Introduction
Hemangiomas (HAs) are benign vascular tumors
affecting approximately 10% of all infants (1,2). They
generally occur within the first few months after birth, usually on
the head and neck. Infantile HAs characteristically increase in
size (proliferative phase) for 4–6 months followed by a rest phase,
before eventually shrinking (involutive phase). The involutive
phase is typically longer than the proliferative phase, and may
take up to 10 years. Severe HAs occasionally result in tissue
damage. Although HAs generally disappear without physical
complications, they can cause significant psychological damage in
school children when considerable disfigurement is present on the
face. Several treatments are available including surgery and
propranolol or oral corticosteroid therapy. The causes and
mechanisms of HA development are currently unknown.
Propranolol, a non-selective β-blocker used to treat
high blood pressure, has been used since 2008 to treat HAs. The
therapeutic action of propranolol is believed to be the inhibition
of blood vessel growth, as well as the constriction of existing
blood vessels (3). This drug
triggers apoptosis by interacting with β-adrenergic receptors and
reducing the release of vascular endothelial growth factor (VEGF)
and basic fibroblast growth factor, both known to stimulate blood
vessel growth (4). Corticosteroid
treatment has been shown to suppress VEGF secretion from HA-derived
stem cells, and VEGF-A silencing in these cells reduced
vasculogenesis in vivo.
The pathways controlling cell proliferation and
apoptosis during the proliferative and involutive phases of HA are
still to be elucidated. Apoptosis may be inhibited during the
proliferative phase, as is the case in other proliferative
diseases. For example, the knockdown of Livin, an inhibitor of
apoptosis, inhibited cell growth and invasion of gastric cancer by
blocking the MAPK pathway in cancerous cells (5). Whereas, during involution, the levels
of apoptosis are thought to increase in HAs (6,7).
Regarding HA proliferation, the role of VEGF has recently been
reported. Knockdown of VEGF in primary HA-derived endothelial cells
(HDEC) inhibited cell viability and induced apoptosis (8). Knockdown of VEGF has also been
reported to decrease expression of p-AKT, p-ERK, p-p38MAPK and
Ki-67 and increase expression of caspase-3 (9). Similarly, knockdown of insulin-like
growth factor-II, a signaling molecule involved in cell growth,
migration and differentiation, reduced proliferation and increased
apoptosis in involuting HA cells, possibly by blocking the cell
cycle regulation of the PI3K/AKT signaling transduction pathway
(10).
It is, therefore, clear that the role and regulation
of proliferation and apoptosis in the proliferating and involuting
phases of HAs is complex. Insight may be provided by the use of
specific pathway inhibitors. p53 has been shown to be involved in
apoptosis, gene transcription, and downstream signaling (6,11,12).
Y27632 is a highly potent ATP-competitive inhibitor of
Rho-associated protein kinase (ROCK), a serine-threonine kinase
implicated in various cellular functions including cytoskeleton
organization and cell motility (13–15)
and has been shown to play a role in tumorigenesis (16) and other diseases.
In this study, Y27632 was used to examine the
effects on gene expression, proliferation and apoptosis in
proliferating and involuting HAs. Our findings provide insight into
the regulation of apoptosis in HA and potentially aid the
development of therapeutic strategies.
Materials and methods
Samples
Freshly resected human HA samples were collected
from the Department of General Surgery Affiliated with Xinhua
Hospital and were classified according to the International Society
for the Study on Vascular Anomalies criteria. Tissues and clinical
information were obtained as part of an approved study at Shanghai
Jiao Tong University School of Medicine. There were 49 cases of
proliferating phase HAs and 47 cases of involuting phase HAs. A
portion of each tissue sample was fixed with 10% formalin for
immunohistochemical examination. All HA tissues were diagnosed by
two independent pathologists.
The primary HDECs used in these experiments were
from the Institute of Biochemistry and Cell Biology (Shanghai,
China). All applicable international, national, and institutional
guidelines for the care and use of animals were followed. All
procedures performed in studies involving human participants were
in accordance with the ethical standards of the institutional and
national research committee and with the 1964 Helsinki declaration
and its later amendments or comparable ethical standards. Informed
consent was obtained from all participants included in the
study.
Cell culture and inhibitor
studies
Proliferating phase HDECs were cultured in
Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific
Inc., Waltham, MA, USA) supplemented with 10% heat-inactivated
fetal bovine serum (FBS, Thermo Fisher Scientific Inc.), 100 U/ml
penicillin (Invitrogen, Carlsbad, CA, USA) and 100 g/ml
streptomycin (Invitrogen). Cultures were maintained in a humidified
atmosphere containing 5% CO2 at 37°C. For inhibitor
studies, 1×105 cells were treated with 2–10 µM Y-27632
(Sigma-Aldrich, St. Louis, MO, USA). Control samples were treated
with phosphate-buffered saline (PBS). Recombinant VEGF-A was from
R&D Systems (Minneapolis, MN, USA). Cells (1×105)
were stimulated with 200 ng/ml VEGF (R&D Systems).VEGF samples
were treated with VEGF. A small interfering RNA (siRNA) that
targeted p53 (siP53) or VEGF (siVEGF), and a negative control
vector (siNC) were from Genechem (Shanghai, China). Cells
(1×105) were transfected with the following constructs
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer's instructions. In some experiments, cells were
transfected with siRNA for 24 h followed by treatment with Y-27632,
a ROCK inhibitor.
Animals and tumor growth
Vascular tumors were established by subcutaneous
injection of HDEC cells into the flanks of 4- to 6-week-old male
nude mice (1×107 cells per site, two sites per mouse,
five mice per group). Mice were monitored daily, and three out of
four mice developed a subcutaneous tumor. When the tumor size
reached ~5 mm in length, it was surgically removed, cut into 1–2
mm3 pieces, and re-seeded individually into other mice
on the right flanks. When tumor size reached ~5 mm in length,
Y27632 was injected intraperitoneally daily. Negative control mice
were injected with PBS. The mice were observed closely every day to
monitor their general condition and to measure tumor size using a
caliper. The mice were euthanized three weeks after treatment.
