Introduction
Esophageal cancer is a multifaceted disease with
high incidence and mortality rates. Functional analysis of
oncogenes or tumor suppressors will help in understanding the
complex interactome and mechanism involved in its development and
progression thereby opening new doors for novel therapeutics and
cancer interventions. Recently, emerging evidence has shed light on
the significant role of microRNAs in tumorigenesis by regulating
key signaling cascades, such as cell cycle, apoptosis,
epithelial-to-mesenchymal transition (EMT), cell migration and
angiogenesis, thereby establishing a landmark in cancer biology.
The expression of miRNAs in esophageal cancer was first studied by
Guo et al (1) in a quest to
identify differentially expressed miRNAs in esophageal cancer.
Microarray analysis revealed a strong correlation between low
expression of hsa-miR-103/107 and an extended overall survival
period. miR-107 has also been implicated in hypoxic signaling and
tumor angiogenesis in colon cancer. Its knockdown resulted in
increased HIF-1β expression and hence an increased hypoxic
signaling (2). He et al
(3) demonstrated a tumor-suppressor
role of miR-107 wherein, it suppressed glioma cell growth by
directly targeting Spalt-like transcription factor 4 (SALL4),
leading to the activation of FADD/caspase-8/caspase-3/−7 signaling
pathway of cell apoptosis. Notably, miR-107 activated the ATR/Chk1
pathway and suppressed cervical cancer invasion by targeting
myeloid cell leukemia 1 (MCL1) (4).
Additionally, p53-induced microRNA-107 inhibited proliferation,
migration and invasion of glioma cells by modulating the expression
of cyclin-dependent kinase 6 (CDK6) and Notch-2 (5). It induced cell cycle G1 arrest and
inhibited invasion by targeting CDK6, thereby inhibiting tumor
progression in pancreatic, gastric, bladder and non-small cell lung
cancer (NSCLC) (6–9). In one study, eukaryotic translation
initiation factor 5 (EIF5) was found to be a direct target of
miR-107 and a high miR-107 level induced cell cycle arrest at the
G2/M phase and retarded tumor growth in nude mice (10). miR-107 has also been shown to
downregulate the expression of brain-derived neurotrophic factor
(BDNF), C-C chemokine receptor type 5 (CCR5), granulin (GRN),
cyclin-dependent kinase 8 (CDK8), let-7 and cyclin-dependent kinase
5 regulatory subunit 1 (CDK5R1)/p35 (11–16).
Furthermore, Zhang et al (17) demonstrated that miR-107 plays a key
role in cisplatin resistance by targeting the CDK8 protein in NSCLC
cell lines. According to a recent report by Datta et al
(18) miR-107 functions as a
candidate tumor-suppressor gene in head and neck squamous cell
carcinoma (HNSCC) by downregulating protein kinase Cε (PKCε). Its
treatment significantly blocked DNA replication, cell
proliferation, colony formation and invasion in HNSCC cell lines.
Moreover, lipid-based nanoparticle delivery of pre-miR-107 was
found to inhibit the tumorigenicity of HNSCC (19). Zhang et al (20) suggested that the
miR-25/miR-107-LATS2 axis may decrease the expression of LATS2,
thereby affecting the growth and invasion of gastric cancer
cells.
These observations suggest a possible role for
miR-107 in the regulation of hypoxia, tumor progression and
invasion (Fig. 1). Previously, we
reported significantly decreased expression of miR-107 in
neoplastic and preneoplastic esophageal tissues and esophageal
cancer serum samples as compared to expression noted in normal
subjects (21). However, its role
in esophageal cancer has not yet been elucidated. Therefore, the
aim of the present study was to analyze the function of miR-107 in
esophageal tumorigenesis. Moreover, in our previous study, we
predicted potential targets of miR-107 using Diana-miRGen that
predicts targets using four widely used target prediction programs
viz. PicTar, miRanda, TargetScanS, DIANA-microT. The targets were
further screened on the basis of their Gene Ontology terms and
Cdc42 was found to be one of the potential targets of miR-107.
Notably, its expression was found to be inversely correlated with
that of miR-107 in 66% cases of esophageal cancer tissues (21). Therefore, we further aimed to
ascertain whether Cdc42 is a direct downstream target of
miR-107.
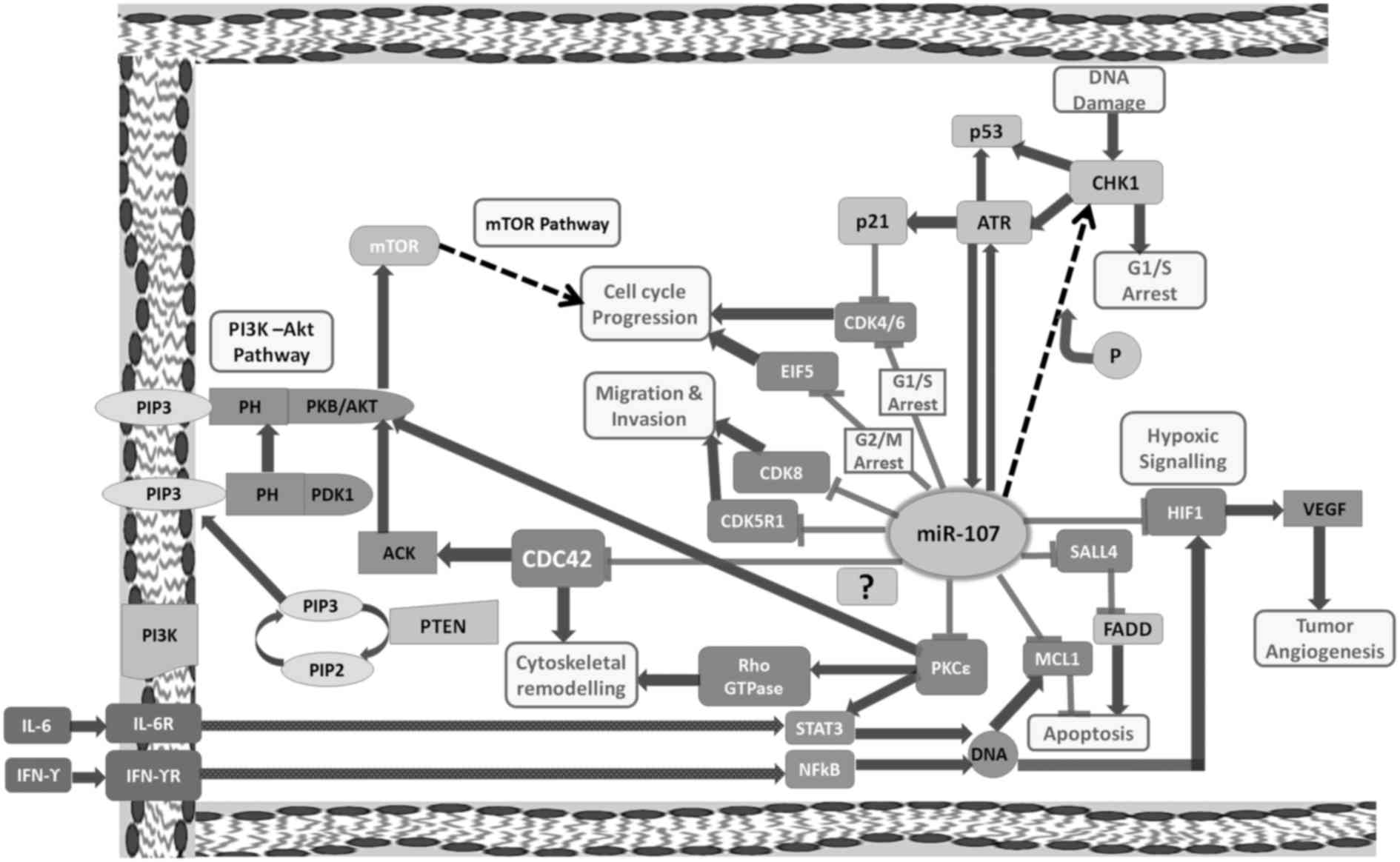 | Figure 1.Schematic representation of
miR-107-mRNA crosstalk in various types of cancers. ACK, activated
Cdc42-associated kinase 1; ATR, ataxia telangiectasia and Rad3
related; CDC42, cell division cycle 42; CDK4, cyclin-dependent
kinase 4; CDK6, cyclin-dependent kinase 6; CDK8, cyclin-dependent
kinase 8; CDK5R1, cyclin-dependent kinase 5 regulatory subunit 1;
CHK1, checkpoint kinase 1; EIF5, eukaryotic translation initiation
factor-5; HIF1, hypoxia-inducible factor 1; IFNγR, interferon γ
receptor 1; IL6R, interleukin 6 receptor; MCL1, myeloid cell
leukemia 1; mTOR, mechanistic target of rapamycin; P,
phosphorylation; PDK1, 3-phosphoinositide dependent protein kinase
1; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3,
phosphatidylinositol (3,4,5)-trisphosphate; PKB, protein kinase B;
PKCε, protein kinase C ε; PTEN, phosphatase and tensin homolog;
SALL4, spalt-like transcription factor 4; STAT3, signal transducer
and activator of transcription 3; NFκB, nuclear factor κ B; VEGF,
vascular endothelial growth factor. CDK8, CDK4/6, EIF5, CDK5R1,
HIF1, SALL4, MCL1, PKCε are the direct targets of miR-107. We aimed
to ascertain whether CDC42 is a direct downstream target of
miR-107. |
Materials and methods
Cell culture
Human esophageal squamous cell carcinoma cell line
KYSE-410 was purchased from the European Collection of
Authenticated Cell Cultures (ECACC), supplied by Sigma-Aldrich
(Bangalore, India). The cell culture was maintained in Roswell Park
Memorial Institute (RPMI)-1640 medium (Sigma-Aldrich) supplemented
with fetal bovine serum (FBS; HiMedia Laboratories Pvt. Ltd.,
Mumbai, India) 10% v/v, 100 U/ml of penicillin and 100 µg/ml of
streptomycin (HiMedia Laboratories). The HEK-293T cell line was a
kind gift by Dr Nimisha Sharma (GGSIPU, New Delhi). It was
maintained in Dulbecco's modified Eagle's medium (DMEM; HiMedia
Laboratories) supplemented with FBS 10% v/v, 100 U/ml of penicillin
and 100 µg/ml of streptomycin. The cell lines were maintained in a
humidified incubator with 5% CO2 and 95% humidity in
25-cm2 culture flasks (Corning Inc., Corning NY, USA) at
37°C. The cells were passaged twice weekly to maintain an
exponential growth phase. Other cell lines, SCC4, SCC9 and MCF-7,
were a kind gift from Dr Shyam S. Chauhan (AIIMS, New Delhi, India)
and Dr S. A. Raju Bagadi from the National Institute of Pathology
(ICMR), New Delhi, India, respectively.
