Introduction
Thiosemicarbazones have received considerable
attention due to their numerous biological activities, such as
antibacterial, antitumor and anti-inflammatory properties (1–3), and
their biological activities are involved in the i) inhibition of
ribonucleoside diphosphate reductase; ii) creation of lesions in
the DNA strand by oxidative rupture; and iii) binding to the
nitrogen bases of nucleic acid, disturbing replication (4–6).
Moreover, the planar thiosemicarbazone derivatives also display an
intercalating function towards DNA and target DNA topoisomerase
(7). Some of them can induce cell
cycle delay (8). It has been
demonstrated that excellent biological activities stem from the
formation of iron (or copper) complexes in vivo that are
redox-active species. Thereby one hypothesis proposes that these
complexes can promote the Fenton-like reaction and produce
significant amounts of hydroxyl radicals that interfere with normal
cell functions (9). However,
details behind this action are still not fully understood.
The unfolded protein response (UPR) is a cellular
stress response related to the endoplasmic reticulum (ER), which
can be triggered by a wide variety of cellular events, such as
accumulation of misfolded/unfolded proteins, or changes in the
redox environment of the ER and disruption of Ca2+
homeostasis (10). It has been
demonstrated that Cu2+ thiosemicarbazone complexes
generate oxidative stress, whereas the ligands do not (11–14).
Similarly this is the case for 2-pyridinecarbaldehyde
N,N-bis(2-pyridinylmethyl)thiosemicarbazone (11). However, the metal-free
thiosemicarbazones 3-AP and 3-AP-Me display a different approach in
the induction of ER stress that is reactive oxygen species (ROS)
independent (15). These studies
revealed that thiosemicarbazones have diverse mechanisms of
action.
Calcium ions are important for cellular signaling
and display allosteric regulatory effects on many enzymes and
proteins. Therefore the intracellular Ca2+ concentration
([Ca2+]i) plays a key role in cellular life
(16).
[Ca2+]i is adjusted by Ca2+ entry
and Ca2+ release from intracellular stores. In
non-excitable cells, the inositol-1,4,5-trisphosphate
(IP3)-sensitive ER Ca2+ stores contribute to
the increase in [Ca2+]i (17). IP3 causes the release of
Ca2+ from the ER. Reverse discharge of Ca2+
stores often triggers Ca2+ entry, refilling
Ca2+ stores. Although the derivatives of
thiosemicarbazone induce ER stress, the effect of related
Ca2+ flux on anti-proliferation has received less
attention. In the present study, the antitumor mechanisms of
2-pyridinecarboxaldehyde thiosemicarbazone (PCT) and its copper
complex (PCT-Cu) were preliminarily investigated, revealing that
ROS production plays an important role in the inhibition of
proliferation, DNA fragmentation and apoptosis. The upregulation of
ER stress molecular marker, GRP78, indicated the involvement of ER
stress in the inhibition of proliferation. Furthermore, the
investigated thiocarbazones had the distinct ability to sensitize
the target, thapsigargin, a Ca2+ ATPase inhibitor
(18). The present study suggests
that the antitumor mechanism of thiosemicarbazones may also be
involved in calcium mobilization, which has not yet been
elucidated.
Materials and methods
Materials
All reactants and solvents were AR grade. MTT,
ethidium bromide (EB), RPMI-1640 and agarose were purchased from
Sigma-Aldrich (St. Louis, MO, USA). LC3 and GRP78 antibodies were
obtained from ProteinTech Group, Inc. (Wuhan, China). Caspase-3 and
−8, β-actin, Bax and Bcl-2 were purchased from Boster Biological
Technology (Wuhan, China).
Preparation of PCT
PCT was prepared as previously described (19). To test whether PCT can react with
copper, we mixed a solution of PCT with a solution of
CuCl2 at 1:1 molar ratio, and subsequently, a marked
color change was observed (20).
Its copper complex was prepared by mixing equivalent amounts of PCT
(in DMSO) and CuCl2 (in water). 1H NMR of PCT
(ppm): 7.41 (t, 1H), 7.87 (t, 1H), 8.18 (s, 1H), 8.30 (d, 1H), 8.37
(s, 1H), 8.59 (d, 1H), 11.67 (s, 1H); m/z, 181.0600.
Cytotoxicity assay (MTT assay)
The stock solution of PCT was prepared in DMSO and
it was diluted to the required concentration with culture medium
when used. Human colorectal carcinoma cell line (HCT-116) and liver
carcinoma cells (HepG2) were cultured in RPMI-1640 medium
supplemented with 10% fetal calf serum (FCS) and antibiotics. The
cells collected from exponential-phase (1×104/ml) were
seeded equivalently into 96-well plates and various amounts of PCT
were added after the cells adhered. Following a 48-h incubation at
37°C in a humidified atmosphere of 5% CO2, 10 µl MTT
solution (1 mg/ml) was added to each well, followed by further
incubation for 4 h. The cell culture was removed by aspiration and
100 µl DMSO was added to each well to dissolve the formazan
crystals. The assessment of the absorbances of the solution that
were related to the number of live cells was performed on a
microplate reader (MK3; Thermo Fisher Scientific) at 570 nm.
Percentage of growth inhibition was defined as the percentage of
absorbance inhibition within an appropriate absorbance in each cell
line. The same assay was performed in triplicate.
ROS detection
H2DCF-AM was converted to
dichlorofluorescein (DCF) according to a previously described
method (21). Briefly, 0.25 ml of 2
mM H2DCF-AM in absolute ethanol was added to 2.0 ml of
10 mM NaOH and allowed to stand at room temperature for 30 min. The
hydrolysate was then neutralized with 10 ml of 25 mM sodium
phosphate buffer (pH 7.2), and kept on ice for use. Reaction
mixtures contained 0.4 µM DCF in 50 mM sodium phosphate buffer (pH
7.4) and the eventual addition of 25 µl of 1 mM
NH4FeSO4 or 1 mM PCT in a total volume of 4.0
ml. After the addition of 0.2 ml of 4 mM
H2O2, the fluorescence was assessed on an
FC-960 spectrofluorimeter (excitation at 488 nm and emission at 525
nm). The measurements were conducted at room temperature.
The intracellular ROS production was assessed as
recommended by the manufacturer (Beyotime Institute of
Biotechnology, Beijing, China). Approximately 1×106
HepG2 cells were collected and washed by PBS. The cell pellets were
re-suspended in DCFH-DA containing serum-free culture medium and
incubated for 30 min. The stained cells were re-collected and
washed with serum-free culture medium. Then 100 µl of the cells
were transferred to PCR tubes and the test compound was added.
Following a 1-h incubation, the cell suspension was used directly
for ROS determination on an FC-960 spectrofluorimeter by excitation
at 488 nm and emission at 525 nm.
Comet assay
The comet assay was adapted from a previous study as
described (22). HepG2 cells were
exposed to the investigated agents (12 and 25 µM for the PCT or 2.5
and 5 µM for PCT-Cu complex) for a 24-h incubation in a humidified
atmosphere of 5% CO2. The cells were harvested by
centrifugation after trypsinization and then embedded in 0.5%
low-melting-point agarose at a final concentration of
1×104 cells/ml. A 20 µl aliquot of this cellular
suspension was then spread onto duplicate frosted slides that had
previously been covered with 1% normal melting point agarose as a
basal layer. Slides were allowed to solidify for 10 min at 4°C
before being placed in lysis buffer for 1 h [2.5 M NaCl, 0.1 M
ethylenediaminetetraacetic acid (EDTA), 0.01 M Tris, 1% Triton
X-100, 10% DMSO, pH 10.0]. After lysis, the slides were transferred
into an alkaline buffer for 40 min (0.001 M EDTA, 0.3 M NaOH, pH
>13.0) to allow the DNA to unwind before migration at 0.66 V/cm
and 300 mA for 30 min. All these steps were performed in the dark.
After neutralization in 0.4 M Tris-HCl pH 7.4, the slides were
stained with EB (20 µg/ml) and covered with a cover-slip. The
images were captured using fluorescence microscopy.