Resulting HAs were measured and subjected to qRT-PCR and western
blot analysis.
Immunohistochemical staining
Tissue sections were processed for
immunohistochemical analysis of proteins as previously described
(8). The following antibodies were
used: anti-ROCK goat polyclonal IgG; anti-p53 goat polyclonal IgG;
anti-VEGF goat polyclonal IgG; anti-Ki67 goat polyclonal IgG;
anti-caspase-3 goat polyclonal IgG; and anti-PCNA goat polyclonal
IgG (Santa Cruz Biotechnology, Dallas, TX, USA). Antigen unmasking
was performed for VEGF and Ki-67 with 10 mM sodium citrate buffer,
pH 6.0, at 90°C for 30 min and was not necessary for the other
proteins. Normal serum or PBS was used instead of antibodies in
negative control samples.
Quantitative real-time PCR
Real-time PCR was performed to determine mRNA
abundance. Total RNA was extracted from 1×105 cells of
proliferating phase HDECs or tumors using the TRIzol reagent
(Invitrogen), according to the manufacturer's instructions. Reverse
transcription was performed with 5 µg total RNA and an M-MLV
reverse transcriptase (Promega, Madison, WI, USA) and cDNA
amplification was performed using the SYBR Green Master Mix kit
(Takara, Otsu, Japan), according to the manufacturer's instructions
and the following primers: VEGF, 5′-ATCCAATCGAGACCCTGGTG-3′ and
5′-ATCTCTCCTATGTGCTGGCC-3′; Ki-67, 5′-GGGTTACCTGGTCTTAGTT-3′ and
5′-ATGGTTGAGGCTGTTCC-3′; PCAN, 5′-TGATGAGGTCCTTGAGTG −3′ and
5′-GAGTGGTCGTTGTCTTTC-3′; and, as a control, β-actin,
5′-AGCGAGCATCCCCCAAAGTT-3′ and 5′-GGGCACGAAGGCTCATCATT-3′.
Real-time PCR assays were performed on an ABI PRISM 7500 system
(Applied Biosystems, Foster City, CA, USA). The expression of each
gene was normalized to the β-actin gene. Relative mRNA levels were
calculated by the ∆∆Ct method. Three separate experiments were
performed for each clone.
Western blot assay
Protein extracts were prepared from 1×105
cells of proliferating phase HDECs or 30 µg of total protein as
previously described (8). Briefly,
cells were harvested, extracts were prepared and separated on
SDS-PAGE gels. Proteins were transferred to membranes, which, after
blocking, were incubated with the diluted antibodies described
above. Horseradish peroxidase-linked secondary antibodies were
added to the membranes at a dilution ratio of 1:1000, and
incubated. After washing, immunoreactive bands were visualized
using the ECL-PLUS kit (Amersham Biosciences, Piscataway, NJ, USA)
according to the manufacturer's instructions. Western blot signals
were quantitated using 1D Image Analysis Software (Eastman Kodak,
Rochester, NY, USA), and the relative protein abundance was
normalized to β-actin levels. Three separate experiments were
performed for each clone.
TUNEL assay
Apoptosis was detected by the TdT-mediated dUTP nick
end labeling (TUNEL) method with 1×105 cells as
previously described (8). Briefly,
sections were dewaxed, incubated with blocking solution and
permeabilized before detecting apoptosis using an in situ
cell death kit (Boehringer Mannheim, Mannheim, Germany). Positive
cells were visualized by fluorescence microscopy. As a control, the
reaction mixture was incubated without enzyme to detect nonspecific
staining. The apoptotic index was calculated from the ratio of the
number of positively stained tumor cells to the total number of
tumor cells counted per section.
Cell viability assay
Cell viability was analyzed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, proliferating phase HDECs treated with 10 µM
Y-27632 or PBS as control were incubated in 96-well-plates to a
density of 1×105 cells per well in DMEM medium
supplemented with 10% FBS. At 0, 24, 48 and 72 h, 20 µl MTT was
added, and cells were subsequently incubated with 150 µl DMSO for 5
min. The color reaction was measured at 570 nm on an enzyme
immunoassay analyzer (Bio-Rad, Hercules, CA, USA). The
proliferation activity was calculated for each clone.
Cell apoptosis analysis
To detect cell apoptosis, proliferating phase HDECs
(1×105 cells) were trypsinized, washed with cold PBS and
resuspended in the binding buffer of the Cell Apoptosis Propidium
Iodide kit (KeyGen Biotech, Nanjing, China). According to the
manufacturer's instructions, fixed cells were incubated in darkness
with Annexin V conjugated to fluorescein isothiocyanate and
propidium iodide for 20 min at room temperature. Annexin V binding
buffer was added to the mixture before the fluorescence was
measured on a FACsort flow cytometer (BD, Quebec, Canada). Cell
apoptosis was analyzed using the Cell Quest software (Becton
Dickinson, Franklin Lakes, NJ, USA). Three separate experiments
were performed for each clone.
Statistical analysis
Statistical analysis was performed using SPSS
version 17.0 software (SPSS Inc., Chicago, IL, USA). One-way
analysis of variance (ANOVA) was used to analyze the differences
between groups. The Fisher's Least Significant Difference method of
multiple comparisons was used when the probability for ANOVA was
statistically significant. Statistical significance was
P<0.05.
