Introduction
Breast cancer is the second most prevalent
neoplastic disease among women and is characterised by a complex
aetiology and chemoresistant behaviour (1,2).
Although most women are treated with tamoxifen, the current
standard adjuvant therapy in women with estrogen receptor
(ER)-positive breast cancer, they develop resistance to the drug
(3). Cediranib (AZD2171, Recentin,
AstraZeneca) is a potent inhibitor of several receptor tyrosine
kinases (RTKs), such as VEGFR, KIT and PDGFRA (4,5).
Significant results have been obtained for patients with advanced
solid tumours, such as glioblastoma, lung and prostate tumours,
after cediranib treatment (6–9).
Recently, novel targets of cediranib have been identified by in
vitro and in vivo studies with glioblastoma cell lines
(10). Furthermore, it was shown
that cediranib alone or in combination with temozolomide is an
effective drug in anti-angiogenic therapies due to its greater
antitumour activity (10). Animal
models of breast cancer revealed that cediranib affected vessel
density and cell proliferation, but not with chronic administration
(11,12). Phase I/II studies have been
conducted with cediranib for breast cancer treatment, especially in
combination with other drugs, such as fulvestrant, olaparib and
RO4929097, a γ-secretase inhibitor (13–15).
The promise of cediranib treatment is an improvement in overall
survival of up to six months in several tumour types (6,7), and
while it has not yet been approved by the FDA, cediranib, alone or
in combination with other drugs, represents a new potential therapy
for breast cancer.
Despite these observations, in breast cancer cell
lines, the effects of cediranib on cellular processes such as
proliferation, migration and invasion are unknown. Therefore,
exploring the biological effect of cediranib on breast cancer cell
lines and revealing the pathways that can be predictive biomarkers
of cediranib response are necessary. MicroRNAs (miRNAs) are small
noncoding RNAs approximately 22–26 nucleotides (nt) long that are
involved in post-transcriptional repression or mRNA degradation in
a sequence-specific manner (16,17).
The role of miRNAs as new regulatory molecules that are involved in
different cancer processes have recently emerged (18). Several studies on miRNA profiling
have shown that these molecules can act as oncogenes and tumour
suppressors (19). Accordingly,
strategies to correct the deficiencies associated with miRNA
dysregulation have been proposed as future clinical interventions
for cancer patients (20). In
breast cancer, several miRNAs have been described in several steps
of tumorigenesis, angiogenesis and metastasis (21–23).
Furthermore, miRNAs have been considered as potential biomarkers in
breast cancer metastasis (24).
miRNAs also play a role in anticancer drug
resistance (25). Several miRNAs
have been associated with chemoresistance in breast cancer,
including miR-34a with docetaxel, miR-125b with anthracycline,
miR-451 and miR-27 with doxorubicin and miR-326 with doxorubicin
and VP-16 resistance (26–31). Several reports have shown that
deletions of miRNA regions can affect the regulation of RTKs and
the response to gefitinib or imatinib (32,33),
reinforcing a possible role for miRNAs in the mechanism of the
response and/or resistance to RTK inhibitors.
Overall, miRNAs can affect breast cancer drug
sensitivity with implications in clinical management and in
understanding the molecular mechanisms that may contribute to drug
susceptibility and resistance (17). Therefore, in the present study, we
aimed to evaluate the miRNA expression profiles of breast cancer
cell lines exposed to cediranib.
Materials and methods
Cell lines and determination of the
half-maximal inhibitory concentration (IC50)
In the present study, the breast carcinoma cell
lines Hs578T, MDA-MB-231 and T47D were obtained from the American
Type Culture Collection (ATCC; Manassas, VA, USA). Cells were
cultured in Dulbeccos modified Eagles medium (DMEM 1X, high
glucose), supplemented with 10% FBS (both from Gibco, Invitrogen
Life Technologies Grand Island, NY, USA) and 1% penicillin and
streptomycin solution (Sigma-Aldrich) at 37°C and 5%
CO2.
Authentication of cell lines was performed by short
tandem repeat (STR) DNA typing according to the International
Reference Standard for Authentication of Human Cell Lines using a
panel of eight (D5S818, D13S317, D7S820, D16S539, vWA, TH01, TPOX
and CSF1P0) STR loci plus gender determination (AMEL), using the
fluorescent labeling primers as reported by Dirks et al
(34). Briefly, 50 ng of DNA was
amplified in multiplex PCR reaction carried out in a total volume
of 10 µl with Qiagen Multiplex PCR kit (Qiagen) comprising 0.5 µM
of all fluorescent primer pairs plus 1 µM of TH01 primer
reinforcement, performed in a Veriti® 96-well Thermal
Cycler with an initial denaturation at 95°C for 15 min, amplified
for 30 cycles of denaturation at 95°C for 30 sec, annealing at 55°C
for 1 min 30 sec, extension at 72°C for 1 sec and a final cycle at
72°C for 30 min. The DNA amplification products were diluted 1:100
in Nuclease-Free Water Ultrapure (USB, Cleveland, OH, USA) and
combined with 0.3 µl of internal size standard GeneScan
500™ ROX™ (Applied Biosystems, Foster City,
CA, USA) in 8.7 µl formamide and loaded automatically in a
capillary electrophoresis using an Genetic Analyzer ABI PRISM 3500
(Applied Biosystems). The analysis was performed in GeneMapper
software version 4.1 (Applied Biosystems). Genotyping confirmed the
identity of all three cell lines.
The drug used, cediranib (Selleck Chemicals,
Houston, TX, USA), was diluted in 1% DMSO. To determine the
IC50, the cells were plated in increasing concentrations
of the drug from 0.1 to 100 µM, and an MTS proliferation assay was
performed. The IC50 concentration was calculated using
the drc package in R (35,36), and the best model for each cell type
was selected according to the Akaike information Criterion
(AIC).
Invasion and migration assays
The invasion assays were performed using BD BioCoat
Matrigel invasion chambers (BD Biosciences) according to the
manufacturers instructions and as previously described (10,37).
Briefly, 2.5×104 cells were plated in the
Matrigel-coated 24-well Transwell inserts in DMEM-0.5% containing
fixed concentrations of the drug. DMEM-10% was used as a
chemoattractant, and the cells that attached to the inserts were
fixed with methanol and stained with haematoxylin. The cells were
photographed at a ×40 magnification level and counted on a
pixel-by-pixel basis using ImageJ software. The invasiveness of the
cells exposed to cediranib (IC50 dose at 72 h) was
expressed in relation to the DMSO control (considered 100%
invasion) as the mean percentage of invasion ± SD. The assays were
performed in triplicate.
The migration capacity of the cells after cediranib
exposure was assessed using a wound-healing assay as previously
described (10,38). The cells were seeded in 6-well
plates and cultured until reaching 95% confluency. The monolayer of
cells was washed with PBS; a wound was created, and the cells were
incubated with the IC50 concentrations of cediranib for
varying times. The selected areas were photographed at ×40
magnification at 0 to 24 h for the Hs578T and MDA-MB-231 cells and
at 0 to 72 h for T47D cells. The relative migration distance was
calculated using the following formula: Percentage of wound closure
(%) = 100(A − B)/A, where A is the width of the cell wound before
incubation and B is the width of the cell wound after different
lengths of cediranib exposure. The assay was performed in
triplicate with four measurements for each well.
Western blot analysis and human
phospho-RTK array
To evaluate the inhibition of the intracellular
signalling pathways, Hs578T, MDA-MB-231 and T47D cells were
cultured in T25 culture flasks in DMEM 10% FBS. After reaching 90%
confluency, the cell lines were starved and exposed to cediranib
(IC50 values) for 12 h. Analysis of apoptosis was
conducted in 6-well culture plates at 70% confluency and incubated
for 12, 24 and 72 h. For all experiments, after time points, the
cell lines were washed and scraped in cold PBS and lysed in buffer,
containing 50 mM Tris, 150 mM NaCl, 5 mM EDTA, 1 mM
Na3VO4, 10 mM NaF, 10 mM sodium
pyrophosphate, 1% NP-40 and protease cocktail inhibitors. Western
blot analysis was performed using standard 10% sodium dodecyl
sulphate polyacrylamide gel electrophoresis (SDS-PAGE) loading 30
µg protein per line. To assess apoptosis and the activation of
intracellular signaling pathways, the antibodies used were the
following: primary antibody PARP total/cleaved [Cell Signaling
Technology (CST), no. 9532]; total p44/42 MAPK (CST, no. 137F5) and
phospho-p44/42 MAPK (CST, no. 8544); pan AKT (CST, no. C67E7) and
phospho-Akt (Ser473) (CST, no. 4060); β-actin (CST, no. 12262).
Secondary antibodies were used according to the manufacturers
instructions. Immune blots were performed using ECL western
blotting detection reagent (GE Healthcare) and finally the bands
were detected and images were captured using an Automatic
ImageQuant Mini LAS 4000 (GE Healthcare).