Flow cytometric assessment of the
mitochondrial membrane permeability
HepG2 cells (1×105) were seeded in a
6-well plate and incubated for 24 h at 37°C (5% CO2),
then the medium was replaced with fresh medium and supplemented or
not (control) with the agents (5 and 10 µM for PCT or 2.5 and 5 µM
for PCT-Cu complex). After 24 h of incubation, the cells were
harvested with trypsin, followed by washing with PBS, fixed in 70%
ethanol and stored at −20°C. The cells were stained using 20 ng/ml
of rhodamine 123 (R123) in PBS for 30 min. Flow cytometry
(Becton-Dickinson, Franklin Lakes, NJ, USA) was employed to perform
analysis of the depolarization of the mitochondrial membrane as
described in a previous study (23).
Cell cycle analysis
HepG2 cells (1×105) were treated as
previously described in the flow cytometric assessment of the
mitochondrial membrane permeability except that they were stained
using a different dye. The cellular nuclear DNA was stained using
propidium iodide (PI). Briefly, after the removal of 70% ethanol,
the cells were washed with PBS and then suspended in 0.5 ml PBS
containing 50 µg/ml PI and 100 µg/ml RNase. The cell suspension was
incubated at 37°C for 30 min. DNA flow cytometry was performed in
duplicate with a FACSCalibur flow cytometer (Becton-Dickinson). For
each sample 10,000 events were collected and fluorescent signal
intensity was recorded and analyzed by CellQuest and Modifit
(Becton-Dickinson).
PCT and PCT-Cu induce regulation of
apoptosis and ER stress-related proteins
Total RNA was extracted from the cells that were
exposed to the investigated agents for 24 h by TRIzol reagent
(Sangon Biotech Co., Ltd., Shanghai, China) according to the
manufacturer's recommendation. Two micrograms of total RNA were
used for reverse transcription in a total volume of 20 µl with the
M-MLV reverse transcriptase system (LifeFeng Biological Technology
Corp., Shanghai, China). One microliter cDNA was subsequently
amplified in a total volume of 20 µl using the 2X Taq PCR kit
(LifeFeng Biological Technology Corp.). The sense and antisense
primers (Generay Bioengineering Co. Ltd., Shanghai, China) for
β-actin were 5′-ACACTGTGCCCATCTACGAGG-3′ and
5′-CGGACTCGTCATACTCCTGCT-3′ (615 bp) and were used as an internal
control; the sense and antisense primers for caspase-8 were
5′-AAGTTCCTGAGCCTGGACTACAT-3′ and 5′-ATTTGAGCCCTGCCTGGTGTCT-3′ (227
bp); for caspase-3 they were 5′-GAAGCGAATCAATGGACTCTGG-3′ and
5′-ACATCACGCATCAATTCCACAA-3′ (241 bp); for Bcl-2 they were
5′-TTACCAAGCAGCCGAAGA-3′ and 5′-TCCCTCCTTTACATTCACAA-3′ (309 bp);
for Bax they were 5′-TTTTGCTTCAGGGTTTCATC-3′ and
5′-GGCCTTGAGCACCAGTTT-3′ (299 bp), respectively. The RT-PCR was
conducted on a Nexus Gradient Mastercycler (Eppendorf AG, Hamburg,
Germany). The cycling conditions were as follows: 94°C for 3 min,
followed by 30 cycles of 94°C for 30 sec, 53–56°C for 30 sec, and
72°C for 1 min and a final extension of 72°C for 10 min. PCR
products were separated on an 1.5% agarose gel viewed by EB
staining. These data were acquired with a Tocan 360 gel imager
(version 3.2.1 software).
The related gene expression at the protein levels
was monitored by western blotting. Briefly, the 1×107
HepG2 cells that were treated with or without the agents were
scraped off in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl,
1.0% NP-40, 10% glycerol and protease inhibitors) and subjected to
further incubation on ice, following spin down by centrifugation at
14,000 × g. The clear supernatant was collected and stored at
−80°C. After determination of the protein concentration by Bio-Rad
DC protein assay, 30 µg of proteins were separated on a 13% sodium
dodecyl sulfate-polyacrylamide gel at 200 V for 2.5 h. Then the
separated proteins were subsequently transferred onto a PVDF
membrane at 60 V for 1 h. The membrane was washed three times with
Tris-buffered saline (TBS) and then blocked for 2 h in TBS
containing 0.1% Tween-20 and 5% nonfat skimmed milk. The membrane
was incubated at 4°C overnight with the primary monoantibody used
at a dilution of 1:300 in TBST (TBS plus 0.1% Tween-20). The
membrane was washed several times with TBST and was subsequently
incubated with an HRP-conjugated secondary antibody (1:2,000 in
TBST) for 1 h at room temperature. Following another wash of the
membrane with TBST, the protein bands were detected using ECL
solution (Boster Biological Technology).
Ca2+ imaging
[Ca2+]i in the cultured HepG2
cells was assessed using the membrane-permeant acetoxymethyl (AM)
ester of Fura-2 (Dojindo Laboratories, Kumamoto, Japan) dissolved
in DMSO. For the assessment of calcium in the static state, HepG2
cells were treated with PCT (6 µM) or PCT-Cu (3 µM) overnight and
then the cells were washed with FCS-free RPMI-1640, and loaded with
1 µM Fura-2/AM for ~30 min at 37°C in a 5% CO2
atmosphere incubator, followed by three washes with PBS. The
calcium signals were then assessed from the selected cells.
Transient calcium signal measurements were conducted as previously
described (24). Briefly the cells
were pre-treated with or without (control) PCT (6 µM) or PCT-Cu
(1.5 or 3 µM) for ~5 min. Thapsigargin (at a final concentration of
1 µM) was added to stimulate calcium release from the ER stores.
Fluorescent images from selected HepG2 cells were observed with a
×40 water-immersion objective lens and an upright microscope (FN1;
Nikon, Tokyo, Japan). The excitation light at 340 and 380 nm for
Fura-2 experiments was provided by a monochromator (Cairn Research,
Ltd.), while the emission filter (510 nm) was placed in the filter
wheel located in front of the camera. Fluorescent images were
obtained (F340 and F380) every second using a cooled CCD camera
(iXON DU-897; Andor Technology Ltd.). All equipment was used in
combination with MetaFluor software (Molecular Devices, Inc.,
Downingtown, PA, USA).
Results
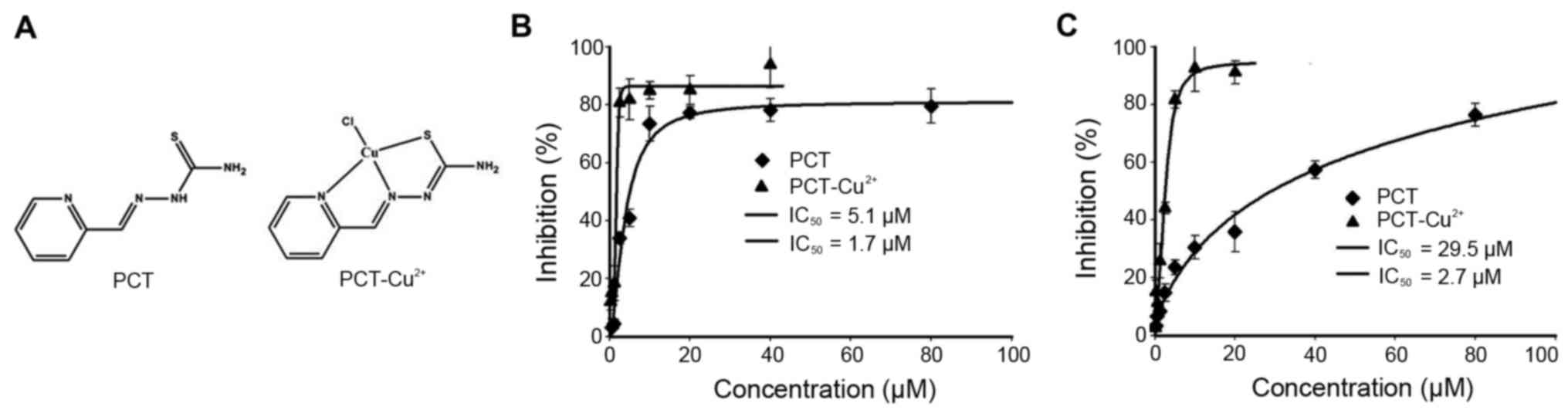
Cytotoxicity of PCT and its copper
complex
It has been shown that derivatives of PCT display
significant inhibition of proliferation against L1210 leukemia cell
lines (25). To elucidate the
underlying mechanism, the inhibitory effects of PCT and its copper
complex (Fig. 1A) against the
proliferation of tumor cell lines was determined. The dose-response
curves of PCT determined against HepG2 and HCT-116 cell lines are
depicted in Fig. 1B and C. As
shown, PCT exhibited significant growth inhibition in HepG2
(IC50, 5.1±1.2 µM) and moderate inhibition for HCT-116
cells (IC50, 29.5±1.5 µM). Due to the chelating ability
of PCT, its copper complex was prepared (see Materials and methods)
and tumor cell proliferation was evaluated. Notably, PCT-Cu
exhibited excellent antitumor activities with an IC50 of
1.7±0.6 µM for HepG2 and 2.7±0.5 µM for HCT-116 cells,
respectively.
