: Inflammation and tumor microenvironments: Defining
the migratory itinerary of mesenchymal stem cells. Gene Ther.
15:730–738. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang J, Ma D, Li Y, Yang Y, Hu X, Zhang W
and Fang Q: Targeted delivery of CYP2E1 recombinant adenovirus to
malignant melanoma by bone marrow-derived mesenchymal stem cells as
vehicles. Anticancer Drugs. 25:303–314. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Jing HX, Duan J, Zhou H, Hu QM and Lei TC:
Adipose derived mesenchymal stem cell facilitated TRAIL expression
in melanoma treatment in vitro. Mol Med Rep. 14:195–201.
2016.PubMed/NCBI
|
|
90
|
Seo KW, Lee HW, Oh YI, Ahn JO, Koh YR, Oh
SH, Kang SK and Youn HY: Anti-tumor effects of canine adipose
tissue-derived mesenchymal stromal cell-based interferon-β gene
therapy and cisplatin in a mouse melanoma model. Cytotherapy.
13:944–955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Tyciakova S, Matuskova M, Bohovic R,
Polakova K, Toro L, Skolekova S and Kucerova L: Genetically
engineered mesenchymal stromal cells producing TNFα have tumour
suppressing effect on human melanoma xenograft. J Gene Med.
17:54–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yi BR, Hwang KA, Aboody KS, Jeung EB, Kim
SU and Choi KC: Selective antitumor effect of neural stem cells
expressing cytosine deaminase and interferon-beta against ductal
breast cancer cells in cellular and xenograft models. Stem Cell Res
(Amst). 12:36–48. 2014. View Article : Google Scholar
|
|
93
|
Yi BR, Kim SU and Choi KC: Additional
effects of engineered stem cells expressing a therapeutic gene and
interferon-β in a xenograft mouse model of endometrial cancer. Int
J Oncol. 47:171–178. 2015.PubMed/NCBI
|
|
94
|
Yi BR, Park MA, Lee HR, Kang NH, Choi KJ,
Kim SU and Choi KC: Suppression of the growth of human colorectal
cancer cells by therapeutic stem cells expressing cytosine
deaminase and interferon-β via their tumor-tropic effect in
cellular and xenograft mouse models. Mol Oncol. 7:543–554. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kim DJ, Yi BR, Lee HR, Kim SU and Choi KC:
Pancreatic tumor mass in a xenograft mouse model is decreased by
treatment with therapeutic stem cells following introduction of
therapeutic genes. Oncol Rep. 30:1129–1136. 2013.PubMed/NCBI
|
|
96
|
Yi BR, Kim SU and Choi KC: Co-treatment
with therapeutic neural stem cells expressing carboxyl esterase and
CPT-11 inhibit growth of primary and metastatic lung cancers in
mice. Oncotarget. 5:12835–12848. 2014. View Article : Google Scholar : PubMed/NCBI
|