Results
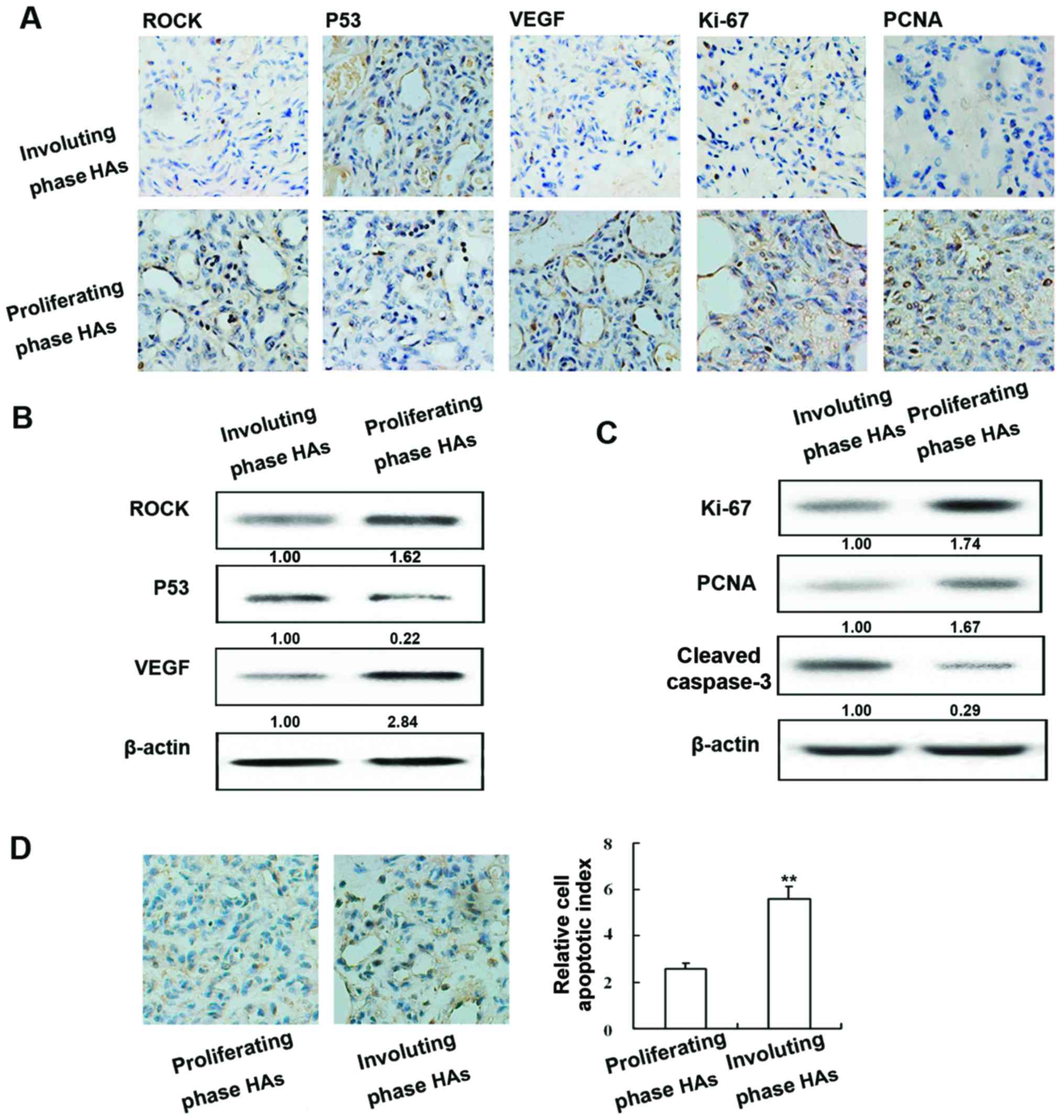
Expression of ROCK, p53, VEGF, Ki-67, PCNA and
caspase-3 in human HAs and apoptotic index of human HAs. The
expression of ROCK, p53, VEGF, Ki-67, PCNA and caspase-3 was
examined in proliferating and involuting HAs by immunohistochemical
staining (Fig. 1A) and western blot
analysis. ROCK and VEGF exhibited similar patterns of protein;
expression was highest in proliferating HA tissue and expression in
involuting HAs was higher than normal tissue. In contrast, p53
protein was most abundant in involuting HAs and its levels were
higher in normal tissue than in proliferating HAs (Fig. 1B).
As shown in Fig. 1C
the expression of Ki-67, PCNA and cleaved caspase-3 was examined in
proliferating and involuting HAs, as well as in normal tissue by
immunohistochemical staining and western blot analysis (Fig. 1C). Positive staining of apoptotic
cells in different phase HAs was examined using the TUNEL assay
(Fig. 1D). The amount of apoptotic
cells was markedly higher in involuting HAs compared to
proliferating HAs.