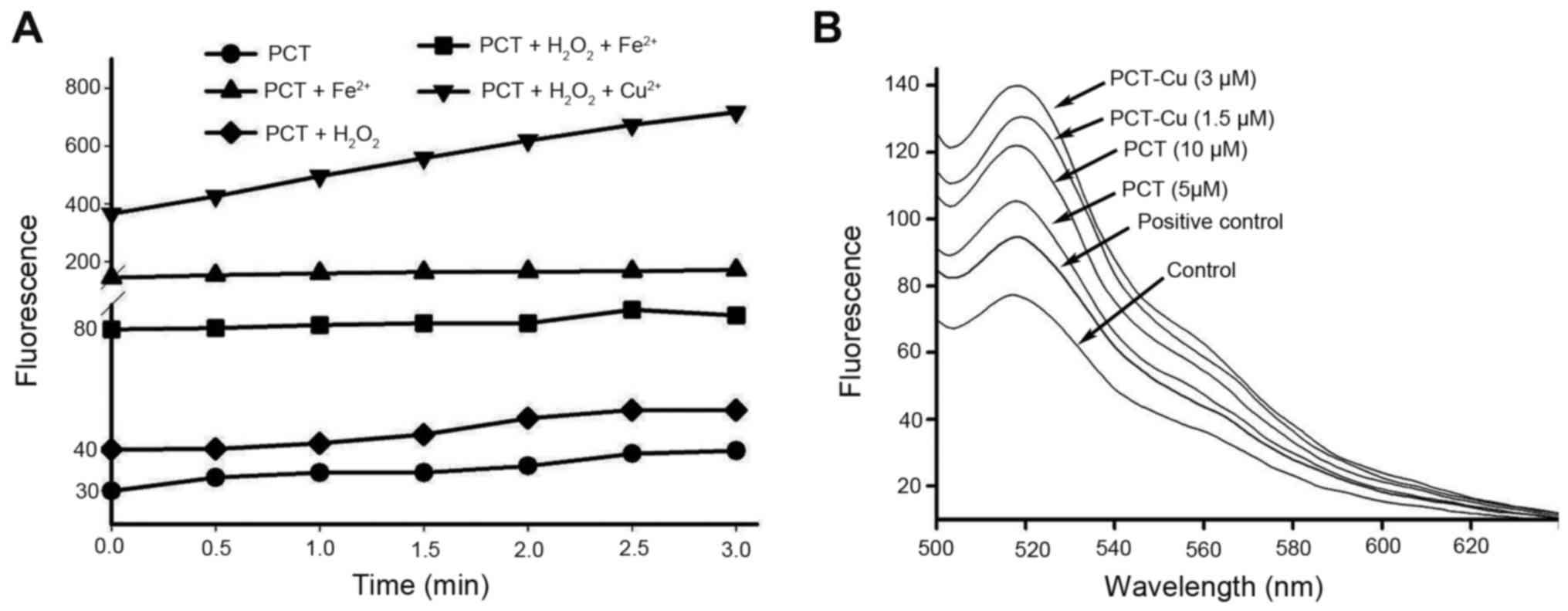
PCT and its copper complex induce ROS
generation
The production of ROS from iron chelators is an
important antitumor mechanism of drugs, and it is correlated with
the cytotoxicity of many drugs. Thus an ROS generation assay was
performed to probe the possibility in vitro and in
vivo. As shown in Fig. 2A, PCT
had the ability to induce ROS formation in a Fenton-like reaction,
indicating that the PCT-Fe2+ complex was a redox-active
species. In contrast to PCT-Fe2+, PCT-Cu2+ in
the presence of H2O2 produced more ROS in
terms of fluorescent intensity, suggesting that PCT-Cu2+
could catalyze hemolysis of H2O2. Based on
the aforementioned results, we speculated that PCT once it crosses
the cell membrane may chelate Fe2+ from labile iron pool
and therefore additional investigation in vivo was
conducted. As expected, both PCT and PCT-Cu induced ROS generation
in a concentration-dependent manner (Fig. 2B). It was noted that the fluorescent
intensity of DCF from the PCT-Cu2+-treated cells was
significantly greater than that of PCT, which could be due to the
redox feature of Cu2+/1+ complexes and the catalyzed
hemolysis of H2O2. PCT-Cu2+ can be
decreased by intracellular reductants, such as glutathione, vitamin
C or other reducing agents to form a PCT-Cu1+ complex
that is able to react with H2O2 (13).
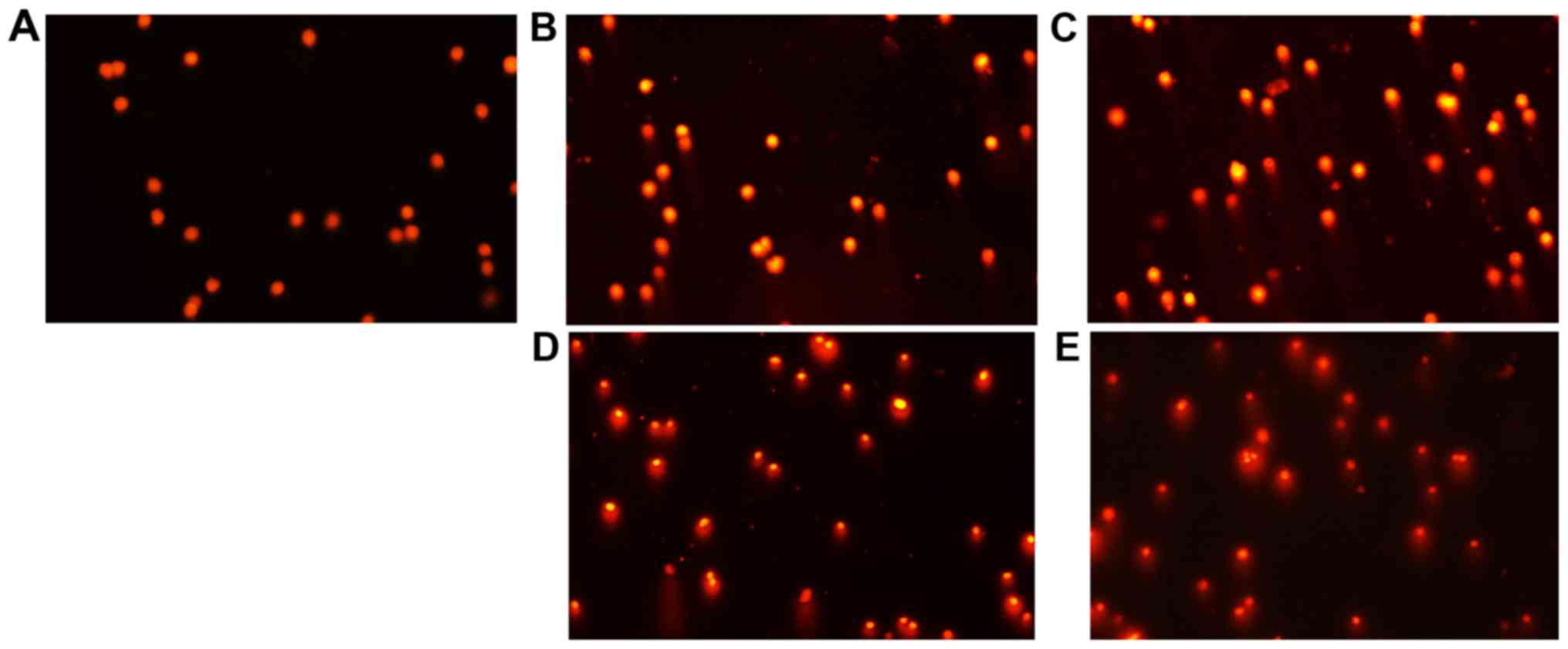
PCT and PCT-Cu induce ROS leading to
cellular DNA fragmentation
It has been well documented that ROS cause genetic
DNA breakage of host cells and induce cytotoxicity. A comet assay
is a widely used technique for the evaluation of cellular DNA
breakage. To further confirm the involvement of ROS in the
inhibition of the proliferation of the agents, DNA integrity of
HepG2 cells in the presence of PCT (or PCT-Cu) was assessed. As
shown in Fig. 3A-E, typical images
of migrated cell nuclei with DNA strand breaks revealed that the
greater portion of the DNA had fragmented in a
concentration-dependent manner following treatment with PCT
compared to the control. It was noted that the PCT-Cu complex
displayed a stronger effect on nuclear DNA damage compared to PCT
(Fig. 3B and D, and C and E),
suggesting that the copper complex caused DNA fragmentation more
rapidly as it required a much lower concentration compared to PCT,
which was consistent with the results from the ROS generation in
vitro and in vivo (Fig. 2A
and B).