Human phospho-RTK array (PN no. 894042, R&D
Systems) was used according to the manufacturers instructions. In
brief, after the blocking step, 750 µg of protein were incubated
overnight at 4°C with nitrocellulose membranes containing 49
different anti-RTK Abs spots in duplicate. Next they were incubated
with anti-phospho-tyrosine-HRP for 2 h and detection was performed
with a Chemi Reagent Mix in Automatic ImageQuant Mini LAS 4000 (GE
Healthcare).
RNA isolation and quality control
Total RNA was isolated using an miRNeasy kit
(Qiagen) according to the manufacturers instructions.
Quantification was performed using a NanoDrop ND-1000
spectrophotometer (NanoDrop Products, Wilmington, DE, USA), and the
RNA quality was assessed using an Agilent Nano RNA chip with a
bioanalyser device (Agilent Technologies), as previously described
(39).
miRNA microarrays
To assess the expression of miRNAs after 24 h of
treatment with an IC50 dose of cediranib in Hs578T,
MDA-MB-231 and T47D cells and the same cell lines without treatment
were considered as controls, the Agilent Human miRNA Microarray
(8×15K-G4471A, Agilent Technologies) was used. Total RNA samples
(200 ng) were hybridised using an miRNA complete labelling and a
Hyb kit (Agilent Technologies) according to the manufacturers
instructions. The reactions followed a two-step preparation; an
initial dephosphorylation and denaturation of the total RNA was
performed, and the Cy3 fluorochrome was then incorporated with T4
ligase. The next steps included standard washing procedures and
hybridisation with the microarray slides. Images were scanned with
an Agilent DNA microarray scanner with SureScan technology (Agilent
Technologies), as previously described (40).
miRNA microarray data analysis
The raw data were obtained using the Feature
Extraction software v.11.0 (Agilent Technologies) and submitted to
R environment version 3.0.1 (36)
for additional analyses. Median signals (gMedianSignal and
gBGMedianSignal) were used as intensity values.
Normalisation was performed using the quantile method with the
Bioconductor ‘aroma.light’ package (41). miRNAs differentially expressed
between the cediranib-treated and control cells were obtained by
rank product analysis considering a P-value and pfp (positive false
predictions) ≤0.05 using the RankProd package (42). Heatmaps of the differentially
expressed miRNAs were constructed using hclust R and the distance
and average Pearson linkage were determined using the gplots R
package (43).
Target prediction and functional
analysis
Target prediction was performed using the mirDIP
interface (44). At least 3 of the
12 available algorithms were selected for prediction. To perform a
functional enrichment analysis, all the targets were separated
according to up- or downregulation and submitted to the Database
for Annotation, Visualization, and Integrated Discovery (DAVID)
(45). This approach was used to
identify significant biological processes in Gene Ontology level 2
that may be shared among the targets of the miRNAs of interest. A
biological process or pathway was considered significant if it
contained a minimum of 3 genes per category that had score values
<0.05 after a Benjamini-Hochberg correction. A compilation of
the categories according to a GoSlim summarisation was performed
using the REVIGO tool (46).
Confirmation by quantitative real-time
PCR (qRT-PCR)
qRT-PCR using TaqMan miRNA assays (Life
Technologies, Foster City, CA, USA) was used to confirm the
expression of five miRNAs from the microarray data that were
differentially expressed between cells exposed to cediranib
compared with the control group. The criterion for selecting the
miRNAs for confirmation was the level of expression. All of the
samples used in the microarray experiments were performed in
technical triplicates for the RT-PCR reactions. A total of 10 ng of
RNA was used in the reverse transcription reaction with
miRNA-specific primers in an Eppendorf Mastercycler (Eppendorf).
The real-time reactions were performed in a 7900 HT Fast Real-time
PCR system (Applied Biosystems).
All of the analysis procedures and graphs were
constructed using the R environment v.3.0.1. The normalisation step
was performed according to the 2−ΔΔCt method (47) using the minimum value of expression
of the untreated group as a calibrator. The cycle threshold (Ct)
values from the selected miRNA targets were subtracted from the Ct
values of the endogenous small noncoding RNA controls RNU44 and
RNU48 (control miRNA assay; Applied Biosystems). The criterion for
selecting the best endogenous RNA was the stability of expression
according to the NormFinder software (48). Differences in miRNA expression
between the treated and non-treated cell lines (Hs578T, MDA-MB-231
and T47D) were evaluated using Mann-Whitney U test, and differences
with a P≤0.05 were considered significant.
Results
Following an initial assessment of the basal
viability conditions for all the three breast cell lines, a total
density of 4×103 cells/well for Hs578T, 6×103
cells/well for MDA-MB-231 and 1×104 cells/well for the
T47D cell line was used. The IC50 was then determined
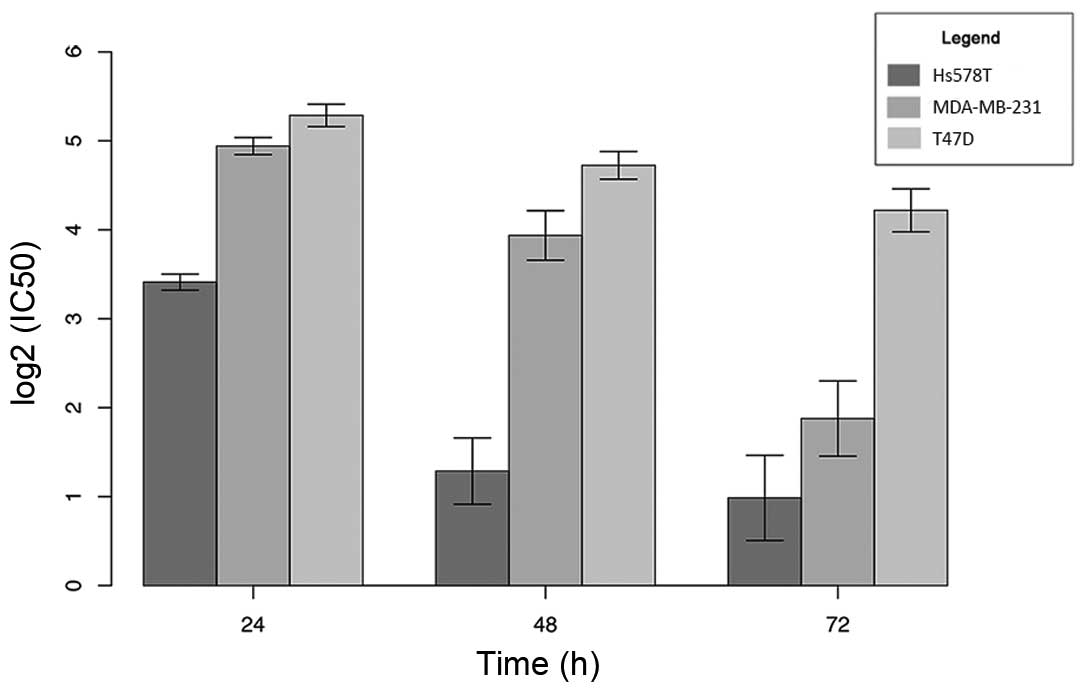
for the breast cancer cells at 24, 48 and 72 h, as shown in
Table I and Fig. 1. Our results showed that the most
sensitive breast cancer cell line was Hs578T, followed by
MDA-MB-231. The T47D cell line was the most resistant, with an
IC50 value 4-fold higher than that of the Hs578T cells
and almost 2-fold higher than that of the MDA-MB-231 cells after a
24-h treatment.
 | Table I.Half-maximal inhibitory concentration
(IC50) at 24, 48 and 72 h of cediranib exposure in the
Hs578T, MDA-MB-231 and T47D cells. |
Table I.
Half-maximal inhibitory concentration
(IC50) at 24, 48 and 72 h of cediranib exposure in the
Hs578T, MDA-MB-231 and T47D cells.
