Introduction
First proposed by C.H. Waddington in 1957,
epigenetics is a branch of genetics that involves the aberrant
methylation of tumor-suppressor genes, abnormal histone
post-translational modification, non-coding RNA interference and
chromatin remodeling. Acute leukemia (AL) often exhibits aberrant
epigenetic modifications, such as inactivation of the methylation
of Wnt antagonists and excessive activation of histone deacetylase.
Therefore, investigating the epigenetic changes in AL is essential
for identifying potential key therapeutic targets.
H4K20 was discovered in 1969 (1), but it was not identified as a major
site of methylation until 2001 (2).
In multicellular organisms, H4K20 may present 3 different levels of
modification: monomethylation, bimethylation and trimethylation.
The methylation status is regulated by specific methyltransferases;
KMT5A/SETD8/Pr-Set7 (SET8) is the only methyltransferase that
catalyzes H4K20 monomethylation (3). H4K20me1 is the precursor of H4K20me2/3
and is involved in various processes such as DNA damage response,
mitosis, chromatin condensation, DNA replication and gene
regulation (4–6). Previously, researchers believed that
H4K20me1 was a transcription-inhibiting histone (7); however, in 2014, studies revealed that
H4K20me1 plays an important role in enhancing the transcription and
elongation of RNA polymerase II (8).
To date, no studies have been conducted to
investigate the relationship between H4K20me1 and SET8, the
expression of tumor suppressor genes, and DNA methylation in
leukemia. Our previous study showed that hypermethylation of the
promoter region of Wnt5a (a Wnt pathway tumor suppressor gene) was
detected in some clinical acute myeloid leukemia (AML) specimens.
However, there was no decrease in Wnt5a expression in these
specimens, suggesting that hypermethylation had no effect on the
expression of suppressor genes. Moreover, the expression of histone
H4K20me1 was higher in these specimens than in those with low Wnt5a
expression. Therefore, we hypothesized that the expression of
H4K20me1 and Wnt5a may be related and that H4K20me1 may play a
larger role in gene transcription than methylation status. This
hypothesis may help explain the discordant hypermethylation and
inhibition of gene expression observed in clinical specimens, thus
providing additional information to the current theory of
methylation-induced gene inactivation.
Materials and methods
Clinical specimens
A total of 95 cases of treatment-naive AL diagnosed
and treated at the Affiliated Union Hospital of Fujian Medical
University between November 2011 and December 2013 were included in
the present study. Among them, 53 patients were male and 42 were
female, with a median age of 37 (7–82) years. Moreover, 62 patients
had AML and 33 had acute lymphocytic leukemia (ALL) [T cell ALL
(ALL-T), n=11; B-cell ALL (ALL-B), n=22]. All cases met the
diagnostic criteria of the French-American-British (FAB)
classification system concerning morphology, immunology,
cytogenetics and molecular biology (MICM). A total of 58 bone
marrow transplant donors were included in the normal control group
(Table I). All patients signed an
informed consent form indicating that they agreed to the use of
their specimens for research. All specimens were collected from
bone marrow.
 | Table I.Age and gender composition of acute
leukemia patients and normal controls. |
Table I.
Age and gender composition of acute
leukemia patients and normal controls.
|
|
| Gender |
|---|
|
|
|
|
|---|
| Group | Age (years) | Male (n) | Female (n) |
|---|
| AML | 32.12±16.21 | 31 | 31 |
| ALL | 42.44±16.64 | 22 | 11 |
| Normal control | 33.65±7.37 | 24 | 34 |
Collection of bone marrow mononuclear
cells
A bone marrow specimen (5 ml, heparin-treated) was
collected, slowly added (with no mixing) to the top of an equal
volume of lymphocyte separation medium in a centrifuge tube and
centrifuged at room temperature at 2,500 rpm for 20 min. After
centrifugation, the specimen was separated into 4 layers (top to
bottom): plasma, mononuclear cells, separation medium, red blood
cells and platelets. The mononuclear cell layer was carefully
collected, subsequently washed with phosphate-buffered saline (PBS)
and centrifuged at 2,000 rpm for 5 min twice. The supernatant was
discarded and the cell pellet was used to extract RNA, proteins or
DNA, or stored at −80̊C for later use.
RNA extraction and fluorescence
qPCR
qRT-PCR was performed to detect Wnt5a, HDPR1, DKK1
and DKK3 expression. TRIzol reagent (Invitrogen Corp., Carlsbad,
CA, USA) was used for one-step total RNA extraction from bone
marrow specimens according to the manufacturer's instructions.
First-strand cDNA was synthesized using an RNA reverse
transcription kit (Fermentas Inc., Glen Burnie, MD, USA) according
to the manufacturer's instructions. An ABI 7500 fluorescence qPCR
instrument and FastStart Universal SYBR-Green Master (ROX) kit were
used for real-time fluorescence qPCR, using GAPDH as an internal
reference; Ct values were compared to analyze differences in the
mRNA expression levels of Wnt5a, and related genes in bone marrow
cells between ALL, AML and the normal control group: ΔCt = Ct value
(target primer) - Ct value (GAPDH), and then the 2−ΔCt
values were compared. The following specific mRNA primers were
used: Wnt5a, 5′-GACCACATGCAGTACATCGGAGAAG-3′ (forward) and
5′-TCCACCTTCGATGTCGGAATTG-3′ (reverse); HDPR1,
5′-AAGAGCACCTGGAGACAGACAG-3′ (forward) and
5′-GCTGGAATGACAACTGGATAAAC-3′ (reverse); DKK1,
5′-CTGCAAAAATGGAATATGTGT-3′ (forward) and
5′-CTTCTTGTCCTTTGGTGTGA-3′ (reverse); DKK3, 5′-GATGCCCTTGTGCCAGT-3′
(forward) and 5′-TGCCAACTTCATACTCATCGG-3′ (reverse); and GAPDH,
5′-GGATGCAGGGATGATGTTCT-3′ (forward) and 5′-TGCCACTCAGAAGACTGTGG-3′
(reverse).
Western blotting
Cell lysis buffer and a protease inhibitor cocktail
were added to the cell pellet, and a bicinchoninic acid (BCA)
protein assay kit (Pierce, Rockford, IL USA) was used to determine
protein concentration.
For SDS-polyacrylamide gel electrophoresis
(SDS-PAGE), the sample volume to be loaded was calculated based on
the protein concentration (30 µg protein). Proteins were separated
via SDS-PAGE, transferred to a polyvinylidene fluoride membrane,
blocked in skim milk at room temperature for 1 h, and incubated
with diluted mouse anti-human Wnt5a monoclonal antibody (1:5,000),
rabbit anti-human H4K20me1 monoclonal antibody (1:10,000) (both
from Millipore, Billerica, MA, USA), or rabbit anti-human GAPDH
monoclonal antibody (1:5,000; Abcam, Cambridge, MA, USA) overnight
at 4̊C. Next, the membrane was washed with Tris-buffered saline
with Tween-20 (TBST) (10 min, ×3) and incubated with horseradish
peroxidase-labeled goat anti-mouse antibody or goat anti-rabbit
secondary antibody for 1 h. An equal volume of reagent A and B of
an ECL luminescent kit was mixed and evenly spread on the membrane.