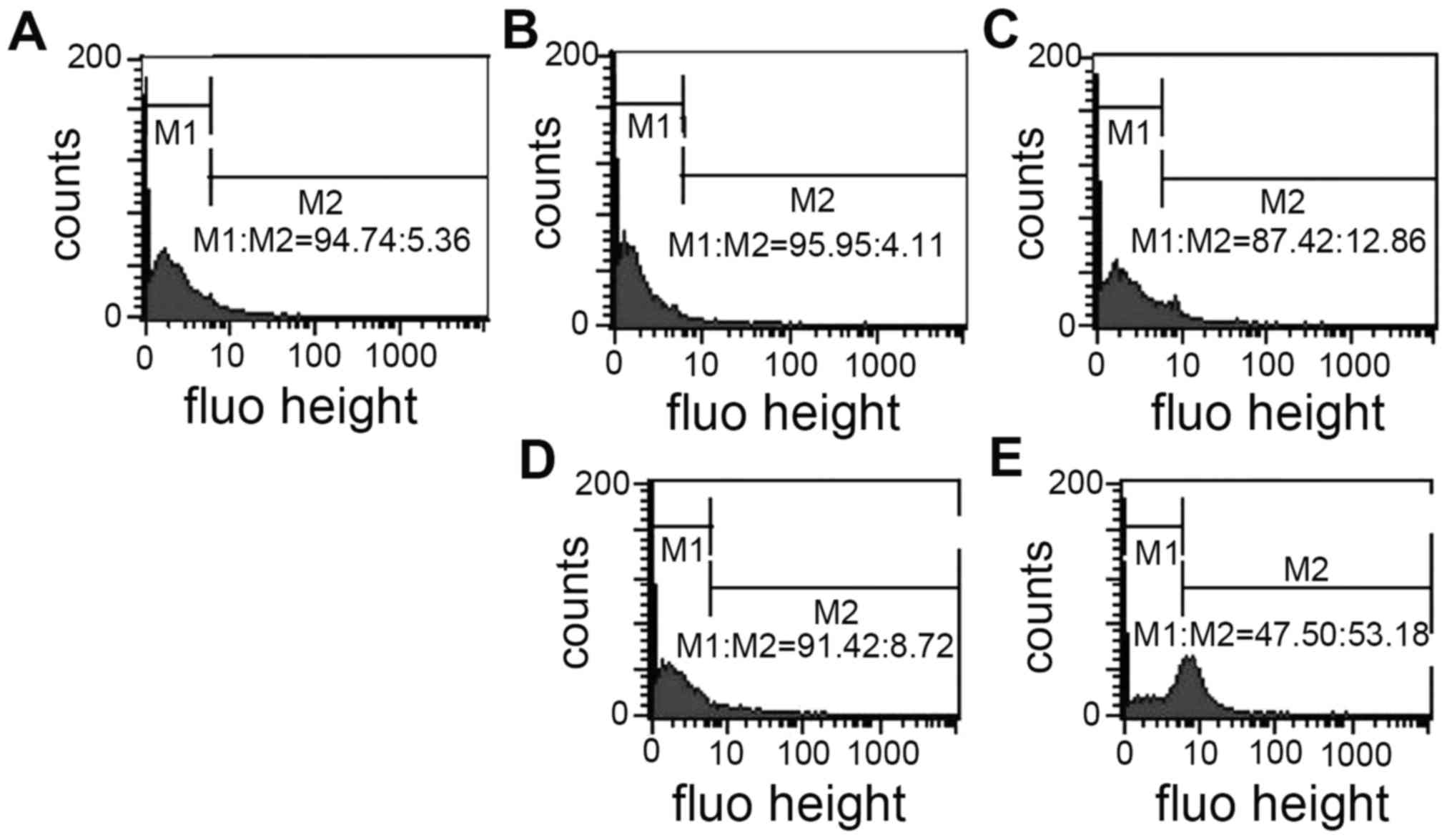
PCT and PCT-Cu induce the
depolarization of the mitochondrial membrane
In vitro ROS generation indicated that the
formed PCT iron or copper complex are redox agents in a reducing
environment and the newly formed reactive species in vivo
could attack other biomolecules except DNA, including the
mitochondrial membrane and depolarize the mitochondrial membrane
and change the mitochondrial membrane potential. R123 is a
fluorescent cationic dye commonly used as a mitochondrial probe.
Thus PCT and PCT-Cu-induced depolarization of the mitochondrial
membrane were evaluated using flow cytometry. As shown in Fig. 4, the mitochondrial fluorescent
intensity from PCT-treated cells was slightly altered compared to
that of the control; a 7% increase in the M2 gate at 10 µM of PCT.
In contrast, 5 µM of PCT-Cu treatment led to a 53% increase at the
M2 gate, an ~50% increase (Fig.
4E), indicating that more cellular mitochondrial membranes were
depolarized. These observations were consistent with the results of
ROS production in vitro and in vivo.
PCT and PCT-Cu exhibit differential
effects on the cell cycle
It has been demonstrated that ROS induce cell cycle
delay and disturb cell cycle progression. We, therefore, evaluated
the effect of PCT and its copper complex on cell cycle distribution
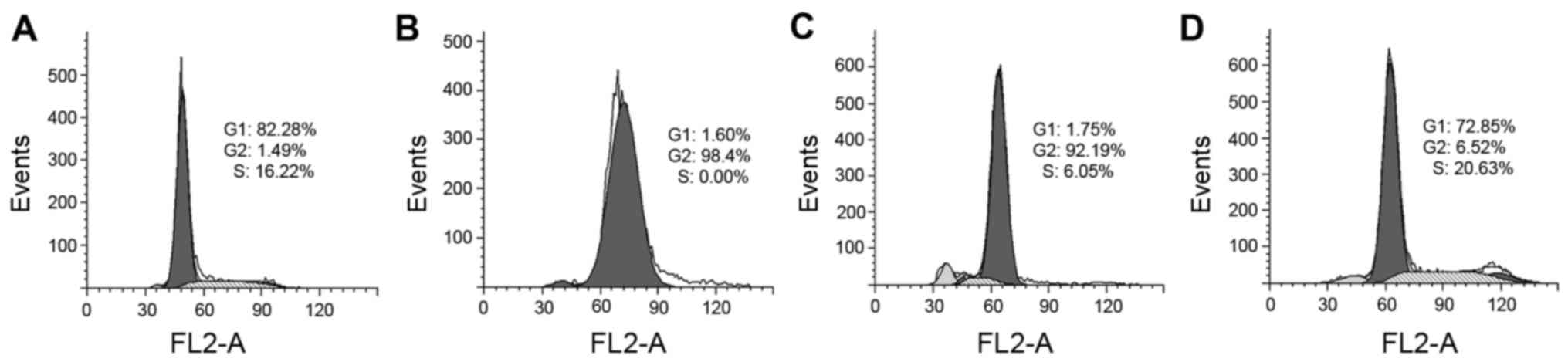
using PI staining and flow cytometry. As shown in Fig. 5, PCT caused an accumulation of cells
in the G2 phase. The percentage of cells at the G2 phase
significantly increased from ~1 to 92% after treatment with PCT.
However, in contrast to PCT, PCT-Cu was less able to disturb the
cell cycle, and led to a slight increase in the percenatge (16–20%)
at the S phase. These results indicated that PCT and PCT-Cu had
differential effects on the cell cycle.