|
| IC50
(µM) |
|---|
|
|
|
|---|
| Cell line | 24 h | 48 h | 72 h |
|---|
| Hs578T | 10.66±0.66 | 2.51±0.62 | 2.08±0.77 |
| MDA-MB-231 | 30.77±2.01 | 15.57±3.03 | 2.52±0.81 |
| T47D | 38.69±2.90 | 26.54±2.79 | 18.85±3.21 |
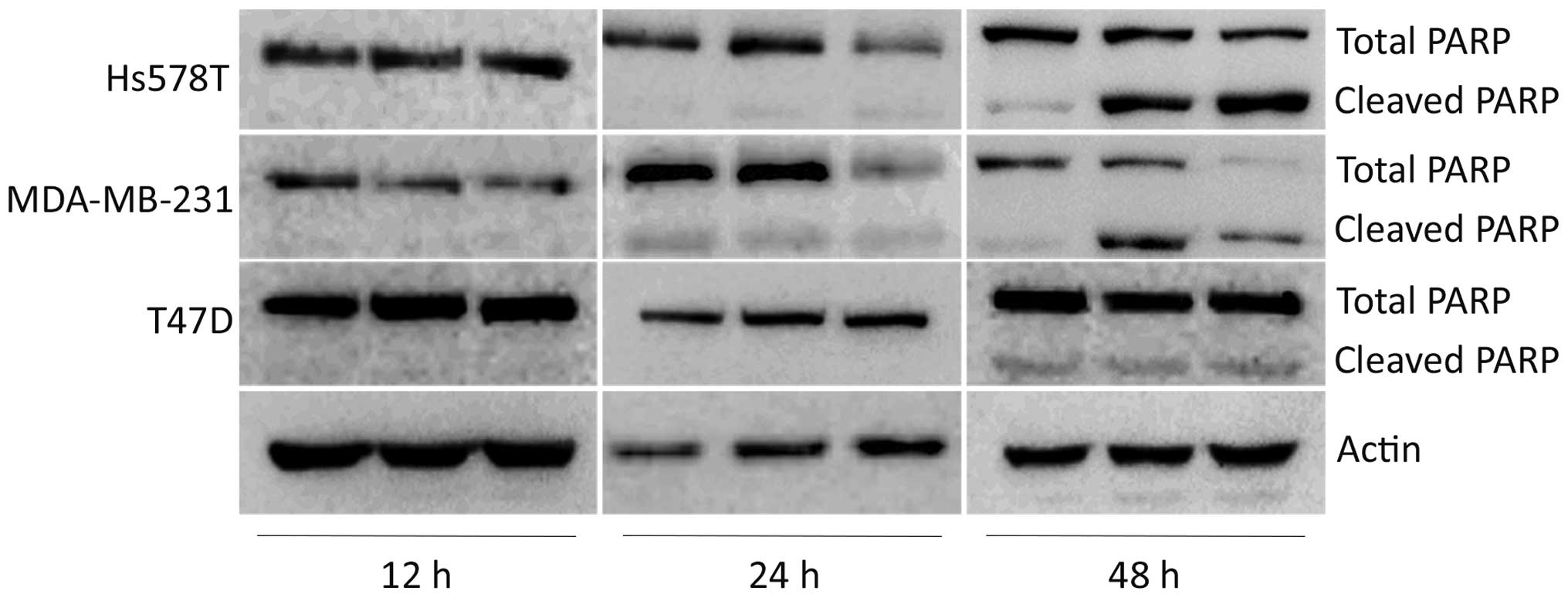
To determine whether decreased viability was due to
cytotoxic effects, we analyzed poly(ADP ribose) polymerase (PARP)
cleavage. We observed a striking effect on PARP cleavage using low
doses (0.5 and 2.0 µM) of cediranib in the Hs578T cell line.
Similar effects were demonstrated in the MDA-MB-231 cell line that
exhibited higher cleaved PARP levels. In contrast, the T47D cell
line showed lower PARP cleaved levels (8 and 18 µM) using a high
dose of cediranib drug, representing a resistant profile. Cytotoxic
effects demonstrated by cleaved PARP were not found at times less
than 24 h for both cell lines (Fig.
2).
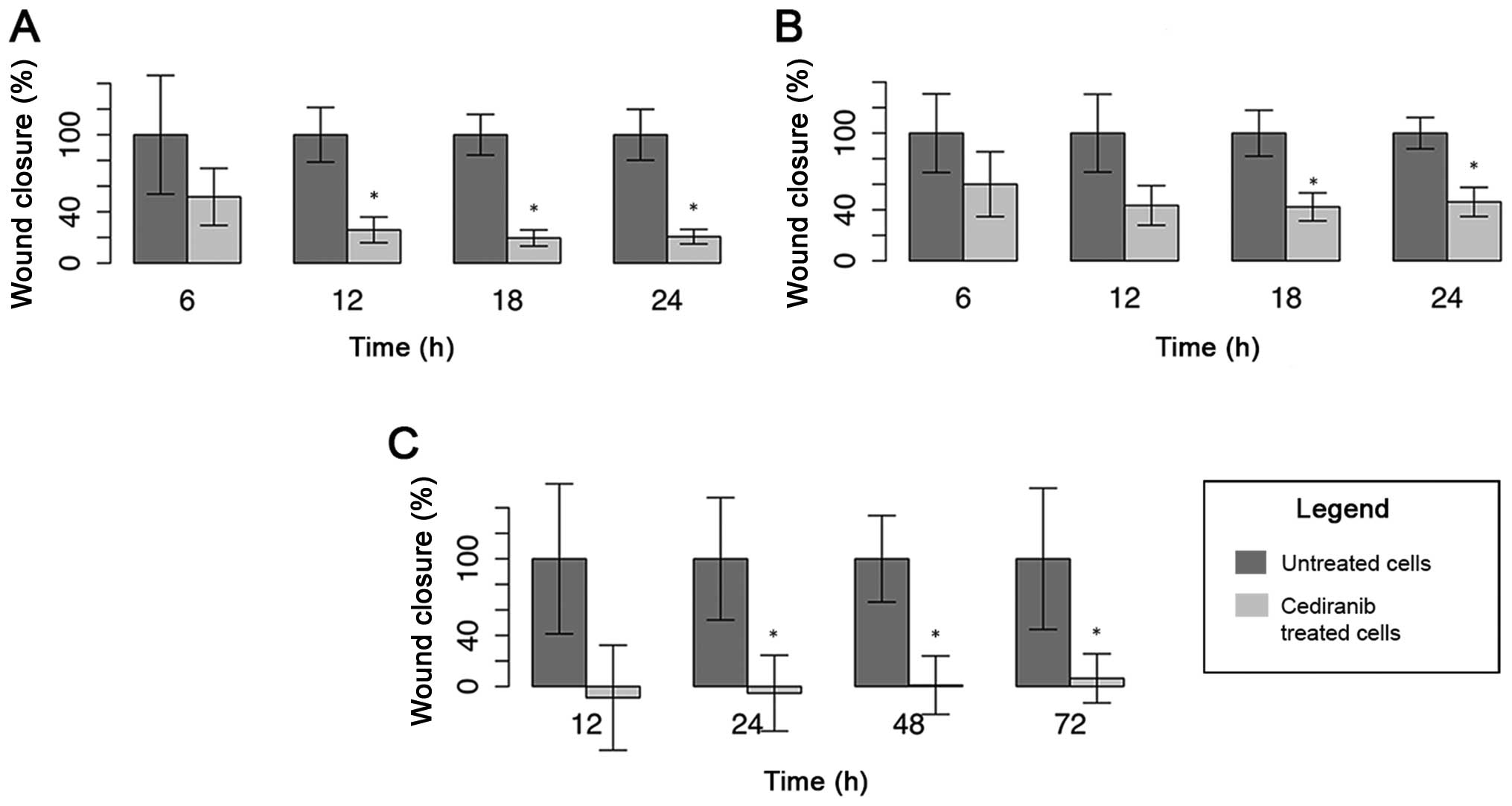
In order to assess the functional impact of
cediranib in migration, we used the wound-healing migration assay,
and observed the inhibition of cell migration in all of the cell
lines. After 24 h of cediranib exposure, the Hs578T cells exhibited
the most inhibition (80%), followed by the T47D cells (70%) and
MDA-MB-231 cells (54%) (Fig. 3).
Even the adhesion of this cell type appeared to be affected by the
drug (data not shown), leading to negative values, i.e., the wound
was extended because of the detachment of these cells. Due to this
behaviour, we studied the T47D migration ability for 72 h.
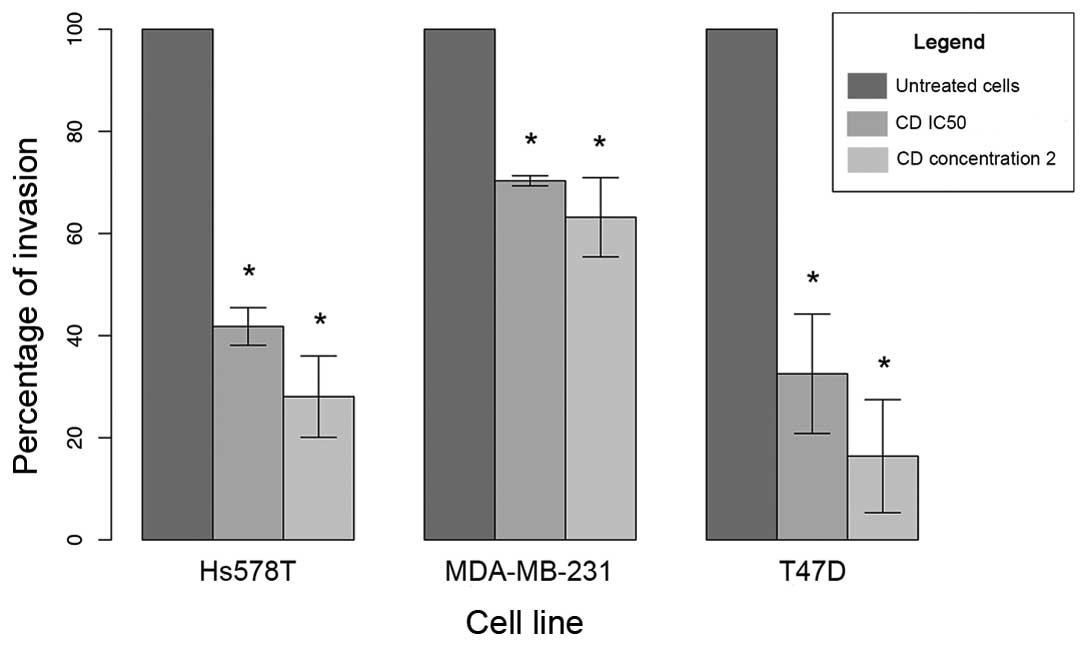
Similarly, cediranib treatment significantly inhibited cell
invasion in all breast cancer cell lines. The reduction in the
percentage of cell invasion at 24 h was ~70% in T47D, 60% in Hs578T
and 30% in the MDA-MB-231 cells (Fig.
