Introduction
Human hepatocellular carcinoma (HCC) is one of the
most common malignant tumors worldwide and ranks third in terms of
global cancer-related mortality (1). Liver cirrhosis is believed to be the
most significant risk factor for 70–90% of HCC patients (2). Moreover, viral infection, liver
cytotoxicity, chronic inflammation, and many other factors have
been implicated in HCC progression (3). Unfortunately, although some
improvement has been achieved in the clinic, overall the prognosis
is poor due to the development of resistance to chemotherapy and
radiotherapy (4). Therefore, the
development of novel targeted anticancer agents is extremely
important to overcome this disease.
Cancerous inhibitor of protein phosphatase 2A
(CIP2A) stabilizes the c-Myc protein via suppressing the protein
phosphatase 2A (PP2A) (5). It has
been reported to be amplified or overexpressed in a wide variety of
human malignancies, such as gastric (6), breast (7), renal cell (8), bladder (9), and lung (10) cancer. The functional roles of CIP2A
involve cell growth, cell invasion, drug resistance, and tumor
formation (10–12). Several compounds from traditional
Chinese medicine have been reported to exhibit anticancer activity
via degradation of CIP2A and subsequent inactivation of AKT
(13,14). These findings indicate that CIP2A
could be a promising target for cancer chemotherapy.
The genus Garcinia is known for its rich
variety of oxygenated and prenylated phenol derivatives. Gambogic
acid is a major active component of gamboge isolated from the resin
of Garcinia hanburyi, which has been shown to have potent
anticancer activity and is authorized to be tested in clinical
trails (15–18). Gambogenic acid (GEA) is another
active component of gamboge which exhibits cytotoxicity and
anti-inflammatory activity (19,20).
The molecular mechanisms that underlie the effects of GEA include
induction of cell cycle arrest (21,22),
apoptosis 21,23–27,
autophagy (22,28), necroptosis (29) and chemosensitivity (29,30).
Herein, we demonstrated that GEA induced rapid proteasome-mediated
degradation of CIP2A. GEA also showed potent anticancer activity
and enhanced the effect of chemotherapeutic agents against HCC.
Materials and methods
Chemicals and reagents
GEA was extracted from gamboges by Dr Quanbin Han as
previously described (20),
dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis,
MO, USA) to make a stock solution (20 mM) and stored at −20°C.
NH4Cl and 3-MA were purchased from Sigma-Aldrich.
Cycloheximide (CHX) and nocodazole were obtained from Beyotime
Institute of Biotechnology (Jiangsu, China). CellTiter 96 AQueous
One Solution Cell Proliferation Assay and Z-VAD-FMK were obtained
from Promega (Madison, WI, USA). Antibodies used in our study were
as follows: anti-β-actin (Sigma-Aldrich); anti-CIP2A, c-Myc,
ubiquitin (Santa Cruz Biotechnology); anti-pAKT (Ser473)
(Cell Signaling Technology); and anti-rabbit and anti-mouse
HRP-conjugated secondary antibodies (Pierce). Detection was
performed using a Chemiluminescent Western Blot Detection kit
(Thermo Fisher Scientific, Rockford, IL, USA).
Cell culture
The human hepatoma cell lines Hep G2 and Bel-7402
were obtained from the American Tissue Culture Collection (ATCC;
Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's
medium (DMEM; Gibco-BRL, Gaithersburg, MD, USA) supplemented with
10% fetal bovine serum (FBS; Biological Industries, Kibbutz
Beit-Haemek, Israel), 100 U/ml penicillin, 100 µg/ml streptomycin,
and 2 mM glutamine (Invitrogen Life Technologies, Carlsbad, CA,
USA). All cells were cultured in a humidified incubator at 37°C
under 5% CO2.
Cell viability assay
Cells were seeded in 96-well plates
(1×103 cells/well) and then exposed to the indicated
agents. After incubation for the indicated time, cell viability
assay was conducted using the cell titer assay. Cell growth curve
was estimated using trypan blue dye exclusion.
Real-time reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from cells by using the
TRIzol reagent (Invitrogen Life Technologies) according to the
manufacturer's instructions. The first strand complementary DNA
(cDNA) was synthesized using PrimeScript™ RT reagent kit with gDNA
Eraser (Takara, Dalian, China). Primers used for RT-PCR analysis of
human CIP2A included sense, 5′-CCATATGCTCACTCAGATGATGT-3′ and
antisense, 5′-GTGTATCATCTCCACAGAGAGTT-3′; and for GAPDH sense,
5′-TCACCAGGGCTGCTTTTA-3′ and antisense, 5′-AAGGTCATCCCTGAGCTGAA-3′.
PCR products were separated on 1.5% agarose gels and stained with
GoldView. To confirm CIP2A mRNA expression, real-time polymerase
chain reaction (qRT-PCR) was performed using the SYBR Premix Ex Taq
(Takara). The primers were as follows: CIP2A sense,
5′-TGCGGCACTTGGAGGTAATTTC-3′ and antisense,
5′-AGCTCTACAAGGCAACTCAAGC-3′; actin sense,
5′-ATCGTCCACCGCAAATGCTTCTA-3′ and antisense,
5′-AGCCATGCCAATCTCATCTTGTT-3′. The amplifications were performed as
follows: 94°C for 10 min and then 40 cycles of 94°C for 15 sec,
60°C for 30 sec and 72°C for 30 sec. Quantified values for gene
expression were generated by the relative quantification
method.
siRNA assays
Cells were transfected with double-stranded siRNA
oligonucleotides (100 nM) in 6-well plates using Lipofectamine 2000
(Invitrogen Life Technologies) according to the manufacturer's
instructions. The sequences of siRNAs are as follows: CIP2A siRNA,
5′-GGUGCACGUUUCAUCAAUU-3′; NC siRNA, 5′-GGUGCACGUUUCAUCAAUU-3′.
Western blotting
Cells were suspended in lysis buffer containing 50
mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP40, 1 mM EDTA, 1 mM
Na3VO4, 1 mM NaF, and a cocktail of 1 mM PMSF
and 1 mM protease inhibitors. The lysates were centrifuged at
12,000 × g for 10 min at 4°C, followed by measurements of protein
concentrations using Pierce BCA Protein Assay Kit (Thermo Fisher
Scientific). The supernatants were collected as NP40-soluble
fractions. The pellets (NP40-insoluble fractions) were lysed in
lysis buffer containing 2% SDS and boiled at 100°C for 7 min and
chilled on ice. Proteins (20 µg) were separated on 10% SDS-PAGE gel
and transferred to PVDF membranes (Millipore Corp., Billerica, MA,
USA). The membranes were incubated overnight with specific primary
antibodies at 4°C after being blocked with 5% non-fat milk. After
being washed three times with PBS containing 0.05% Tween-20 (PBST),
the membranes were incubated with HRP-conjugated secondary
antibodies for 1 h at room temperature, followed by 3-times washing
with 0.05% Tween-20/PBS and then detected using chemiluminescent
substrate.