Transient transfection of miR-107
mimic using Lipofectamine 3000
KYSE-410 cells were cultured in a 25-cm2
culture flask to ~80% density and collected by digestion and
centrifugation, and then seeded into 96-well plates
(1.2×104 cells/well), 24-well plate (5×104
cells/well) or 6-well plate (3×105 cells/well).
hsa-miR-107 mimic (100 nM) (Ambion, Inc., Foster City, CA, USA) was
transfected into the cells using Lipofectamine 3000 (Invitrogen,
Carlsbad, CA, USA) and Opti-MEM medium (Invitrogen) according to
the manufacturers instructions. Negative control #1 (Ambion) was
used as the negative control.
miRNA extraction and quantitative
RT-PCR
Total RNA was extracted using TRIzol (Invitrogen).
First-strand cDNA synthesis was carried out using universal cDNA
synthesis kit (Exiqon A/S, Vedbaek, Denmark) according to the
manufacturers protocol. qRT-PCR analysis of miR-107 was performed
using SYBR-Green Master Mix (Exiqon A/S) and predesigned miR-107
specific LNA™ PCR primer sets (Exiqon A/S).
All PCR reactions were performed in aliquots of 20
µl containing 8 µl diluted cDNA template, 2 µl of 10X Primer Mix
and 10 µl of 2X SYBR-Green Master Mix on Opticon2 real-time PCR
system (Bio-Rad Laboratories, Hercules, CA, USA) (21). Thermal cycling parameters included a
first denaturation step at 95°C for 10 min, followed by 40 cycles
of 95°C for 10 sec and 60°C for 1 min. The cycle threshold (Ct) was
recorded for each sample.
qRT-PCR of Cdc42 was carried out using gene-specific
primers (Table I) and KAPA SYBR
FAST real-time PCR kit (Kapa Biosystems, Inc., Wilmington, MA, USA)
following the manufacturers protocol. Its expression was analyzed
at 48 and 72 h post miR-107 transfection using qRT-PCR. The
expression of Cdc42 mRNAs in the mimic-treated cells was normalized
to that of the cells treated with NC. Thus, ΔΔCt =
ΔCt(miR-107) - ΔCt(negative control).
 | Table I.Primers used for expression analysis
of Cdc42 by qRT-PCR. |
Table I.
Primers used for expression analysis
of Cdc42 by qRT-PCR.
| S.No. | Gene | Sequence | Product length
(bp) |
|---|
| 1) | Cdc42 | Forward:
5-TGACAGATTACGACCGCTGAGTT-3 |
|
|
|
| Reverse:
5-GGAGTCTTTGGACAGTGGTGAG-3 | 134 |
| 2) | 5S rRNA | Forward:
5-GTCTACGGCCATACCACCCTG-3 |
|
|
|
| Reverse:
5-AAAGCCTACAGCACCCGGTAT-3 | 121 |
A small RNA, 5S rRNA (Table I) was used as the endogenous control
for data normalization. The 2−ΔΔCT method was used to
calculate the fold-change where, Ct is the cycle number at which
the fluorescence signal of the amplification plot passes a fixed
threshold. ΔCt = Ct(miR-107) - Ct(5s), ΔΔCt =
ΔCt(miR-107) - ΔCt(scrambled or untreated). A
negative control without a template was run in parallel to assess
the overall specificity of the reaction.
The qRT-PCR amplification products were analyzed by
melting curve analysis and confirmed by agarose gel
electrophoresis. Single dissociation peak in the melting curve was
indicative of specific amplification of the PCR product.
MTT assay
KYSE-410 cells were treated with 100 nM miR-107
mimic and the MTT assay was performed at time intervals of 24, 48
and 72 h post transfection. A total of 20 µl MTT (HiMedia
Laboratories) at the concentration of 5 mg/ml was added to
1.2×104 cells suspended in 200 µl RPMI-1640 medium and
incubated for 4 h at 37°C in dark. Then, 100 µl dimethyl sulfoxide
(DMSO; Amresco, Solon, OH, USA) was added to each well and was
shaken for 20 min to dissolve the crystals. Blank samples were
prepared using the same procedure. Absorbance was measured by using
the SpectraMax spectrophotometer (Molecular Devices, Sunnyvale, CA,
USA) at 570 nm. Each reading was converted to the percentage of
inhibition which was calculated as following: Inhibition rate (%) =
(OD value of the control group - OD value of the experimental
group)/OD value of the control group×100%.
Colony formation assay
KYSE-410 cells were transfected with 100 nM miR-107
mimic or NC. Twenty-four hours post transfection, the cells were
trypsinized into single-cell suspension and added to a 6-well plate
at a density of 2×103 cells/well. The plates were then
incubated at 37°C for 7 days and the medium was changed every two
days. After 7 days the supernatant was discarded. The colonies were
washed twice with phosphate-buffered saline (PBS) and fixed with
pre-chilled methanol for 20 min. The colonies were then stained
with crystal violet (HiMedia Laboratories) for 20 min. Images of
the stained tumor cell colonies were then recorded with a digital
camera and colonies containing at least 50 cells were quantified
using imaging analysis tool ImageJ.
Cell cycle analysis
Cell cycle analysis was performed by flow cytometry
on FACSCalibur (BD Biosciences, San Jose, CA, USA). Briefly, cells
were fixed in 70% ethanol overnight at −20°C. For fixing
mimic/NC-treated and untreated cells were harvested and resuspended
in 300 µl of PBS and then 700 µl of chilled 100% ethanol was added.
After fixing, the cells were stained with propidium iodide (10
µg/ml) and RNaseA (100 µg/ml). The stained cells were then
subjected to flow cytometry and data was acquired. Analysis was
carried out using BD CellQuest (BD Biosciences).
Apoptosis assay
Effect of miR-107 overexpression on apoptosis of the
EC cells was analyzed by flow cytometry using the Annexin V/7-AAD
apoptosis detection kit (BD Biosciences) on LSRII
(Becton-Dickinson). Briefly, 2.5×105 cells were treated
with 100 nM miR-107 or NC and the cells were harvested at 72 h
post-transfection. The cell pellet was washed twice with cold PBS
and resuspended in 100 µl of 1X binding buffer. To the cell
suspension, 5 µl of PE Annexin V and 5 µl of 7-AAD were added. The
cells were gently vortexed and incubated for 15 min at room
temperature (25°C) in the dark. After incubation, 400 µl of 1X
binding buffer was added to each tube and analyzed by flow
cytometry using FACSDiva version 8.1.3 (BD Biosciences). The
untreated population was used to define the basal level of
apoptotic or dead cells.
Wound healing assay
KYSE-410 cells (3×105 cells/well) were
seeded in a 6-well plate overnight to obtain 90% confluency. At 24
h post-transfection with 100 nM miRNA mimic, a scratch was made
through the center of each well using a 1000-µl pipette tip,
creating an open ‘wound’ that was clear of cells. The dislodged
cells were removed by two washes with PBS, fresh media was added
and plates were cultured. Migration into the open area was observed
at 24, 48 and 72 h post-scratching using ImageJ software. The
percentage of wound closure was calculated as percentage of wound
area covered at a given time compared to the initial wound
surface.
Transwell-Matrigel invasion assay
Transwell-Matrigel invasion assay was performed at
48 and 72 h after miR-107 transfection, using Transwell inserts
(Corning Inc.) coated with Matrigel (BD Biosciences). Cells
(4×104) in PBS were seeded to the upper chamber and RPMI
medium containing 20% FBS was added to the lower chamber as the
chemoattractant. After incubation at 37°C for 24 h, the non-invaded
cells were removed from the upper chamber using a cotton swab. The
invaded cells were then fixed in methanol, stained with DAPI and
photographed in at least five fields using an inverted fluorescence
microscope (Nikon, Tokyo, Japan) under a 10X objective. The cells
were counted using Nikon imaging software NIS-Elements BR
Ver4.40.00.
Western blot analysis
The cells were washed twice with ice cold PBS and
scraped with RIPA buffer (Invitrogen) supplemented with a protease
inhibitor cocktail (Invitrogen). Cells were further lysed with two
pulses of sonication for 9 sec at 39%. The protein concentration
was measured using the Bradford assay (Bio-Rad Laboratories).
Equivalent quantities (70 µg) of protein were separated by 15%
sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred to polyvinylidene fluoride microporous
(PVDF) membranes (Microdevices, Inc., New Delhi, India). The
membranes were blocked with 5% non-fat milk in PBS overnight at
4°C. Membranes were then incubated for 1 h with primary antibodies
of Cdc42 and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA)
at dilutions of 1:50 and 1:200, respectively. The membranes were
washed 3 times in 0.2% PBS-Tween followed by one wash with PBS and
incubated with the horseradish peroxidase (HRP)-conjugated
anti-rabbit secondary antibody for 1 h. After washing, bound
secondary antibody was detected using an enhanced chemiluminescence
(ECL) system (Pierce Biotechnology Rockford, IL, USA). Western blot
results were analyzed quantitatively by ImageJ software.