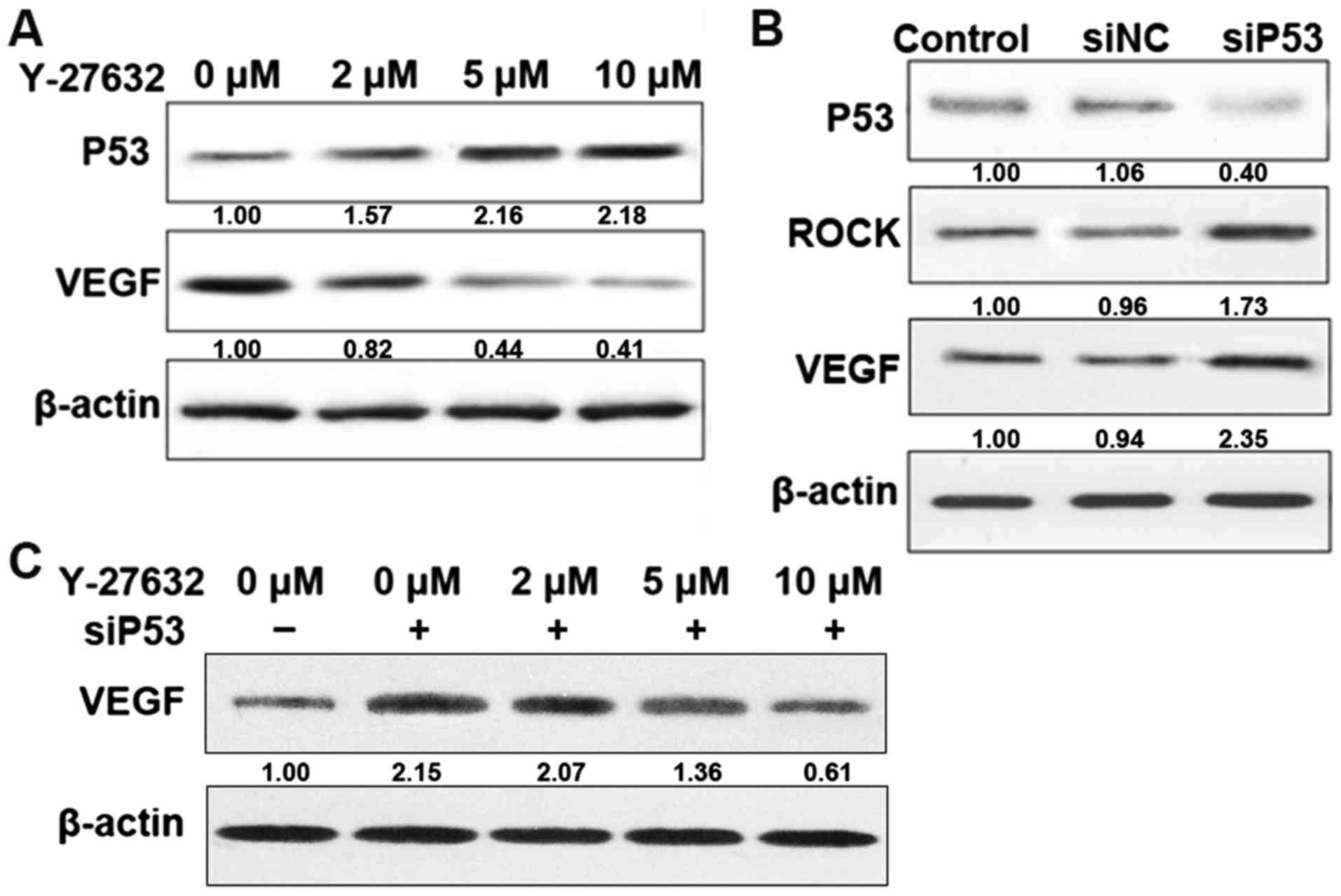
ROCK regulates p53 and VEGF
expression
To confirm the effects of various ROCK inhibitors on
p53 and VEGF expression in HDECs, protein levels were determined in
untreated cells and cells treated with Y-27632 or transfected with
siRNA. Treatment with increasing concentrations of Y-27632 (0, 2,
5, 10 µM), a potent inhibitor of ROCK, resulted in a dose-dependent
decrease in the expression of VEGF and a dose-dependent increase in
p53 (Fig. 2A). Cell transfected
with siP53 resulted in an increase in ROCK and VEGF expression.
However, cell transfected with siP53 for 24 h followed by treatment
with increasing concentrations of Y-27632, decreased VEGF
expression in a dose-dependent manner (Fig. 2C). This result indicated that
crosstalk between ROCK and p53 regulates VEGF expression, and the
effect of ROCK on VEGF is mediated at least in part by p53.
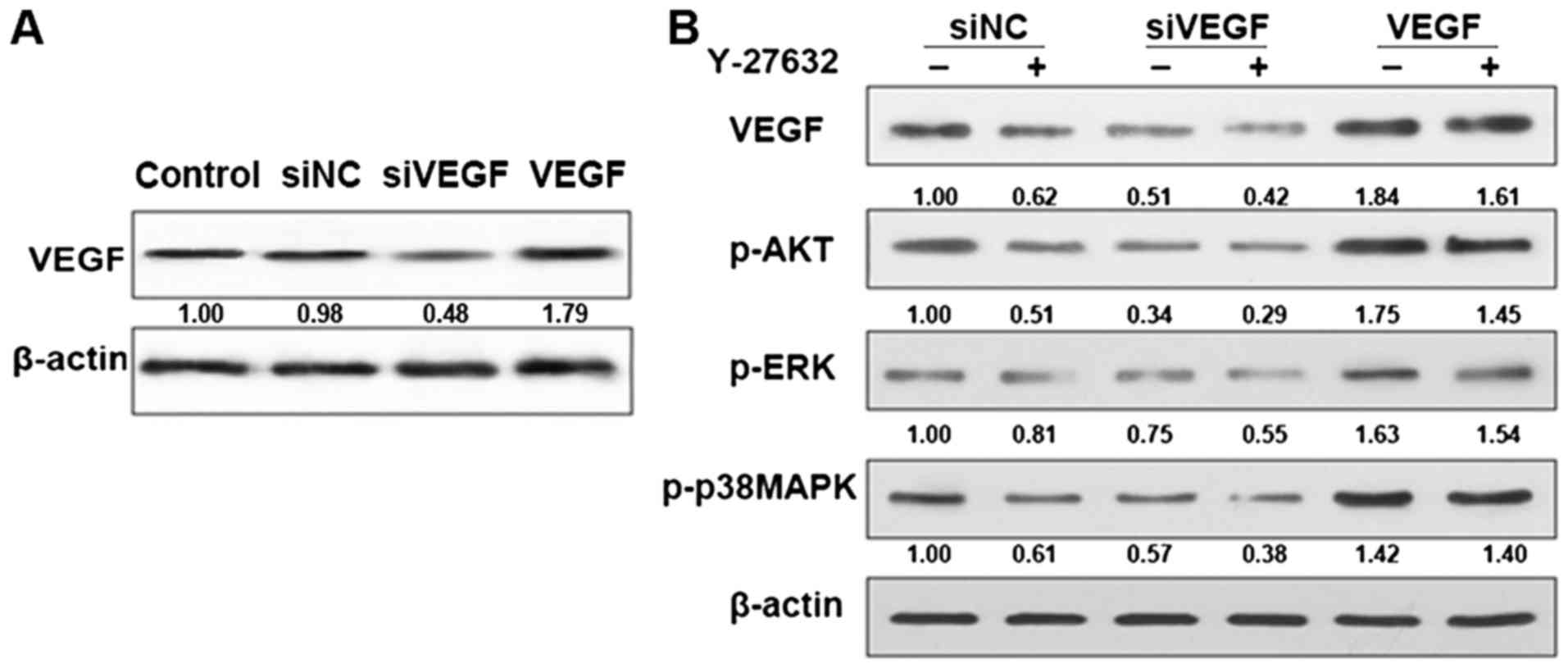
Effect of ROCK inhibitors on AKT and
ERK pathways
The fact that Y-27632 reduced VEGF levels in cells
supports the idea that ROCK is involved in AKT and ERK signaling
which has a close relationship with VEGF signaling. To this end, we
treated control and VEGF knockdown cells with or without Y-27632
and monitored the levels of pAKT, pERK and p-p38MAPK. As shown in
Fig. 3, pAKT, pERK and p-p38MAPK
levels were decreased in control cells treated with Y-27632
indicating that the pathway is inhibited. However, in the VEGF
knockdown cell levels of pAKT, pERK and p-p38MAPK were induced to a
much lower level. In the VEGF cells, levels of pAKT, pERK and
p-p38MAPK was the opposite. This finding supports the idea that
ROCK contributes to AKT and ERK pathway activation in HDECs.