Statistical analysis
Quantitative data are presented as means ± SED from
triplicate experiments. Comparison between groups was performed by
ANOVA and P≤0.05 was considered to indicate a statistically
significant difference.
Results
GEA triggers degradation of CIP2A
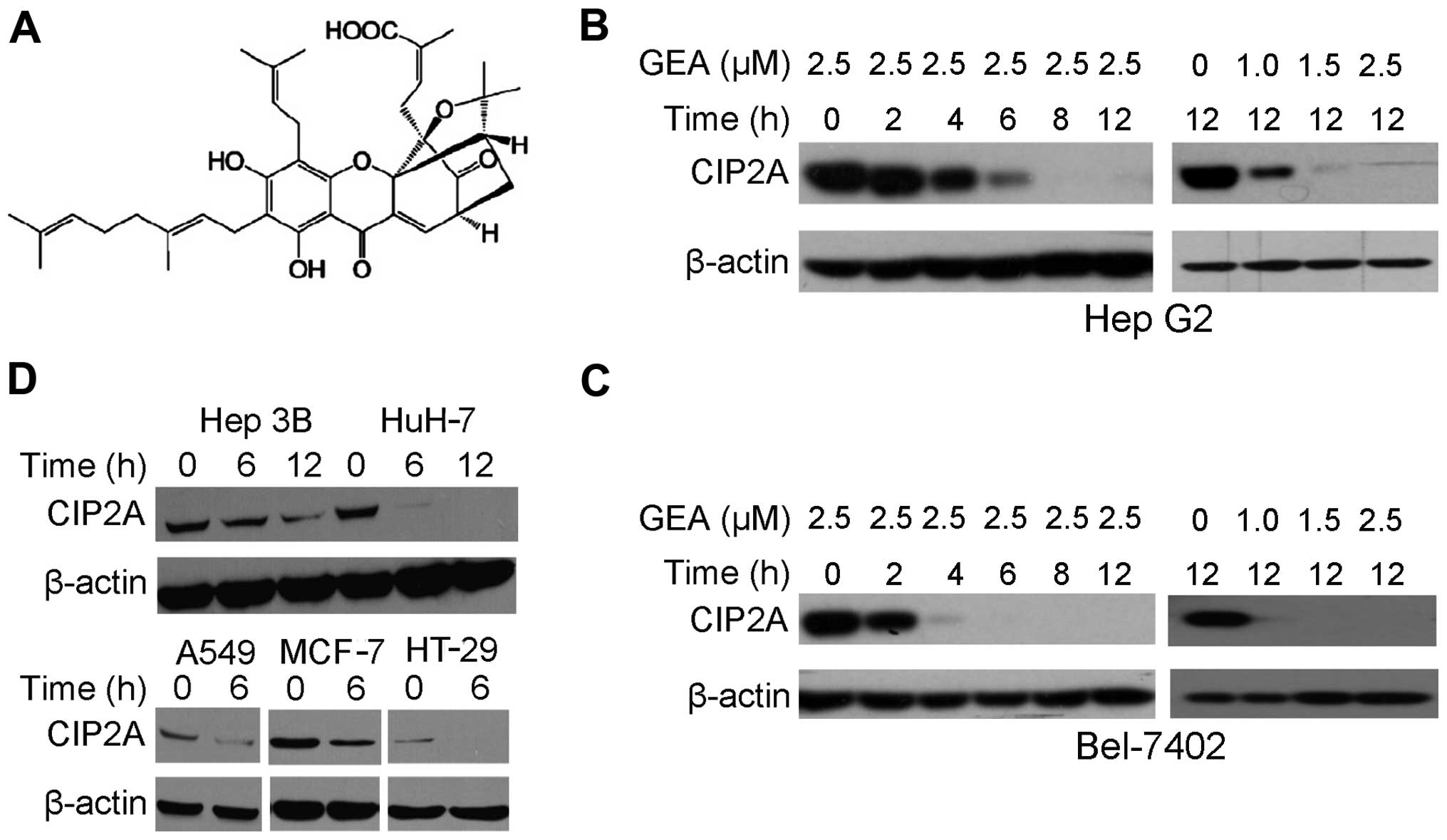
We aimed to ascertain whether GEA (Fig. 1A) affects CIP2A protein expression.
We demonstrated that following treatment with GEA at 2.5 µM for
2–12 h, a dramatic decrease in CIP2A expression in Hep G2 cells was
observed (Fig. 1B). Similarly,
treatment with GEA at 1.0–2.5 µM for 12 h decreased the CIP2A
expression in a dose-dependent manner (Fig. 1B). These observations were further
confirmed in Bel-7402 cells (Fig.
1C). Treatment with GEA at 2.5 µM for 6–12 h also suppressed
CIP2A in other hepatocellular carcinomas (Hep 3B, HuH-7), lung
(A549), breast (MCF-7) and colon (HT-29) cancer cell lines,
indicating that GEA-induced CIP2A downregulation is not cell-type
specific (Fig. 1D).