Cdc42-3UTR reporter construct
Cdc42 has three splice variants. Splice variant 1
(NM_001791) and splice variant 3 have a similar 3UTR while splice
variant 1 and splice variant 2 (NM_044472) have a similar 5UTR and
coding sequence but differ at 3UTR. RNA hybrid was used to
calculate minimum free energy of hybridization for target sites
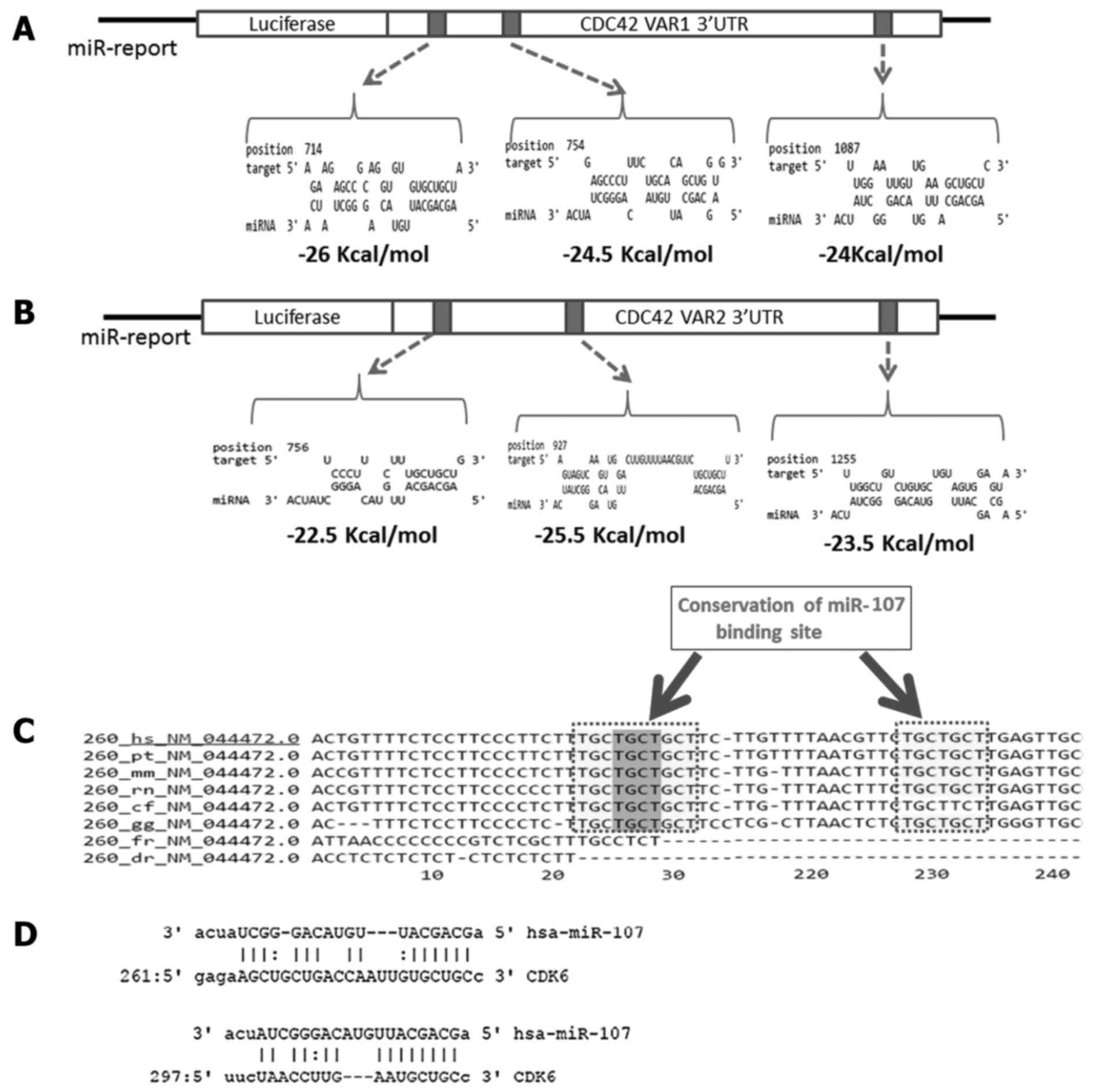
present at the 3UTR. Cdc42 variant 1 has three target sites at
positions 714 (MFE=−26 Kcal/mol), 754 (MFE=−24.5 Kcal/mol) and 1087
(MFE=−24 Kcal/mol) (Fig. 2A).
Target sites are present at positions 722, 756 and 927 in variant 2
with MFE of −21.126, −22.526 and −25.526 Kcal/mol (Fig. 2B and C). Therefore, the 3UTRs of
variant 1 (757 bp) and variant 2 (473 bp) containing the predicted
miR-107 target sites were amplified and cloned into the pMIR-REPORT
luciferase vector (Promega) between the Mlu1 and
HindIII restriction sites, downstream to the luciferase
reporter gene using primers as described in Table II.
| Table II.Primers used for preparing Cdc42
pMIR-REPORT construct. |
Table II.
Primers used for preparing Cdc42
pMIR-REPORT construct.
|
| Sequence | Position |
|---|
| Cdc42 Var1/Var2
(3UTR) forward primer |
5-GCCACGCGTGCCTATCACTCCAGAGACTGC-3 | 571–591 |
| Cdc42 Var1 (3UTR)
reverse primer |
5-CGCAAGCTTGAGCACCAGATGGGGAACAT-3 | 1308–1327 |
| Cdc42 Var2 (3UTR)
reverse primer |
5-CGCAAGCTTGAGGACATTCTTAAAGCCAGACC-3 | 1021-1043 |
CDK6-3UTR- pMIR-REPORT construct
To generate the 3UTR-REPORT construct, a 340-bp
region of CDK6 3UTR (NM_001259.6) containing the predicted miR-107
binding site (Fig. 2D) was
amplified and cloned into the pMIR-REPORT luciferase vector
(Promega) between the Mlu1 and HindIII restriction
sites, downstream to the luciferase reporter gene using the primers
as described in Table III.
| Table III.Primers used for preparing CDK6
pMIR-REPORT construct. |
Table III.
Primers used for preparing CDK6
pMIR-REPORT construct.
|
| Sequence | Position |
|---|
| CDK6 (3UTR) forward
primer |
5-CTTACGCGTTGTCTTCTGGACAGGCTCTG-3 | 1511–1531 |
| CDK6 (3UTR) reverse
primer |
5-CGCAAGCTTAGAATCTCTCACATACACAC-3 | 1831-1850 |
Luciferase reporter assay
Post-transcriptional inhibition of the luciferase
reporter gene by miR-107 was assayed in HEK-293T cells. Briefly,
HEK-293T cells were seeded into 24-well plates and cultured until
80% confluent. The cells were then co-transfected with either
miR-107 mimic or negative control at a 100 nM final concentration
and with 100 ng of pMIR-REPORT construct containing Cdc42 3UTR
along with 10 ng Renilla luciferase vector using
Lipofectamine 3000 transfection reagent according to the
manufacturers recommendations (Invitrogen). Relative firefly
luciferase activity, which was normalized with Renilla
luciferase, was measured using a Dual-luciferase reporter gene
assay system (Promega) and the results were plotted as the
percentage of change over the respective control.
Statistical analysis
All experiments were repeated at least three times
and the Student's t-test was used to analyze the statistical
significance between two groups while one-way ANOVA was used to
determine the significance of differences among multiple groups.
P<0.05 was considered to indicate a statistically significant
difference. When the difference between the groups was found to be
statistically significant, then multiple comparisons were performed
between treatments within each time period by Tukey's HSD test. All
the statistical analysis were performed using GraphPad Prism
software version 6.00 (GraphPad Software, Inc., San Diego, CA, USA)
and SPSS software 16.0 (SPSS, Inc., Chicago, IL, USA).