Subsequently, the membrane was exposed, developed and fixed to
Kodak film in the darkroom; Quantity One software (Bio-Rad
Laboratories, Hercules, CA USA) was used for the quantitative
analysis, using GAPDH as an internal reference. The experiment was
repeated 3 times.
Pyrophosphate sequencing
Pyrophosphate sequencing was performed to determine
the methylation rate of the CpG island of the Wnt5a promoter in
bone marrow cells. PyroMark Assay Design 2.0 was used to design
pyrophosphate sequencing primers, which were synthesized by BGI
(Shenzhen, China). The following primer sequences were used: Wnt5a,
5′-ATATTTGGGGTTGGAAAGTTTTAA-3′ (forward) and
5′-ACCCACAACAAAAACAAAACCTAATC-3′ (reverse); Wnt5a-sequence,
GGGTTGGAAAGTTTTAATTAT.
Bone marrow mononuclear cells were collected, and
genomic DNA was extracted using phenol-chloroform. An EpiTect
Bisulfite kit (Qiagen, Valencia, CA, USA) was used for bisulfite
modification and purification of genomic DNA in strict accordance
with the manufacturer's instructions. For amplification of sulfated
genomic DNA, the total reaction volume was 50 µl, including 0.2 µl
of Taq (5 U/µl), 2 µl of template, 1 µl of dNTPs (10 mM
each), 1 µl of forward and reverse primer (50 pmol/µl), 10 µl of 5X
buffer GC (KAPA), and 34.8 µl of nuclease-free water. The reaction
conditions were as follows: 95̊C pre-denaturation for 3 min; 94̊C
denaturation for 30 sec, 50̊C annealing for 30 sec, and 72̊C
extension for 60 sec for 40 cycles; and 72̊C extension for 7 min.
Then, 2 µl of reactant haptoglobin, 38 µl of binding buffer and 40
µl of PCR product were added into a 96-well PCR reaction plate and
mixed at room temperature for 10 min. A vacuum pump was used to
draw the haptoglobin and PCR product suspension, which was then
successively immersed in 70% ethanol, 0.2 M NaOH, and washing
buffer for 5 sec each. Next, the vacuum pump was turned off and
haptoglobin and the PCR product on the probe were placed into 40 µl
of annealing buffer (containing 1.5 µl of sequencing primers),
denatured at 85̊C for 2 min, and cooled to room temperature,
allowing annealing and hybridization between the primers and the
template. Pyrosequencing design software was used to calculate the
dose, and the substrate mixture, enzyme mixture and 4 types of
dNTPs were successively added into the reagent compartment, which,
along with the 96-well reaction plate, was placed into a
pyrosequencing detector (PyroMark Q96 ID) (both from Qiagen) for
reaction; Pyro Q-CpG software was used for automatic analysis of
the methylation status of each site.
Chromatin immunoprecipitation
(ChIP)
Three donor bone marrow specimens were collected.
Moreover, 3 AML specimens with methylation of the Wnt5a promoter,
and 3 AML specimens each with high or low Wnt5a expression were
selected. The EZ-Magna ChIP™ A/G One-Day Chromatin
Immunoprecipitation kit (cat no. 17-10086; Millipore) was used for
ChIP of bone marrow cells. Cell isolation, sonication, ChIP,
eluting and de-crosslinking of protein complexes by
immunoprecipitation were performed according to the manufacturer's
instructions. Immunoprecipitated DNA was detected with real-time
PCR; primers were designed for different regions of the Wnt5a gene,
using GAPDH as an internal reference. The following primer
sequences were used: Wnt5a promoter region,
5′-GGTCTTTTGCACAATCACGCC-3′ (forward) and
5′-TTTCCAACGTCCATCAGCGAC-3′ (reverse); Wnt5a coding region,
5′-GATGGCTGGAAGTGCAATGTCT-3′ (forward) and
5′-ACCTGGGCGAAGGAGAAAAA-3′ (reverse); and GAPDH,
5′-TACTAGCGGTTTTACGGGCG-3′ (forward) and
5′-TCGAACAGGAGGAGCAGAGAGCGA-3′ (reverse). The following antibodies
were used: CHIPAb + monomethyl-histone H4 (Lys20) (cat no. 17-651;
Millipore), and anti-KMT5A/SETD8/Pr-SET7 antibody, (ab3798;
Abcam).
Fluorescence qPCR was performed using DNA fragments
from each immunoprecipitation and from each input sample, and the
data were analyzed using % of input and fold-change. Standardized
amount of DNA: ΔCtnormalized ChIP = CtChIP -
[CtInput - Log2(Input Dilution Factor)];
Input Dilution Factor = (fraction of the input
chromatin)−1 = (1%)−1 = 100; % of Input =
2−ΔCt normalized ChIP×100%; Fold-Change =
2−ΔΔCt, where ΔΔCt = ΔCt normalized ChIPtarget group -
ΔCt normalized ChIPcontrol group.
Statistical analysis
The data are expressed as the mean ± SD and were
analyzed using SPSS 19.0 software. The data were non-normally
distributed. A Mann-Whitney U test was performed for between-group
comparisons, and a Chi-square test was performed to compare
percentages between 2 groups. P<0.05 was considered to indicate
a statistically significant result.
Results
Wnt5a, HDPR1, DKK1 and DKK3 expression
is lower in AML patients than in normal controls
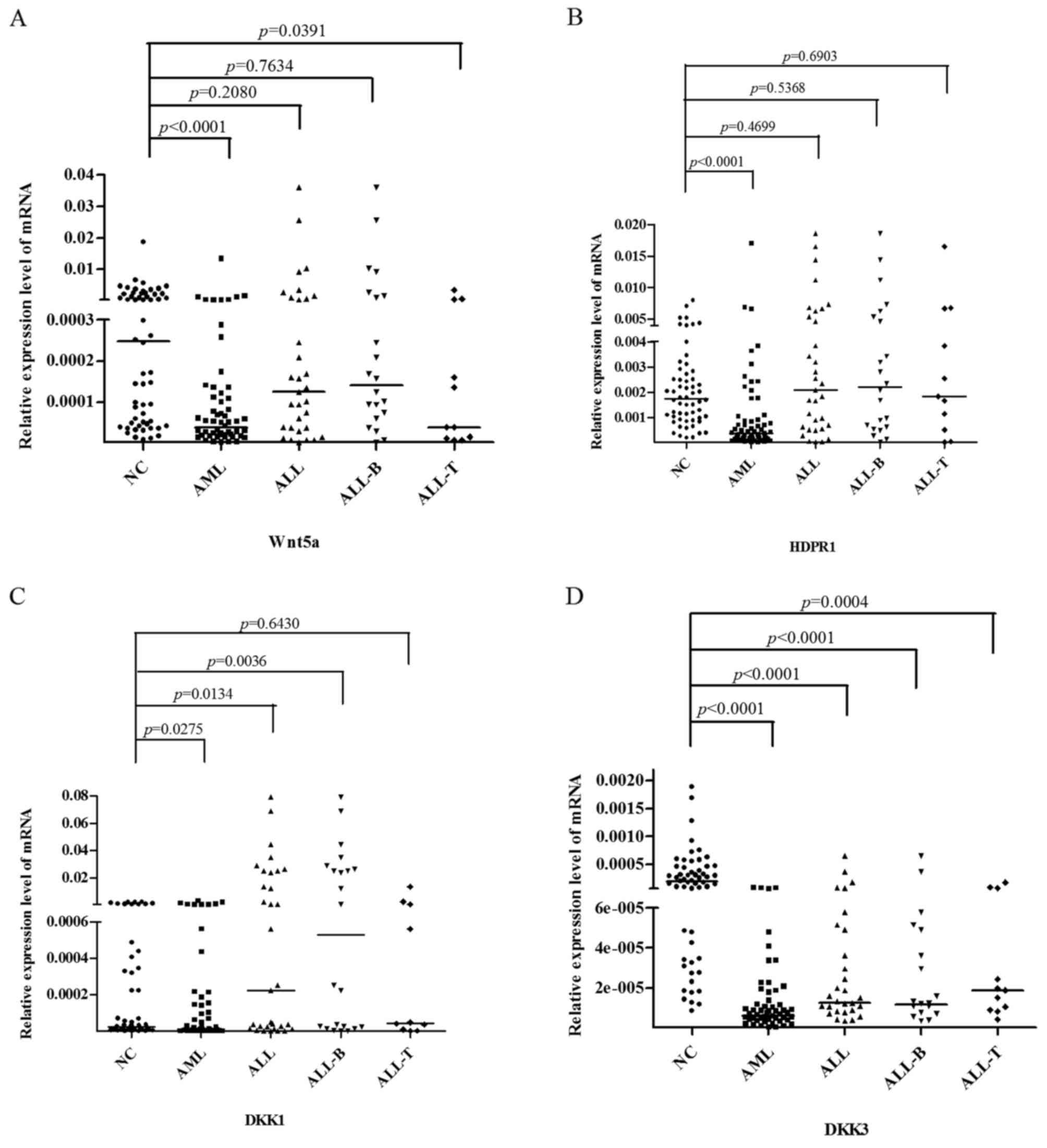
qRT-PCR showed that the relative expression of Wnt5a
was lower in bone marrow cells from the AML and ALL (T cell)
patients than that from the normal controls [AML patients vs.
