Introduction
Brain glioblastoma is a commonly occurred tumor with
aggressive features. The therapeutic way to treat this disease is
to use a surgical resection followed by a combination of
radiotherapy and/or temozolomide (TMZ) chemotherapy (1). However, glioblastoma cells often
generate resistance to TMZ treatment and the average survival time
for patients after general treatment is ~15 months (2,3).
Therefore, it is essential to find an effective agent to treat
glioblastoma, especially drug-resistant glioblastoma.
Numerous studies have suggested that sulforaphane
(SFN) obtained from cruciferous vegetables induced apoptosis in a
variety of tumors (4–7). In vivo, SFN is metabolized to
produce sulforaphane-glutathione (SFN-GSH),
sulforaphanecysteine-glycine (SFN-CG), sulforaphane-cysteine
(SFN-Cys), and sulforaphane-N-acetylcysteine (SFN-NAC) via
mercapturic acid pathway (5,8,9).
Further studies showed that SFN induced cell apoptosis, inhibited
cell proliferation, invasion and angiogenesis (10–14).
In human glioblastoma cells, it has been reported that SFN induced
apoptosis (15) and our previous
study showed that SFN inhibited cell invasion via ERK1/2 signaling
pathway (12). Compared with SFN,
SFN-Cys inhibits histone deacetylase (HDAC) more efficiently and
has longer half-life and retention time in vivo (8,16).
HDAC is highly related to cell growth. Hence we assume that SFN-Cys
inhibits cell growth and induces apoptosis with higher efficiency,
it is important to investigate the underlying mechanisms. These
results will provide new insight into the SFN analog anticancer
effect, so that we might develop new anticancer agents.
The extracellular signal-regulated kinases (ERK1/2)
regulate many cellular responses by phosphorylating a number of
downstream effectors (12,13,17–20).
Transient phosphorylation of ERK1/2 (5–15-min stimulation)
contributes to cell growth (18)
while sustained phosphorylation of ERK1/2 (>15 min stimulation)
causes cell apoptosis (19). Our
previous studies demonstrated that SFN inhibited invasion by
sustained activation of ERK1/2 to regulate E-cadherin and CD44v6 in
human prostate cancer DU145 cells (13) and SFN-Cys suppressed invasion by
sustained phosphorylation of ERK1/2 and downregulating galectin-1
in human prostate cancer DU145 and PC3 cells (20). Moreover, SFN inhibited invasion via
activating ERK1/2 signaling in human glioblastoma U87MG and U373MG
cells. Therefore, we hypothesized that SFN-Cys might induce
apoptosis by activating ERK1/2 and the apoptosis-related
proteins.
Both Bax and Bcl-2 are members of the Bcl-2 family
regulating the apoptosis of glioblastoma cells (21). Bax contributes to the apoptotic
response of glioblastoma cells and the overexpression of Bax
increases survival of glioblastoma patients (22). Downregulation of Bcl-2 effectively
induced apoptosis in glioblastoma cells (23), while overexpression of Bcl-2
inhibited apoptosis and decreased the effect of radiotherapy or
chemotherapy in many other cancers (24). The apoptosis-inducing ability of Bax
is suppressed by Bcl-2 binding to its homologous C-terminal domain
(25–27). Thus, the increasing Bax/Bcl-2 ratio
results in apoptosis in human glioblastoma cells while the
decreasing Bax/Bcl-2 ratio represses cell apoptosis and contributes
to the resistance of glioblastoma cells to chemotherapeutic agent
(28). It is reported that the
upregulation of Bax/Bcl-2 resulted in the loss of mitochondrial
membrane potential (MMP), inducing cell apoptosis via the intrinsic
pathway in glioma cells (29).
Since the ratio of Bax/Bcl-2 is regulated by ERK1/2 signaling
pathway (30), we hypothesized that
SFN-Cys might induce intrinsic apoptosis by upregulating Bax/Bcl-2
ratio mediated via the activated ERK1/2.
Cysteine-aspartic proteases (caspases) constitute a
family of proteolytic enzymes and are largely known for their
functions in apoptosis (31). The
caspases are divided into two groups, the initiator caspases such
as caspase 8 and 9 and the effector caspases such as caspase 3
(32,33). Caspase 3 exists as an inactive
proenzyme while it is activated after cleaved by initiator caspases
(34). Cleaved caspase 3 in turn
cleaves multiple cell substrates such as poly(ADP-ribose)
polymerase (PARP), leading to cell apoptosis (35). It is reported that SFN activated
caspase 3 via ERK1/2 pathway, leading to cell apoptosis in human
glioblastoma cells (36).
Additionally, caspase 3 is a downstream protein of Bax/Bcl-2, and
the upregulation of Bax/Bcl-2 ratio leads to the activation of
caspase 3 (28). Taken together, we
thought that SFN-Cys might activate ERK1/2, increasing the ratio of
Bax/Bcl-2 and upregulating cleaved caspase 3, which led to cell
apoptosis in U373MG and U87MG cells. Altogether, our results
provide evidence for SFN-Cys inducing apoptosis in glioblastoma,
and further explore the underlying mechanisms, facilitating finding
more natural products for treating glioblastoma.
Materials and methods
Reagents
D, L-Sulforaphane-L-cysteine (SFN-Cys) and caspase 3
antibody were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
DMEM/HIGH glucose culture medium was from Hyclone (Logan, UT, USA).
Fetal bovine serum (FBS) was from Zhejiang Tianhang Biological
Technology Co., Ltd. (Zhejiang, China). Penicillin-streptomycin was
from Invitrogen (Carlsbad, CA, USA). Dimethyl sulfoxide (DMSO) was
acquired from AppliChem GmbH (Ottoweg4, D-64291 Darmstadt,
Germany). β-actin antibody was from Proteintech Group, Inc.
(Chicago, IL, USA). CCK8 assay kit was from Dojindo Laboratories
(Shanghai, China). Annexin V-FITC Apoptosis assay kit was acquired
from Genstar (Beijing, China). Mitochondrial membrane potential
assay kit with JC-1 was from Beyotime Biotechnology (Shanghai,
China). pERK1/2 antibody, ERK1/2 antibody and PD98059 were acquired
from Cell Signaling Technology, Inc. (Shanghai, China). The
antibodies against Bax and Bcl-2 were from Sangon Biotech
(Shanghai, China).