Results
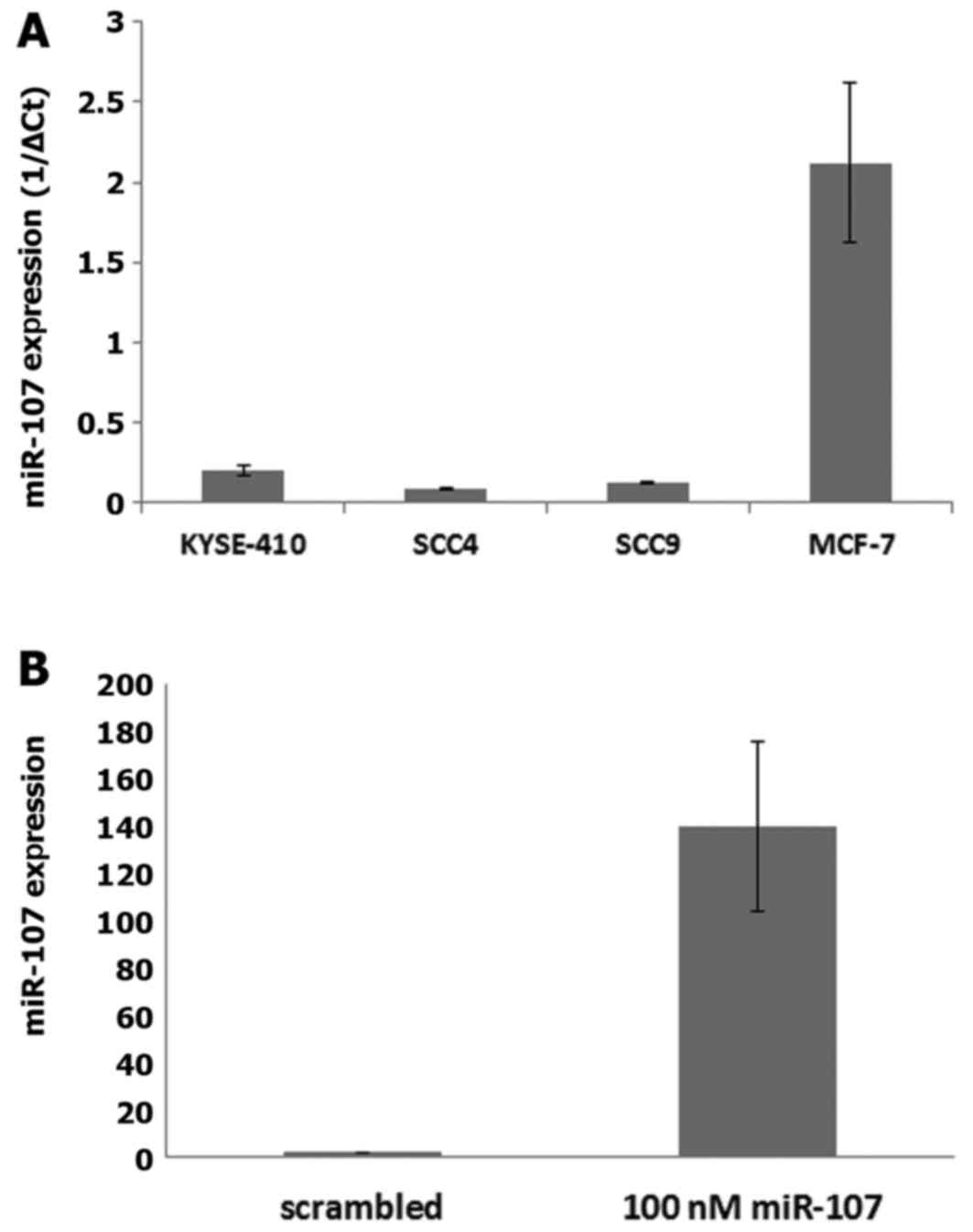
miR-107 expression in the cell
lines
The expression of miR-107 was assessed in four
different cell lines: esophageal squamous cell carcinoma
(KYSE-410), tongue squamous cell carcinoma (SCC-4 and SCC-9) and
breast adenocarcinoma (MCF-7) cell lines. The results showed that
the level of miR-107 was relatively lower in the KYSE-410, SCC-4
and SCC-9 cell lines as compared to the level noted in the MCF-7
cells (Fig. 3A).
Transfection efficiency
Real-time PCR analysis revealed an increase in
miR-107 expression by 160-fold at 48 h post miR-107 mimic
transfection in the KYSE-410 cells as compared to the cells treated
with negative control (NC) indicating that miR-107 was successfully
transfected (Fig. 3B).
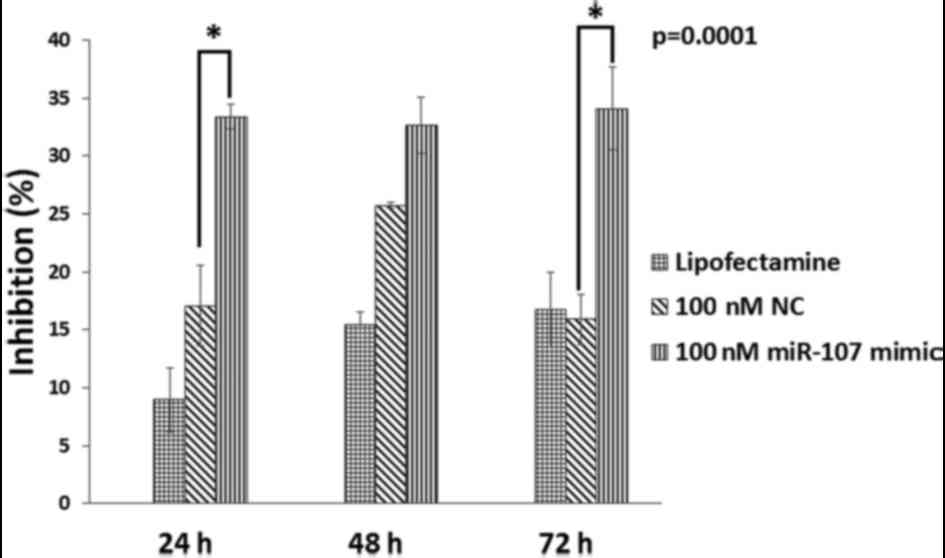
Overexpression of miR-107 suppresses
cell proliferation of esophageal cancer cells
Compared with the control group, cell proliferation
in the miR-107-treated group was inhibited in a dose- and
time-dependent manner. Cell proliferation was significantly
(P=0.0001) inhibited when cells were treated with 100 nM miR-107
mimic for 72 h with the inhibition rate of 34.079% (Fig. 4). Notably, Tukey's multiple
comparisons test revealed that cell proliferation of KYSE-410 cells
was significantly inhibited at 24 and 72 h after transfection with
100 nM miR-107 mimic as compared to the NC. Whereas, at 48 h the
difference in cell proliferation of the two groups was not found to
be statistically significant.
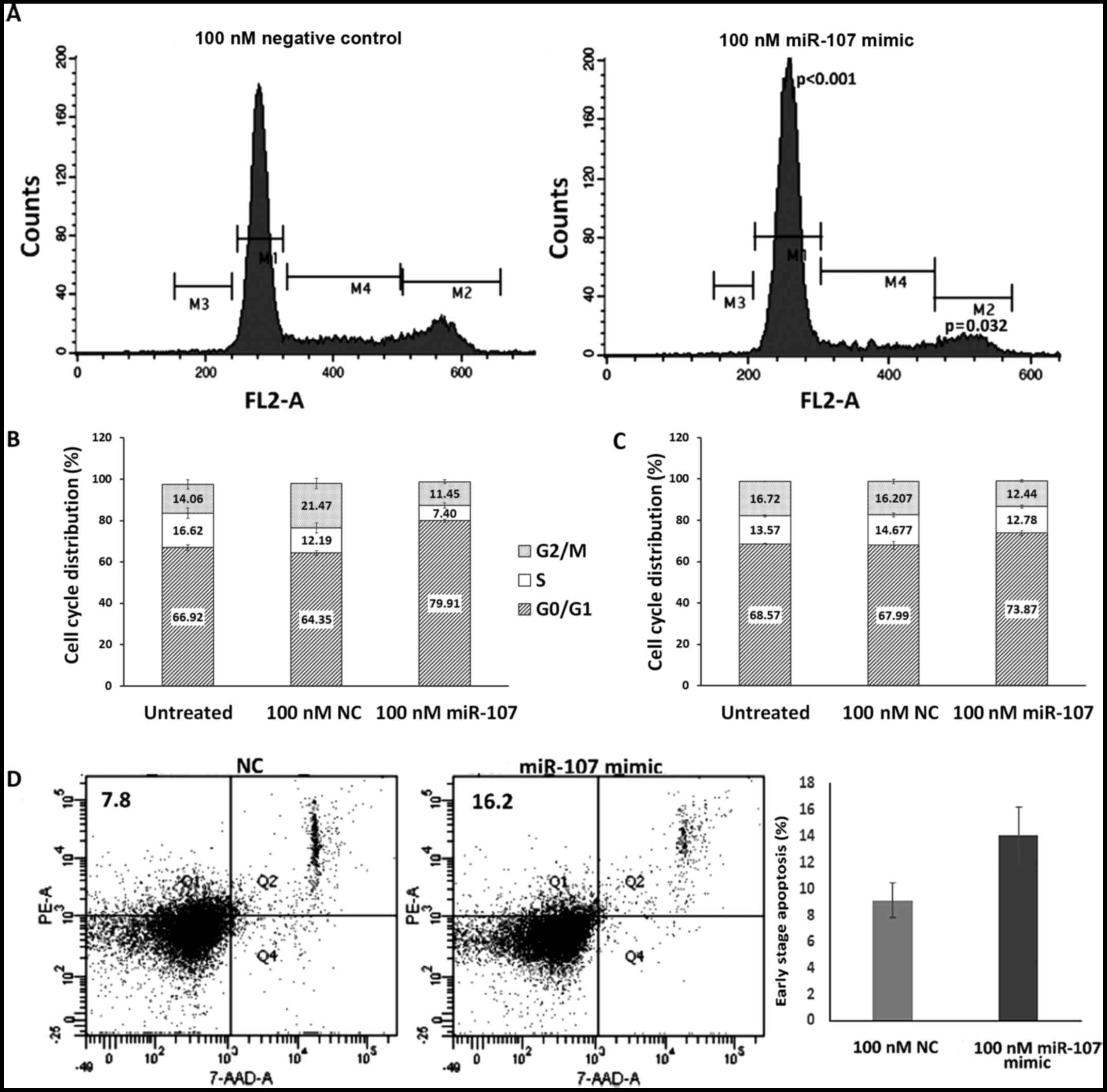
Overexpression of miR-107 causes cell
cycle arrest in G1/S phase
DNA content analysis by flow cytometry showed a
significantly (P<0.001) increased percentage of KYSE-410 cells
in the G1 phase (from 64.35±1.16 to 79.91±0.77%) of the cell cycle,
at 48 h post-miR-107 transfection as compared to the NC-treated
cells (Fig. 5A and B). While a
substantial decrease in S phase (from 12.19±2.48 to 7.4±1.3%) and a
significant decrease in G2/M phase (from 21.47±2.56 to 11.45±0.98%,
P=0.032) populations was observed in the miR-107-treated cells as
compared to the NC.