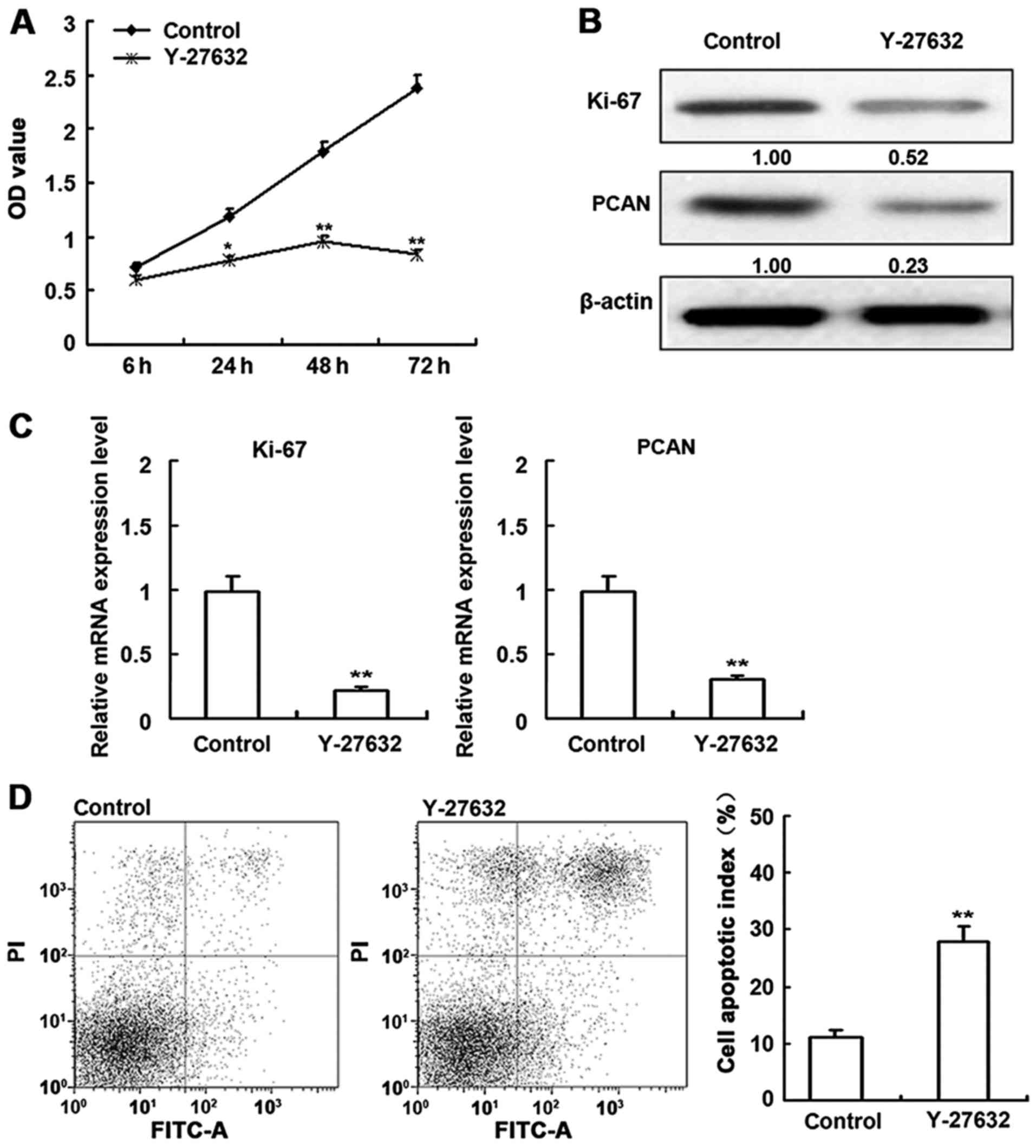
Effect of ROCK inhibitors on cell
viability
To examine the effect of the inhibitors on
proliferation, HDECs were subjected to an MTT assay 6, 24, 48 and
72 h after treatment (Fig. 4A).