Cell culture
Human glioblastoma U87MG cell line was purchased
from the Cell Resource Center, Peking Union Medical College
(CRC/PUMC) and U373MG cell line was purchased from American Type
Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured
in DMEM/HIGH glucose culture medium supplemented with 10% FBS, 100
U/ml penicillin and 100 U/ml streptomycin in a standard humidified
incubator with 5% CO2 at 37°C. The cells were treated
with SFN-Cys for 24 h and pretreated with ERK1/2 inhibitor PD98059
(25 µM) for 30 min.
Morphological observation
U373MG and U87MG cells were seeded in 6-well culture
plates and not exposed to SFN-Cys with different concentrations (0,
15, 30, and 45 µM) for 24 h until the cells were grown to 70%
confluence. Cell morphology was observed with phase-contrast
microscope at ×100 magnification (Leica, Mannheim, Germany) and
documented by the digital camera (Olympus, Tokyo, Japan) connected
to the microscope.
Cell viability assay
The cell viability was evaluated by utilizing the
Cell Counting Kit-8 (CCK-8) assay. Cells were seeded in 96-well
plates at 5×103 cells/well and treated with various
doses of SFN-Cys for 24 h. Then 20 µl CCK-8 reagents were added to
each well and the 96-well plate was incubated at 37°C for an
additional 30 min. Then the absorbance was measured at 450 nm by a
BioTek microplate reader (Synergy™ HT, BioTek Instruments, Inc.,
Winooski, VT, USA).
Flow cytometry analysis of
apoptosis
Apoptosis was assessed by utilizing Annexin
V-FITC/propidium iodide (PI) staining followed by flow cytometry.
The cells were plated in 6-well plates and incubated with various
concentrations of SFN-Cys (0, 15, 30 and 45 µM). After incubated
for 24 h, cells were collected and centrifuged at 1000 rpm for 5
min. Then cells were washed twice with cold PBS to remove excessive
medium. Next, the cells were re-suspended at a concentration of
1×106 cells/ml in binding buffer and incubated with
annexin for 5 min V-FITC and PI was added afterwards at room
temperature in the dark. All samples were analyzed on flow
cytometry (FACSAria, BD Biosciences, San Jose, CA, USA) to
determine the cell apoptotic rate.
Western blot analysis
Cells were collected and lysed with RIPA (Thermo
Scientific, Waltham, MA, USA) combined with protease inhibitors
(Roche, Mannheim, Germany) for 30 min. The cell lysate was
centrifuged at 12,000 rpm for 15 min. Samples were separated on
SDS-PAGE gels and then transferred to nitrocellulose membranes.
Membranes were blocked with 1.5% BSA in TBS Tween-20 (TBS-T) buffer
for 1 h and then incubated with primary antibody at 4°C overnight.
Afterwards, the membrane was washed with TBS-T for 30 min and then
incubated with the fluorescence-labeled secondary antibody (LI-COR
Bioscience, Lincoln, NE, USA) for 1 h. After washing, the membrane