GEA downregulates CIP2A at the
post-transcriptional level
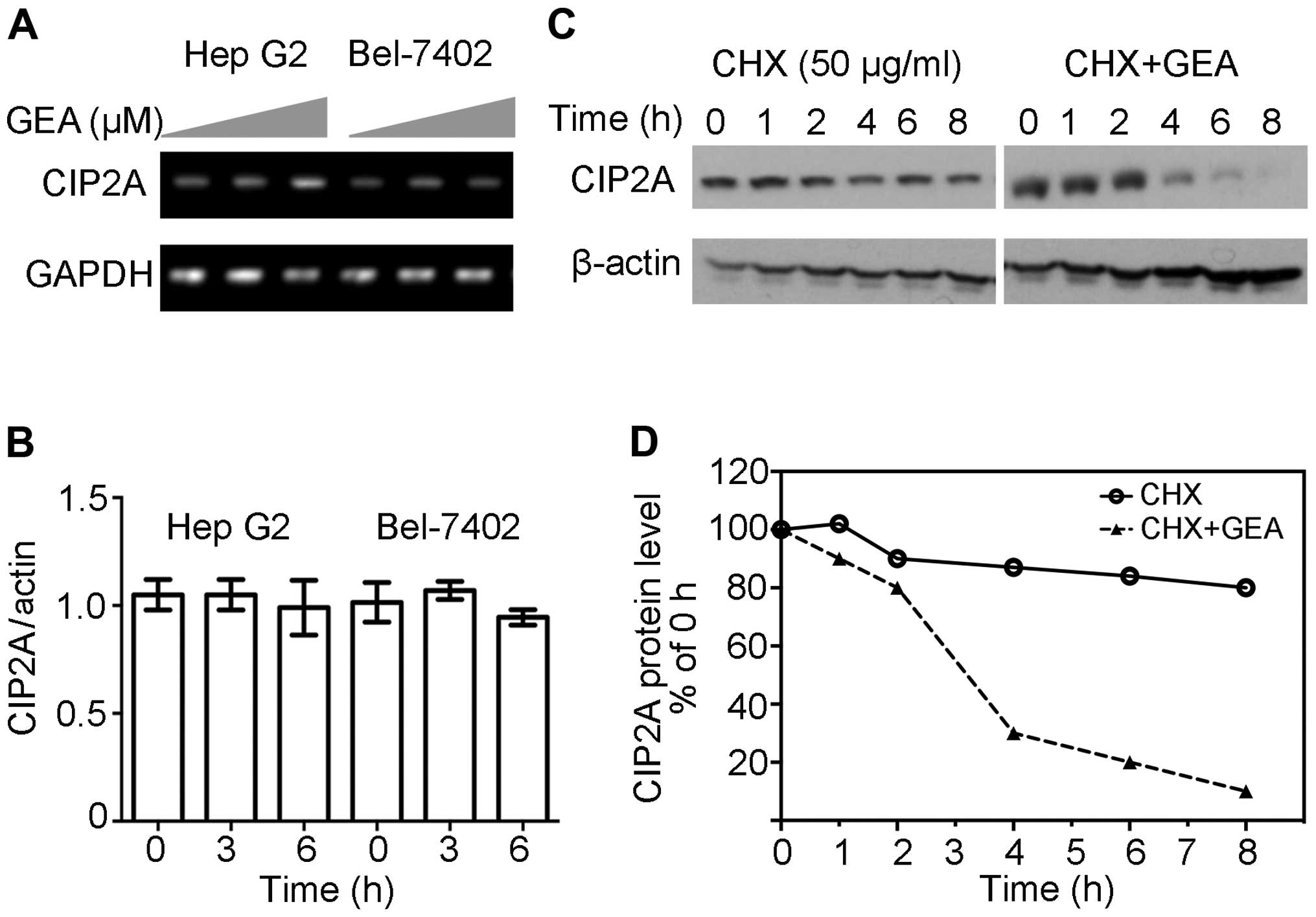
In order to clarify the underlying mechanism
involved in CIP2A downregulation, RT-PCR assays were performed and
revealed that GEA at 2.5 µM for 6 h did not exhibit inhibitory
effects on the expression of CIP2A mRNA in Hep G2 cells (Fig. 2A). These similar observations were
confirmed by quantitative RT-PCR (Fig.
2B). Since GEA-mediated downregulation of CIP2A is not
associated with transcription, we hypothesized that the reduction
in CIP2A might be due to protein stabilization and degradation. As
shown in Fig. 2C and D, protein
synthesis inhibitor CHX barely reduced the expression of CIP2A
within 8 h, however, the combination of GEA and CHX resulted in a
marked reduction of CIP2A at the protein level within 4 h. These
results indicate that GEA decreased CIP2A at the post-transcription
level.
GEA triggers
ubiquitin-proteasome-mediated degradation of CIP2A
Four major proteolytic systems mediate protein
stability: caspase, calpain, lysosome and proteasome (31). The caspase family of cysteine
proteases is involved in cell death and cleavage of substrate
proteins (32). Hep G2 cells were
pre-treated with pan-caspase inhibitor Z-VAD-fmk (Z-VAD) for 2 h,
followed by treatment with or without GEA for 6 h. However, no
significant reversal effect was observed in the presence of Z-VAD
(Fig. 3A). Calpains represent a
well-conserved family of calcium-dependent cysteine proteases
(33). We then pre-treated Hep G2
cells with calpain inhibitor III MDL-28170 (MDL), and the
degradation of CIP2A was not reversed (Fig. 3B). Lysosomes and autophagosomes are
organelles which play a central role in the control of cell fate
(34). The Hep G2 cells were
pretreated with a lysosomal protease inhibitor (NH4Cl)
and an autophagy inhibitor (3-MA) and then treated with GEA. The
two inhibitors did not prevent GEA-induced CIP2A degradation
(Fig. 3C and D). The
ubiquitin-proteasome pathway plays an important role in
intracellular proteolysis (35,36).
Interestingly, we found that following treatment with the
proteasome inhibitor MG132 or PSI alone, the expression of CIP2A in
the Hep G2 cells was not affected within 6 h (Fig. 3E). However, the combination of
MG132/PSI and GEA markedly impaired CIP2A degradation (Fig. 3E). Previous studies have reported
that the proteasome inhibitor promotes accumulation of
ubiquitinated proteins and shifts them into detergent-insoluble
cellular fractions, suggestive for aggresomes (37). To determine the distribution of
CIP2A, Hep G2 cells were pre-treated with MG132 followed by GEA
treatment. The results showed that accumulation of CIP2A in NP40
was induced in the insoluble fractions, while it had minimal effect
on GEA-treated controls (Fig. 3F).
Similar results were noted with GEA in combination with PSI in the
Hep G2 cells (Fig. 3F). To further
elucidate the molecular mechanism underlying the above process, we
immunoprecipitated CIP2A from the NP40-insoluble fraction after
treatment with GEA alone, MG132 alone, or their combinations.
Notably, GEA alone was able to accumulate the ubiquitinated CIP2A
(Fig. 3G, lane 3). Most
importantly, these effects were enhanced following treatment with
the combination of MG132 and GEA (Fig.
3G, lane 5). Aggresome formation is related to redistribution
of the intermediate filament protein and blocked by microtubule
depolymerizing (38,39). We showed that microtubule
depolymerizing agent nocodazole prevented the levels of CIP2A in
the NP40 insoluble fraction (Fig.
3H, lane 6 vs. 4). Expectedly, an increase in CIP2A levels was
detected in the NP40-soluble fraction (Fig. 3H, lane 6 vs. 4). These results
suggest that GEA stimulates ubiquitin proteasome-mediated
degradation of CIP2A.