Y-27632 significantly diminished proliferation in a time-dependent
manner. The effects of Y-27632 on Ki-67 and PCAN protein and mRNA
levels were also determined (Fig. 4B
and C). Compared to untreated cells, Y-27632 decreased Ki-67
and PCAN expression. Assays performed on HDECs (Fig. 4D) indicated that cell proliferation
was decreased and apoptosis was increased by Y-27632 treatment.
Taken together, these data indicate that ROCK inhibition induces
apoptosis. The corresponding effects on Ki-67 and PCAN supports
this conclusion.
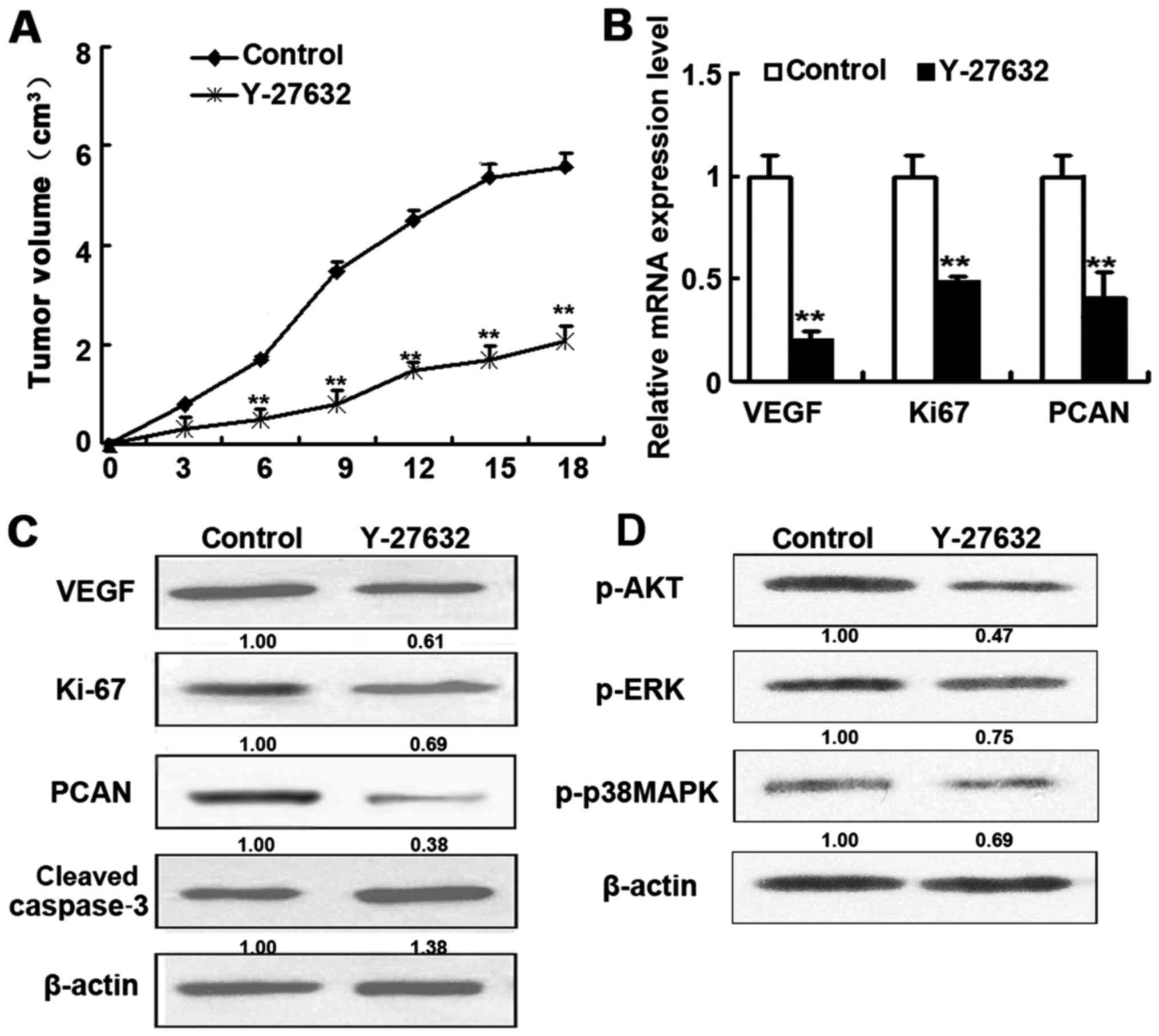
Effect of ROCK inhibitors in vivo
To confirm the in vivo effects of ROCK
inhibitor treatment, nude mice injected with HDECs (to induce HAs)
were treated with Y-27632 for 18 days before HAs were resected and
subjected to RT-PCR and western blot analysis (Fig. 5). Mice treated with Y-27632
displayed considerably smaller tumors and higher levels of cleaved
caspase-3 as well as lower levels of VEGF, Ki-67 and PCAN
expression than in control mice (Fig.
5B and C). In addition, treatment with Y-27632 also inhibited
the AKT and ERK signaling (Fig.
5D). These results are in agreement with the results obtained
with HDECs.
Discussion
Understanding the switch from the proliferative
phase to the involutive phase is essential for the management and
treatment of HAs. As apoptosis plays a major role in HA involution,
we examined the expression of several genes involved in apoptosis
and cell proliferation in proliferating and involuting HAs.
Expression in HAs was confirmed in HDEC lines, for which
proliferation and apoptosis could be conveniently measured, and the
differential responses to inhibitors were determined.
Our results indicated that the examined genes
exhibited two distinct patterns of protein and mRNA accumulation.
Consistent with their roles in cancer and cell proliferation, ROCK,
VEGF, Ki-67 and PCNA exhibited the highest levels in proliferating
HA tissue. ROCK expression is associated with cancer progression
and is elevated in several types of cancer (17); Ki-67 and PCNA are well established
markers for cell proliferation [see, for example (18)]; and VEGF has been shown to control
endothelial cell proliferation and migration (19). The inverse expression pattern (i.e.
higher levels in the involutive phase than in the proliferative
phase) was observed for p53 and caspase-3. p53 is a known cancer
suppressor, and mutations in this gene contribute to malignant
progression of cancer (20).