was scanned by Odyssey Infrared Imaging System (LI-COR Bioscience).
The same membrane was stripped and incubated with β-actin antibody
for equal loading and normalization.
Mitochondrial membrane potential
assay
Mitochondrial membrane potential (MMP) was examined
by using the mitochondrial membrane potential kit with JC-1. The
cells were plated in 24-well plate and treated with drugs for 24 h.
Then, cells were washed with PBS three times and incubated with 250
µl culture medium and 250 µl JC-1 working solution for 20 min in
the dark at 37°C. After that, the cells were washed three times (3
min each time) with ice cold JC-1 washing buffer. Images were
captured by a fluorescence microscope at ×200 magnification (Axio
Imager A2, Zeiss, Jena, Germany). Fluorescent intensity was
analyzed by Image-Pro Plus and the level of MMP was calculated as
the JC-1 aggregate/monomer ratio.
Statistical analysis
Data are expressed as mean ± SD and the differences
among the groups were analyzed by one-way ANOVA. P<0.05 was
considered to be statistically significant.
Results
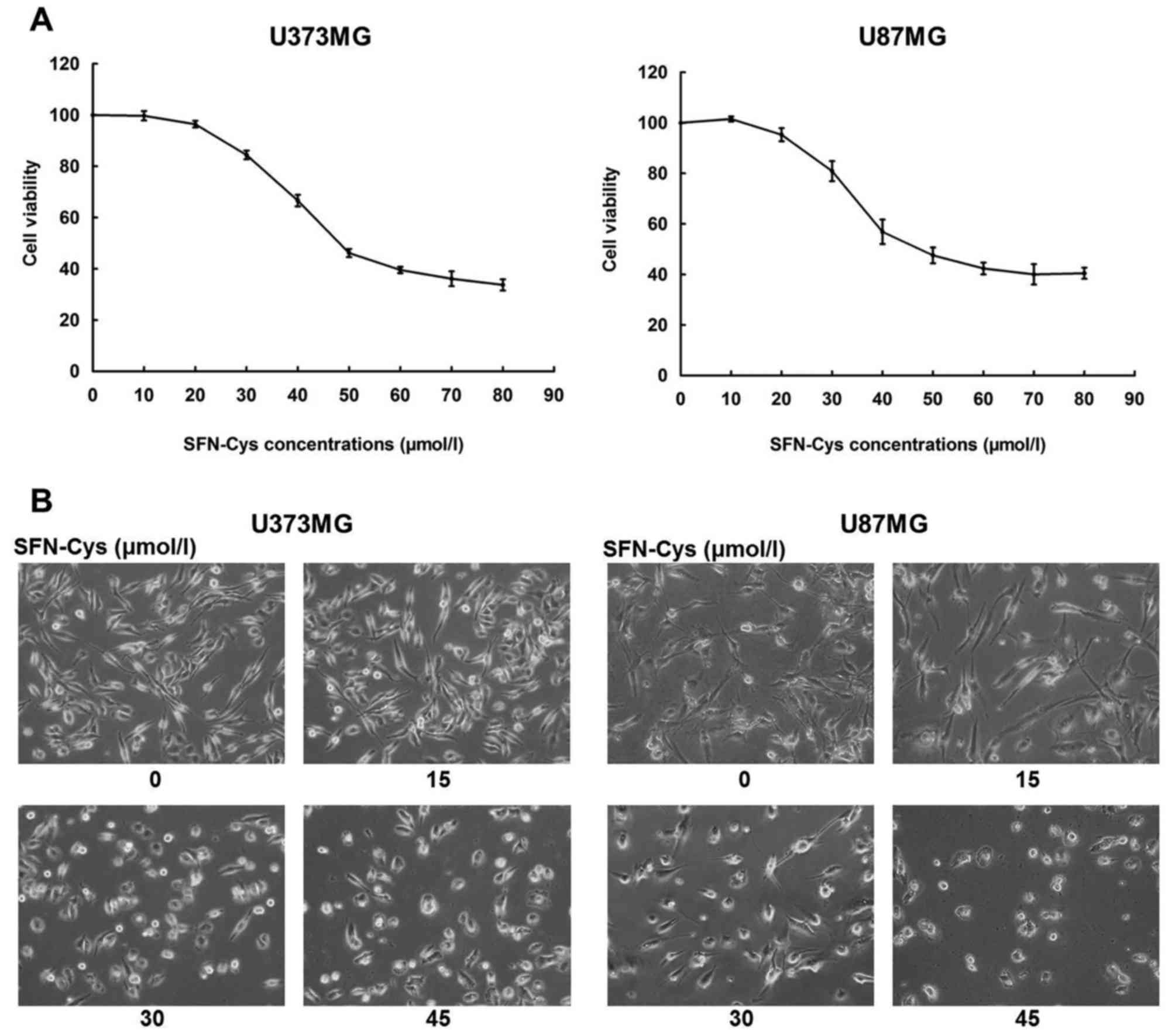
SFN-Cys dose-dependently decreases
cell viability and changed cell morphology
CCK-8 assay was utilized to examine the reduction of
cell viability after cells were incubated with SFN-Cys in U373MG
and U87MG cells. The cells were exposed to 0, 10, 20, 30, 40, 50,
60, 70 and 80 µM SFN-Cys for 24 h. SFN-Cys was found to decrease
cell viability in a dose-dependent manner (Fig. 1). Moreover, we found that SFN-Cys
decreased cell viability remarkably when cells were exposed to 30
µM SFN-Cys for 24 h (Fig. 1A).
Cells treated with SFN-Cys of 0, 15, 30 and 45 µM for 24 h were
observed by the light microscope. The cells treated by SFN-Cys were
shrunken and exhibited rounded shape compared with the untreated
cells. Microscopic images in Fig.
1B showed that the cell morphology exposed to 30 µM SFN-Cys for
24 h began to change.
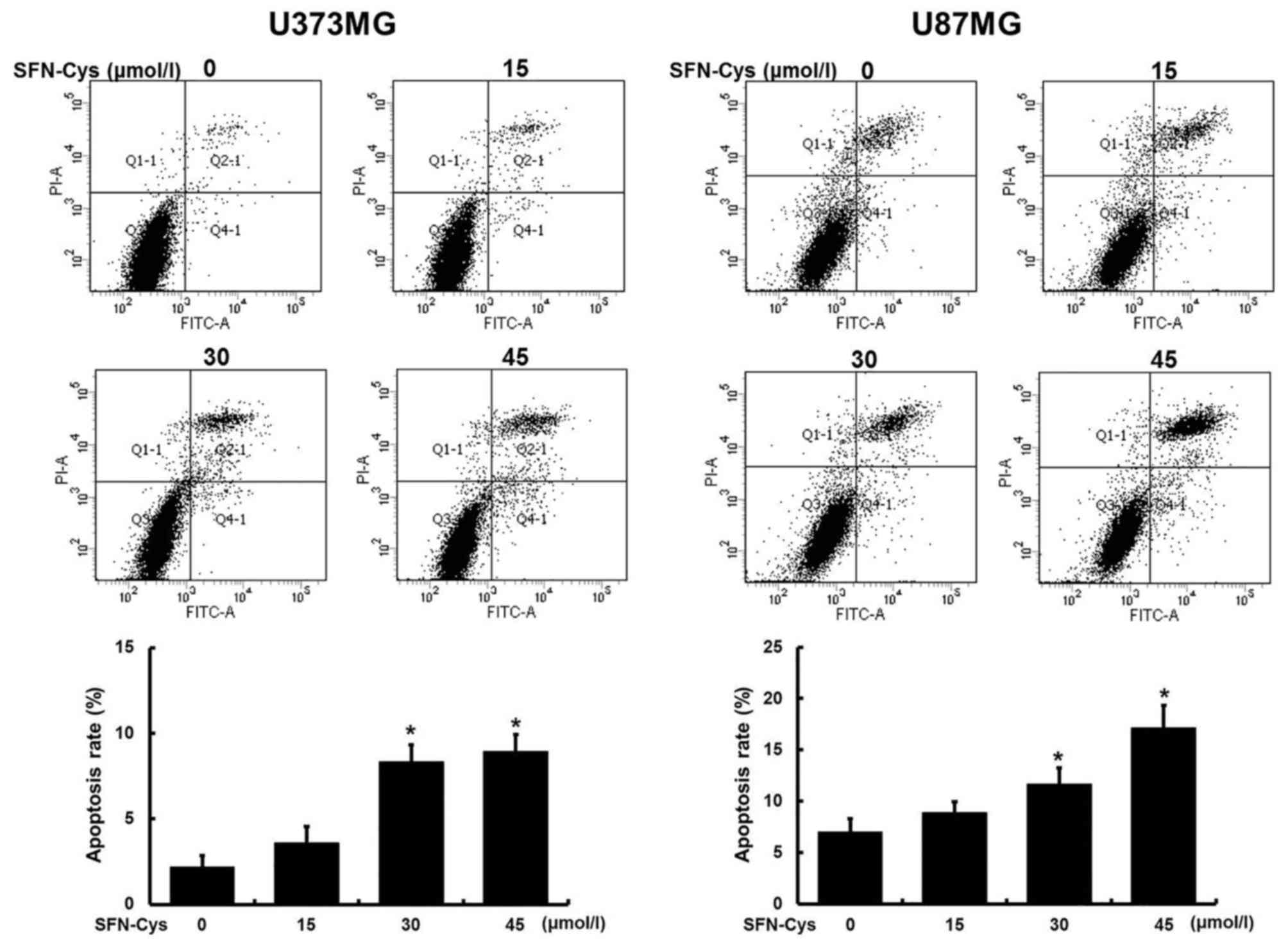
SFN-Cys dose-dependently induces cell
apoptosis
Flow cytometry was utilized to prove whether the
decrease of cell viability and the changes of morphology (Fig. 1) were caused by cell apoptosis. The
cells were treated with 0, 15, 30 and 45 µM SFN-Cys for 24 h before
flow cytometry analysis. The cell apoptosis rates are displayed in
Fig. 2. We found that SFN-Cys
induced cell apoptosis in a dose-dependent manner in both U373MG
and U87MG cells. In addition, compared with the untreated control,
a significant increase of apoptosis rate was shown in 30 µM SFN-Cys
treated cells. Moreover, apoptosis rate in U87MG cells was higher
than that in U373MG cells, suggesting the distinct sensitivity to
SFN-Cys between U87MG and U373MG cells. Taken together, we chose 30
µM SFN-Cys as an optimal concentration for the following
studies.