At 72 h after transfection, the percentage of cells
in the G1 phase was significantly (P=0.044) increased from
67.99±1.89 to 73.87±1.23% when transfected with 100 nM miR-107 as
compared to the NC-transfected cells (Fig. 5C). A significant decrease in the
G2/M (P=0.024) cell population was also observed in the
miR-107-treated cells as compared to the NC-treated cells.
Effect of miR-107 overexpression on
the apoptosis of ESCC cells
In addition to the cell cycle distribution, we also
analyzed the effect of miR-107 overexpression on the apoptosis of
ESCC cells. An increase in the percentage of early apoptotic cells
was observed at 72 h post transfection in the miR-107-transfected
group (from 9.1±1.3 to 14.05±2.15%) as compared to the NC group
(Fig. 5D).
Overexpression of miR-107 inhibits
ESCC colony formation
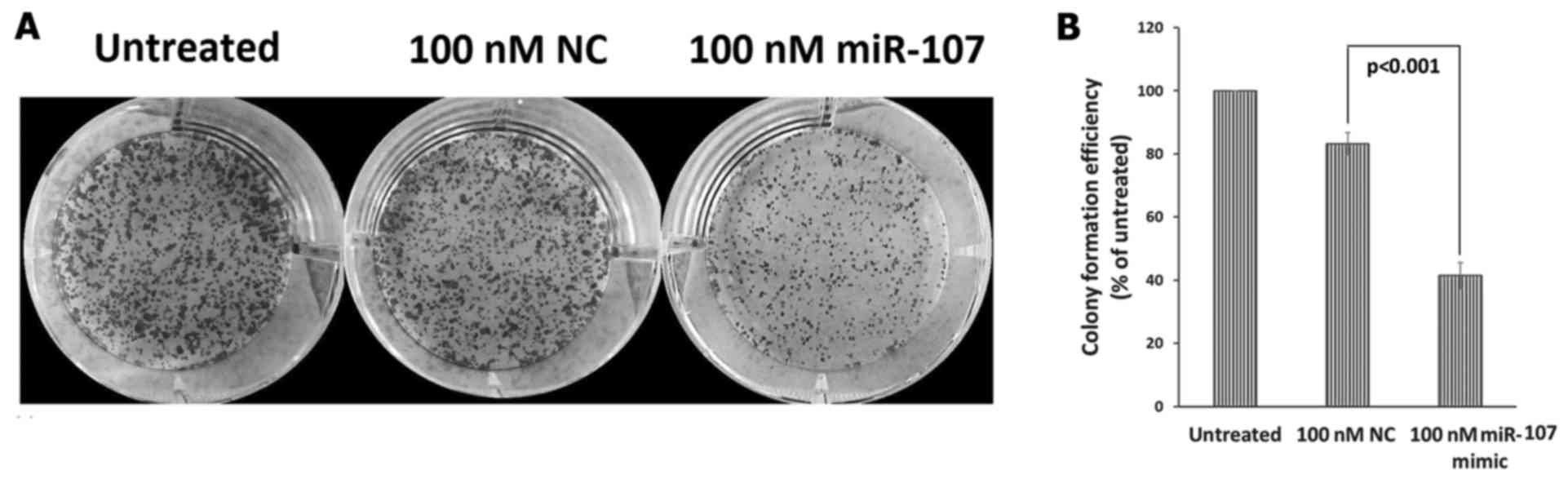
We also investigated the role of miR-107 on ESCC
clonogenic survival, and the results demonstrated a significant
(P<0.001) decrease in the clonogenic survival of the
miR-107-treated cells (colony count, 265±22.9), as compared to the
NC-treated cells (colony count, 529±10.17) and untreated ESCC cells
(colony count, 637±27.79) (Fig.
6A). When normalized to untreated cells, the colony formation
efficiency of the miR-107-treated cells was significantly reduced
to 41.44±4.23% as compared to the NC-treated cells where the colony
formation efficiency was 83.27±3.51% (Fig. 6B).
Increased expression of miR-107
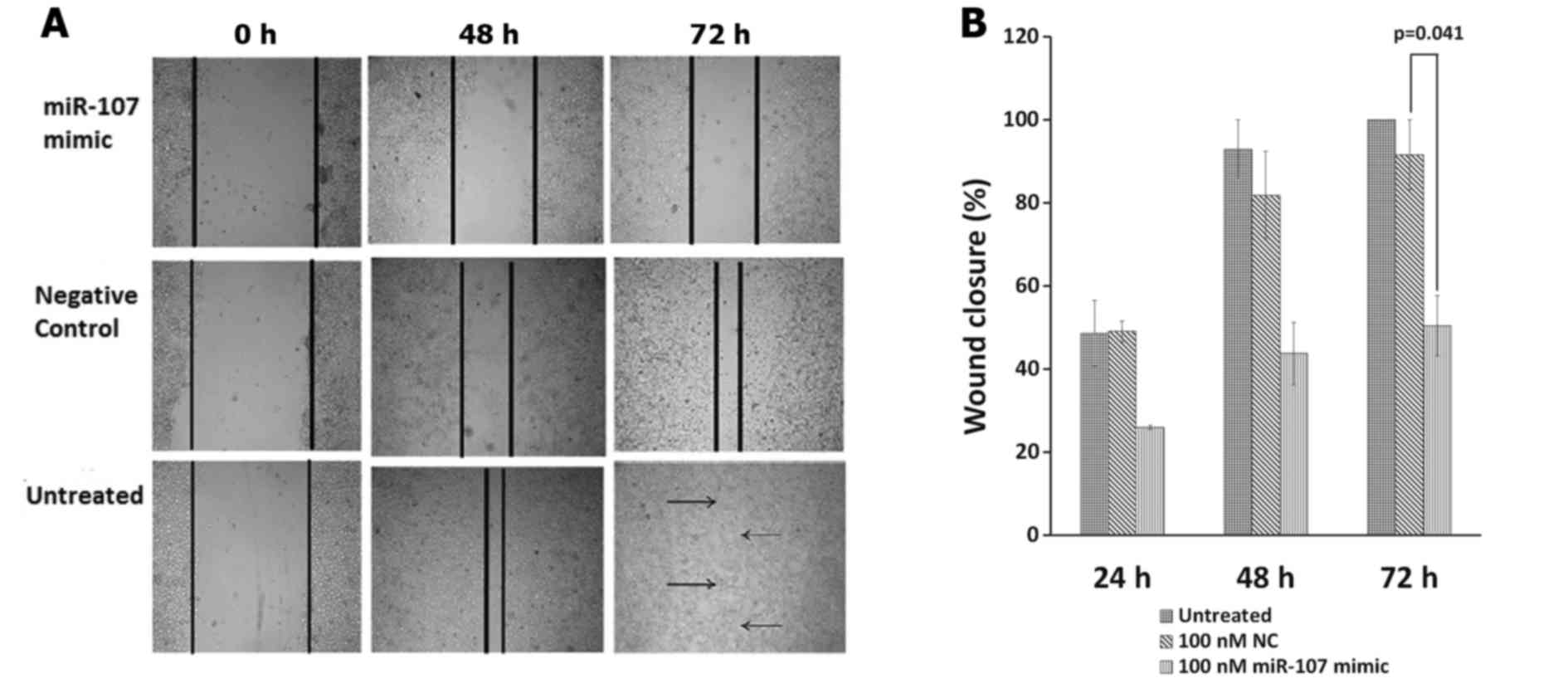
suppresses wound healing in the ESCC cells
Scratch assay was used to detect the effect of
miR-107 on the migration of KYSE-410 cells. Wound closure was
evaluated at different time-points, 24, 48 and 72 h post
scratching. Seventy-two hours were required for the wound to
completely close in the untreated cells. At 72 h post miR-107
transfection, a significant difference was found between the groups
(P=0.023). Tukey's multiple comparisons test revealed that cell
migration was significantly inhibited at 72 h after transfection of
100 nM miR-107 as compared to the NC group (P=0.041). miR-107
treatment inhibited cell migration in a time-dependent manner
reducing the wound closure to only 50.41±7.23% at 72 h as compared
to the NC where the scratch wound healed up to 91.535±8.465%
(Fig. 7A and B). However, at 24 or
48 h, there was no significant difference in cell migration of the
two groups.