Caspases are a family of endoproteases that provide critical links
in cell regulatory networks controlling inflammation and cell
death, and caspase-3 plays a specific role in apoptosis (21). Thus, the high levels of expression
of these two genes in involuting phase HAs was also expected.
There is accumulating data for the essential role of
ROCK in VEGF function; VEGF mediates angiogenesis (2), venular permeability (22) and neovascularization (23) and is therefore upregulated in
expression in the proliferative phase in HAs. p53 is also a
regulator of VEGF function, typically by modifying VEGF
transcription, and disruption of this interaction plays a role in
cancer progression (24–27). p53 was found to be downregulated in
expression in the involutive phase in HAs. p53 knockdown by siRNA
increased expression of VEGF, indicating that p53 contributes to
VEGF inhibition in cells. It may, therefore, be a fine balance and
interactions between ROCK, p53 and VEGF that modulate the switch
between the proliferative and involutive phases in HAs. In
addition, the fact that ROCK inhibitor stimulated apoptosis in
proliferating HA suggests that the corresponding pathways act
upstream of p53-mediated apoptosis.
VEGF signaling is thought to promote tumor
angiogenesis via regulation of the PI3K/AKT and ERK pathways
(28). There is growing evidence to
suggest the significant role of the activated PI3K/AKT and ERK
pathways in cancer, and one study reported that the AKT and ERK
signaling pathways were involved in the pathogenesis of canine HAs
(29), while another confirmed the
involvement of the PI3K/AKT pathway in the development of human HAs
(30). Upregulation of Ki67 and
PCNA in our study is indicative of activation of the PI3K/AKT
pathway (31). Similar genetic
regulatory pathways has been reported previously [see for example
Ma et al (32), Takeba et
al (25)]. In the current
study, ROCK inhibition decreased the VEGF expression in
vitro and in vivo. We found this coincided with
decreased pAKT, pERK and p-p38MAPK expression, suggesting ROCK
downregulation may inhibit the AKT and ERK pathways in part by
decreasing VEGF expression. Clarifying the interactions that
orchestrate the switch from the proliferating to the involuting
phase may provide future therapeutic targets.
The fact that similar ROCK inhibitor effects were
observed in both HDECs and HAs from HDEC-injected nude mice
confirms that HDEC lines are useful for modeling HA development. In
addition, the inhibitory effect of Y-27632 on HA development was
confirmed in situ with live animals. In a recent study, low
concentrations of rapamycin, a known inhibitor of mTOR, were shown
to inhibit HA growth in nude mice (33). Although the toxicity of rapamycin
prevents it from being used in an unmodified form for HA therapy,
these results, together with our findings, suggest that inhibitors,
such as Y-27632, could potentially be developed as effective
therapies for the treatment of HAs. As these results were obtained
in immunodeficient mice, further work is necessary to determine the
role of the nude mutation in the observed mediation of HA
development.
Acknowledgements
This study was funded by the National Natural
Science Foundation of China (grant no. 81572673), the Science and
Technology Commission Foundation of Shanghai, China (grant no.
13140903802 and 15140901600) and the Medicine and Engineering Cross
Foundation of Shanghai Jiaotong University (grant no. YG2012MS33
and YG2015MS66).
References
|
1
|
Amir J, Metzker A, Krikler R and Reisner
SH: Strawberry hemangioma in preterm infants. Pediatr Dermatol.
3:331–332. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bryan BA, Dennstedt E, Mitchell DC, Walshe
TE, Noma K, Loureiro R, Saint-Geniez M, Campaigniac JP, Liao JK and
D'Amore PA: RhoA/ROCK signaling is essential for multiple aspects
of VEGF-mediated angiogenesis. FASEB J. 24:3186–3195. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hogeling M, Adams S and Wargon O: A
randomized controlled trial of propranolol for infantile
hemangiomas. Pediatrics. 128:e259–e266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sans V, de la Roque ED, Berge J, Grenier
N, Boralevi F, Mazereeuw-Hautier J, Lipsker D, Dupuis E, Ezzedine
K, Vergnes P, et al: Propranolol for severe infantile hemangiomas:
Follow-up report. Pediatrics. 124:e423–e431. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ou JM, Ye B, Qiu MK, Dai YX, Dong Q, Shen
J, Dong P, Wang XF, Liu YB, Quan ZW, et al: Knockdown of Livin
inhibits growth and invasion of gastric cancer cells through
blockade of the MAPK pathway in vitro and in vivo.
Int J Oncol. 44:276–284. 2014.PubMed/NCBI
|
|
6
|
Farah IO, Begum RA and Ishaque AB:
Differential protection and transactivation of P53, P21, Bcl2,
PCNA, cyclin G, and MDM2 genes in rat liver and the HepG2 cell line
upon exposure to pifithrin. Biomed Sci Instrum. 43:116–121.
2007.PubMed/NCBI
|
|
7
|
Razon MJ, Kräling BM, Mulliken JB and
Bischoff J: Increased apoptosis coincides with onset of involution
in infantile hemangioma. Microcirculation. 5:189–195. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ou JM, Yu ZY, Qiu MK, Dai YX, Dong Q, Shen
J, Wang XF, Liu YB, Quan ZW and Fei ZW: Knockdown of VEGFR2
inhibits proliferation and induces apoptosis in hemangioma-derived
endothelial cells. Eur J Histochem. 58:22632014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ou JM, Qui MK, Dai YX, Dong Q, Shen J,
Dong P, Wang XF, Liu YB and Fei ZW: Combined blockade of AKT/mTOR
pathway inhibits growth of human hemangioma via downregulation of
proliferating cell nuclear antigen. Int J Immunopathol Pharmacol.