normal controls, P<0.0001; ALL (T cell) patients vs. normal
controls, P=0.0162]. Furthermore, the relative expression of HDPR1
was lower in bone marrow cells from the AML patients than that from
the normal controls (AML patients vs. normal controls, P<0.0001)
and was higher in the ALL patients than that in the normal controls
(P=0.00362). The relative expression of DKK1 was lower in bone
marrow cells from the AML patients than that from the normal
controls (P=0.0430) and was higher in the ALL patients than that in
the normal controls (P=0.0016). The relative expression of DKK3 was
significantly lower in bone marrow cells from the AML and ALL
patients than that from the normal controls (AML patients vs.
normal controls, P<0.0001; ALL patients vs. normal controls,
P<0.0001) (Fig. 1).
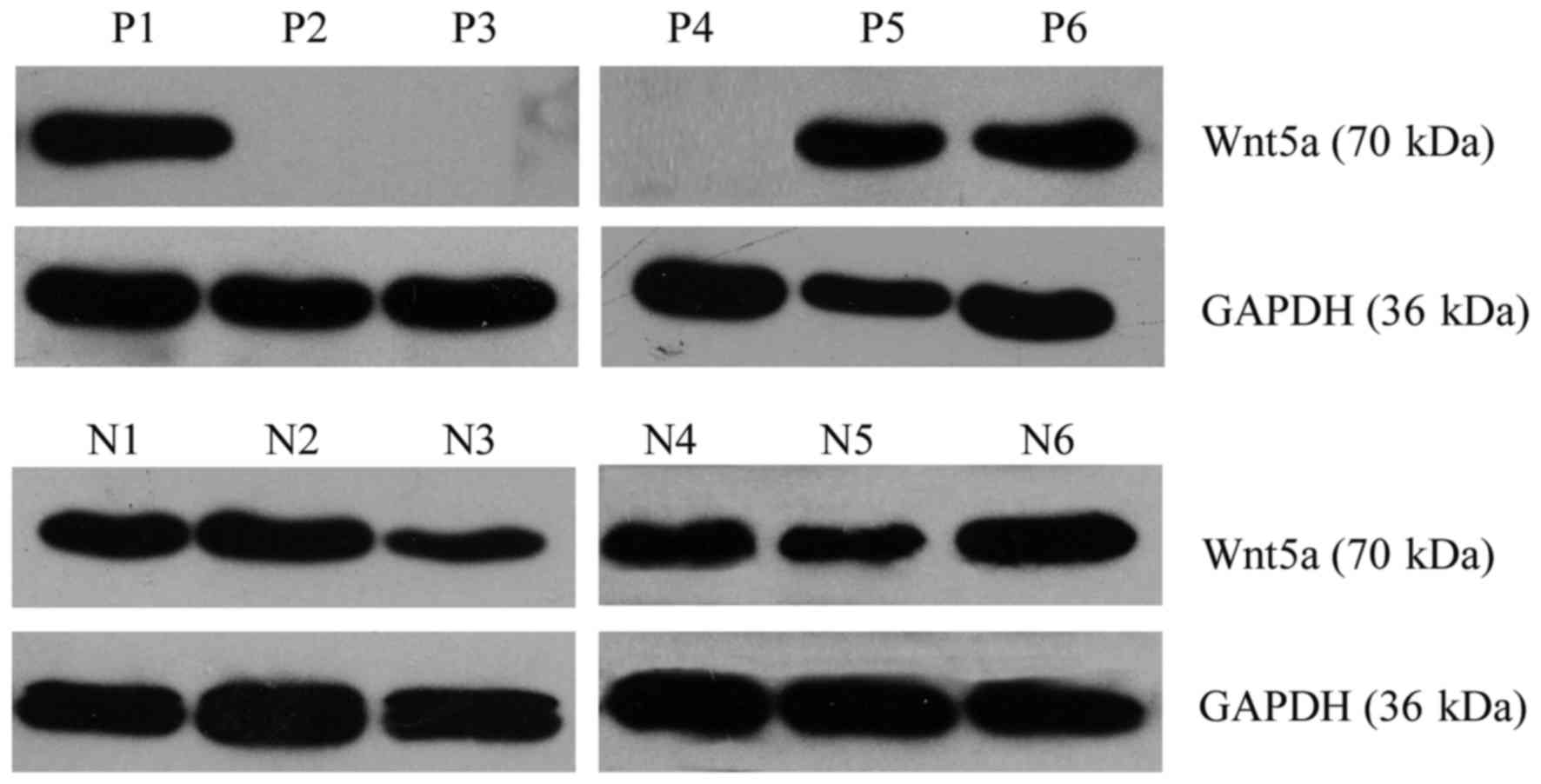
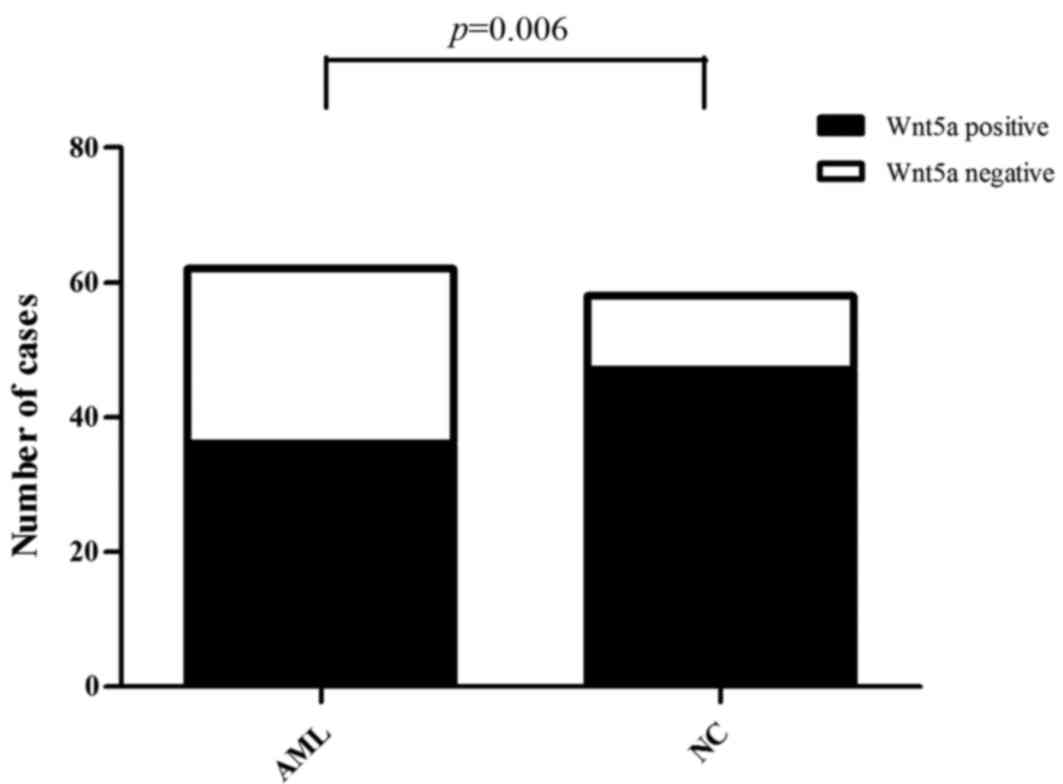
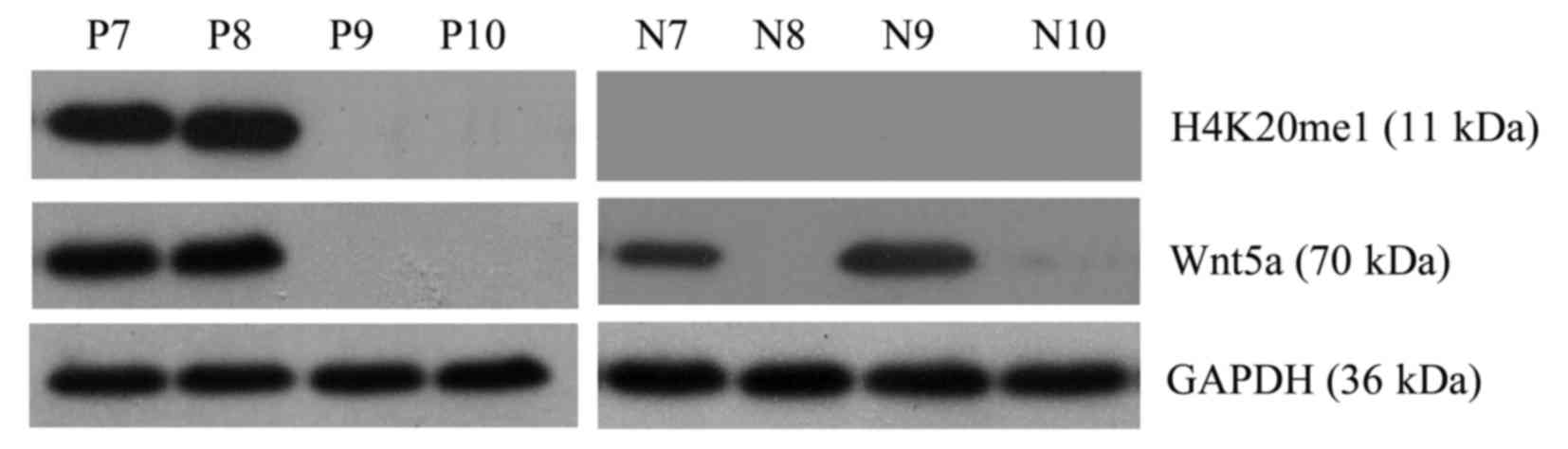
Western blotting was performed to detect the
expression level of the Wnt5a protein. Of the 62 AML cases, Wnt5a
expression was detected in 36 cases, and thus, the positive
expression rate was 58.06%. Of the 58 normal control samples, Wnt5a
expression was detected in 47 cases, and thus, the positive