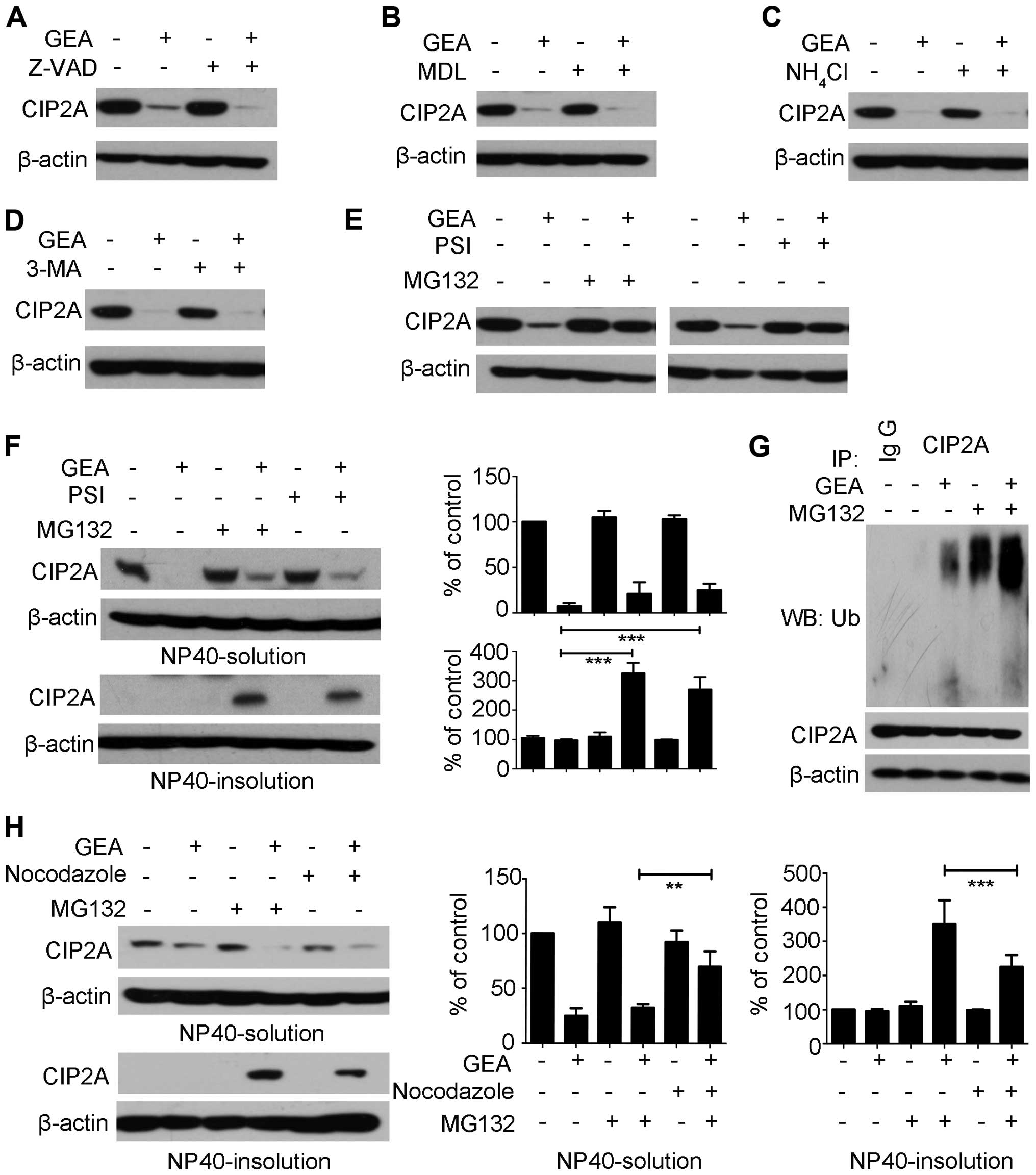 | Figure 3.GEA triggers ubiquitin
proteasome-mediated degradation of CIP2A. (A-D) Hep G2 cells were
pre-treated with (A) Z-VAD (20 µM), (B) MDL (40 µM), (C)
NH4Cl (20 mM), (D) 3-MA (10 mM) for 2 h, followed by GEA
treatment (2.5 µM) for 6 h and the protein level of CIP2A was
detected. (E) Hep G2 cells were pre-treated with MG132 (10 µM) or
PSI (10 µM) for 2 h, followed by GEA treatment (2.5 µM) for 6 h.
Cell lysates were subjected to western blotting using the
anti-CIP2A antibody. (F) Hep G2 cells were treated with MG132 (10
µM) or PSI (10 µM) for 2 h prior to GEA treatment for 6 h. Cells
were harvested and lysed in non-NP40 lysis buffer. The soluble and
insoluble fractions were separated, boiled in SDS buffer for 10
min, and analyzed. Protein expression was quantified by
densitometric analysis and normalized against β-actin expression.
(G) Effect of GEA on CIP2A ubiquitination. Hep G2 cells were
pre-treated with MG132 (10 µM) for 2 h, followed by GEA treatment
for 1 h. NP40-insoluble fractions were extracted as described above
and subjected to immunoprecipitation with the CIP2A antibody,
followed by western blotting using ubiquitin antibody against
ubiquitinated CIP2A. (H) Effect of GEA on aggresome formation in
the presence of proteasome inhibitor MG132. Hep G2 cells were
pre-treated with MG132 (10 µM) and/or nocodazole (20 µg/ml) for 2
h, followed by GEA incubation (2.5 µM) for 6 h. The soluble and
insoluble fractions were separated in cell lysis, and then analyzed
by western blotting. Protein expression was quantified by
densitometric analysis and normalized against β-actin expression.