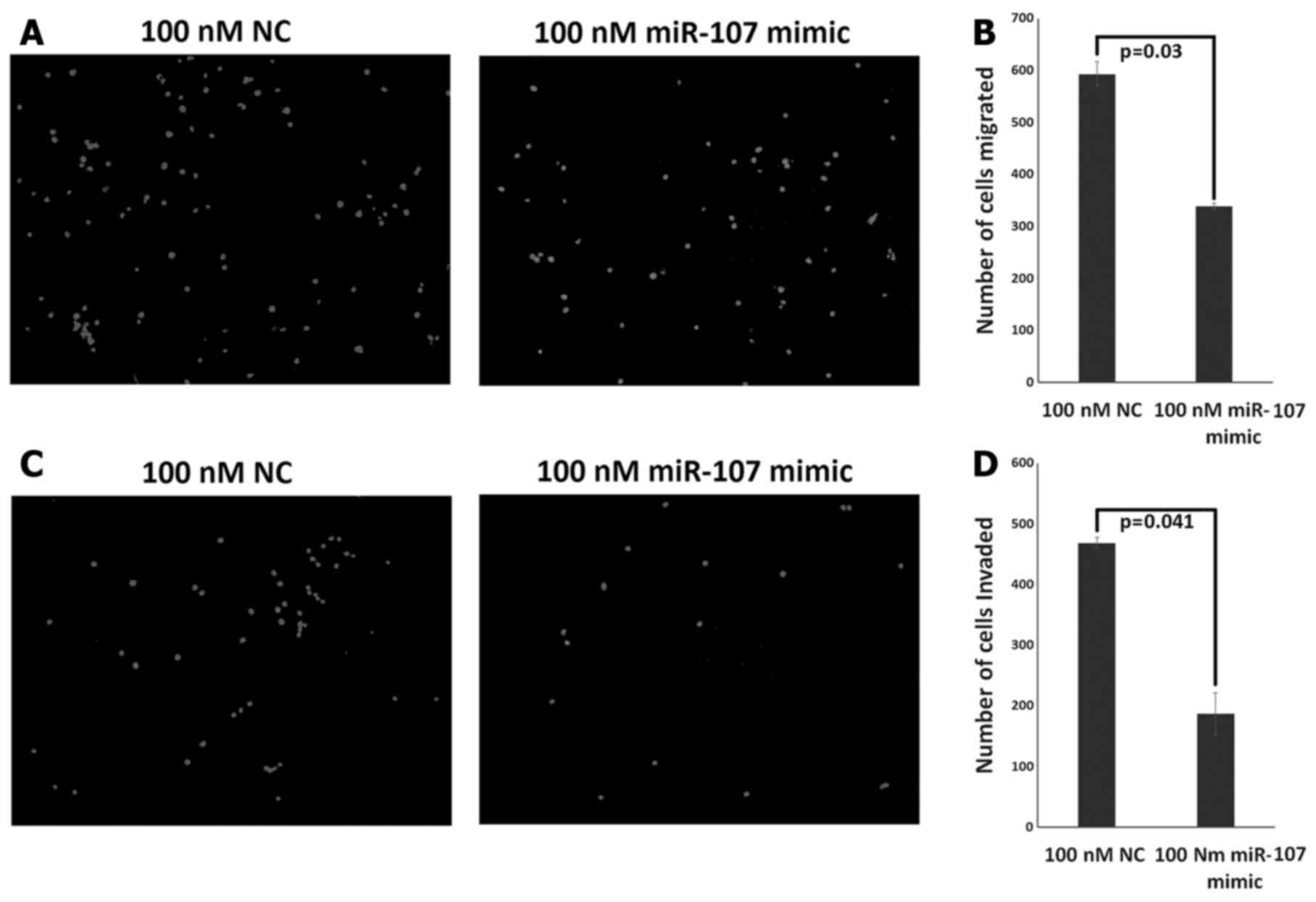
Overexpression of miR-107 results in
decreased migration and invasion potential of the ESCC cells
We further evaluated the effect of miR-107 on the
migration and invasion abilities of the KYSE-410 cells using
Transwell assay. Overexpression of miR-107 resulted in
significantly decreased migratory ability of the KYSE-410 cells at
72 h post transfection (P=0.03) (Fig.
8A). The number of KYSE-410 cells that had migrated through the
chamber was 338±6 and 592±23 in the miR-107 mimic-treated and
NC-treated group, respectively (Fig.
8B). Notably, the invasive ability of the KYSE-410 cells was
significantly decreased at 72 h post miR-107 transfection as
compared to the NC group (P=0.041) (Fig. 8C). The number of invasive cells was
468±9 in the NC-treated group as compared to the mimic-treated
group where only 187±34 cells could invade the chamber (Fig. 8D). Out of all the migrated cells
55±9% of the cells invaded in the mimic-treated group while 79±1.5%
of the cells invaded in the NC-treated group suggesting relatively
weaker invasive ability of the KYSE-410 cells after miR-107
overexpression. However, at 48 h post miR-107 transfection, there
was no significant difference in migration or invasion potential of
the two groups (data not shown).
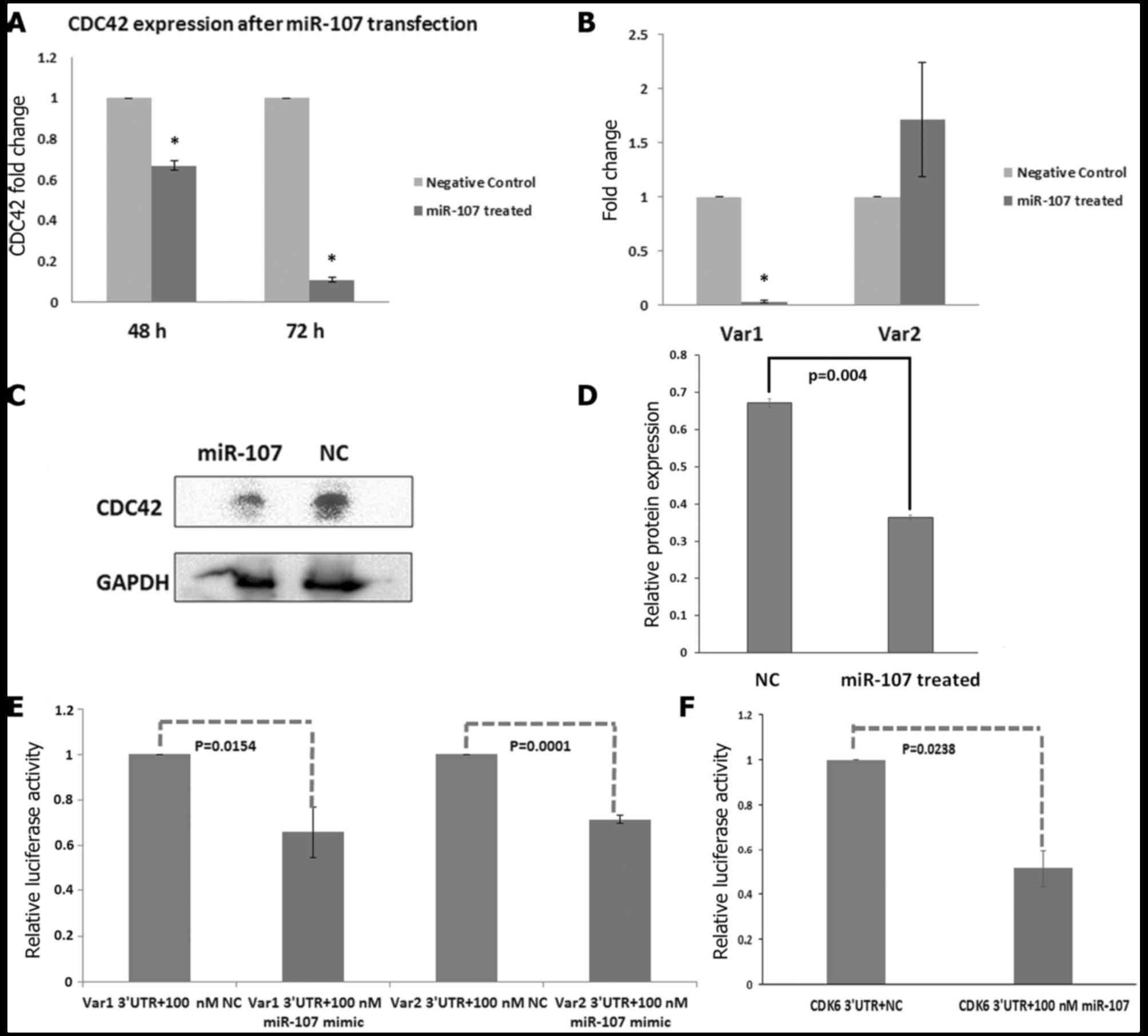
miR-107 overexpression results in
decreased expression of Cdc42 at the mRNA level
Notably, overexpression of miR-107 resulted in
significantly decreased expression of Cdc42 at the mRNA level in
the miR-107-treated cells (fold-change, 0.67 at 48 h, P=0.0074 and
0.11-fold at 72 h, P=0.0016) as compared to the NC-treated cells,
suggesting that miR-107 may suppress the expression of Cdc42 at the
transcriptional level (Fig.
9A).
Furthermore, the expression of Cdc42 variants was
analysed at 48 and 72 h after transfection of the miR-107 mimic.
Notably, overexpression of miR-107 resulted in significantly
(P=0.0041) decreased expression of Cdc42 variant 1 at the mRNA
level in the miR-107-treated cells (fold-change, 0.033) as compared
to the NC-treated cells at 72 h post-transfection, suggesting that
miR-107 may regulate the expression of Cdc42 variant 1 at the
transcriptional level (Fig. 9B).
However, no decrease in expression of Cdc42 variant 2 was observed
in the miR-107 mimic-treated cells as compared to the NC-treated
cells (Fig. 9B).
Enforced expression of miR-107 results
in decreased expression of Cdc42 at the protein level
To verify that miR-107 acts as a Cdc42 suppressor,
KYSE-410 cells were transfected with 100 nM miR-107 and the NC. The
relative expression of Cdc42 protein was calculated as band
intensity of Cdc42/band intensity of GAPDH. Densitometry analysis
showed that at 48 h post-transfection, the overexpression of
miR-107 decreased the Cdc42 protein level by 45.45% (P=0.004) as
compared to the NC (Fig. 9C and
D).
miR-107 targets Cdc42-3UTR
directly
Three sites in Cdc42 variant 1 and variant 2 were
predicted to be the potential target sites of miR-107 by RNAhybrid
software. Comparing the human sequence for interspecies homology,
we found that, out of these three sites, two were conserved across
species such as the mouse, rat, dog and chicken (Fig. 2C).
To further examine whether, Cdc42 is a direct target
of miR-107, we cloned the 3UTR of Cdc42 variants into the
pMIR-REPORT vector. Co-transfection of miR-107 and
pMIR-Cdc42-3UTR-Var1 in the HEK293T cells resulted in 35.41%
(P=0.0154) decrease in the luciferase activity as compared to the
NC (Fig. 9E). Co-transfection of
miR-107 and pMIR-Cdc42-3UTR-Var2 in HEK293T cells resulted in
decreased luciferase activity by 28.688% (P=0.0001) as compared to
the NC-treated cells (Fig. 9E).