expression rate was 81.03% (Fig.
2). Consequently, Wnt5a protein expression was lower in bone
marrow cells from the AML patients than that from the normal
controls (Pearson Chi-square value=7.414; P=0.006) (Fig. 3).
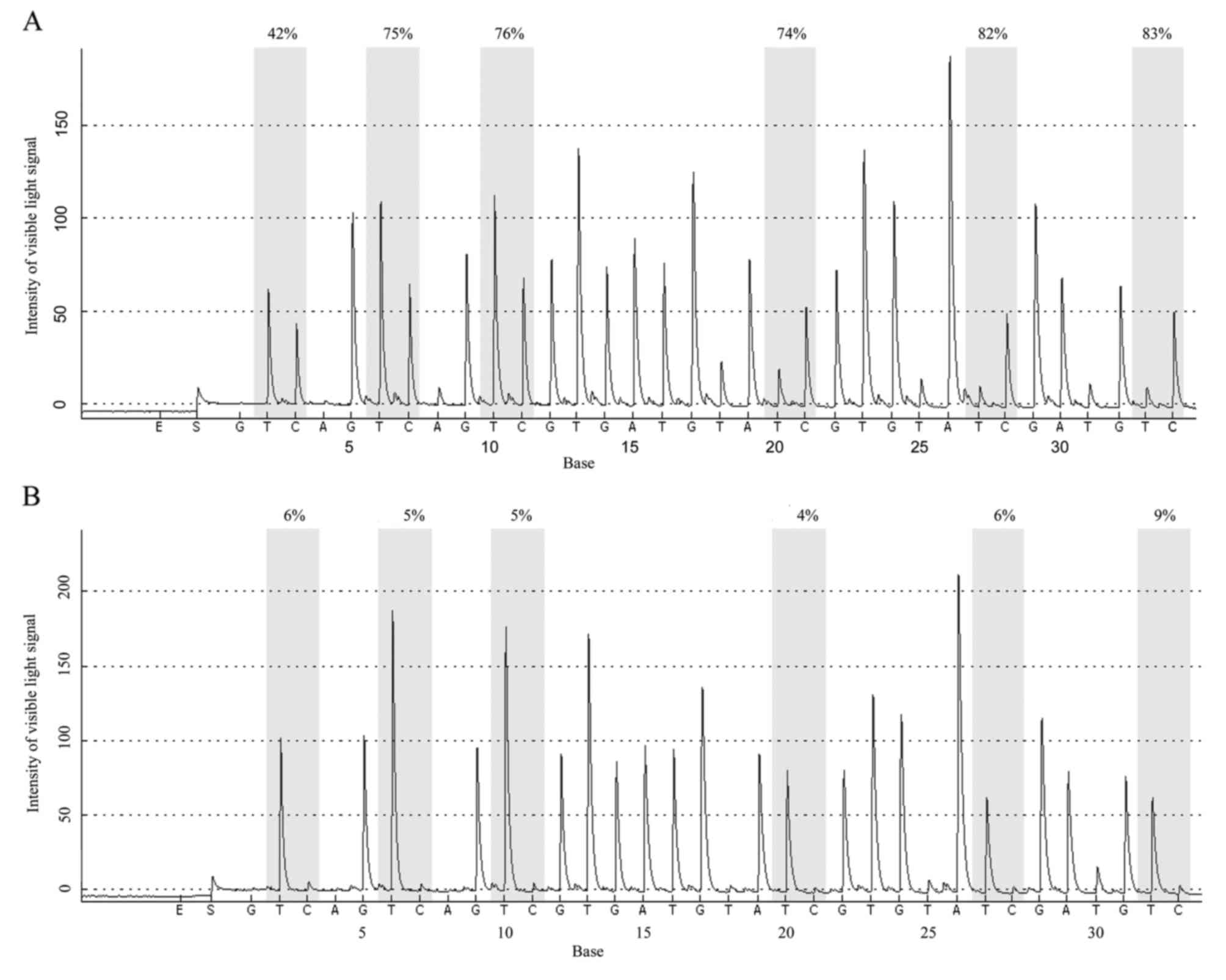
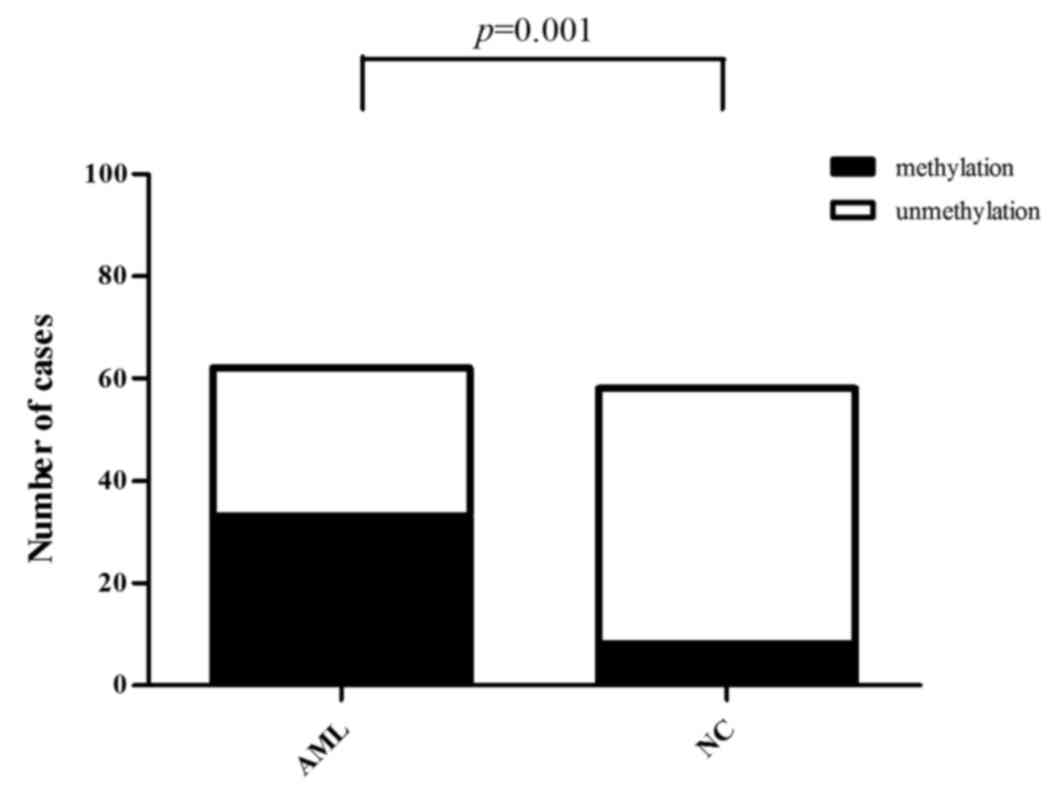
DNA methylation rate of the Wnt5a
promoter is higher in AML patients than in normal controls
The mean methylation rate of 6 CpG islands in the
Wnt5a promoter was determined using 10% as the cut-off value; that
is, methylation was negative when the mean methylation rate was
0–10% and positive when the mean methylation rate was ≥10%. The
results revealed that among the 62 AML cases, 33 were positive and
29 were negative for methylation of the Wnt5a promoter (53.23%
positive rate). Among the 58 normal controls, 8 were positive and
50 were negative for methylation of the Wnt5a promoter (13.79%
positive rate). Statistical analysis revealed that the positive
methylation rate of the Wnt5a promoter was significantly higher in
the AML patients than that in the normal controls (Fig. 4A and B) (Pearson Chi-square
value=20.716, P<0.01; Fig. 5,
Table II).
| Table II.The methylation rate of CpG islands in