GEA, gambogenic acid; CIP2A, cancerous inhibitor of protein
phosphatase 2A. Data are presented as means ± SEM. **P<0.01,
***P<0.001. |
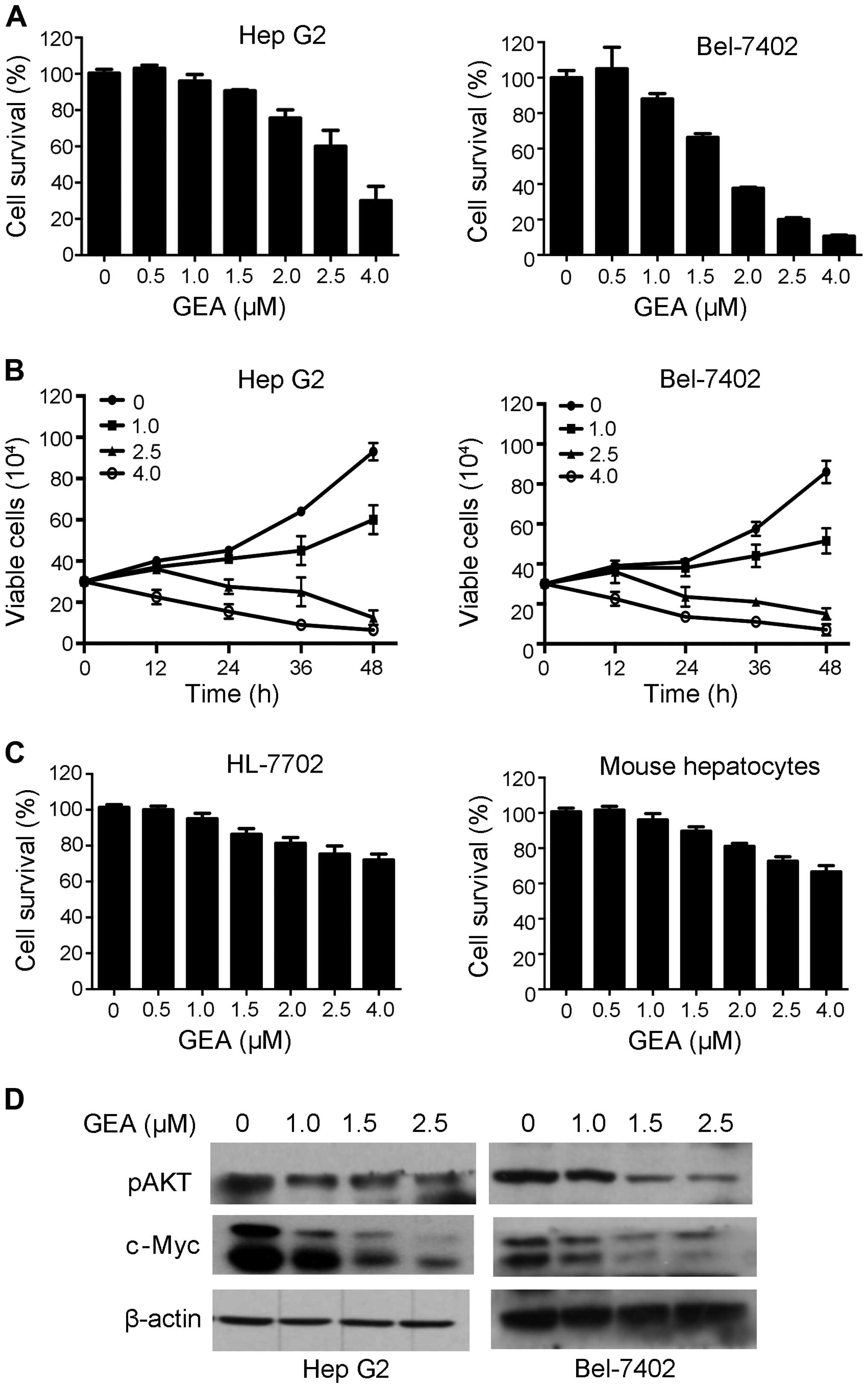
GEA suppresses cell proliferation and
downregulates CIP2A-downstream molecules
CIP2A is a candidate therapeutic target and
inhibition of its activity has potent anticancer effects (40). We therefore tested the effects of
GEA on HCC cells, and found that GEA inhibited cell proliferation
in a dose- and time-dependent manner (Fig. 4A and B). To determine whether GEA is
more sensitive to tumor than normal cells, we examined the effect
of GEA on normal human hepatocyte HL-7702 and mouse primary
hepatocyte cells. We found that its cytotoxic effects on normal
liver cells was weak (Fig. 4C),
suggesting that GEA selectively affects tumor cells. Previous
studies have demonstrated that CIP2A may activate AKT and modulate
c-Myc stability (41). We therefore
detected the effects of GEA on c-Myc and pAKT. We found that GEA
markedly dowregulated c-Myc and pAKT in the Hep G2 and Bel-7402
cell lines (Fig. 4D). These results
indicate that CIP2A-pAKT may play an important role in the
inhibitory effect of GEA against tumor cells.
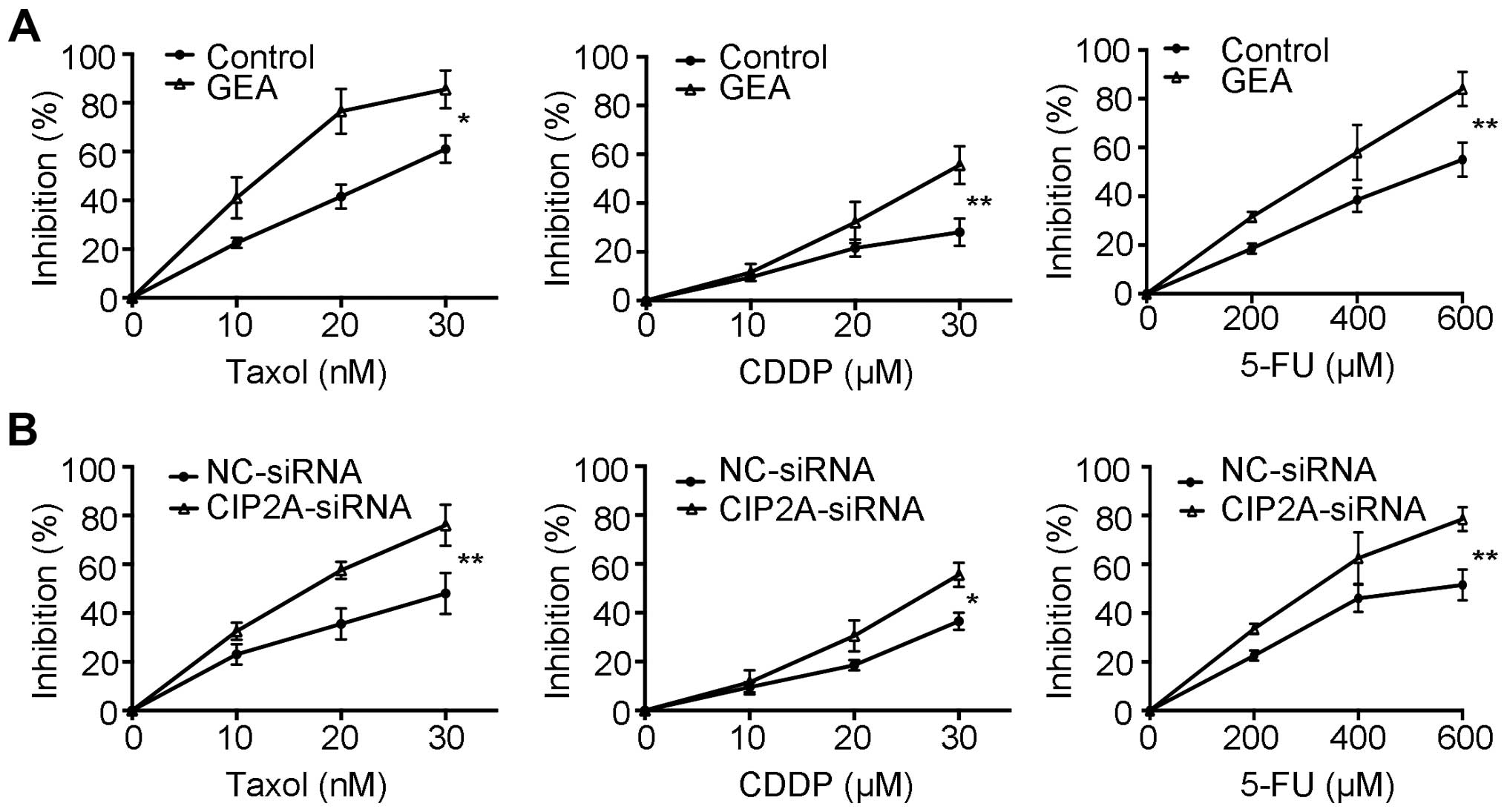
GEA and CIP2A silencing enhance the
sensitivity to chemotherapeutic agents
Previous studies demonstrated that CIP2A
overexpression is associated with drug resistance (12,42).