4).
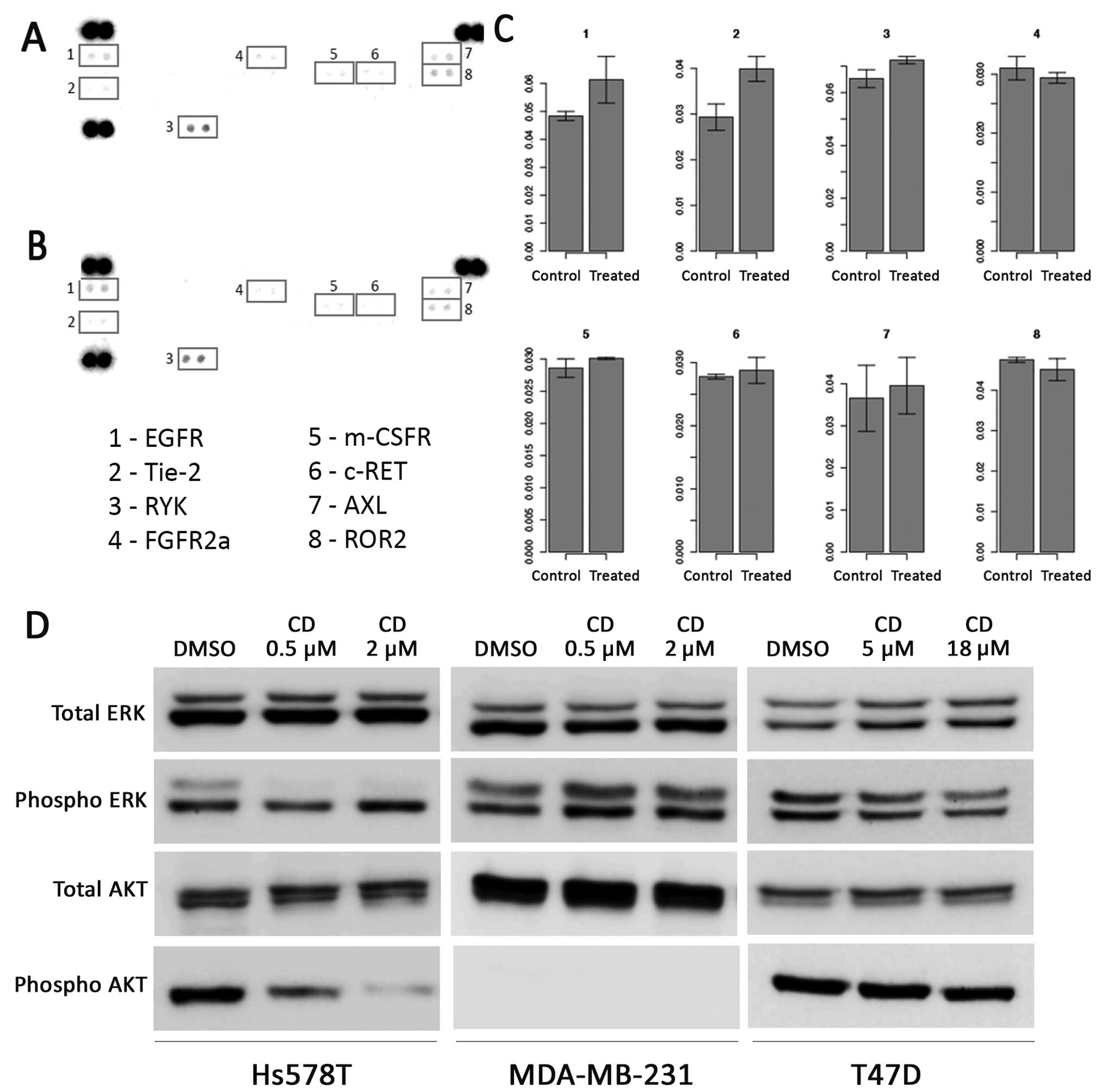
To identify the RTKs that are targets of cediranib,
we used a phospho-RTK array that assesses the levels of 49 RTKs, in
the most sensitive cell line, Hs578T, which was exposed to 2 µM
cediranib. Under basal levels, we observed the presence of the
active forms of EGFR, Tie-2, RYK, FGFR2α, m-CSFR, c-RET, AXL and
ROR2 (RTK-like orphan receptor 2) tyrosine kinase receptors
(Fig. 5A). Following cediranib
treatment, we found a slight increase in EGFR and Tie-2
phosphorylation levels (Fig. 5A-C)
and diminished phosphorylation levels of FGFR2α and ROR2. We did
not detect any significant changes in the phosphorylation of the
other RTKs after treatment with cediranib (Fig. 5C).
We further addressed the inhibitory effect of
cediranib in intracellular pathways, using higher concentrations of
the drug for 12 h in both cell lines (Fig. 5D). We observed a decrease in ERK
phosphorylation at the concentrations analyzed in the Hs578T cells.
However, this cell line showed that the inhibition of AKT seemed to
be dose-dependent (2 µM) after cediranib treatment. Both cell
lines, HS578T and MDA-MB-231, were exposed to equal cediranib
concentrations (0.5 and 2 µM), and only the MDA-MB-231 cell line
did not exhibit decreased ERK phosphorylation levels. The absence
of AKT phosphorylation was detected in the MDA-MB-231 cell line.
The phosphorylation levels of ERK and AKT were unchanged in the
T47D cell line after exposure to cediranib (Fig. 5D).