25:945–953. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ou JM, Lian WS, Qiu MK, Dai YX, Dong Q,
Shen J, Dong P, Wang XF, Liu YB, Quan ZW, et al: Knockdown of IGF2R
suppresses proliferation and induces apoptosis in hemangioma cells
in vitro and in vivo. Int J Oncol. 45:1241–1249.
2014.PubMed/NCBI
|
|
11
|
Fridman JS and Lowe SW: Control of
apoptosis by p53. Oncogene. 22:9030–9040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Murphy PJ, Galigniana MD, Morishima Y,
Harrell JM, Kwok RP, Ljungman M and Pratt WB: Pifithrin-alpha
inhibits p53 signaling after interaction of the tumor suppressor
protein with hsp90 and its nuclear translocation. J Biol Chem.
279:30195–30201. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Uehata M, Ishizaki T, Satoh H, Ono T,
Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M,
et al: Calcium sensitization of smooth muscle mediated by a
Rho-associated protein kinase in hypertension. Nature. 389:990–994.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takahara A, Sugiyama A, Satoh Y, Yoneyama
M and Hashimoto K: Cardiovascular effects of Y-27632, a selective
Rho-associated kinase inhibitor, assessed in the
halothane-anesthetized canine model. Eur J Pharmacol. 460:51–57.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Z, Han S, Wang X, Han F, Zhu X, Zheng
Z, Wang H, Zhou Q, Wang Y, Su L, et al: Rho kinase inhibitor
Y-27632 promotes the differentiation of human bone marrow
mesenchymal stem cells into keratinocyte-like cells in xeno-free
conditioned medium. Stem Cell Res Ther. 6:172015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rath N and Olson MF: Rho-associated
kinases in tumorigenesis: Re-considering ROCK inhibition for cancer
therapy. EMBO Rep. 13:900–908. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morgan-Fisher M, Wewer UM and Yoneda A:
Regulation of ROCK activity in cancer. J Histochem Cytochem.
61:185–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kordek R, Biernat W, Alwasiak J and
Liberski PP: Proliferating cell nuclear antigen (PCNA) and Ki-67
immunopositivity in human astrocytic tumours. Acta Neurochir
(Wien). 138:509–512; discussion 513. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang S, Li X, Parra M, Verdin E,
Bassel-Duby R and Olson EN: Control of endothelial cell
proliferation and migration by VEGF signaling to histone
deacetylase 7. Proc Natl Acad Sci USA. 105:7738–7743. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 5:a0086562013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun H, Breslin JW, Zhu J, Yuan SY and Wu
MH: Rho and ROCK signaling in VEGF-induced microvascular
endothelial hyperpermeability. Microcirculation. 13:237–247. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kroll J, Epting D, Kern K, Dietz CT, Feng
Y, Hammes HP, Wieland T and Augustin HG: Inhibition of
Rho-dependent kinases ROCK I/II activates VEGF-driven retinal
neovascularization and sprouting angiogenesis. Am J Physiol Heart
Circ Physiol. 296:H893–H899. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu YF, Zhang Y, Shen N, Zhang RY and Lu
XQ: Effect of VEGF, P53 and telomerase on angiogenesis of gastric
carcinoma tissue. Asian Pac J Trop Med. 7:293–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takeba Y, Matsumoto N, Watanabe M,
Takenoshita-Nakaya S, Ohta Y, Kumai T, Takagi M, Koizumi S, Asakura
T and Otsubo T: The Rho kinase inhibitor fasudil is involved in
p53-mediated apoptosis in human hepatocellular carcinoma cells.
Cancer Chemother Pharmacol. 69:1545–1555. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ghahremani Farhang M, Radaelli E, Haigh K,
Bartunkova S, Haenebalcke L, Marine JC, Goossens S and Haigh JJ:
Loss of autocrine endothelial-derived VEGF significantly reduces
hemangiosarcoma development in conditional p53-deficient mice. Cell
Cycle. 13:1501–1507. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ghahremani Farhang M, Goossens S and Haigh
JJ: The p53 family and VEGF regulation: ‘It's complicated’. Cell
Cycle. 12:1331–1332. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang L, Ji Q, Liu X, Chen X, Chen Z, Qiu
Y, Sun J, Cai J, Zhu H and Li Q: Norcantharidin inhibits tumor
angiogenesis via blocking VEGFR2/MEK/ERK signaling pathways. Cancer
Sci. 104:604–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Murai A, Asa Abou S, Kodama A, Sakai H,
Hirata A and Yanai T: Immunohistochemical analysis of the
Akt/mTOR/4E-BP1 signalling pathway in canine haemangiomas and
haemangiosarcomas. J Comp Pathol. 147:430–440. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ji Y, Chen S, Li K, Xiao X, Xu T and Zheng
S: Upregulated autocrine vascular endothelial growth factor
(VEGF)/VEGF receptor-2 loop prevents apoptosis in
haemangioma-derived endothelial cells. Br J Dermatol. 170:78–86.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fu Y, Zhang Q, Kang C, Zhang K, Zhang J,
Pu P, Wang G and Wang T: Inhibitory effects of adenovirus mediated
COX-2, Akt1 and PIK3R1 shRNA on the growth of malignant tumor cells
in vitro and in vivo. Int J Oncol. 35:583–591.
2009.PubMed/NCBI
|
|
32
|
Ma X and Bai Y: IGF-1 activates the
P13K/AKT signaling pathway via upregulation of secretory clusterin.
Mol Med Rep. 6:1433–1437. 2012.PubMed/NCBI
|
|
33
|
Jahn SC, Law ME, Corsino PE, Davis BJ,
Harrison JK and Law BK: Signaling mechanisms that suppress the
cytostatic actions of rapamycin. PLoS One. 9:e999272014. View Article : Google Scholar : PubMed/NCBI
|