AKT is a key mediator of cell survival and resistance to
chemotherapy (43). We then
proceeded to evaluate whether GEA enhances chemosensitivity. Our
data showed that pre-treatment with GEA increased the cytotoxic
effects compared to treatment with Taxol, cisplatin (CDDP) and
5-fluorouracil (5-FU) alone, respectively (Fig. 5A). We next explored the role of
CIP2A in GEA-induced sensitivity in HCC. We showed that knockdown
of CIP2A also enhanced the sensitivity of HCC to chemotherapy
agents (Fig. 5B). These results
imply that GEA sensitizes HCC to chemotherapy, and this effect is
associated with CIP2A expression.
Discussion
CIP2A is strongly implicated in carcinogenesis which
is associated with cigarette smoking and Helicobacter pylori
infection, suggesting that CIP2A can serve as an indicator for
assessing the potential carcinogenic risk (10,44).
Furthermore, overexpression of CIP2A is associated with drug
resistance and tumor formation. Moreover, overexpression of CIP2A
is frequently noted in most human cancers, and implies an indicator
of poor clinical outcomes (5,12).
CIP2A-targeting compounds such as bortezomib (12), ethoxysanguinarine (13), and celastrol (14) exert chemopreventive effects. Herein,
we report that GEA is a new CIP2A inhibitor which enhances the
chemosensitivity of HCC to chemotherapeutic agents. We further
showed that GEA triggered degradation of CIP2A in HCC, lung, breast
and colon cancer cell lines (Fig.
1). Since CIP2A can be regulated at the transcriptional and
post-transcription levels, subsequent data demonstrated that GEA
did not inhibit CIP2A mRNA, but decreased the half-life of CIP2A
protein (Fig. 2). These results
suggest that GEA induced proteolytic degradation of CIP2A.
We further explored the mechanisms underlying the
GEA-induced CIP2A degradation. Protein instability and degradation,
such as caspase or calpain cleavage, lysosomal or autophagic
protein degradation, and proteasome-mediated degradation, play a
critical role in proteolysis (31).
We focused on these and found that proteasome inhibitors impaired
the GEA-induced CIP2A degradation, while inhibitors of caspases,
calpain, lysosomes and autophagosomes had no effect on GEA-induced
CIP2A degradation (Fig. 3A-E).
Emerging evidence previously showed that proteasome inhibitors
promote the accumulation of ubiquitinated CIP2A and aggresome
formation (14). We observe herein
that the proteasome inhibitors were able to protect CIP2A from
GEA-mediated degradation (Fig. 3F).
This phenomenon may be associated with accumulation of insoluble
ubiquitinated CIP2A, leading to their aggregation and deposition in
detergent-insoluble fractions (12). Indeed, treatment with GEA or MG132
induced accumulation of ubiquitinated CIP2A (Fig. 3G). Importantly, a significant
increase was observed following co-treatment with GEA and MG132
(Fig. 3G). These results further
confirmed that GEA is a proteasome inhibitor and GEA treatment
promotes CIP2A ubiquitination and accumulation. Previous evidence
revealed that aggresome formation is associated with microtubule
disruption (38). Consistent with
this, our study found that microtubule depolymerizing agent
nocodazole suppressed CIP2A aggregation in NP40-insoluble fractions
(Fig. 3H). In addition, the levels
of CIP2A were significantly enhanced in the NP40-soluble fractions,
suggesting that aggresome formation is an important process in
GEA-triggered degradation of CIP2A (Fig. 3H). Collectively, we speculate that
proteasome inhibition may cause protein misfolding and aggregation,
leading to accumulation of ubiquitinated CIP2A in NP40-insoluble
fractions. These findings partially indicate that GEA-induced CIP2A
degradation is mediated by the ubiquitin-proteasome pathway, but
the detailed mechanisms, including the binding type (directly or
indirectly), sites and the E3 ligase remain unclear and warrant
further investigation.
Several studies have shown that GEA has a potent
anticancer effect in various cancer cells in vitro and in
vivo. However, the mechanisms underlying the anticancer
activity remain unclear. Herein, we showed that GEA suppressed the
proliferation and cell growth in HCC cell lines, but had a mild
effect on normal liver cells (Fig.
4A-C). Interestingly, the expression of CIP2A-downstream
molecules c-Myc and pAKT were impaired in cells following GEA
treatment (Fig. 4D). These findings
suggest that GEA potentiates the inhibitory effect via affecting
the CIP2A-pAKT signaling pathway. Recent evidence suggests that
bortezomib serves as a CIP2A inhibitor and enhances the effect of
radiotherapy dependent on the CIP2A-PP2A-AKT signaling network,
indicating that CIP2A may be associated with drug resistance
(12). GEA was found to synergize
with the cytotoxicity of chemotherapeutic drugs (Fig. 5A). Importantly, CIP2A-silenced cells
were also more sensitive to chemotherapeutic agents (Fig. 5B). These results indicate that GEA
as a CIP2A-targeting inhibitor modulates CIP2A and enhances the
sensitivity of HCC cells to multiple chemotherapeutic agents.