the Wnt5a promoter in AML patients (A-C) and normal controls
(D-F). |
Table II.
The methylation rate of CpG islands in
the Wnt5a promoter in AML patients (A-C) and normal controls
(D-F).
|
| Methylation (%) |
|
|
|---|
|
|
|
|
|
|---|
| Sample ID | Pos. 1 | Pos. 2 | Pos. 3 | Pos. 4 | Pos. 5 | Pos. 6 | No. of included
CpGs | Mean (%) |
|---|
| A | 45.34 | 51.39 | 49.64 | 82.35 | 71.12 | 81 | 6 | 63.47 |
| B | 56.97 | 34.83 | 42.72 | 80.22 | 69.23 | 64.79 | 6 | 58.13 |
| C | 71.64 | 76.2 | 73.98 | 71.66 | 70.77 | 72.6 | 6 | 72.81 |
| D | 5.47 | 5.04 | 0 | 0 | 0 | 5.3 | 6 | 2.64 |
| E | 4.55 | 5.54 | 4.34 | 3.85 | 5.26 | 6.15 | 6 | 4.95 |
| F | 3.79 | 4.07 | 3.22 | 4.52 | 5.09 | 7.23 | 6 | 4.65 |
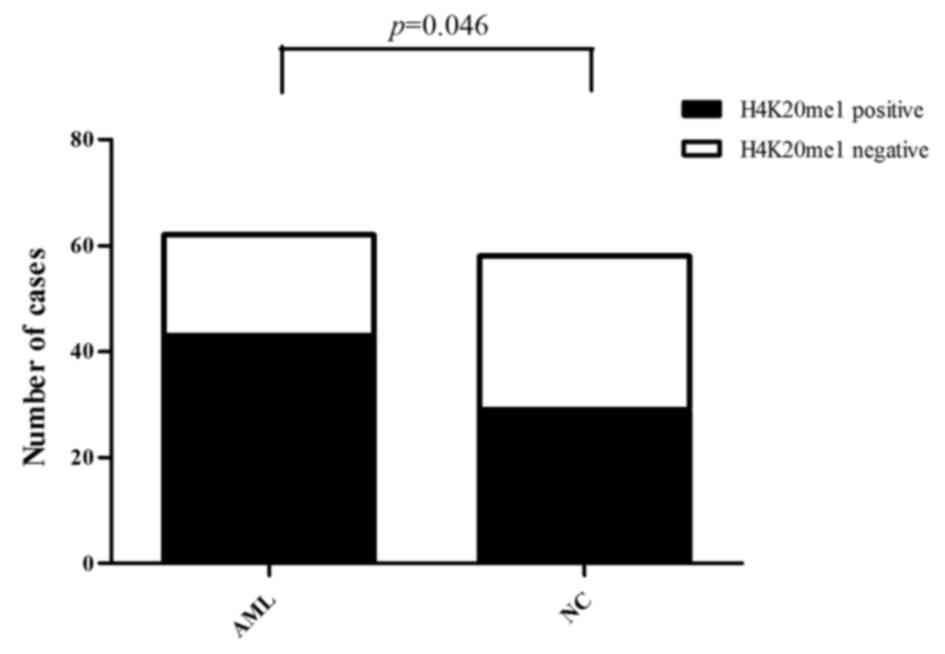
Overall H4K20me1 expression is higher
in AML patients than in normal controls
Of the 62 AML cases, H4K20me1 expression was
positive in 43 cases and negative in 19 cases (69.35% positive
expression rate). Of the 58 normal controls, H4K20me1 expression
was positive in 29 cases and negative in 29 cases (50.00% positive
expression rate). Consequently, the expression of H4K20me1 was
significantly higher in the AML patients than that in the normal
controls (Pearson Chi-square value=4.677, P<0.05) (Fig. 6).