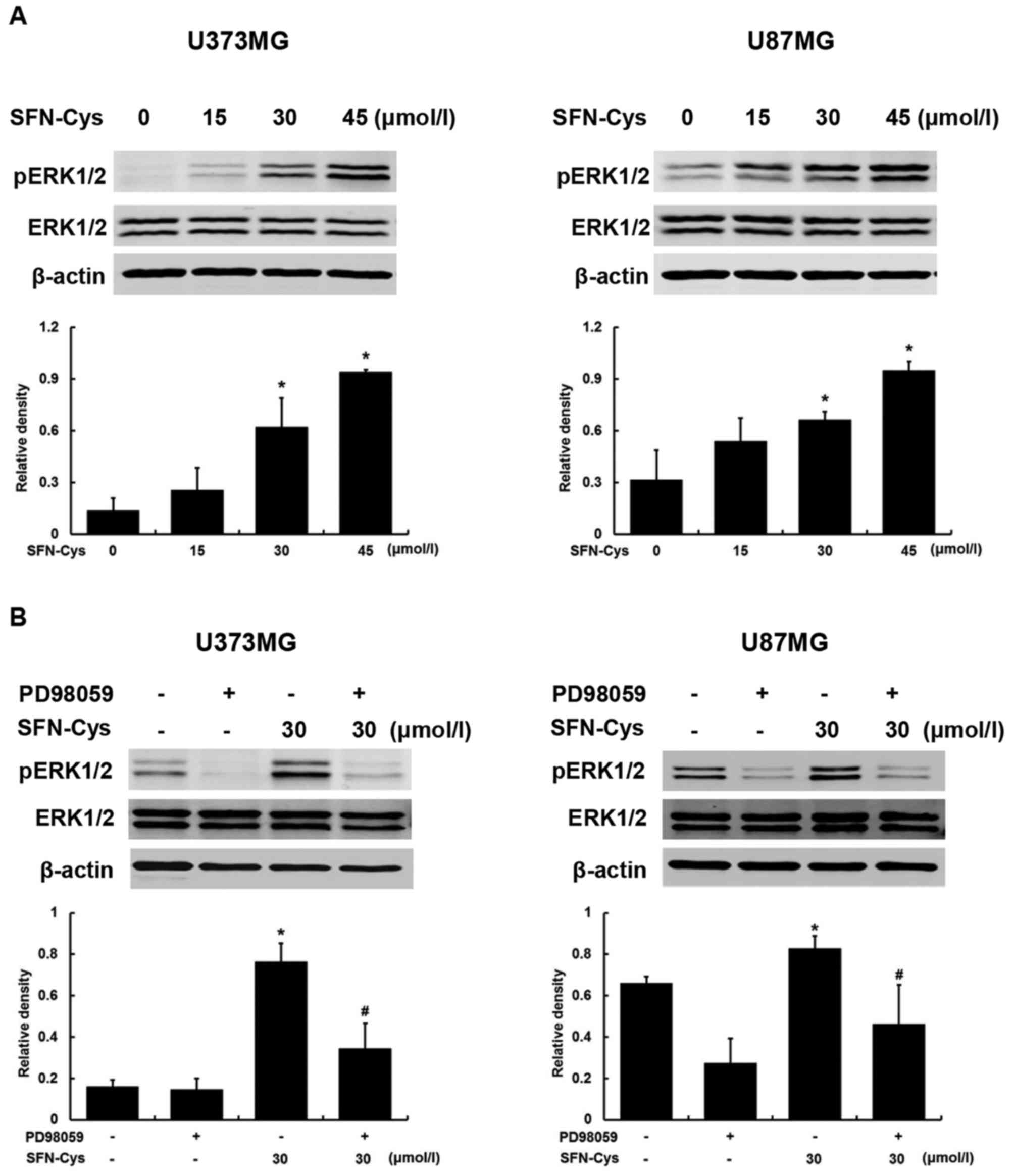
SFN-Cys dose-dependently activates
ERK1/2
U373MG and U87MG cells were treated with increasing
doses of SFN-Cys (0, 15, 30 and 45 µM) for 24 h. Western blot
analysis showed that SFN-Cys activated ERK1/2 (Thr202/Tyr204) in a
dose-dependent way and pERK1/2 was significantly increased at 30 µM
of SFN-Cys (Fig. 3A), which was in
agreement with our previous studies that SFN and SFN-Cys
contributed to the phosphorylation of ERK1/2 in prostate cancer
cells (13,20). PD98059, ERK1/2 inhibitor, was
utilized to investigate the role of ERK1/2 in SFN-Cys-induced
apoptosis. Fig. 3B shows that the
phosphorylation of ERK1/2 was significantly diminished in cells
treated with both SFN-Cys (30 µM) and PD98059 (25 µM) compared with
the cells treated with SFN-Cys alone.