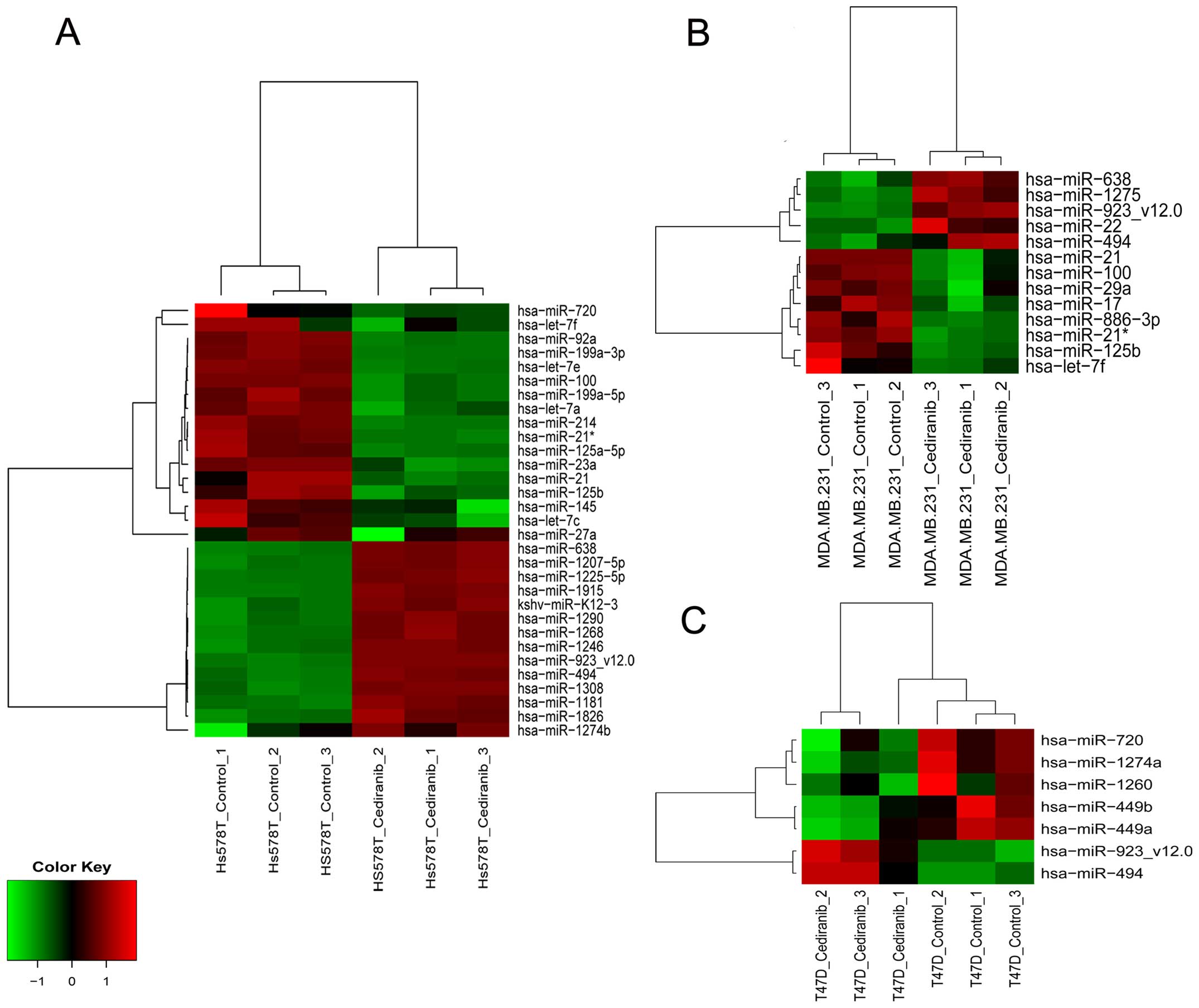
To further identify potential miRNAs in response to
cediranib, we performed miRNA expression profiles in each breast
cancer cell line and compared them with the controls. The results
revealed 31 differentially expressed miRNAs in the Hs578T cells, 13
miRNAs in the MDA-MB-231 cells and 7 miRNAs in the T74D cell line
(Fig. 6). The targets of these
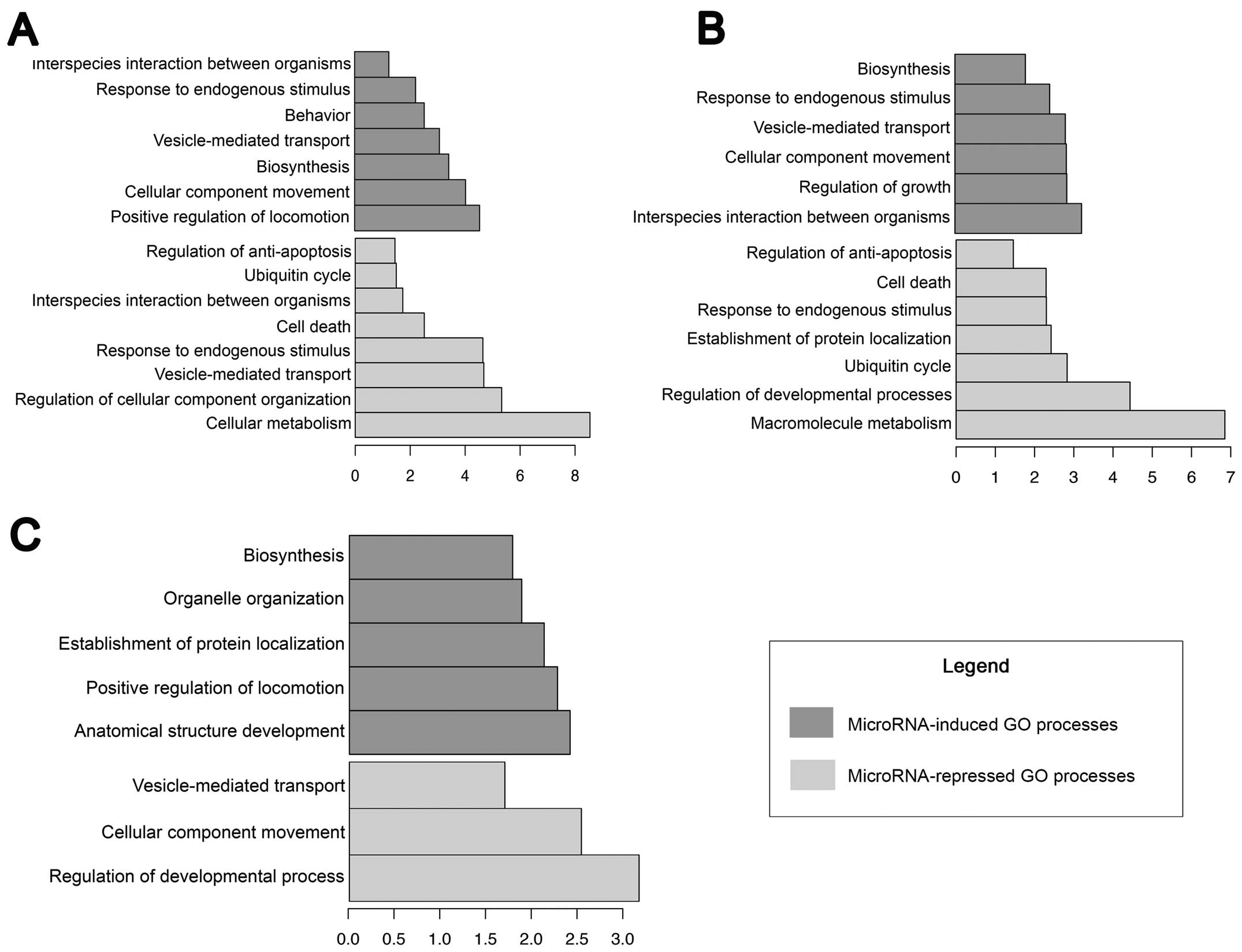
identified miRNAs are summarised according to their biological
processes. The Hs578T cell line shared several processes with the
MDA-MB-231 cells, including the induction of a response to
endogenous/chemical stimuli, biosynthesis, vesicle-mediated
transport, the movement of cellular components, the repression of
the ubiquitin cycle, and the regulation of anti-apoptosis and cell
death (Fig. 7). The T74D cell line
presented specific biological processes; only biosynthesis was
upregulated in the other cell lines in a similar manner, while
vesicle-mediated transport and cellular component movement were
repressed (Fig. 7).
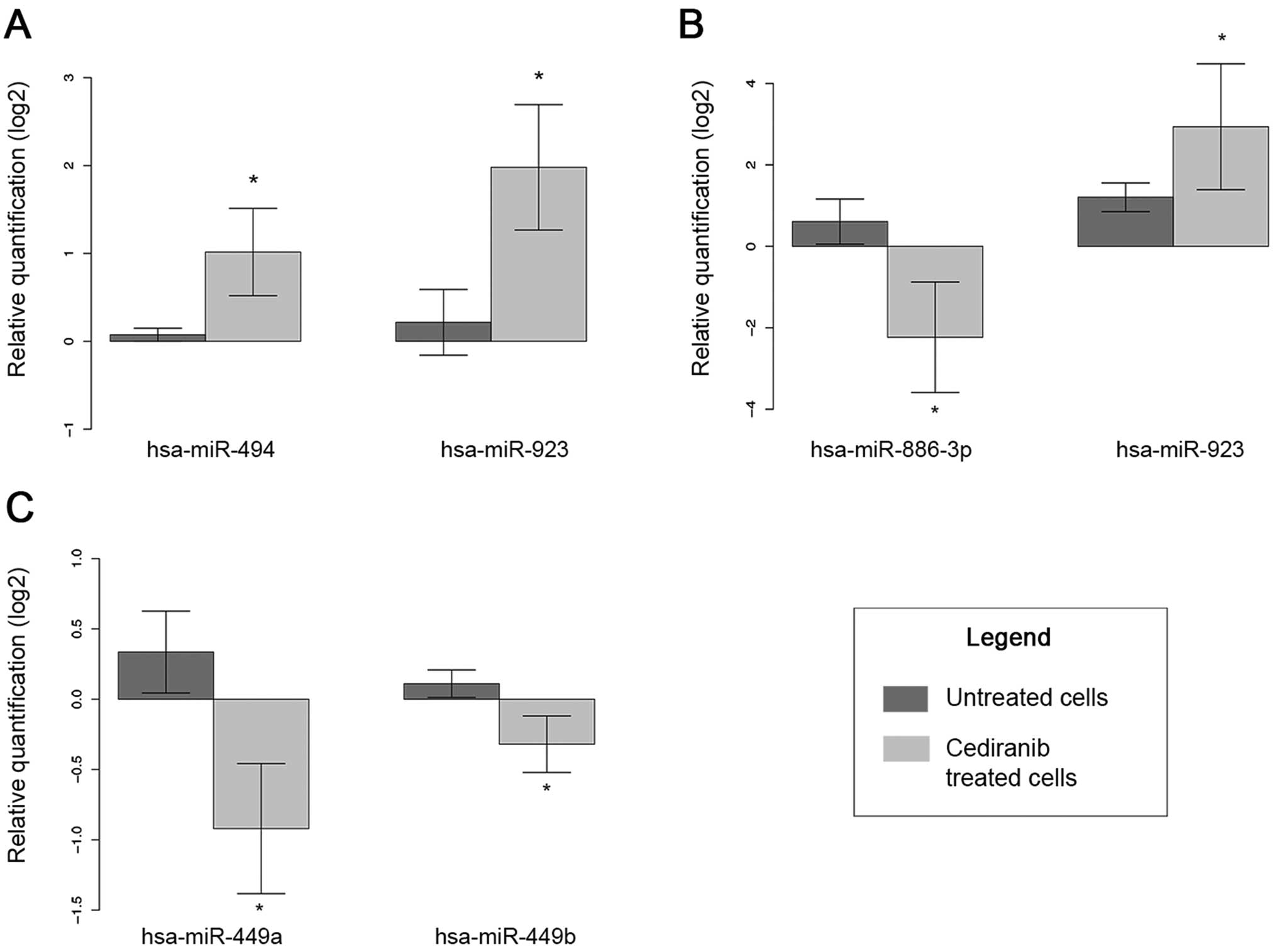
Among the miRNAs identified, five miRNAs, miR-494,
miR-923, miR-449a, miR-449b and miR-886-3p, were selected for
further confirmation of expression due to the greater change in
expression and the biological relevance of these miRNAs (Fig. 8). The endogenous miRNA RNU-48 was
the most stable in our results, and this miRNA was thus used for
all the analyses. The results confirmed the overexpression of
miR-494 and miR-923 in the Hs578T cells exposed to cediranib
compared to the control cells. In the MDA-MB-231 cells, the
overexpression of miR-923 and the decreased expression of
miR-886-3p after exposure to cediranib were confirmed. Both
miR-449a and miR-449b were dowregulated in the T47D cell line after
cediranib treatment.