Correlation of Wnt5a and H4K20me1
expression, and the methylation rate of the Wnt5a promoter in bone
marrow mononuclear cells from AML patients
Correlation analysis of the gray-scale values of
H4K20me1 and Wnt5a bands revealed that Wnt5a expression was
positively correlated with H4K20me1 expression in the AML samples;
that is, high H4K20me1 expression was associated with high Wnt5a
expression (Pearson correlation coefficient=0.843; P<0.05). In
the normal controls, Wnt5a expression was unrelated to H4K20me1
expression (Pearson correlation coefficient=−0.239; P>0.05)
(Fig. 7).
Correlation analysis of the methylation rate of the
Wnt5a promoter (pyrophosphate sequencing) and Wnt5a and H4K20me1
protein expression (western blotting) revealed that Wnt5a and
H4K20me1 expression was highly concordant and positively correlated
in the AML samples. However, Wnt5a expression and promoter
methylation was not necessarily concordant; Wnt5a expression was
detected in all AML samples with a negative methylation status
(methylation rate <10%), but in AML samples with a positive
methylation status (methylation rate ≥10%), Wnt5a expression (and
H4K20me1 expression) was negative in samples with an inactivation
of promoter methylation and was positive in samples with promoter
methylation (Table III).
| Table III.The relationship among the
methylation rate of the Wnt5a promoter and Wnt5a and H4K20me1
expression. |
Table III.
The relationship among the
methylation rate of the Wnt5a promoter and Wnt5a and H4K20me1
expression.
| ID | Disease type | Mean methylation
rate (%) | Wnt5A | H4K20me1 |
|---|
| P13 | M5 | 0.00 | + | + |
| P14 | M2 | 3.19 | + | + |
| P15 | M1 | 3.42 | + | + |
| P16 | M5 | 4.25 | + | + |
| P17 | M5b | 5.04 | + | + |
| P18 | M3 | 7.70 | + | + |
| P19 | M3 | 10.69 | − | − |
| P20 | M5b | 14.54 | + | + |
| P21 | M3 | 18.29 | − | − |
| P22 | M1 | 19.84 | + | + |
| P23 | M2 | 20.27 | − | − |
| P24 | M5 | 26.58 | + | + |
| P25 | M3 | 30.31 | − | − |
| P26 | M2 | 50.53 | + | + |
| P27 | M5 | 55.18 | + | + |
| P28 | M0 | 72.07 | + | + |
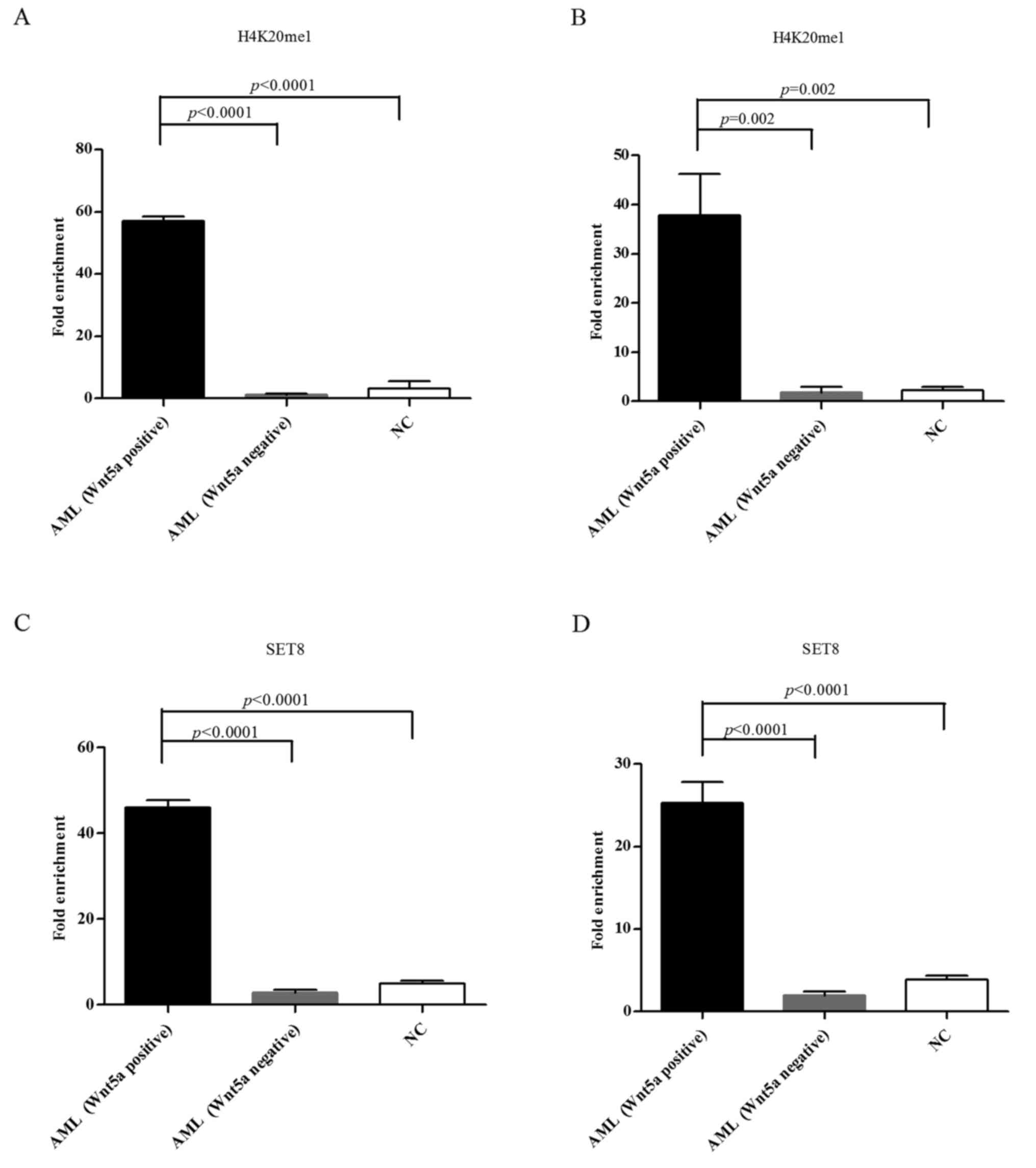
H4K20me1 and SET8 enrichment in the
Wnt5a promoter in bone marrow cells from AML patients compared with
normal controls
ChIP-qPCR was performed to assess H4K20me1 and SET8
enrichment in the Wnt5a promoter and coding regions of normal
controls and the AML samples that displayed methylation of the
Wnt5a promoter. The results showed that the H4K20me1 and SET8
enrichment degree was significantly higher in the Wnt5a promoter
and coding regions of AML samples with a relatively high Wnt5a
expression (Wnt5a band on western blotting) compared with both
normal controls and AML cases with a relatively low Wnt5a
expression (no Wnt5a western blot band); these differences were
significant (both P<0.05) (Fig.