PCT and its copper complex induce
cellular apoptosis
ROS play a crucial role in cell growth and
apoptosis. ROS production induced by PCT and PCT-Cu prompted us to
investigate the underlying molecular mechanism. Generally excess
ROS induce apoptosis, thus changes in the levels of
apoptotic-related genes were investigated. First we assessed
whether the apoptotic genes at the transcription level were
affected when the cells were exposed to the agents. As shown in
Fig. 6A, caspase-3, −8, Bcl-2 and
Bax displayed no evident changes, indicating that the cytotoxicity
(or growth inhibition) exhibited by the agents was not via the
disruption of the transcription of the investigated genes. To
further seek evidence that their cytotoxicity may involve
apoptosis, western blotting was performed to determine the changes
in levels of the corresponding proteins. As shown in Fig. 6B, the changes in the levels of
molecular markers of apoptosis, such as Bcl-2 and Bax were observed
when HepG2 cells were exposed to PCT. The downregulation of Bcl-2
and upregulation of Bax clearly indicated that apoptosis involved
cell growth inhibition or death. Notably in the PCT-Cu-treated
cells, this situation was not evident for Bcl-2 and Bax, but the
caspases (3 and 8) were upregulated, showing that caspase-dependent
initiation of apoptosis may be involved. In both cases, cytoplasmic
cytochrome c exhibited almost no change except with the
PCT-Cu treatment at a higher concentration. These results
demonstrated that PCT and it copper complex displayed distinct
apoptosis induction.
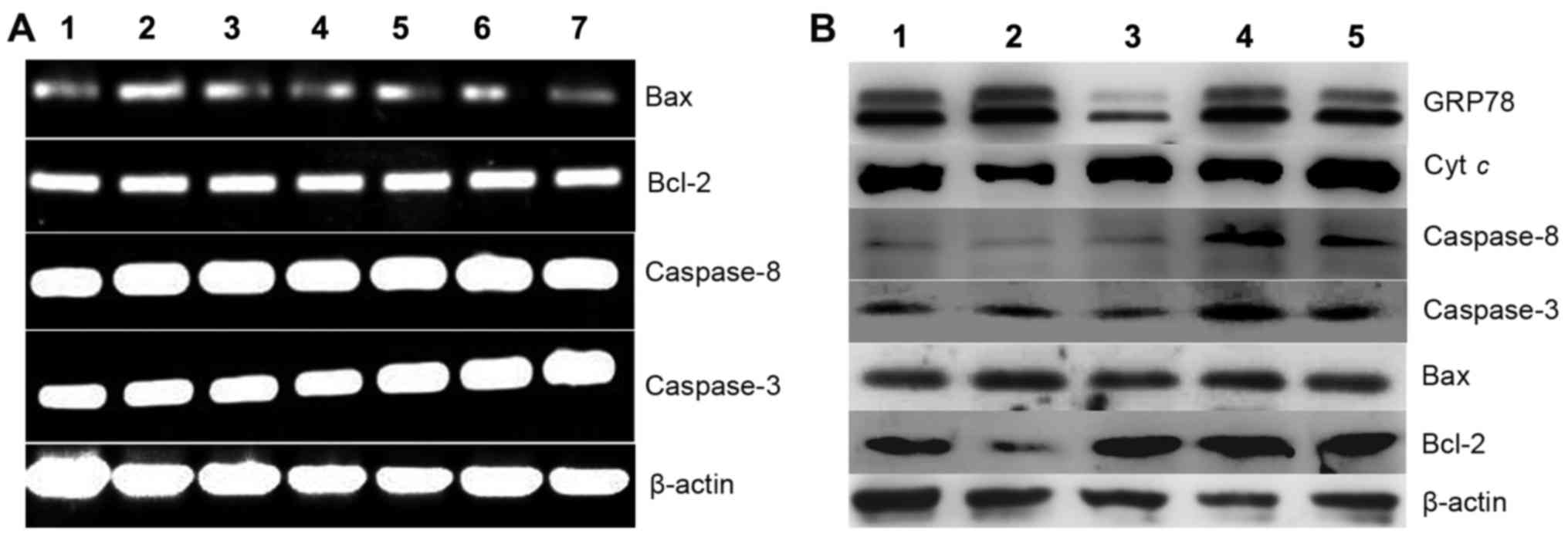 | Figure 6.Gene regulation is induced by PCT and
its copper complex (PCT-Cu) after 24-h treatment of HepG2 cells.
(A) Gene regulation at the mRNA level: lane 1, 10 µM PCT; lane 2, 5
µM PCT; lane 3, 2.5 µM PCT; lane 4, control; lane 5, 0.6 µM PCT-Cu;
lane 6, 1.2 µM PCT-Cu; lane 7, 2.5 µM PCT-Cu. (B) Gene regulation
at the protein level: lane 1, 10 µM PCT; lane 2, 5 µM PCT; lane 3,
control; lane 4, 1.5 µM PCT-Cu; lane 5, 3 µM PCT-Cu. PCT,
2-pyridinecarboxaldehyde thiosemicarbazone. |
Oxidative stress causes calcium
mobilization in the ER
Excess ROS production leads to oxidative stress, ER
stress and inflammatory response, and there is cross-talk between
the cellular responses (26). ER is
an organelle responsible for folding and assembly of membrane and
secreted proteins, synthesis of lipids and sterols, and storage of
free calcium (27). GRP78 is a
molecular marker of ER stress and its upregulation indicated that
ER stress was induced by the agents (Fig. 6B). It has been demonstrated that ER
stress can trigger calcium release from the ER and disrupt
homeostasis. In order to evaluate the possibility of calcium
mobilization involved in the cellular response, the cytoplasmic
calcium was determined by Fura-2/AM fluorescence method. Calcium
release from the ER in the static state and transient state were
assessed to gain insight, as calcium signals from the static state
indicate overall results, while those from the transient state
indicate initial responses. Fig. 7
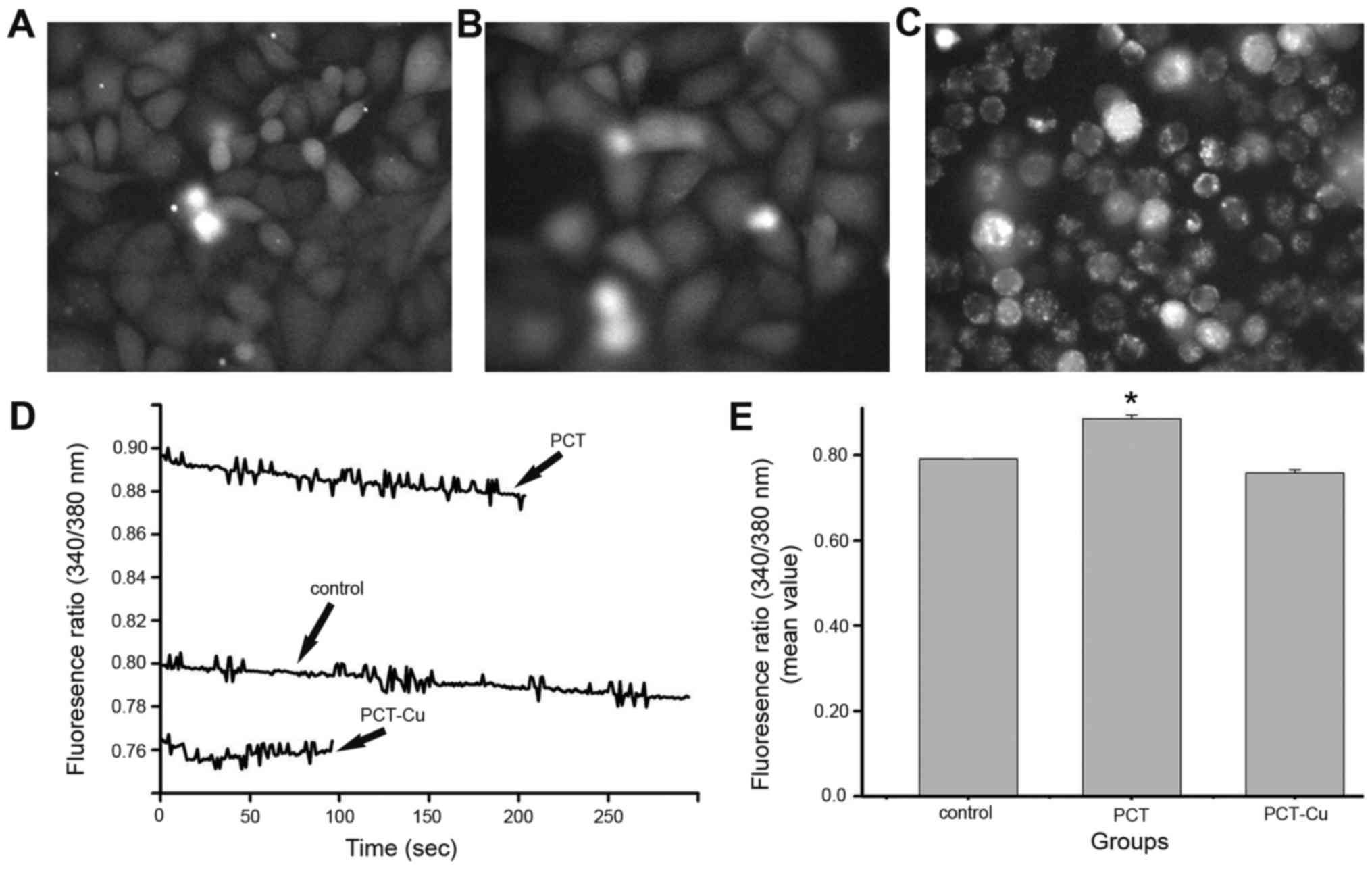
shows the calcium changes in the static state after overnight
exposure to the agents. The calcium signals were greater than that
of the control for PCT, but weaker for the PCT-Cu-treated cells
(Fig. 7D). Further investigation
indicated that the decreased calcium signals after PCT-Cu treatment