In summary, we provide initial evidence that GEA
triggered the degradation of CIP2A in various cancer cell lines.
This rapid degradation may be associated with the
ubiquitin-proteasome pathway. In addition, GEA suppressed the
CIP2A-pAKT pathway and enhanced sensitivity to chemotherapeutic
agents. Our findings indicate that GEA is a novel CIP2A inhibitor
that may have therapeutic potential in HCC. Admittedly, the
detailed mechanisms of GEA-induced CIP2A degradation remain unclear
and warrant further investigation.
Acknowledgements
We thank Dr Haibing Zhang and Dr Quanbin Han for the
long-term support. This study was supported by grants from the
National Natural Science Foundation of China (81502548), the
Natural Science Foundation of Hubei Province of China (2015CFB210),
the Natural Science Foundation of Hubei Provincial Department of
Education (Q20152101), the Young Scientist Innovation Team Project
of Hubei Colleges (T201510), the Scientific and Technological
Project of Shiyan City of Hubei Province (15Y08), the Initial
Project for Post-Graduates of Hubei University of Medicine
(2014QDJZR09), the Foundation for Innovative Research Team of Hubei
University of Medicine (2014 CXX02) and Thousand Young Talents
Program of the Chinese government.
References
|
1
|
Wang X, Zhang A and Sun H: Power of
metabolomics in diagnosis and biomarker discovery of hepatocellular
carcinoma. Hepatology. 57:2072–2077. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fattovich G, Giustina G, Christensen E,
Pantalena M, Zagni I, Realdi G and Schalm SW: Influence of
hepatitis delta virus infection on morbidity and mortality in
compensated cirrhosis type B. The European Concerted Action on
Viral Hepatitis (Eurohep). Gut. 46:420–426. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El Tayebi HM, Omar K, Hegy S, El Maghrabi
M, El Brolosy M, Hosny KA, Esmat G and Abdelaziz AI: Repression of
miR-17-5p with elevated expression of E2F-1 and c-MYC in
non-metastatic hepatocellular carcinoma and enhancement of cell
growth upon reversing this expression pattern. Biochem Biophys Res
Commun. 434:421–427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fujino H, Kimura T, Aikata H, Miyaki D,
Kawaoka T, Kan H, Fukuhara T, Kobayashi T, Naeshiro N, Honda Y, et
al: Role of 3-D conformal radiotherapy for major portal vein tumor
thrombosis combined with hepatic arterial infusion chemotherapy for
advanced hepatocellular carcinoma. Hepatol Res. 45:607–617. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Junttila MR, Puustinen P, Niemelä M, Ahola
R, Arnold H, Böttzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et
al: CIP2A inhibits PP2A in human malignancies. Cell. 130:51–62.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li W, Ge Z, Liu C, Liu Z, Björkholm M, Jia
J and Xu D: CIP2A is overexpressed in gastric cancer and its
depletion leads to impaired clonogenicity, senescence, or
differentiation of tumor cells. Clin Cancer Res. 14:3722–3728.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Côme C, Laine A, Chanrion M, Edgren H,
Mattila E, Liu X, Jonkers J, Ivaska J, Isola J, Darbon JM, et al:
CIP2A is associated with human breast cancer aggressivity. Clin
Cancer Res. 15:5092–5100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Basile JR and Czerninski R: The role of
CIP2A in oral squamous cell carcinoma. Cancer Biol Ther.
10:700–702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang LP, Savoly D, Sidi AA, Adelson ME,
Mordechai E and Trama JP: CIP2A protein expression in high-grade,
high-stage bladder cancer. Cancer Med. 1:76–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma L, Wen ZS, Liu Z, Hu Z, Ma J, Chen XQ,
Liu YQ, Pu JX, Xiao WL, Sun HD, et al: Overexpression and small
molecule-triggered downregulation of CIP2A in lung cancer. PLoS
One. 6:e201592011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ren J, Li W, Yan L, Jiao W, Tian S, Li D,
Tang Y, Gu G, Liu H and Xu Z: Expression of CIP2A in renal cell
carcinomas correlates with tumour invasion, metastasis and
patients' survival. Br J Cancer. 105:1905–1911. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang CY, Wei CC, Chen KC, Chen HJ, Cheng
AL and Chen KF: Bortezomib enhances radiation-induced apoptosis in
solid tumors by inhibiting CIP2A. Cancer Lett. 317:9–15. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Z, Ma L, Wen ZS, Cheng YX and Zhou GB:
Ethoxysanguinarine induces inhibitory effects and downregulates
CIP2A in lung cancer cells. ACS Med Chem Lett. 5:113–118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Z, Ma L, Wen ZS, Hu Z, Wu FQ, Li W,
Liu J and Zhou GB: Cancerous inhibitor of PP2A is targeted by
natural compound celastrol for degradation in non-small-cell lung
cancer. Carcinogenesis. 35:905–914. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pandey MK, Sung B, Ahn KS, Kunnumakkara
AB, Chaturvedi MM and Aggarwal BB: Gambogic acid, a novel ligand
for transferrin receptor, potentiates TNF-induced apoptosis through
modulation of the nuclear factor-kappaB signaling pathway. Blood.
110:3517–3525. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang X, Chen Y, Han QB, Chan CY, Wang H,
Liu Z, Cheng CH, Yew DT, Lin MC, He ML, et al: Proteomic
identification of molecular targets of gambogic acid: Role of
stathmin in hepatocellular carcinoma. Proteomics. 9:242–253. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang J, Chi Y, Zhan XK, Xie GR, Wang ZZ,
Xiao W, Wang YG, Hu JF, Yu H, Yang L, et al: An open-labeled,
randomized, multicentered, phase IIa study for advanced cancer
treatment by gambogic acid injection (THS). J Clin Oncol.