8).
Discussion
Acute leukemia (AL) is a malignant hematopoietic
tumor that ranks as the sixth most common tumor in China. The cause
of AL is unknown. Studies have shown that in addition to
cytogenetic changes, the expression of one or more tumor-suppressor
genes related to DNA methylation, histone modifications or
RNA-related silencing is often absent in leukemia. Epigenetic
silencing decreases or eliminates the expression of
tumor-suppressor genes in leukemia cells, which may be an
alternative mechanism for genetic variation.
Methylation of CpG islands in the promoters of Wnt
antagonists contributes to the development of hematologic
malignancies, such as ALL, AML, chronic lymphocytic leukemia and
myeloproliferative neoplasms (9–13).
Tolwinski and Wieschaus (10)
examined samples from a group of young patients with de novo
AML and a moderate-risk karyotype and found that the methylation of
6 Wnt antagonists (including sFRP1, sFRP2, sFRP4, sFRP5, DKK1 and
DKK3) was associated with poor outcomes in these patients. Given
the role of unusual hypermethylation of the Wnt pathway signaling
molecules and the absence of their expression in the development of
AL, we studied the Wnt pathway in the present study.
Wnt5a can activate or inhibit downstream pathways in
both classical and non-canonical Wnt signaling. These dichotomous
roles may be attributed to the protumor or antitumor effect of
Wnt5a, tumor type and stage, and expression of cell surface
receptors (14,15). For example, Wnt5a is considered an
oncogene in breast cancer and melanoma. However, in colorectal and
thyroid cancer, ALL and AML, Wnt5a is considered a tumor-suppressor
gene (16,17). Perner et al (4) determined the methylation status of the
Wnt5a promoter to the first exon in 252 AML samples using MSP and
sequencing and found that the methylation rate was 43%. Low Wnt5a
expression was correlated with methylation and Wnt5a
hypermethylation was correlated with high cyclin D1 expression. For
patients with negative methylation, the relapse and mortality rates
were low, the disease-free survival (DFS) rate was 60%, and the 6-
to 7-year overall survival (OS) was 27%. For patients with
hypermethylation, the DFS and OS rates were 20 and 0%,
respectively.
Studies in colorectal cancer showed that in addition
to Wnt5a methylation, more histone modifications, including low
expression of histone H3 and H4, H3K4me2 acetylation and high
H3K27me3 expression in the promoter region, were observed in the
colorectal cancer cell line SW620, which has low Wnt5a expression,
compared with SW480 cells (17).
However, the relationship between Wnt5a and histone modifications
during the development of AL has not been studied. Our previous
study showed that H3K9me3 was enriched in the Wnt5a promoter in the
ALL cell line Jurkat. This histone modification inhibited gene
transcription and provided the rationale for us to further
investigate histone modifications of the Wnt5a promoter.
The present study showed that the mRNA expression
levels of Wnt antagonists (Wnt5a, HDPR1, DKK1 and DKK3) were
significantly lower in the AML patients than those in the normal
controls and had a differential expression in the ALL patients (low
Wnt5a expression in ALL-T, but normal Wnt5a expression in ALL-B).
The expression of DKK3 was lower in the ALL patients than that in
the normal controls. The expression of HDPR1 was comparable between
the ALL patients and the normal controls. The expression of DKK1
was significantly higher in the ALL patients than that in the
normal controls. The differential expression in ALL may be used to
diagnose different types of leukemia; in particular, the high DKK1
expression in the ALL patients and the low DKK1 expression in the
AML patients may be used as alternative diagnostic biomarkers.
Pyrophosphate sequencing revealed that the
methylation rate of the Wnt5a promoter was significantly higher in
the AML patients compared with that in the normal controls. Western
blotting showed that overall, Wnt5a expression was significantly
lower in the AML patients than in the normal controls, which was
consistent with the real-time fluorescence qPCR results and largely
consistent with pyrophosphate sequencing results. Most samples with
a high methylation rate exhibited low protein expression. These
results suggest that promoter methylation is the main cause of the
low expression of Wnt antagonists and that inactivation of the
methylation of tumor suppressor genes may be closely related to the
development of AML, which is consistent with the results of other
international studies. However, some samples with hypermethylation
of the Wnt5a promoter did not exhibit low protein expression,
refuting the aforementioned theory.
Furthermore, western blotting confirmed that
overall, the expression of H4K20me1 was significantly higher in the
AML patients than that in the normal controls and that H4K20me1 and
Wnt5a expression were positively correlated in AML patients (no
such correlation was observed in the normal controls). Analysis of
the methylation rate of the Wnt5a promoter and H4K20me1 and Wnt5a
protein expression in the AML patients revealed that all patients
with a negative methylation status (<10%) exhibited Wnt5a and
H4K20me1 expression, whereas in patients with positive methylation
(≥10%), some were negative for both Wnt5a and H4K20me1 expression
and some were positive for both Wnt5a and H4K20me1 expression.
ChIP-qPCR was performed to determine H4K20me1 and SET8 enrichment
in the Wnt5a promoter and coding regions in the AML samples that
exhibited methylation of the Wnt5a promoter. The results revealed
that the H4K20me1 and SET8 enrichment factor was significantly
higher in the AML samples with a relatively high Wnt5a expression
than in both the normal controls and AML samples with a relatively
low Wnt5a expression. These data suggest that Wnt5a expression is
affected by more than one methylation mechanism in AML. In the
absence of H4K20me1 and SET8, promoter methylation is associated
with a low expression level; with H4K20me1 and SET8 enrichment in
the promoter and coding regions, transcription is activated in
patients with methylation, as SET8 promotes H4K20 methylation and
H4K20me1 promotes transcription, offsetting the inhibitory effect
of methylation on transcription. Furthermore, a higher level of
H4K20me1 enrichment was associated with higher Wnt5a expression;
this correlation is consistent with the theory that H4K20me1
promotes transcription initiation and elongation and provides
additional information to the conventional theory that methylation
inactivates gene expression. Therefore, although promoter
methylation inhibits overall gene expression, H4K20me1 may play a
larger role in gene expression than methylation. Collectively, we
propose a new theory: Wnt5a (a Wnt pathway antagonist gene)
expression is correlated with H4K20me1 expression, but not
necessarily with methylation, in AML patients. Further research is
needed to verify this theory. Moreover, in-depth research is needed
to investigate why some patients with Wnt5a methylation exhibit
Wnt5a expression while others do not, whether H4K20me1 binding is
selective, and whether these results can be applied to the
methylation of other promoters. We did not observe these trends in
normal controls, likely since the overall H4K20me1 expression was
low in these samples and since H4K20me1 is not a key regulatory
histone of Wnt5a expression in normal tissues. Due to the
significant differential expression of H4K20me1 between normal
controls and AML patients, H4K20me1 may be used as a diagnostic
biomarker.