Discussion
Cediranib is a tyrosine kinase inhibitor that
represents promise in the treatment of several tumours (6). The main targets of this
pharmacological drug are the VEGFR proteins, which regulate blood
vessel formation in tumours and are associated with the
anti-angiogenic effects of cediranib (4). In the present study, we showed that
cediranib exhibited an effect on breast cancer cells, affecting
cell migration and invasion, and the molecular processes associated
with the miRNA expression altered in response to treatment was also
studied.
Regarding the sensitivity to the agent, the
IC50 value varied according to the cell line used. The
most sensitive cell line (Hs578T) had IC50 values (~2
µM) similar to those of glioma cells after 72 h of cediranib
exposure (10). Notably, PARP is an
abundant, chromatin-associated enzyme which responds to DNA damage
(49). Our results showed that when
exposed to cediranib (according to the IC50 values) the
cell line Hs578T displayed cleavage of PARP at 48 h of exposure,
followed by MDA-MB-231 and T47D cell lines. Based on this result,
we demonstrated that the Hs578T cell line had greater sensitivity
to cediranib. ERK inhibition was detected only in sensitive cell
line Hs578T, showing the efficient inhibition in the conserved
RAS-mitogen activated protein kinase (MAPK) signalling pathway
which may affect the cellular growth, survival, and differentiation
(50). In the MDA-MB-231 and T47D
cell lines, the phosphorylation of ERK was not significantly
blocked. A recent study showed similar results in another breast
cancer cell line, which presented a resistance profile when exposed
to cediranib at 1–10 µM/l (51).
The persistent AKT activation in some solid cancers, such as
non-small cell lung carcinomas (NSCLCs), is associated with TKI
response (52). We observed a total
blockade of the AKT phosphorylation in the Hs578T cell line when
exposed to different cediranib concentrations. However the
MDA-MB-231 cell line showed absence of phosphorylated proteins and
T47D displayed no significant changes. Therefore, we may
hypothesized that AKT inhibition may be related with cediranib
response in breast cancer cell lines.
The proteome profiler of phospho-RTKs showed that
although Hs578T cells did not express the described cediranib
targets under baseline conditions, such as VGFRs, KIT or PDGFRA,
this cell line exhibited other important RTKs, such as EGFR, Tie-2,
RYK, FGFR2α, m-CSFR, c-RET, AXL and ROR2, and upon cediranib
exposure we observed reduction of the ROR2 and FGFR2α RTKs. ROR2 is
a novel Wnt receptor recently discovered and is associated with
progression of solid tumours such as melanoma, osteosarcoma and
prostate cancer (53). An
immunohistochemistry study conducted in patients with breast cancer
showed ROR2 overexpression in 87% of the cases, and its association
with a worse outcome (54). Further
studies are needed to validate our findings, and more importantly
to determine whether the expression levels of ROR2 and FGFR2α in
human breast cancer tissue may predict the response to
cediranib.
Regarding the expression of miRNAs, the Hs578T cell
line presented a greater number of modulated miRNAs than the
MDA-MB-231 and T47D cell lines, as an indicator of sensitivity to
the drug. Although the Hs578T cell line presented 22 specific
miRNAs, the most significant biological processes, such as
biological regulation, development, metabolic processes, cell
motility and homeostasis, were related to targets that were shared
with the MDA-MB-231 cell line, including VEGFA,
VEGFC, and PDGFRA. Several studies support the roles
of these molecules in these processes, especially roles in tissue
metabolism and homeostasis in breast cancer (55,56).
Of the targets shared between the Hs578T and
MDA-MB-231 cell lines, miR-923 upregulation was observed. Moreover,
few miRNAs were upregulated in our findings. miR-923 dysregulation
was highly associated with drug resistance. The upregulation of
this miRNA was also observed in breast cancer cells that are
resistant to Taxol (57). In the
same study, miR-125b was upregulated, which was associated with
Bcl-2 antagonist killer 1 (Bak1) expression. In our findings,
miR-125b was also modulated but was downregulated. The interaction
between specific targets may further be explored. miR-923 and
miR-886-3p, which were also validated in the present study, were
shown to be involved in cisplatin resistance in bladder cancer;
changes in expression were also correlated with survival (58). The upregulation of miR-923 was also
associated with multidrug resistance in the HEp-2 cell line
(59).
Among the specific miRNAs found in the MDA-MB-231
cell line, miR-886 has been recently proposed as a vault RNA
(vtRNA2-1), which may form a complex that is implicated in cancer
drug resistance. Recently, Lee et al (60) identified pre-miR-886 as a 102-nt,
abundant cytoplasmic RNA that is neither a pre-miRNA nor a vault
RNA but is a noncoding molecule (nc886). Some evidence indicates
that nc886 in an immature state is physically associated with PKR
(protein kinase RNA-activated), a double-stranded RNA-dependent
kinase (61).
The involvement of miR-886 in the drug response and
the modulation of this molecule by cediranib in the sensitive cell
line MDA-MB-231 supports evidence that some mechanisms related to
apoptosis could be activated in this context and affect the
sensitivity of these cells, as observed in the present study. In
addition, miR-886-3p repression was recently described as being
mediated by methylation processes in lung cancer cell lines
(62) and can affect cell
proliferation, migration, and invasion in lung and non-medullary
thyroid cancer (63,64). This fact is in concordance with our
findings because we determined that migration and invasion
processes were highly affected by cediranib in MDA-MB-231
cells.
In contrast to the other cell lines studied, few
miRNAs were modulated in the T47D cell line. The T47D-specific
miRNAs miR-449a and miR-449b were highly downregulated, and these
results were confirmed via qRT-PCR. The miRNA miR-449 has been
identified in a wide range of tumours (65–69)
and has several identified targets, including c-Myc and
c-Met (66,67). This miRNA induces cell cycle
progression and apoptosis by regulating CDK6 and CDC25A, direct
targets that lead to the phosphorylation of E2F1 (70–72).
In contrast to the Hs578T and MDA-MB-231 cells, no target
associated with VEGFR or PDGFRA was identified in the
T47D cells with the target prediction. Considering the difference
in the T47D resistance to cediranib, this finding suggests that
other RTK targets are possibly affected by cediranib, in addition
to VEGFR, the most potent RTK affected, and these other targets may
activate other downstream pathways that affect the invasion and
migration processes, instead of cell death. Further studies are
necessary to confirm this hypothesis.
The crosstalk of the EGFR pathways and certain
miRNAs has been described; for example, EGFR mutations can
regulate miR-21 in lung cancer and glioblastoma cell lines
(73). Similarly, miR-145 was found
to inhibit cell proliferation by targeting EGFR in lung
adenocarcinoma and in a model of colon cancer (74,75).
In the present study, both miRNAs were modulated in the Hs578T
cells, and only miR-21 was downregulated in MDA-MB-231 cells.
However, some different processes seem to occur in the T47D cells.
Considering that miR-449 (specific to T47D) can also modulate
EGFR, the ambiguities of this process should be further
explored.
The present study assessed for the first time the
anti-neoplastic effects of cediranib in breast cancer cell lines.
We observed a distinct degree of response in all the cell lines.
Cediranib impaired mechanisms leading to cancer cell invasion and
migration. The present study revealed an anti-neoplastic role of
cediranib in human breast cancer cell lines. Importantly, we
identified ROR2 and FGFR2α as novel RTK targets of cediranib. The
miRNA expression profiling revealed the effect of this drug on
several miRNAs involved in several molecular processes. Finally,
more studies are necessary to validate these miRNA signatures as
indicators of the therapeutic response in human breast cancer, and
these signatures may lead to the selection of patients who would be
most effectively treated with cediranib.