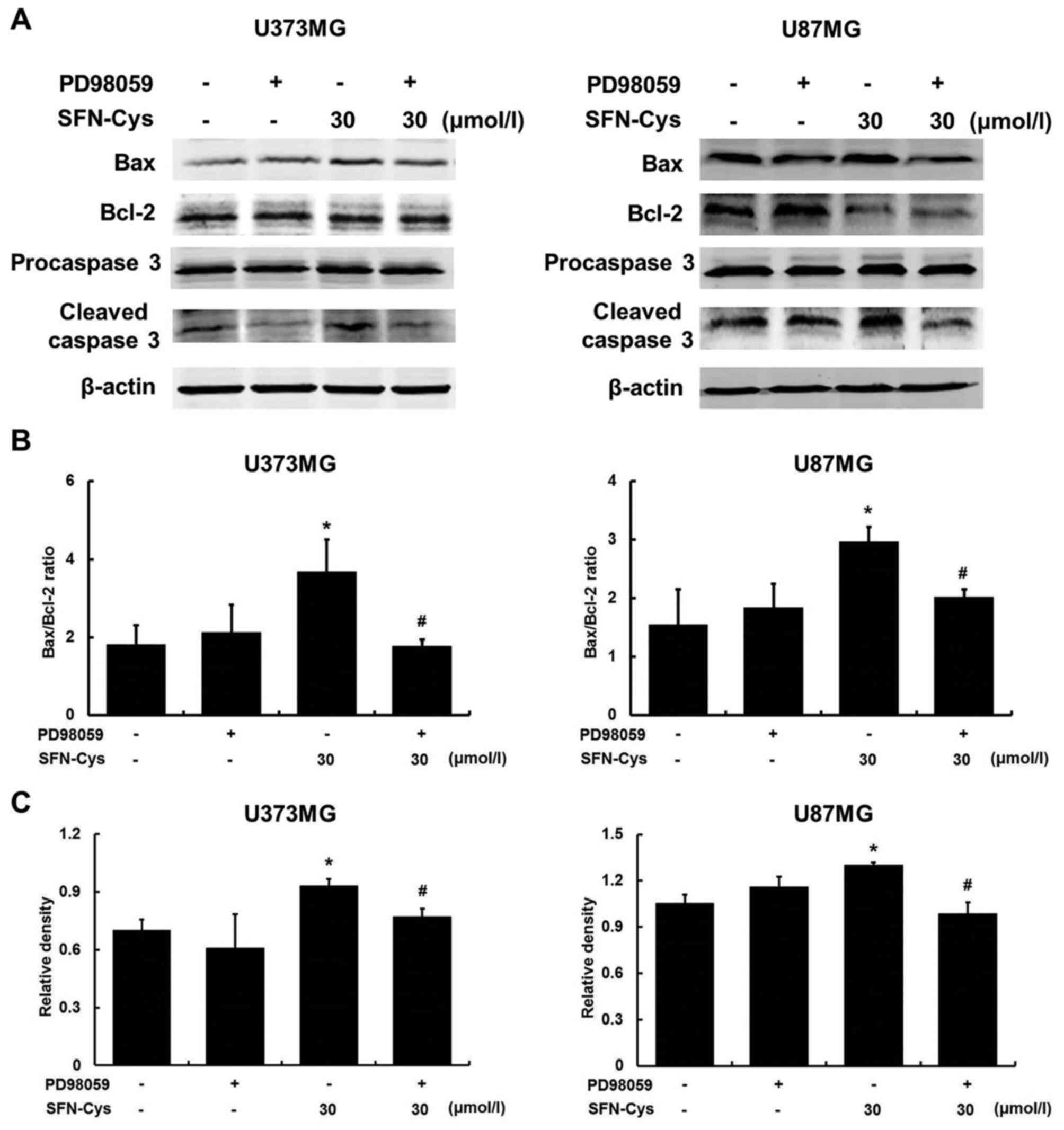
SFN-Cys increases the ratio of
Bax/Bcl-2 and upregulates cleaved caspase 3 by activating
ERK1/2
Western blot analysis was utilized to evaluate the
expression of the apoptosis regulatory proteins. We have already
demonstrated that PD98059 blocked the ERK1/2 signaling pathway
activated by SFN-Cys (Fig. 3B). As
shown in Fig. 4, the ratio of
Bax/Bcl-2 was increased in SFN-Cys-treated cells while the
increased ratio was reversed by PD98059 in SFN-Cys with the PD98059
treated cells, indicating that Bax and Bcl-2 were the downstream
effectors of ERK1/2. After cells were treated with SFN-Cys for 24
h, the expression of cleaved caspase 3 was significantly increased
compared with the untreated cells. PD98059 reversed the expression
of cleaved caspase 3 in both PD98059 and SFN-Cys treated cells
compared with SFN-Cys only treated cells, suggesting that SFN-Cys
induced cell apoptosis through sustained activation of ERK1/2 and
subsequently activating caspase 3 in U373MG and U87MG cells.
Altogether, these results indicated that SFN-Cys activated ERK1/2,
increasing the ratio of Bax/Bcl-2 and upregulating cleaved caspase
3 in U373MG and U87MG cells.