were due to serious apoptosis or death when comparing the
morphology of the treated cells with the control (Fig. 7A-C). The quantitative analysis of
calcium is shown in Fig. 7E. In
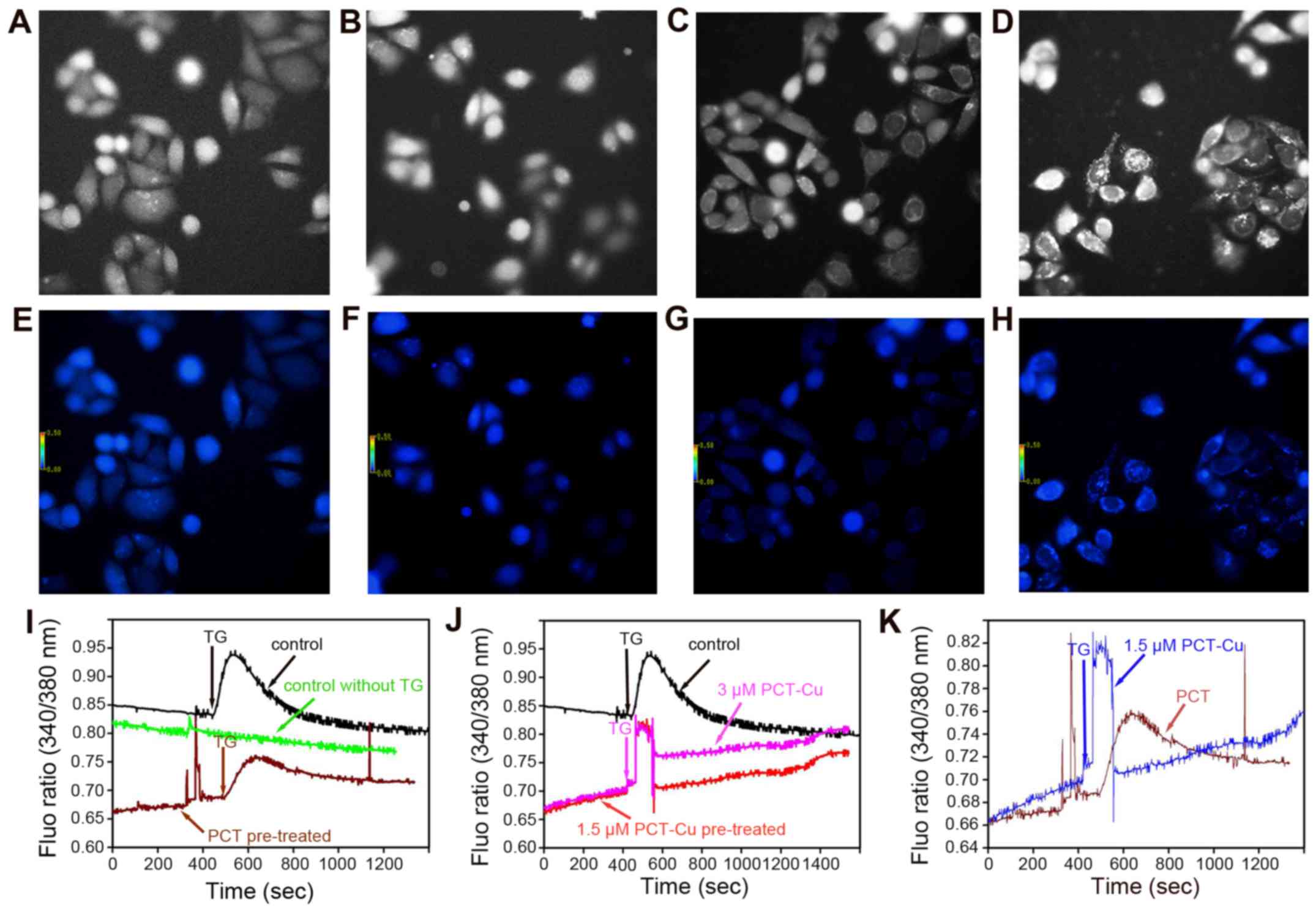
addition, the transient changes of the calcium signals were further
investigated when the cells were exposed to the agents for a short
time period. As we speculated, the calcium signals from both the
PCT- and PCT-Cu-treated cells continuously increased upon exposure
to the agents (first period), and it appeared that more calcium was
released in the PCT-Cu-treated cells (Fig. 8I and J). In contrast, the calcium
signal from the control group decreased even though the fluorescent
intensity was higher than that of the PCT or PCT-Cu group. The
increase of calcium signals upon stimulation of the agents prompted
us to probe whether the agents sensitize the cells to,
thapsigargin, a Ca2+ ATPase inhibitor which stimulates
calcium mobilization in the ER. Thereby, after HepG2 cells were
pretreated for 5 min thapsigargin was added. As shown in Fig. 8I, PCT treatment decreased the
sensitivity of thapsigargin stimuli. In contrast to PCT, PCT-Cu
enhanced the sensitivity of thapsigargin stimuli. The difference
induced by PCT and PCT-Cu in the sensitivity of thapsigargin
stimuli is shown in Fig. 8K.
Clearly, both PCT and PCT-Cu induced transient calcium release, but
the latter was stronger, in correlation to their ROS
production.
Discussion
Anticancer drug treatment frequently causes
caspase-dependent apoptosis, ROS generation and mitochondrial
damage (28). Induced apoptosis in
most of the cancer cells is correlated with excess ROS formation
(29,30). Thiosemicarbazones have potential
metal chelation ability, and the iron chelates are redox-active and
produce ROS in most cases (31).
PCT as the Dp44mT analog, induced ROS formation in vitro and
in vivo (Fig. 2), implying
the possibility that PCT participated in a Fenton-like reaction
chelating iron from the labile iron pool (32). The PCT copper complex (PCT-Cu) also
exhibited significant and potent ROS generation in vivo
(Fig. 2B) due to the easy
transition of Cu2+/1+ in the cellular environment, which
produced a redox cycle to generate ROS as previously described
(12). In addition, the catalytic
activity in hemolysis of H2O2 may be a
contributive factor to ROS (Fig.
2A). In this study, the ROS output induced by PCT and PCT-Cu
appeared to be correlated to their growth inhibition, a situation
which has been observed in other studies (33,34).
Cell cycle arrest is another mechanism exhibited by most anticancer
drugs. PCT caused HCT-116 cell delay at the G2/M phase in an
ROS-dependent manner (35,36), but PCT-Cu had a different effect on
cell cycle progression. It induced an increase in the S phase
population (Fig. 5), suggesting
that chelation altered their effect. Excess ROS production disrupts
homeostasis of cellular redox, causing oxidative stress leading to
cellular component-damage. The DNA fragmentation in the present
study was correlated to their ability in ROS generation (Fig. 3). Cell death may occur in diverse
manners. Apoptosis and necrosis, and the evaluation of apoptotic
genes both at the mRNA and protein level provide important
information concerning the mechanism (37). PCT and PCT-Cu did not disturb the
transcription of apoptotic genes (Fig.