Acknowledgements
We would like to thank Olga Martinho and Celine
Pinheiro for assisting in the cellular experiments. This study
received financial support from Fundação de Amparo à Pesquisa do
Estado de São Paulo (FAPESP Proc. no. 2010/16796-0, São Paulo,
Brazil).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lønning PE: Molecular basis for therapy
resistance. Mol Oncol. 4:284–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arpino G, Green SJ, Allred DC, Lew D,
Martino S, Osborne CK and Elledge RM: HER-2 amplification, HER-1
expression, and tamoxifen response in estrogen receptor-positive
metastatic breast cancer: A southwest oncology group study. Clin
Cancer Res. 10:5670–5676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wedge SR, Kendrew J, Hennequin LF,
Valentine PJ, Barry ST, Brave SR, Smith NR, James NH, Dukes M,
Curwen JO, et al: AZD2171: A highly potent, orally bioavailable,
vascular endothelial growth factor receptor-2 tyrosine kinase
inhibitor for the treatment of cancer. Cancer Res. 65:4389–4400.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brave SR, Ratcliffe K, Wilson Z, James NH,
Ashton S, Wainwright A, Kendrew J, Dudley P, Broadbent N, Sproat G,
et al: Assessing the activity of cediranib, a VEGFR-2/3 tyrosine
kinase inhibitor, against VEGFR-1 and members of the structurally
related PDGFR family. Mol Cancer Ther. 10:861–873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Drevs J, Siegert P, Medinger M, Mross K,
Strecker R, Zirrgiebel U, Harder J, Blum H, Robertson J,
Jürgensmeier JM, et al: Phase I clinical study of AZD2171, an oral
vascular endothelial growth factor signaling inhibitor, in patients
with advanced solid tumors. J Clin Oncol. 25:3045–3054. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Batchelor TT, Duda DG, di Tomaso E,
Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J,
Hochberg FH, Benner T, et al: Phase II study of cediranib, an oral
pan-vascular endothelial growth factor receptor tyrosine kinase
inhibitor, in patients with recurrent glioblastoma. J Clin Oncol.
28:2817–2823. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goss GD, Arnold A, Shepherd FA, Dediu M,
Ciuleanu TE, Fenton D, Zukin M, Walde D, Laberge F, Vincent MD, et
al: Randomized, double-blind trial of carboplatin and paclitaxel
with either daily oral cediranib or placebo in advanced
non-small-cell lung cancer: NCIC clinical trials group BR24 study.
J Clin Oncol. 28:49–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dahut WL, Madan RA, Karakunnel JJ,
Adelberg D, Gulley JL, Turkbey IB, Chau CH, Spencer SD, Mulquin M,
Wright J, et al: Phase II clinical trial of cediranib in patients
with metastatic castration-resistant prostate cancer. BJU Int.
111:1269–1280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martinho O, Silva-Oliveira R,
Miranda-Gonçalves V, Clara C, Almeida JR, Carvalho AL, Barata JT
and Reis RM: In vitro and in vivo analysis of RTK inhibitor
efficacy and identification of its novel targets in glioblastomas.
Transl Oncol. 6:187–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miller KD, Miller M, Mehrotra S, Agarwal
B, Mock BH, Zheng QH, Badve S, Hutchins GD and Sledge GW Jr: A
physiologic imaging pilot study of breast cancer treated with
AZD2171. Clin Cancer Res. 12:281–288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Denduluri N, Tan AR, Walshe J, Berman A,
Yang SX, Chow CK and Swain SM: A pilot study to evaluate the
vascular endothelial growth factor receptor tyrosine kinase
inhibitor AZD2171 and chemotherapy in locally advanced and
inflammatory breast cancer. Clin Breast Cancer. 6:460–463. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hyams DM, Chan A, de Oliveira C, Snyder R,
Vinholes J, Audeh MW, Alencar VM, Lombard J, Mookerjee B, Xu J, et
al: Cediranib in combination with fulvestrant in hormone-sensitive
metastatic breast cancer: A randomized Phase II study. Invest New
Drugs. 31:1345–1354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu JF, Tolaney SM, Birrer M, Fleming GF,
Buss MK, Dahlberg SE, Lee H, Whalen C, Tyburski K, Winer E, et al:
A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor
olaparib (AZD2281) in combination with the anti-angiogenic
cediranib (AZD2171) in recurrent epithelial ovarian or
triple-negative breast cancer. Eur J Cancer. 49:2972–2978. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sahebjam S, Bedard PL, Castonguay V, Chen
Z, Reedijk M, Liu G, Cohen B, Zhang WJ, Clarke B, Zhang T, et al: A
phase I study of the combination of ro4929097 and cediranib in
patients with advanced solid tumours (PJC-004/NCI 8503). Br J
Cancer. 109:943–949. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Almeida MI, Reis RM and Calin GA: MicroRNA
history: Discovery, recent applications, and next frontiers. Mutat
Res. 717:1–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Calin GA, Sevignani C, Dumitru CD, Hyslop
T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M,
et al: Human microRNA genes are frequently located at fragile sites
and genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Garzon R, Marcucci G and Croce CM:
Targeting microRNAs in cancer: Rationale, strategies and
challenges. Nat Rev Drug Discov. 9:775–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harquail J, Benzina S and Robichaud GA:
MicroRNAs and breast cancer malignancy: An overview of
miRNA-regulated cancer processes leading to metastasis. Cancer
Biomark. 11:269–280. 2012.PubMed/NCBI
|
|
22
|
Singh R and Mo YY: Role of microRNAs in
breast cancer. Cancer Biol Ther. 14:201–212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Valastyan S: Roles of microRNAs and other
non-coding RNAs in breast cancer metastasis. J Mammary Gland Biol
Neoplasia. 17:23–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marino ALF, Evangelista AF, Vieira RAC,
Macedo T, Kerr LM, Abrahão-Machado LF, Longatto-Filho A, Silveira
HC and Marques MM: MicroRNA expression as risk biomarker of breast
cancer metastasis: A pilot retrospective case-cohort study. BMC
Cancer. 14:7392014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wiemer EAC: Role of microRNAs in
anti-cancer drug resistanceMicroRNAs in Cancer Translational
Research. Cho WCS: Springer; Netherlands: pp. 449–483. 2011,
http://link.springer.com/chapter/10.1007/978-94-007-0298-1_19Accessed
November 20, 2013. View Article : Google Scholar
|
|
26
|
Kastl L, Brown I and Schofield AC:
miRNA-34a is associated with docetaxel resistance in human breast
cancer cells. Breast Cancer Res Treat. 131:445–454. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Climent J, Dimitrow P, Fridlyand J,
Palacios J, Siebert R, Albertson DG, Gray JW, Pinkel D, Lluch A and
Martinez-Climent JA: Deletion of chromosome 11q predicts response
to anthracycline-based chemotherapy in early breast cancer. Cancer
Res. 67:818–826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu H, Wu H, Liu X, Evans BR, Medina DJ,
Liu CG and Yang JM: Role of MicroRNA miR-27a and miR-451 in the
regulation of MDR1/P-glycoprotein expression in human cancer cells.
Biochem Pharmacol. 76:582–588. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kovalchuk O, Filkowski J, Meservy J,
Ilnytskyy Y, Tryndyak VP, Chekhun VF and Pogribny IP: Involvement
of microRNA-451 in resistance of the MCF-7 breast cancer cells to
chemotherapeutic drug doxorubicin. Mol Cancer Ther. 7:2152–2159.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang Z, Wu H, Xia J, Li Y, Zhang Y, Huang
K, Wagar N, Yoon Y, Cho HT, Scala S, et al: Involvement of miR-326
in chemotherapy resistance of breast cancer through modulating
expression of multidrug resistance-associated protein 1. Biochem
Pharmacol. 79:817–824. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Andorfer CA, Necela BM, Thompson EA and
Perez EA: MicroRNA signatures: Clinical biomarkers for the
diagnosis and treatment of breast cancer. Trends Mol Med.
17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mahadevan D, Cooke L, Riley C, Swart R,
Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H,
et al: A novel tyrosine kinase switch is a mechanism of imatinib
resistance in gastrointestinal stromal tumors. Oncogene.
26:3909–3919. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weiss GJ, Bemis LT, Nakajima E, Sugita M,
Birks DK, Robinson WA, Varella-Garcia M, Bunn PA Jr, Haney J,
Helfrich BA, et al: EGFR regulation by microRNA in lung cancer:
Correlation with clinical response and survival to gefitinib and
EGFR expression in cell lines. Ann Oncol. 19:1053–1059. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dirks WG, Faehnrich S, Estella IAJ and
Drexler HG: Short tandem repeat DNA typing provides an
international reference standard for authentication of human cell
lines. ALTEX. 22:103–109. 2005.PubMed/NCBI
|
|
35
|
drc R Package. http://cran.r-project.org/web/packages/drc/
|
|
36
|
The R project for statistical computing.
http://www.r-project.org
|
|
37
|
Pinto F, Pértega-Gomes N, Pereira MS,
Vizcaíno JR, Monteiro P, Henrique RM, Baltazar F, Andrade RP and
Reis RM: T-box transcription factor brachyury is associated with
prostate cancer progression and aggressiveness. Clin Cancer Res.