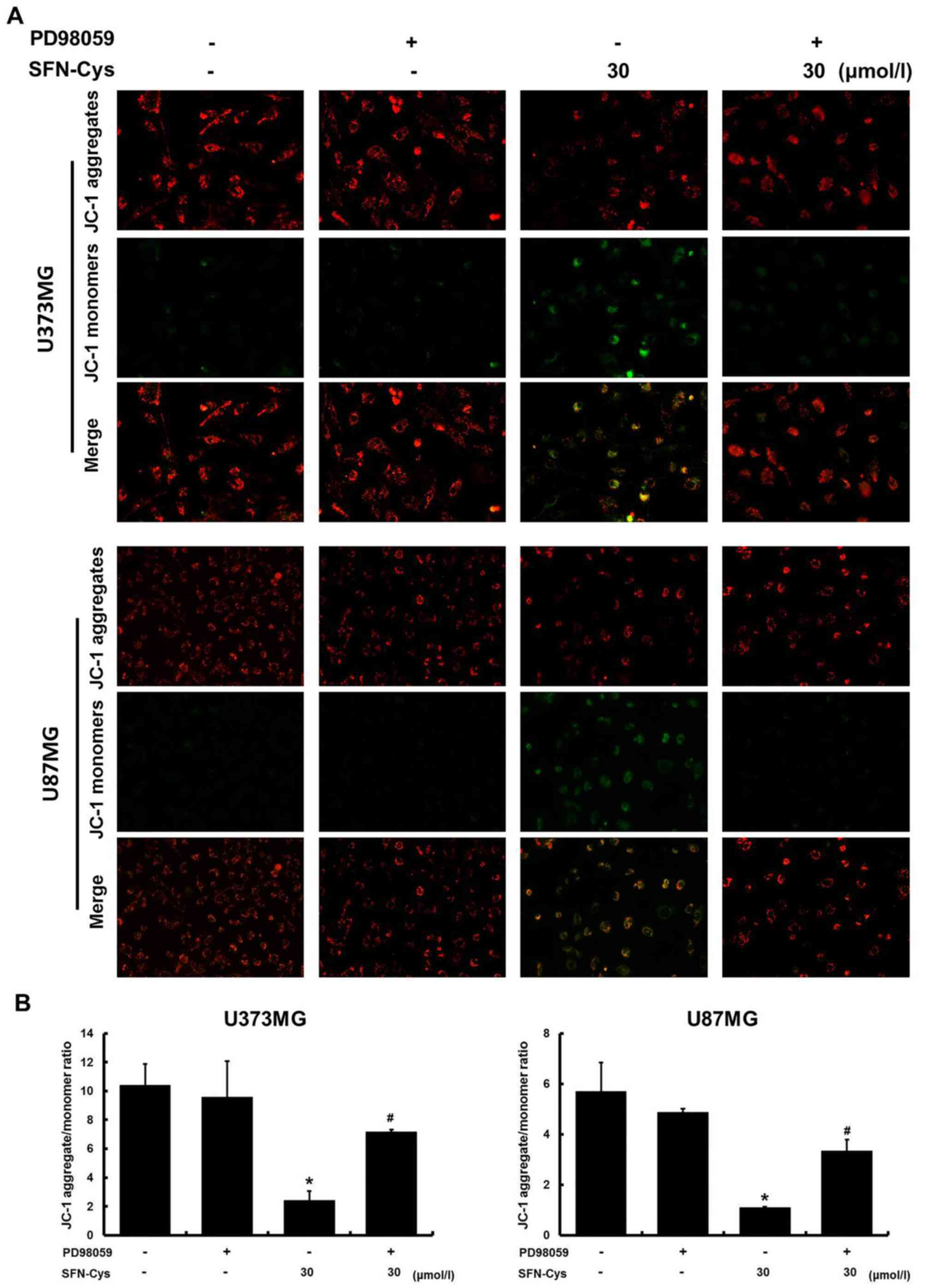
SFN-Cys induces the loss of
mitochondrial membrane potential (MMP) by activating ERK1/2
In order to confirm the intrinsic apoptosis pathway
which was induced by SFN-Cys, JC-1 mitochondrial membrane potential
assay was implemented to analyze mitochondrial membrane potential.
Red fluorescence indicated JC-1 aggregated in cells with high MMP,
while green fluorescence indicated that JC-1 was presented as a
monomer in cells with depolarized mitochondria. In Fig. 5A, the green fluorescence was
increased and the red fluorescence was decreased in SFN-Cys only
treated cells compared with that in untreated cells. Compared with
SFN-Cys only treated cells, both PD98059 and SFN-Cys treated cells
exhibited decreased green fluorescence and the restoration of the
red fluorescence. As shown in Fig.
5B, the level of MMP in SFN-Cys only treated cells was
significantly decreased compared with the untreated cells. PD98059
reversed the level of MMP in both SFN-Cys and PD98059 treated cells
compared with SFN-Cys only treated cells, thus indicating that
SFN-Cys induced the loss of MMP which could be reversed by PD98059.
Taken together, SFN-Cys induced the loss of MMP by activating
ERK1/2.
Discussion
Dietary bioactive components of natural products
have been extensively studied for cancer prevention and treatment
with low toxicity (37). Of all,
sulforaphane (SFN) was demonstrated to be the safest for human
cancer therapy (38). Here we
demonstrated that sulforaphane-cysteine (SFN-Cys), as a metabolite
of SFN in vivo, has potentiality to induce apoptosis. By
treating mice with 20 µmoles SFN, SFN was undetectable in brain by
2 h, while SFN-Cys was abundantly detectable in brain by 6 h,
indicating that SFN-Cys could penetrate the blood-brain barrier
(BBB) to target the tumor with a longer half-life compared with SFN
in vivo (8).
The specific hydroxamic acid group of SFN-Cys has
greater affinity for Zn2+ of histone deacetylase (HDAC)
while SFN has little effect, suggesting that SFN-Cys has the most
potentiality to inhibit HDAC (16).
Our previous results showed that SFN-Cys inhibited invasion in
human prostate cancer cells (20),
herein the underlying mechanisms by which SFN-Cys induced cell
apoptosis in human glioblastoma cells is demonstrated.