6A), but did disturb the transcription of the related proteins.
It has been demonstrated that the Bcl-2-family of proteins plays
central roles in cell death regulation that encompass apoptosis,
necrosis and autophagy. Bax as a pro-apoptotic protein induces
mitochondrial outer membrane permeabilization, causing caspase
activation, whereas Bcl-2 as an antiapoptotic protein has the
opposing function of Bax (38). In
PCT-treated HepG2 cells, the increased Bax and decreased Bcl-2
levels compared to that of the control were observed, indicating
that apoptosis was involved. Notably, caspase-3 and −8 exhibited no
significant changes in the treated and control groups (Fig. 6B), implying that PCT was less
effective on mitochondrial outer membrane permeabilization, a
finding which was further supported by the assay of mitochondrial
membrane permeabilization (Fig.
4A-C). A similar situation was also observed in another study
(39). In contrast, in
PCT-Cu-treated cells, Bax and Bcl-2 displayed no evident change,
while caspase-3 and −8 were significantly increased, indicating
that PCT-Cu has the ability to affect mitochondrial membrane
permeabilization, a fact which was further supported by the assay
of mitochondrial membrane permeabilization (Fig. 4A, D and E). The aforementioned data
indicated that there was a distinct difference in the induction of
apoptosis between PCT and PCT-Cu. Generally apoptosis is not linked
with total caspase-3 and −8 because their activity can be affected
by phosphorylation of specific kinases, such as the p38
mitogen-activated protein kinase (40,41).
Therefore PCT-Cu induced caspase-8 and −3 activation possibly via
the mitochondrial pathway (42).
This was supported by an increase of mitochondrial cytochrome
c (cyt c) as the depolarization of the mitochondrial
membrane (or changes in permeability), causes cytochrome c
to leak into the cytosol and initiate activation of a cascade of
caspases (43). In our experiment,
the increase of cytochrome c in the PCT-Cu-treated HepG2
cells was correlated with the increase of caspase-3 and −8.
Protein folding and generation of ROS in the ER are
closely linked events (44). PCT-
and PCT-Cu-induced ROS increase in vivo implied that
oxidative stress occurred. Persistent oxidative stress led to the
oxidation of proteins or protein misfolding, that initiated
activation of the UPR and ER stress. GRP78 is a molecular marker of
ER stress and its overexpression further confirmed that ER stress
occured in a ROS-dependent manner (Fig.
6B). The ER is both a unique place for protein folding and is
also the primary Ca2+-storage organelle in the cell.
Protein-folding and protein chaperone all require higher levels of
ER intraluminal calcium. Thus, calcium release leads to the
malfunction of the ER. The antitumor mechanism of the involvement
of iron chelators in ER stress has been demonstrated (45,46).
But in ER stress, how iron chelators influence calcium storage in
the ER has received less attention. In this study, we preliminarily
explored the relationship between ER stress and cytoplasmic
calcium. Upon exposure to the investigated agents, our data showed
that the transient stimuli of PCT or PCT-Cu did not cause
significant calcium release, but gradually increased (Fig. 8I and J). This may reflect that the
uptake of the agents was slow, or the absence of a specific
receptor and channel because extended cell exposure to these
agents, revealed that the calcium signal significantly increased
(Fig. 7D). Both PCT and PCT-Cu
induce calcium release as reported in curcumin-induced cell death
(47). To examine whether PCT (or
PCT-Cu) affected the target thapsigargin, the HepG2 cells were
pretreated with the agents for 5 min. Then thapsigargin stimulated
the Fura-2/AM loaded cells. Notably PCT attenuated the sensitivity
of thapsigargin (Fig. 8I), but
PCT-Cu had the reverse action and enhanced calcium release
(Fig. 8J), indicating that the
increased calcium flux may contribute to their cytotoxicity.
However caution should be used when drawing this conclusion because
there was ER stress in the present study and whether the calcium
mobilization from the ER can inhibit cell growth without ER stress
is unknown and requires further research.
In conclusion, in this study we evaluated the
cytotoxicity and mechanism of PCT and PCT-Cu. The IC50
differences in the investigated cell lines were indicative of drug
selectivity, but the copper chelate displayed enhanced antitumor
behavior. DNA fragmentation, ER stress and apoptosis were all
indications that ROS production was the most significant factor.
Notably our findings suggested that the calcium mobilization in the
ER was also involved in the cytotoxicity exhibited by the agents.
Cell cycle analysis demonstrated that PCT and PCT-Cu had
differential effects on cell cycle progression, supporting that PCT
was not involved in the inhibition of ribonucleic reductase.
Therefore, the proliferation inhibition of PCT and PCT-Cu may
involve diverse mechanisms.
Acknowledgements
The present study was supported by grants awarded by
the Natural Science Foundation of China (no. 21571153), the Henan
Science and Technology Agency (nos. 114300510012, 132102310250 and
152300410118) and the Henan Educational Administration
(15A416009).
References
|
1
|
Husain K, Bhat AR and Azam A: New Pd(II)
complexes of the synthesized 1-N-substituted thiosemicarbazones of
3-indole carboxaldehyde: Characterization and antiamoebic
assessment against E. histolytica. Eur J Med Chem. 43:2016–2028.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rodríguez-Argüelles MC, Tourón-Touceda P,
Cao R, García-Deibe AM, Pelagatti P, Pelizzi C and Zani F:
Complexes of 2-acetyl-γ-butyrolactone and 2-furancarbaldehyde
thiosemicarbazones: Antibacterial and antifungal activity. J Inorg
Biochem. 103:35–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Quach P, Gutierrez E, Basha MT, Kalinowski
DS, Sharpe PC, Lovejoy DB, Bernhardt PV, Jansson PJ and Richardson
DR: Methemoglobin formation by triapine,
di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT), and
other anticancer thiosemicarbazones: Identification of novel
thiosemicarbazones and therapeutics that prevent this effect. Mol
Pharmacol. 82:105–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
García-Tojal J, García-Jaca J, Cortés R,
Rojo T, Urtiaga KM and Arriortua IM: Synthesis and spectroscopic
properties of two pyridine-2-carbaldehyde
thiosemicarbazonecopper(II) compounds: [CuX2(C7H8N4S)]·H2O (X= Br,
Cl). Crystal structure of the bromo complex. Inorg Chim Acta.
249:25–32. 1996. View Article : Google Scholar
|
|
5
|
Chandra S, Parmar S and Kumar Y:
Synthesis, spectroscopic, and antimicrobial studies on bivalent
zinc and mercury complexes of 2-formylpyridine thiosemicarbazone.
Bioinorg Chem Appl. 2009:8513162009. View Article : Google Scholar :
|
|
6
|
Baldini M, Belicchi-Ferrari M, Bisceglie
F, Capacchi S, Pelosi G and Tarasconi P: Zinc complexes with cyclic
derivatives of alpha-ketoglutaric acid thiosemicarbazone:
Synthesis, X-ray structures and DNA interactions. J Inorg Biochem.
99:1504–1513. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pelosi G: Thiosemicarbazone metal
complexes: from structure to activity. Open Crystallogr J. 3:16–28.