20:4949–4961. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Martinho O, Zucca LE and Reis RM: AXL as a
modulator of sunitinib response in glioblastoma cell lines. Exp
Cell Res. 332:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Viana CR, Neto CS, Kerr LM, Palmero EI,
Marques MMC, Colaiacovo T, de Queiroz Junior AF, Carvalho AL and
Siqueira SA: The interference of cold ischemia time in the quality
of total RNA from frozen tumor samples. Cell Tissue Bank.
14:167–173. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Macedo C, Evangelista AF, Marques MM,
Octacílio-Silva S, Donadi EA, Sakamoto-Hojo ET and Passos GA:
Autoimmune regulator (Aire) controls the expression of microRNAs in
medullary thymic epithelial cells. Immunobiology. 218:554–560.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aroma-light. http://www.bioconductor.org/packages/2.12/bioc/html/aroma.light.html
|
|
42
|
Hong F, Breitling R, McEntee CW, Wittner
BS, Nemhauser JL and Chory J: RankProd: A bioconductor package for
detecting differentially expressed genes in meta-analysis.
Bioinformatics. 22:2825–2827. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
gplots: Various R programming tools for
plotting data. http://cran.r-project.org/web/packages/gplots/index.html
|
|
44
|
mirDIP: microRNA data integration portal.
http://ophid.utoronto.ca/mirDIP/
|
|
45
|
Bioinformatics Resouces DAVID: 6.7.
http://david.abcc.ncifcrf.gov
|
|
46
|
Supek F, Bošnjak M, Škunca N and Šmuc T:
REVIGO summarizes and visualizes long lists of gene ontology terms.
PLoS One. 6:e218002011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Andersen CL, Jensen JL and Ørntoft TF:
Normalization of real-time quantitative reverse transcription-PCR
data: A model-based variance estimation approach to identify genes
suited for normalization, applied to bladder and colon cancer data
sets. Cancer Res. 64:5245–5250. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lindahl T, Satoh MS, Poirier GG and
Klungland A: Post-translational modification of poly(ADP-ribose)
polymerase induced by DNA strand breaks. Trends Biochem Sci.
20:405–411. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tao LY, Liang YJ, Wang F, Chen LM, Yan YY,
Dai CL and Fu LW: Cediranib (recentin, AZD2171) reverses ABCB1- and
ABCC1-mediated multidrug resistance by inhibition of their
transport function. Cancer Chemother Pharmacol. 64:961–969. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tetsu O, Phuchareon J, Eisele DW, Hangauer
MJ and McCormick F: AKT inactivation causes persistent drug
tolerance to EGFR inhibitors. Pharmacol Res. 102:132–137. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ford CE, Ma SS Qian, Quadir A and Ward RL:
The dual role of the novel Wnt receptor tyrosine kinase, ROR2, in
human carcinogenesis. Int J Cancer. 133:779–787. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Henry C, Quadir A, Hawkins NJ, Jary E,
Llamosas E, Kumar D, Daniels B, Ward RL and Ford CE: Expression of
the novel Wnt receptor ROR2 is increased in breast cancer and may
regulate both β-catenin dependent and independent Wnt signalling. J
Cancer Res Clin Oncol. 141:243–254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Malavaki CJ, Roussidis AE, Gialeli C,
Kletsas D, Tsegenidis T, Theocharis AD, Tzanakakis GN and Karamanos
NK: Imatinib as a key inhibitor of the platelet-derived growth
factor receptor mediated expression of cell surface heparan sulfate
proteoglycans and functional properties of breast cancer cells.
FEBS J. 280:2477–2489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Thanigaimani S, Kichenadasse G and Mangoni
AA: The emerging role of vascular endothelial growth factor (VEGF)
in vascular homeostasis: Lessons from recent trials with anti-VEGF
drugs. Curr Vasc Pharmacol. 9:358–380. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi
Y, Xiong W, Li G, Lu J, Fodstad O, et al: MicroRNA-125b confers the
resistance of breast cancer cells to paclitaxel through suppression
of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J
Biol Chem. 285:21496–21507. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nordentoft I, Birkenkamp-Demtroder K,
Agerbæk M, Theodorescu D, Ostenfeld MS, Hartmann A, Borre M,
Ørntoft TF and Dyrskjøt L: miRNAs associated with chemo-sensitivity
in cell lines and in advanced bladder cancer. BMC Med Genomics.
5:402012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yin W, Wang P, Wang X, Song W, Cui X, Yu H
and Zhu W: Identification of microRNAs and mRNAs associated with
multidrug resistance of human laryngeal cancer Hep-2 cells. Braz J
Med Biol Res. 46:546–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lee K, Kunkeaw N, Jeon SH, Lee I, Johnson
BH, Kang GY, Bang JY, Park HS, Leelayuwat C and Lee YS: Precursor
miR-886, a novel noncoding RNA repressed in cancer, associates with
PKR and modulates its activity. RNA. 17:1076–1089. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jeon SH, Lee K, Lee KS, Kunkeaw N, Johnson
BH, Holthauzen LMF, Gong B, Leelayuwat C and Lee YS:
Characterization of the direct physical interaction of nc886, a
cellular non-coding RNA, and PKR. FEBS Lett. 586:3477–3484. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jeon SH, Johnson BH and Lee YS: A tumor
surveillance model: A non-coding RNA senses neoplastic cells and
its protein partner signals cell death. Int J Mol Sci.
13:13134–13139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cao J, Song Y, Bi N, Shen J, Liu W, Fan J,
Sun G, Tong T, He J, Shi Y, et al: DNA methylation-mediated
repression of miR-886-3p predicts poor outcome of human small cell
lung cancer. Cancer Res. 73:3326–3335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xiong Y, Zhang L, Holloway AK, Wu X, Su L
and Kebebew E: MiR-886-3p regulates cell proliferation and
migration, and is dysregulated in familial non-medullary thyroid
cancer. PLoS One. 6:e247172011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Noonan EJ, Place RF, Basak S, Pookot D and
Li LC: miR-449a causes Rb-dependent cell cycle arrest and
senescence in prostate cancer cells. Oncotarget. 1:349–358. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Miao LJ, Huang SF, Sun ZT, Gao ZY, Zhang
RX, Liu Y and Wang J: MiR-449c targets c-Myc and inhibits NSCLC
cell progression. FEBS Lett. 587:1359–1365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Luo W, Huang B, Li Z, Li H, Sun L, Zhang
Q, Qiu X and Wang E: MicroRNA-449a is downregulated in non-small
cell lung cancer and inhibits migration and invasion by targeting
c-Met. PLoS One. 8:e647592013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fang Y, Gu X, Li Z, Xiang J and Chen Z:
miR-449b inhibits the proliferation of SW1116 colon cancer stem
cells through downregulation of CCND1 and E2F3 expression. Oncol
Rep. 30:399–406. 2013.PubMed/NCBI
|
|
69
|
Bou Kheir T, Futoma-Kazmierczak E,
Jacobsen A, Krogh A, Bardram L, Hother C, Grønbæk K, Federspiel B,
Lund AH and Friis-Hansen L: miR-449 inhibits cell proliferation and
is down-regulated in gastric cancer. Mol Cancer. 10:292011.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Feng M and Yu Q: miR-449 regulates
CDK-Rb-E2F1 through an auto-regulatory feedback circuit. Cell
Cycle. 9:213–214. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yan F, Liu H, Hao J and Liu Z: Dynamical
behaviors of Rb-E2F pathway including negative feedback loops
involving miR449. PLoS One. 7:e439082012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yang X, Feng M, Jiang X, Wu Z, Li Z, Aau M
and Yu Q: miR-449a and miR-449b are direct transcriptional targets
of E2F1 and negatively regulate pRb-E2F1 activity through a
feedback loop by targeting CDK6 and CDC25A. Genes Dev.
23:2388–2393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhou X, Ren Y, Moore L, Mei M, You Y, Xu
P, Wang B, Wang G, Jia Z, Pu P, et al: Downregulation of miR-21
inhibits EGFR pathway and suppresses the growth of human
glioblastoma cells independent of PTEN status. Lab Invest.
90:144–155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cho WCS, Chow ASC and Au JSK: MiR-145
inhibits cell proliferation of human lung adenocarcinoma by
targeting EGFR and NUDT1. RNA Biol. 8:125–131. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhu H, Dougherty U, Robinson V, Mustafi R,
Pekow J, Kupfer S, Li YC, Hart J, Goss K, Fichera A, et al: EGFR
signals downregulate tumor suppressors miR-143 and miR-145 in
Western diet-promoted murine colon cancer: Role of G1 regulators.
Mol Cancer Res. 9:960–975. 2011. View Article : Google Scholar : PubMed/NCBI
|