CDK6 3UTR was used as a positive control and co-transfection of
miR-107 and pMIR-CDK6 3UTR in HEK293T cells resulted in decreased
luciferase activity by 48.4% (P=0.0238) as compared to the
NC-treated cells (Fig. 9F).
Discussion
Understanding the precise molecular mechanism
underlying esophageal tumorigenesis is imperative for better
management of this disease. In the present study, we unraveled the
tumor suppressor role of miR-107 in esophageal carcinogenesis and
further validated one of its newly identified targets i.e.
Cdc42.
miR-107, a member of the miR-15/107 superfamily, has
been shown to be downregulated in glioma, gastric cancer,
pancreatic cancer, NSCLS and cervical cancer (5–7,9,12).
We previously showed miR-107 to be significantly (P=0.004)
downregulated in 72% of esophageal cancer tissues as compared to
the matched distant non-malignant tissues (21). Moreover, increasing evidence
suggests a tumor-suppressor role for miR-107. It has been
implicated in the regulation of cell proliferation, cell cycle
arrest, migration and invasion in multiple cancers (5,6,9,16).
Mechanistic studies revealed that miR-107 functions as a
tumor-suppressor miRNA by targeting CDK6, CDK8, let-7, CDK5R1/p35,
protein kinase Cε and MCL1 in various cancers (4,5,12,15,16,18).
Notably, overexpression of miR-107 slightly delayed c-Myc induced
liver tumor formation (22) and
induced cell cycle arrest at the G2/M phase thereby retarding tumor
growth in nude mice (10). Our
results are in concordance with the above mentioned studies
suggesting a similar function of miR-107 in EC. However, our
findings are in contradiction to the observations of Martello et
al (23) wherein overexpression
of miR-107 in MCF10A mammary epithelial cells promoted
epithelial-to-mesenchymal transition resulting in a highly
metastatic phenotype thorough modulation of Dicer1. In line with
this report, knockdown of miR-107 lead to an increase in Dicer1 and
inhibition of cell invasion and migration in gastric cancer cells
(24). Taken together, these
results indicate that the biology of miR-107 is complex and highly
cell-type dependent. The role of miR-107 in esophageal
carcinogenesis is still elusive, thus, warranting further
investigation.
The above-mentioned facts thus point towards a need
for in-depth functional analysis of miR-107 in ESCC to shed light
on its role in tumorigenesis. Therefore, to characterize the
functional importance of miR-107 in ESCC tumorigenesis, we examined
the effect of miR-107 on the proliferation of KYSE-410 cells. We
observed that overexpression of miR-107 significantly suppressed
the proliferation of KYSE-410 cells at 72 h post transfection.
Cell cycle analysis revealed that the overexpression
of miR-107 in ESCC cells significantly induced cell cycle arrest at
the G0/G1 phase thereby contributing to suppression of
proliferation as observed by MTT assay. The anti-proliferative role
of miR-107 observed herein is consistent with the previously
suggested growth inhibitory function of miR-107 in gastric cancer
cells and lung carcinoma (6,9). Its
downregulation by c-Myc has been shown to cause G2/M cell cycle
progression by activating long non-coding RNA H19 in NSCLC cells
(25). In line to these reports,
Chen et al (5) showed that
p53-induced miR-107 inhibited proliferation and arrested the cell
cycle at the G0/G1 phase in glioma cells, supporting its
tumor-suppressor role. Moreover, it targets CDK6 and induces cell
cycle G1 arrest thereby inhibiting invasion in gastric cancer cells
(6) and NSCLS (9).
Most of the cancer deaths occur due to metastasis
rather than the original tumor; therefore, inhibiting cancer cell
migration is an important aspect of cancer therapy and management.
Previous studies have shown that miR-107 reduced cell migration and
invasion, possibly through CDK-8 (26) and CDK5R1 (16) in breast cancer cells and
neuroblastoma, respectively. Stückrath et al (27) showed that overexpression of miR-107
decreased migration and invasion of MCF-7 and MDA-MB-231 cells. It
inhibited glioma cell migration and invasion by targeting CDK6 and
NOTCH2 (5) and suppressed cervical
cancer invasion by targeting MCL1 (4). To access the role of miR-107 in
migration, we evaluated the effect of miR-107 on wound closure in
an ESCC cell line using scratch assay. miR-107 treatment
significantly inhibited cell migration in a time-dependent manner
reducing the wound closure to only 50.41±7.23% at 72 h as compared
to the negative control where cells had almost completely closed
the wounds. Further investigation by Transwell-Matrigel invasion
assay demonstrated that the invasive ability of KYSE-410 cells was
markedly decreased after miR-107 transfection as compared to the
cells transfected with NC.
Next, we investigated the effect of miR-107
overexpression on colony formation efficiency of ESCC cells and our
results indicated an inhibitory effect of miR-107 on the colony
formation potential of ESCC cells. The present study thus further
supports the previous findings, wherein, miR-107 treatment
inhibited the colony formation efficiency of cervical cancer and
HNSCC cells (12,18).
To explore the molecular mechanism underlying the
tumor-suppressor function of miR-107, we validated one of the
bioinformatically predicted targets of miR-107. We earlier
predicted Cdc42 to be one of the putative targets of miR-107 using
bioinformatics tools. Moreover, expression analysis in esophageal
cancer tissues carried out in our previous study revealed an
inverse correlation between expression of miR-107 and the Cdc42
transcript (21). Cdc42 is a member
of the Rho family of small GTPases and its activation induces key
signaling pathways such as cytoskeletal remodeling, cell cycle
progression, establishment of cell polarity, cellular
transformation and cell migration (28,29).
Overexpression of Cdc42 has been reported in several types of human
cancers including NSCLC, colorectal adenocarcinoma, melanoma,
breast and testicular cancer (30–34).
In ESCC its overexpression was correlated with lymph node
metastasis and pathological differentiation (35,36).
In addition to the cell cycle, Cdc42 also regulates the cell
cytoskeleton and adhesion, cell functions that are important for
cell migration and invasion in several types of cancers (37). In the present study, overexpression
of miR-107 in the ESCC cell line KYSE-410 resulted in significantly
decreased expression of Cdc42 at the mRNA level in the
miR-107-treated cells as compared to the NC-treated cells,
suggesting that miR-107 may regulate the expression of Cdc42 at the
transcriptional level. A significant decrease in expression of
Cdc42 protein was also observed in the miR-107-treated cells as
compared to the NC-treated cells. Further validation by luciferase
reporter assay confirmed Cdc42 as a direct downstream target of
miR-107. Interestingly, previous studies have reported that Cdc42
is involved in proliferation, cell cycle regulation and migration
as well as it increases the activity of matrix metalloproteinases
(MMPs). In a positive feedback loop, Cdc42 induces the accumulation
of its upstream activator i.e. EGFR thereby contributing to
cellular proliferation and transformation (38,39).
Additionally, Cdc42-associated tyrosine kinase 1 (ACK1), a
downstream effector of Cdc42, was found to positively regulate Akt
and other prosurvival factors thus promoting cell survival and
growth (40). Tu et al
(41) demonstrated that FasL
activated caspase-3 and caspase-8 catalyzes the cleavage of Cdc42.
Notably, in the same study, caspase-insensitive Cdc42 mutants
provided strong antiapoptotic effects. Furthermore, under hypoxic
conditions, Cdc42 indirectly phosphorylates JNK resulting in its
nuclear translocation. When translocated to the nucleus, JNK binds
and activates the AP-1/MMP9 axis leading to metastatic processes
(42). MMPs are critical enzymes
involved in degradation of basement membrane and extracellular
matrix thus helping in cancer invasion and metastasis (29). Taken together, the above mentioned
facts thus indicate that Cdc42 plays an important role in the
progression of a wide array of cancers. In the present study, we
demonstrated that miR-107 was involved in the regulation of
proliferation, migration and cell cycle of ESCC cells.
Additionally, we identified Cdc42 as a novel direct target of
miR-107 and showed that enforced expression of miR107 in ESCC cells
led to reduced expression of Cdc42. However, further in-depth
analysis is warranted to establish the fact that the inhibition of
Cdc42 expression may be a key mechanism by which miR-107 impairs
esophageal cancer cell growth.
In conclusion, our results herein, document a key
tumor-suppressor role of miR-107 in esophageal carcinogenesis by
inhibiting proliferation, migration and causing cell cycle arrest
in ESCC cells. We further identified and validated Cdc42 as a
direct downstream target of miR-107. Future challenges include
identifying additional targets of miR-107 to further discern its
function and access its applicability in the treatment of
esophageal cancer.
Acknowledgements
The present study was funded by the Department of
Biotechnology; Ministry of Science and Technology; Government of
India (grant no. BT/PR13311/GBD/27/246/2009). We are thankful to
the Council of Scientific and Industrial Research for funding the
fellowship to P.S. (grant no. 09/806(0021)/2011-EMR-I).
References
|
1
|
Guo Y, Chen Z, Zhang L, Zhou F, Shi S,
Feng X, Li B, Meng X, Ma X, Luo M, et al: Distinctive microRNA
profiles relating to patient survival in esophageal squamous cell
carcinoma. Cancer Res. 68:26–33. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yamakuchi M, Lotterman CD, Bao C, Hruban
RH, Karim B, Mendell JT, Huso D and Lowenstein CJ: P53-induced
microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad
Sci USA. 107:6334–6339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
He J, Zhang W, Zhou Q, Zhao T, Song Y,
Chai L and Li Y: Low-expression of microRNA-107 inhibits cell
apoptosis in glioma by upregulation of SALL4. Int J Biochem Cell
Biol. 45:1962–1973. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou C, Li G, Zhou J, Han N, Liu Z and Yin
J: miR-107 activates ATR/Chk1 pathway and suppress cervical cancer
invasion by targeting MCL1. PLoS One. 9:e1118602014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen L, Zhang R, Li P, Liu Y, Qin K, Fa
ZQ, Liu YJ, Ke YQ and Jiang XD: P53-induced microRNA-107 inhibits
proliferation of glioma cells and down-regulates the expression of
CDK6 and Notch-2. Neurosci Lett. 534:327–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feng L, Xie Y, Zhang H and Wu Y: miR-107
targets cyclin-dependent kinase 6 expression, induces cell cycle G1
arrest and inhibits invasion in gastric cancer cells. Med Oncol.