2010. View Article : Google Scholar
|
|
8
|
Cabrera M, Gomez N, Lenicov F Remes,
Echeverría E, Shayo C, Moglioni A, Fernández N and Davio C: G2/M
cell cycle arrest and tumor selective apoptosis of acute leukemia
cells by a promising benzophenone thiosemicarbazone compound. PLoS
One. 10:e01368782015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shao J, Zhou B, Di Bilio AJ, Zhu L, Wang
T, Qi C, Shih J and Yen Y: A Ferrous-Triapine complex mediates
formation of reactive oxygen species that inactivate human
ribonucleotide reductase. Mol Cancer Ther. 5:586–592. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cao SS and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress in cell fate decision and
human disease. Antioxid Redox Signal. 21:396–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hancock CN, Stockwin LH, Han B, Divelbiss
RD, Jun JH, Malhotra SV, Hollingshead MG and Newton DL: A copper
chelate of thiosemicarbazone NSC 689534 induces oxidative/ER stress
and inhibits tumor growth in vitro and in vivo. Free Radic Biol
Med. 50:110–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lovejoy DB, Jansson PJ, Brunk UT, Wong J,
Ponka P and Richardson DR: Antitumor activity of metal-chelating
compound Dp44mT is mediated by formation of a redox-active copper
complex that accumulates in lysosomes. Cancer Res. 71:5871–5880.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kowol CR, Heffeter P, Miklos W, Gille L,
Trondl R, Cappellacci L, Berger W and Keppler BK: Mechanisms
underlying reductant-induced reactive oxygen species formation by
anticancer copper(II) compounds. J Biol Inorg Chem. 17:409–423.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kowol CR, Trondl R, Arion VB, Jakupec MA,
Lichtscheidl I and Keppler BK: Fluorescence properties and cellular
distribution of the investigational anticancer drug triapine
(3-aminopyridine-2-carboxaldehyde thiosemicarbazone) and its
zinc(II) complex. Dalton Trans. 39:704–706. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trondl R, Flocke LS, Kowol CR, Heffeter P,
Jungwirth U, Mair GE, Steinborn R, Enyedy ÉA, Jakupec MA, Berger W,
et al: Triapine and a more potent dimethyl derivative induce
endoplasmic reticulum stress in cancer cells. Mol Pharmacol.
85:451–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berridge MJ: Elementary and global aspects
of calcium signalling. J Physiol. 499:291–306. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Berridge MJ: Inositol trisphosphate and
calcium signalling. Nature. 361:315–325. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rogers TB, Inesi G, Wade R and Lederer WJ:
Use of thapsigargin to study Ca2+ homeostasis in cardiac cells.
Biosci Rep. 15:341–349. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chandra S, Raizada S, Tyagi M and Sharma
PK: Spectroscopic and biological approach of Ni(II) and Cu(II)
complexes of 2-pyridinecarboxaldehyde thiosemicarbazone.
Spectrochim Acta A Mol Biomol Spectrosc. 69:816–821. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen D, Cui QC, Yang H, Barrea RA, Sarkar
FH, Sheng S, Yan B, Reddy GP and Dou QP: Clioquinol, a therapeutic
agent for Alzheimer's disease, has proteasome-inhibitory, androgen
receptor-suppressing, apoptosis-inducing, and antitumor activities
in human prostate cancer cells and xenografts. Cancer Res.
67:1636–1644. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang T and Li C, Sun X, Zhu Z, Fu Y, Liu
Y, Yuan Y, Li S and Li C: The antitumor mechanism of
di-2-pyridylketone 2-pyridine carboxylic acid hydrazone and its
copper complex in ROS generation and topoisomerase inhibition, and
hydrazone involvement in oxygen-catalytic iron mobilization. Int J
Oncol. 47:1854–1862. 2015.PubMed/NCBI
|
|
22
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fu Y, Zhang Y, Zhou SF, Liu Y, Wang J,
Wang Y, Lu C and Li C: Effects of substitution of carboxyl with
hydrazide group on position 3 of ciprofloxacin on its antimicrobial
and antitumor activity. Int J Pharmacol. 9:416–429. 2013.
View Article : Google Scholar
|
|
24
|
Wang J, Wang Y, Wang Y, Wang R, Zhang Y,
Zhang Q and Lu C: Contribution of α4β2 nAChR in nicotine-induced
intracellular calcium response and excitability of MSDB neurons.
Brain Res. 1592:1–10. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu MC, Lin TS and Sartorelli AC:
Synthesis and antitumor activity of amino derivatives of
pyridine-2-carboxaldehyde thiosemicarbazone. J Med Chem.
35:3672–3677. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang K: Integration of ER stress,
oxidative stress and the inflammatory response in health and
disease. Int J Clin Exp Med. 3:33–40. 2010.PubMed/NCBI
|
|
27
|
Gething MJ and Sambrook J: Protein folding
in the cell. Nature. 355:33–45. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brodská B and Holoubek A: Generation of
reactive oxygen species during apoptosis induced by DNA-damaging
agents and/or histone deacetylase inhibitors. Oxid Med Cell Longev.
2011:2535292011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun Y, Huang L, Mackenzie GG and Rigas B:
Oxidative stress mediates through apoptosis the anticancer effect
of phospho-nonsteroidal anti-inflammatory drugs: Implications for
the role of oxidative stress in the action of anticancer agents. J
Pharmacol Exp Ther. 338:775–783. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rigas B and Sun Y: Induction of oxidative
stress as a mechanism of action of chemopreventive agents against
cancer. Br J Cancer. 98:1157–1160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jansson PJ, Hawkins CL, Lovejoy DB and
Richardson DR: The iron complex of Dp44mT is redox-active and
induces hydroxyl radical formation: An EPR study. J Inorg Biochem.
104:1224–1228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kakhlon O and Cabantchik ZI: The labile
iron pool: Characterization, measurement, and participation in
cellular processes(1). Free Radic Biol Med. 33:1037–1046. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Preza AM, Jaramillo ME, Puebla AM, Mateos
JC, Hernández R and Lugo E: Antitumor activity against murine
lymphoma L5178Y model of proteins from cacao (Theobroma cacao L.)
seeds in relation with in vitro antioxidant activity. BMC
Complement Altern Med. 10:612010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vandamme M, Robert E, Lerondel S, Sarron
V, Ries D, Dozias S, Sobilo J, Gosset D, Kieda C, Legrain B, et al:
ROS implication in a new antitumor strategy based on non-thermal
plasma. Int J Cancer. 130:2185–2194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang X, Chen M, Zou P, Kanchana K, Weng
Q, Chen W, Zhong P, Ji J, Zhou H, He L, et al: Curcumin analog WZ35
induced cell death via ROS-dependent ER stress and G2/M cell cycle
arrest in human prostate cancer cells. BMC Cancer. 15:8662015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo J, Wu G, Bao J, Hao W, Lu J and Chen
X: Cucurbitacin B induced ATM-mediated DNA damage causes G2/M cell
cycle arrest in a ROS-dependent manner. PLoS One. 9:e881402014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vandaele L, Goossens K, Peelman L and Van
Soom A: mRNA expression of Bcl-2, Bax, caspase-3 and −7 cannot be
used as a marker for apoptosis in bovine blastocysts. Anim Reprod
Sci. 106:168–173. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liang Y, Yan C and Schor NF: Apoptosis in
the absence of caspase 3. Oncogene. 20:6570–6578. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Alvarado-Kristensson M, Melander F,
Leandersson K, Rönnstrand L, Wernstedt C and Andersson T: p38-MAPK
signals survival by phosphorylation of caspase-8 and caspase-3 in
human neutrophils. J Exp Med. 199:449–458. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Voss OH, Kim S, Wewers MD and Doseff AI:
Regulation of monocyte apoptosis by the protein kinase
Cdelta-dependent phosphorylation of caspase-3. J Biol Chem.
280:17371–17379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu J, Uematsu H, Tsuchida N and Ikeda MA:
Essential role of caspase-8 in p53/p73-dependent apoptosis induced
by etoposide in head and neck carcinoma cells. Mol Cancer.
10:952011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cai J, Yang J and Jones D: Mitochondrial
control of apoptosis: The role of cytochrome c. Biochim Biophys
Acta. 1366:139–149. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim JL, Lee DH, Na YJ, Kim BR, Jeong YA,
Lee SI, Kang S, Joung SY, Lee SY, Oh SC, et al: Iron
chelator-induced apoptosis via the ER stress pathway in gastric
cancer cells. Tumour Biol. 37:9709–9719. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lane DJ, Mills TM, Shafie NH, Merlot AM,
Moussa R Saleh, Kalinowski DS, Kovacevic Z and Richardson DR:
Expanding horizons in iron chelation and the treatment of cancer:
Role of iron in the regulation of ER stress and the
epithelial-mesenchymal transition. Biochim Biophys Acta.
1845:166–181. 2014.PubMed/NCBI
|
|
47
|
Moustapha A, Pérétout PA, Rainey NE,
Sureau F, Geze M, Petit JM, Dewailly E, Slomianny C and Petit PX:
Curcumin induces crosstalk between autophagy and apoptosis mediated
by calcium release from the endoplasmic reticulum, lysosomal
destabilization and mitochondrial events. Cell Death Dis.
1:150172015. View Article : Google Scholar
|