These results confirmed widespread changes in the
methylation of Wnt antagonists in AML patients and leukemia cell
lines, which may be an important mechanism for the development of
leukemia. Wnt5a, DKK1 and DKK3 may be potential treatment targets
of malignant hematological tumors, including AL, and may also be
useful as diagnostic biomarkers for the diagnosis of and efficacy
screening in these diseases. Moreover, H4K20me1 and Wnt5a
expression levels were positively correlated, and this relationship
was not affected by changes in promoter methylation, refuting the
conventional theory that promoter methylation inhibits gene
expression. Further research is needed to investigate the
relationship between H4K20me1 and gene expression.
Acknowledgements
The present study was supported by the Fujian
Provincial Health Bureau Youth Research Project (2010–1-12), the
National Natural Science Foundation of China (nos. 81370629 and
81300428), the Fujian Key Laboratory of Hematology Fund
(2009J1004), the National and Fujian Clinical Key Specialist
Construction Project, and the Youth Innovation Project of Natural
Science Foundation of Fujian Province (2017J05132).
Glossary
Abbreviations
Abbreviations:
|
AML
|
acute myeloid leukemia
|
|
ALL
|
acute lymphocytic leukemia
|
|
AL
|
acute leukemia
|
|
M0
|
acute myeloid leukemia with minimal
differentiation
|
|
M1
|
AML with partial differentiation
|
|
M2
|
AML with maturation
|
|
M3
|
acute promyelocytic leukemia
|
|
M4
|
acute myelomonocytic leukemia with
granules
|
|
M5
|
acute monocytic leukemia
|
|
M6
|
acute erythroleukemia
|
|
NC
|
normal control
|
|
qRT-PCR
|
quantitative reverse
transcription-polymerase chain reaction
|
|
ChIP-qPCR
|
chromatin immunoprecipitation-qPCR
|
|
SET
|
KMT5A/SETD8/Pr-Set7
|
|
PBS
|
phosphate-buffered saline
|
|
BCA
|
bicinchoninic acid
|
|
SDS-PAGE
|
sodium dodecyl sulphate-polyacrylamide
gel electrophoresis
|
References
|
1
|
DeLange RJ, Fambrough DM, Smith EL and
Bonner J: Calf and pea histone IV. II. The complete amino acid
sequence of calf thymus histone IV; presence of
epsilon-N-acetyllysine. J Biol Chem. 244:319–334. 1969.PubMed/NCBI
|
|
2
|
Zhang Y and Reinberg D: Transcription
regulation by histone methylation: Interplay between different
covalent modifications of the core histone tails. Genes Dev.
15:2343–2360. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oda H, Okamoto I, Murphy N, Chu J, Price
SM, Shen MM, Torres-Padilla ME, Heard E and Reinberg D:
Monomethylation of histone H4-lysine 20 is involved in chromosome
structure and stability and is essential for mouse development. Mol
Cell Biol. 29:2278–2295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perner J, Lasserre J, Kinkley S, Vingron M
and Chung HR: Inference of interactions between chromatin modifiers
and histone modifications: From ChIP-Seq data to
chromatin-signaling. Nucleic Acids Res. 42:13689–13695. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hori T, Shang WH, Toyoda A, Misu S, Monma
N, Ikeo K, Molina O, Vargiu G, Fujiyama A, Kimura H, et al: Histone
H4 Lys 20 monomethylation of the CENP-A nucleosome is essential for
kinetochore assembly. Dev Cell. 29:740–749. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yao L, Li Y, Du F, Han X, Li X, Niu Y, Ren
S and Sun Y: Histone H4 Lys 20 methyltransferase SET8 promotes
androgen receptor-mediated transcription activation in prostate
cancer. Biochem Biophys Res Commun. 450:692–696. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishioka K, Rice JC, Sarma K,
Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P,
Tempst P, Steward R, et al: PR-Set7 is a nucleosome-specific
methyltransferase that modifies lysine 20 of histone H4 and is
associated with silent chromatin. Mol Cell. 9:1201–1213. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Veloso A, Kirkconnell KS, Magnuson B,
Biewen B, Paulsen MT, Wilson TE and Ljungman M: Rate of elongation
by RNA polymerase II is associated with specific gene features and
epigenetic modifications. Genome Res. 24:896–905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Figueroa ME, Skrabanek L, Li Y, Jiemjit A,
Fandy TE, Paietta E, Fernandez H, Tallman MS, Greally JM, Carraway
H, et al: MDS and secondary AML display unique patterns and
abundance of aberrant DNA methylation. Blood. 114:3448–3458. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tolwinski NS and Wieschaus E: A nuclear
function for armadillo/β-catenin. PLoS Biol. 2:E952004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Claus R, Almstedt M and Lübbert M:
Epigenetic treatment of hematopoietic malignancies: In vivo targets
of demethylating agents. Semi Oncol. 32:511–520. 2005. View Article : Google Scholar
|
|
12
|
Picard F, Cadoret JC, Audit B, Arneodo A,
Alberti A, Battail C, Duret L and Prioleau MN: The spatiotemporal
program of DNA replication is associated with specific combinations
of chromatin marks in human cells. PLoS Genet. 10:e10042822014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bannister AJ, Schneider R and Kouzarides
T: Histone methylation: Dynamic or static? Cell. 109:801–806. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schübeler D, MacAlpine DM, Scalzo D,
Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O'Neill
LP, Turner BM, Delrow J, et al: The histone modification pattern of
active genes revealed through genome-wide chromatin analysis of a
higher eukaryote. Genes Dev. 18:1263–1271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang G, Lin JCY, Wei V, Yoo C, Cheng JC,
Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, et al:
Distinct localization of histone H3 acetylation and H3-K4
methylation to the transcription start sites in the human genome.
Proc Natl Acad Sci USA. 101:7357–7362. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ysebaert L, Chicanne G, Demur C, De Toni
F, Prade-Houdellier N, Ruidavets JB, Mansat-De Mas V, Rigal-Huguet
F, Laurent G, Payrastre B, et al: Expression of β-catenin by acute
myeloid leukemia cells predicts enhanced clonogenic capacities and
poor prognosis. Leukemia. 20:1211–1216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi Y, Lan F, Matson C, Mulligan P,
Whetstine JR, Cole PA, Casero RA and Shi Y: Histone demethylation
mediated by the nuclear amine oxidase homolog LSD1. Cell.
119:941–953. 2004. View Article : Google Scholar : PubMed/NCBI
|