29:856–863. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee KH, Lotterman C, Karikari C, Omura N,
Feldmann G, Habbe N, Goggins MG, Mendell JT and Maitra A:
Epigenetic silencing of microRNA miR-107 regulates cyclin-dependent
kinase 6 expression in pancreatic cancer. Pancreatology. 9:293–301.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhong Z, Lv M and Chen J: Screening
differential circular RNA expression profiles reveals the
regulatory role of circTCF25-miR-103a-3p/miR-107-CDK6 pathway in
bladder carcinoma. Sci Rep. 6:309192016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takahashi Y, Forrest A, Maeno E, Hashimoto
T, Daub CO and Yasuda J: MiR-107 and MiR-185 can induce cell cycle
arrest in human non small cell lung cancer cell lines. PLoS One.
4:e66772009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song N, Ma X, Li H, Zhang Y, Wang X, Zhou
P and Zhang X: microRNA-107 functions as a candidate tumor
suppressor gene in renal clear cell carcinoma involving multiple
genes. Urol Oncol. 33:205.e1–205.e11. 2015. View Article : Google Scholar
|
|
11
|
Xia H, Li Y and Lv X: MicroRNA-107
inhibits tumor growth and metastasis by targeting the BDNF-mediated
PI3K/AKT pathway in human non-small cell lung cancer. Int J Oncol.
49:1325–1333. 2016.PubMed/NCBI
|
|
12
|
Che LF, Shao SF and Wang LX:
Downregulation of CCR5 inhibits the proliferation and invasion of
cervical cancer cells and is regulated by microRNA-107. Exp Ther
Med. 11:503–509. 2016.PubMed/NCBI
|
|
13
|
Wang WX, Kyprianou N, Wang X and Nelson
PT: Dysregulation of the mitogen granulin in human cancer through
the miR-15/107 microRNA gene group. Cancer Res. 70:9137–9142. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song YQ, Ma XH, Ma GL, Lin B, Liu C, Deng
QJ and Lv WP: MicroRNA-107 promotes proliferation of gastric cancer
cells by targeting cyclin dependent kinase 8. Diagn Pathol.
9:1642014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen PS, Su JL, Cha ST, Tarn WY, Wang MY,
Hsu HC, Lin MT, Chu CY, Hua KT, Chen CN, et al: miR-107 promotes
tumor progression by targeting the let-7 microRNA in mice and
humans. J Clin Invest. 121:3442–3455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moncini S, Salvi A, Zuccotti P, Viero G,
Quattrone A, Barlati S, De Petro G, Venturin M and Riva P: The role
of miR-103 and miR-107 in regulation of CDK5R1 expression and in
cellular migration. PLoS One. 6:e200382011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Z, Zhang L, Yin ZY, Fan XL, Hu B,
Wang LQ and Zhang D: miR-107 regulates cisplatin chemosensitivity
of A549 non-small cell lung cancer cell line by targeting cyclin
dependent kinase 8. Int J Clin Exp Pathol. 7:7236–7241.
2014.PubMed/NCBI
|
|
18
|
Datta J, Smith A, Lang JC, Islam M, Dutt
D, Teknos TN and Pan Q: microRNA-107 functions as a candidate
tumor-suppressor gene in head and neck squamous cell carcinoma by
downregulation of protein kinase Cε. Oncogene. 31:4045–4053. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Piao L, Zhang M, Datta J, Xie X, Su T, Li
H, Teknos TN and Pan Q: Lipid-based nanoparticle delivery of
Pre-miR-107 inhibits the tumorigenicity of head and neck squamous
cell carcinoma. Mol Ther. 20:1261–1269. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang M, Wang X, Li W and Cui Y: miR-107
and miR-25 simultaneously target LATS2 and regulate proliferation
and invasion of gastric adenocarcinoma (GAC) cells. Biochem Biophys
Res Commun. 460:806–812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sharma P, Saraya A, Gupta P and Sharma R:
Decreased levels of circulating and tissue miR-107 in human
esophageal cancer. Biomarkers. 18:322–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tao J, Ji J, Li X, Ding N, Wu H, Liu Y,
Wang XW, Calvisi DF, Song G and Chen X: Distinct anti-oncogenic
effect of various microRNAs in different mouse models of liver
cancer. Oncotarget. 6:6977–6988. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martello G, Rosato A, Ferrari F, Manfrin
A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T,
et al: A microRNA targeting Dicer for metastasis control. Cell.
141:1195–1207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Zhang Y, Shi Y, Dong G, Liang J, Han
Y, Wang X, Zhao Q, Ding J, Wu K, et al: MicroRNA-107, an oncogene
microRNA that regulates tumour invasion and metastasis by targeting
DICER1 in gastric cancer. J Cell Mol Med. 15:1887–1895. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cui J, Mo J, Luo M, Yu Q, Zhou S, Li T,
Zhang Y and Luo W: c-Myc-activated long non-coding RNA H19
downregulates miR-107 and promotes cell cycle progression of
non-small cell lung cancer. Int J Clin Exp Pathol. 8:12400–12409.
2015.PubMed/NCBI
|
|
26
|
Li XY, Luo QF, Wei CK, Li DF, Li J and
Fang L: MiRNA-107 inhibits proliferation and migration by targeting
CDK8 in breast cancer. Int J Clin Exp Med. 7:32–40. 2014.PubMed/NCBI
|
|
27
|
Stückrath I, Rack B, Janni W, Jäger B,
Pantel K and Schwarzenbach H: Aberrant plasma levels of circulating
miR-16, miR-107, miR-130a and miR-146a are associated with lymph
node metastasis and receptor status of breast cancer patients.
Oncotarget. 6:13387–13401. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Orgaz JL, Herraiz C and Sanz-Moreno V: Rho
GTPases modulate malignant transformation of tumor cells. Small
GTPases. 5:e290192014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stengel K and Zheng Y: Cdc42 in oncogenic
transformation, invasion, and tumorigenesis. Cell Signal.
23:1415–1423. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Y, Wang Y, Zhang Y, Miao Y, Zhao Y,
Zhang PX, Jiang GY, Zhang JY, Han Y, Lin XY, et al: Abnormal
expression of p120-catenin, E-cadherin, and small GTPases is
significantly associated with malignant phenotype of human lung
cancer. Lung Cancer. 63:375–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Del Gómez Pulgar T, Valdés-Mora F, Bandrés
E, Pérez-Palacios R, Espina C, Cejas P, García-Cabezas MA, Nistal
M, Casado E, González-Barón M, et al: Cdc42 is highly expressed in
colorectal adenocarcinoma and downregulates ID4 through an
epigenetic mechanism. Int J Oncol. 33:185–193. 2008.PubMed/NCBI
|
|
32
|
Tucci MG, Lucarini G, Brancorsini D, Zizzi
A, Pugnaloni A, Giacchetti A, Ricotti G and Biagini G: Involvement
of E-cadherin, beta-catenin, Cdc42 and CXCR4 in the progression and
prognosis of cutaneous melanoma. Br J Dermatol. 157:1212–1216.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fritz G, Brachetti C, Bahlmann F, Schmidt
M and Kaina B: Rho GTPases in human breast tumours: Expression and
mutation analyses and correlation with clinical parameters. Br J
Cancer. 87:635–644. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kamai T, Yamanishi T, Shirataki H, Takagi
K, Asami H, Ito Y and Yoshida K: Overexpression of RhoA, Rac1, and
Cdc42 GTPases is associated with progression in testicular cancer.
Clin Cancer Res. 10:4799–4805. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng JG, Liu Q, Qin X, Geng YH, Zheng ST,
Liu T, Sheyhidin I and Lu XM: Clinicopathological pattern and
Annexin A2 and Cdc42 status in patients presenting with
differentiation and lymph node metastasis of esophageal squamous
cell carcinomas. Mol Biol Rep. 39:1267–1274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Z, Feng JG, Tuersun A, Liu T, Liu H,
Liu Q, Zheng ST, Huang CG, Lv GD, Sheyhidin I, et al: Proteomic
identification of differentially-expressed proteins in esophageal
cancer in three ethnic groups in Xinjiang. Mol Biol Rep.
38:3261–3269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ridley AJ: Rho GTPase signalling in cell
migration. Curr Opin Cell Biol. 36:103–112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang XY, Gan MX, Li Y, Zhan WH, Han TY,
Han XJ, Cheng JQ and Wang JB: Cdc42 induces EGF receptor protein
accumulation and promotes EGF receptor nuclear transport and
cellular transformation. FEBS Lett. 589:255–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hirsch DS, Shen Y and Wu WJ: Growth and
motility inhibition of breast cancer cells by epidermal growth
factor receptor degradation is correlated with inactivation of
Cdc42. Cancer Res. 66:3523–3530. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mahajan K and Mahajan NP: Shepherding AKT
and androgen receptor by Ack1 tyrosine kinase. J Cell Physiol.
224:327–333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tu S and Cerione RA: Cdc42 is a substrate
for caspases and influences Fas-induced apoptosis. J Biol Chem.
276:19656–19663. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xiao B, Chen D, Luo S, Hao W, Jing F, Liu
T, Wang S, Geng Y, Li L, Xu W, et al: Extracellular translationally
controlled tumor protein promotes colorectal cancer invasion and
metastasis through Cdc42/JNK/MMP9 signaling. Oncotarget.
7:50057–50073. 2016.PubMed/NCBI
|