Both SFN-Cys and SFN could significantly decrease
cell viability and induce apoptosis at 30 µM as an optimal
concentration in U373MG cells and U87MG cells. The IC50
of SFN-Cys was approximately 45 µM (Fig. 1A) while the IC50 of SFN
was ~60 µM (12), suggesting that
SFN-Cys was a more effective anticarcinogen than SFN. In previous
studies, we demonstrated that ERK1/2 was sustainedly activated by
SFN-Cys (20 µM) within 48 h in U373MG and U87MG cell lines (data
not shown). Sustained activation of ERK1/2 mediated by SFN-Cys is a
key trigger to induce apoptosis. The downstream effectors might be
involved in apoptosis-related signaling pathways, such as the death
receptor pathway and the mitochondria-related pathways. We further
demonstrated that SFN-Cys (30 µM) induced cell apoptosis by
upregulating Bax/Bcl-2 ratio and causing the loss of mitochondrial
membrane potential (MMP) through activating ERK1/2. Apoptosis might
also involve multiple transcription factors, including early growth
response 1 (EGR-1) and nuclear factor-κB (NF-κB) (39–41).
EGR-1 is known to be activated by ERK1/2 and it can bind to the Bax
gene promoter to induce the expression of Bax (42,43).
Given that SFN activates EGR-1 in glioblastoma (44), SFN-Cys might upregulate Bax by
activating EGR-1 mediated by the ERK1/2 signaling pathway. NF-κB
can be downregulated by ERK1/2 and the activation of NF-κB
contributes to the upregulation of Bcl-2 (45,46).
Besides, it was shown that SFN suppressed the activation of NF-κB
to induce cell apoptosis, suggesting that SFN-Cys may downregulate
Bcl-2 by suppressing the activation of NF-κB mediated by
phosphorylating ERK1/2. We also demonstrated that SFN-Cys activated
caspase 3 via the ERK1/2 signaling pathway, however, whether
SFN-Cys activates caspase 3 by upregulating Bax/Bcl-2 ratio
mediated by ERK1/2 is still unclear. The possible cause of the
activation of caspase 3 is the release of cytochrome c from
the mitochondria regulated by Bax/Bcl-2 (22,47).
Bax, Bcl-2, caspase 3 and the disruption of MMP are the hallmarks
of intrinsic apoptosis pathway, suggesting that SFN-Cys induced
cell apoptosis in human glioblastoma cells via the intrinsic
apoptosis pathway. Therefore, the underlying mechanism of caspase 3
activation is likely to be that the SFN-Cys-mediated increase of
Bax/Bcl-2 ratio acts as an apoptotic stimulus resulting in MMP
disruption, triggering the release of cytochrome c into the
cytosol which then activates caspase 9 and activates caspase 3
subsequently via the ERK1/2 signaling pathway. Alternatively, in
addition to cytochrome c, second mitochondria-derived
activator of caspases (Smac) can be released into the cytosol as
well and bind to the inhibitor of apoptosis (IAP) proteins to
deactivate them, resulting in caspase 3 activation and cell death
eventually (48). It can be
inferred that SFN-Cys may also induce cell apoptosis by triggering
the release of Smac and subsequently deactivating the IAP proteins
or just regulating the IAP proteins to deactivate them directly.
Apart from the intrinsic apoptosis pathway, apoptosis is also
sub-classified into two other types of death pathways; the death
receptor-mediated (extrinsic) pathway and the endoplasmic-reticulum
(ER) stress-mediated apoptosis pathway. The extrinsic pathway
involves death receptors, from the tumor necrosis factor (TNF)
superfamily, to transmit death signal from the surface to the
intracellular signaling pathways, leading to the activation of
caspase 8 and the subsequent activation of caspase 3 (49–51).
Also, the ER stress apoptosis pathway is triggered by the disturb
folding proteins in the ER, leading to the activation of caspase 3
eventually (52). Since these two
pathways could both be activated through the ERK1/2 signaling
pathway (52,53) and lead to the activation of caspase
3, we thought that SFN-Cys might induce cell apoptosis not only via
the intrinsic pathway but via the extrinsic pathway and the ER
stress-mediated pathway as well in human glioblastoma cells.
It is reported that caspase 3 executes cell
apoptosis, to demonstrate which, Abdi et al administered
z-VAD fmk (a pan-caspase inhibitor) and z-DEVD fmk (a selective
caspase 3 inhibitor) to cells, respectively, and both of the
inhibitors resulted in complete inhibition of apoptosis (54). It can be inferred that activated
caspase 3 was the executioner to cell apoptosis. Studies also
showed that caspase 3 could cleave poly (ADP-ribose) polymerase
(PARP) to deprive its abilities of detecting and repairing DNA
damage, leading to cell apoptosis ultimately (35,55).
The above suggested that the activated caspase 3 which was induced
by SFN-Cys could cleave the specific substrates as a proteolytic
enzyme, executing cell apoptosis. Thus, caspase 3 might cleave some
other substrates in cancer cells as well, such as microtubule
associated proteins which play a crucial role in cell apoptosis. In
separate studies, we found that caspase 3 cleaved α-tubulin
resulting in cell apoptosis (data not shown). The proteolytic
activity of caspase 3 is also closely related to the
ubiquitin-proteasome system (UPS): caspase 3 cleavage produces
substrates such as actomyosin resulting in an increase of
proteasome-mediated proteolysis (56). Given that SFN-Cys-activated caspase
3 might cleave α-tubulin to increase proteolysis of UPS, this
excessive proteolysis will result in microtubule depolymerization
and apoptosis (57). Taxol was used
as a microtubule-stabilizing agent to prevent cancers, however, the
glioblastoma cells often resist it (58). Luckily, it is reported that SFN
could induce cell apoptosis in Taxol-resistant cells (59). These studies suggested that SFN-Cys
might be a much more efficient microtubule-targeting agent compared
with the traditional agent such as Taxol due to its versatile
anticarcinogenic abilities. In addition, SFN can modulate
epigenetic events. Previous studies showed that SFN induced cell
apoptosis by regulating the level of epigenetic regulators such as
polycomb group (PcG) proteins which contribute to chromatin
structure modification and gene expression suppression. The
SFN-mediated reduction of PcG proteins in a proteasome-dependent
way led to an increase of cleaved caspase 3 and cell apoptosis
(60). This was consistent with our
hypothesis that SFN-Cys-mediated proteolysis of UPS led to cell
apoptosis. Thus, we inferred that SFN-Cys might also activate
caspase 3 and induce cell apoptosis by modulating the epigenetic
regulators mediated via UPS. Moreover, HDAC inhibitor increased the
level of caspase 3 mRNA (61). This
evidence suggested that the expression of caspase 3 could be
epigenetically regulated by HDAC. Recently, HDAC inhibitors are
used in epigenetic glioblastoma therapies (62). Given that SFN-Cys was the most
potential HDAC inhibitor among SFN and its metabolites, we think
that SFN-Cys might induce cell apoptosis through the epigenetic
regulation of caspase 3. Therefore, there are multiple ways for
SFN-Cys to suppress the tumor progression and further studies
needed to be done.