29:e130952011.
|
|
18
|
Chi Y, Wang J, Zhan X, Xie G, Wang Z, Xiao
W, Wang Y, Hu J, Yu H, Yang L, et al: p53 open-label, randomised,
multicentre, phase 2a study of gambogic acid injection (THS) for
treatment of advanced cancer. EJC Suppl. 9:212011. View Article : Google Scholar
|
|
19
|
Yu X, Zhao Q, Zhang H, Fan C, Zhang X, Xie
Q, Xu C, Liu Y, Wu X, Han Q, et al: Gambogenic acid inhibits
LPS-simulated inflammatory response by suppressing NF-κB and MAPK
in macrophages. Acta Biochim Biophys Sin (Shanghai). 48:454–461.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han QB, Wang YL, Yang L, Tso TF, Qiao CF,
Song JZ, Xu LJ, Chen SL, Yang DJ and Xu HX: Cytotoxic
polyprenylated xanthones from the resin of Garcinia hanburyi. Chem
Pharm Bull (Tokyo). 54:265–267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Q, Cheng H, Zhu G, Yang L, Zhou A, Wang
X, Fang N, Xia L, Su J, Wang M, et al: Gambogenic acid inhibits
proliferation of A549 cells through apoptosis-inducing and cell
cycle arresting. Biol Pharm Bull. 33:415–420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu XJ, Han QB, Wen ZS, Ma L, Gao J and
Zhou GB: Gambogenic acid induces G1 arrest via GSK3β-dependent
cyclin D1 degradation and triggers autophagy in lung cancer cells.
Cancer Lett. 322:185–194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheng H, Su JJ, Peng JY, Wang M, Wang XC,
Yan FG, Wang XS and Li QL: Gambogenic acid inhibits proliferation
of A549 cells through apoptosis inducing through up-regulation of
the p38 MAPK cascade. J Asian Nat Prod Res. 13:993–1002. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan F, Wang M, Chen H, Su J, Wang X, Wang
F, Xia L and Li Q: Gambogenic acid mediated apoptosis through the
mitochondrial oxidative stress and inactivation of Akt signaling
pathway in human nasopharyngeal carcinoma CNE-1 cells. Eur J
Pharmacol. 652:23–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen HB, Zhou LZ, Mei L, Shi XJ, Wang XS,
Li QL and Huang L: Gambogenic acid-induced time- and dose-dependent
growth inhibition and apoptosis involving Akt pathway inactivation
in U251 glioblastoma cells. J Nat Med. 66:62–69. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yan F, Wang M, Li J, Cheng H, Su J, Wang
X, Wu H, Xia L, Li X, Chang HC, et al: Gambogenic acid induced
mitochondrial-dependent apoptosis and referred to phospho-Erk1/2
and phospho-p38 MAPK in human hepatoma HepG2 cells. Environ Toxicol
Pharmacol. 33:181–190. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou J, Luo YH, Wang JR, Lu BB, Wang KM
and Tian Y: Gambogenic acid induction of apoptosis in a breast
cancer cell line. Asian Pac J Cancer Prev. 14:7601–7605. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mei W, Dong C, Hui C, Bin L, Fenggen Y,
Jingjing S, Cheng P, Meiling S, Yawen H, Xiaoshan W, et al:
Gambogenic acid kills lung cancer cells through aberrant autophagy.
PLoS One. 9:e836042014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Su J, Cheng H, Zhang D, Wang M, Xie C, Hu
Y, Chang HC and Li Q: Synergistic effects of 5-fluorouracil and
gambogenic acid on A549 cells: Activation of cell death caused by
apoptotic and necroptotic mechanisms via the ROS-mitochondria
pathway. Biol Pharm Bull. 37:1259–1268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He Y, Ding J, Lin Y, Li J, Shi Y, Wang J,
Zhu Y, Wang K and Hu X: Gambogenic acid alters chemosensitivity of
breast cancer cells to Adriamycin. BMC Complement Altern Med.
15:1812015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y and Zhang Y: Regulation of TET
protein stability by calpains. Cell Reports. 6:278–284. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cohen GM: Caspases: The executioners of
apoptosis. Biochem J. 326:1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Storr SJ, Carragher NO, Frame MC, Parr T
and Martin SG: The calpain system and cancer. Nat Rev Cancer.
11:364–374. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Medina DL, Di Paola S, Peluso I, Armani A,
De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso
C, Forrester A, et al: Lysosomal calcium signalling regulates
autophagy through calcineurin and TFEB. Nat Cell Biol. 17:288–299.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shen M, Schmitt S, Buac D and Dou QP:
Targeting the ubiquitin-proteasome system for cancer therapy.
Expert Opin Ther Targets. 17:1091–1108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ciechanover A: Proteolysis: From the
lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol.
6:79–87. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, Zhao Q, Qi Q, Gu HY, Rong JJ, Mu
R, Zou MJ, Tao L, You QD and Guo QL: Gambogic acid-induced
degradation of mutant p53 is mediated by proteasome and related to
CHIP. J Cell Biochem. 112:509–519. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Johnston JA, Ward CL and Kopito RR:
Aggresomes: A cellular response to misfolded proteins. J Cell Biol.
143:1883–1898. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bence NF, Sampat RM and Kopito RR:
Impairment of the ubiquitin-proteasome system by protein
aggregation. Science. 292:1552–1555. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Khanna A, Rane JK, Kivinummi KK, Urbanucci
A, Helenius MA, Tolonen TT, Saramäki OR, Latonen L, Manni V,
Pimanda JE, et al: CIP2A is a candidate therapeutic target in
clinically challenging prostate cancer cell populations.
Oncotarget. 6:19661–19670. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Khanna A, Böckelman C, Hemmes A, Junttila
MR, Wiksten JP, Lundin M, Junnila S, Murphy DJ, Evan GI, Haglund C,
et al: MYC-dependent regulation and prognostic role of CIP2A in
gastric cancer. J Natl Cancer Inst. 101:793–805. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Choi YA, Park JS, Park MY, Oh KS, Lee MS,
Lim JS, Kim KI, Kim KY, Kwon J, Yoon DY, et al: Increase in CIP2A
expression is associated with doxorubicin resistance. FEBS Lett.
585:755–760. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiao M and Nan KJ: Activation of PI3
kinase/Akt/HIF-1α pathway contributes to hypoxia-induced
epithelial-mesenchymal transition and chemoresistance in
hepatocellular carcinoma. Int J Oncol. 40:461–468. 2012.PubMed/NCBI
|
|
44
|
Zhao D, Liu Z, Ding J, Li W, Sun Y, Yu H,
Zhou Y, Zeng J, Chen C and Jia J: Helicobacter pylori CagA
upregulation of CIP2A is dependent on the Src and MEK/ERK pathways.
J Med Microbiol. 59:259–265. 2010. View Article : Google Scholar : PubMed/NCBI
|