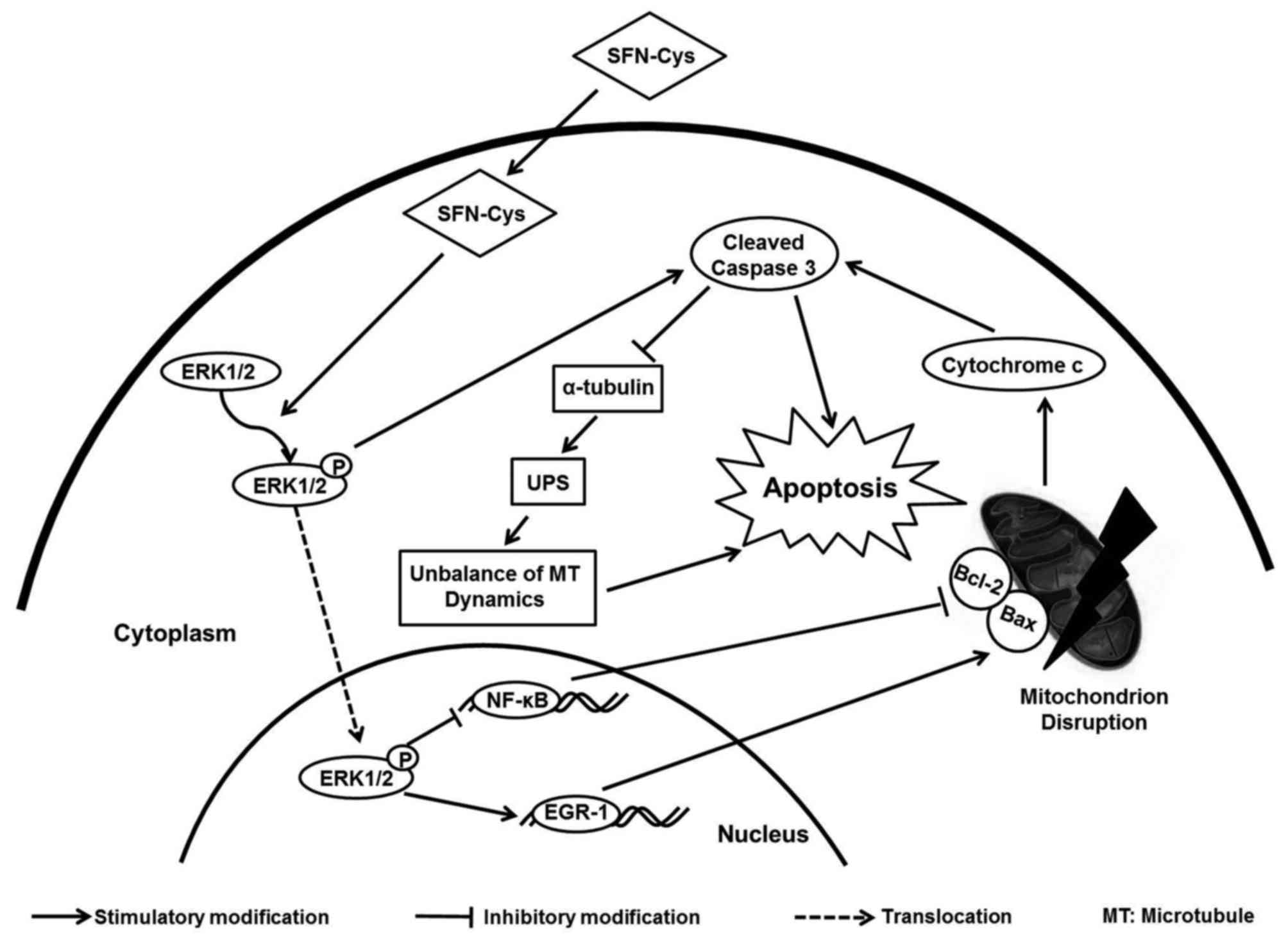
In conclusion, SFN-Cys upregulated Bax/Bcl-2 ratio
and activated caspase 3 subsequently via sustained activating
ERK1/2 signaling, leading to intrinsic apoptosis in human
glioblastoma U373MG and U87MG cells (Fig. 6). The underlying mechanisms that
SFN-Cys triggered provided us with more thoughts and information to
develop new drugs to treat glioblastoma with high efficacy.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81272843 and 81601993).
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gielen PR, Aftab Q, Ma N, Chen VC, Hong X,
Lozinsky S, Naus CC and Sin WC: Connexin43 confers Temozolomide
resistance in human glioma cells by modulating the mitochondrial
apoptosis pathway. Neuropharmacology. 75:539–548. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alifieris C and Trafalis DT: Glioblastoma
multiforme: Pathogenesis and treatment. Pharmacol Ther. 152:63–82.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Razis Abdull AF and Noor NM: Cruciferous
vegetables: Dietary phytochemicals for cancer prevention. Asian Pac
J Cancer Prev. 14:1565–1570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clarke JD, Hsu A, Yu Z, Dashwood RH and Ho
E: Differential effects of sulforaphane on histone deacetylases,
cell cycle arrest and apoptosis in normal prostate cells versus
hyperplastic and cancerous prostate cells. Mol Nutr Food Res.
55:999–1009. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pham NA, Jacobberger JW, Schimmer AD, Cao
P, Gronda M and Hedley DW: The dietary isothiocyanate sulforaphane
targets pathways of apoptosis, cell cycle arrest, and oxidative
stress in human pancreatic cancer cells and inhibits tumor growth
in severe combined immunodeficient mice. Mol Cancer Ther.
3:1239–1248. 2004.PubMed/NCBI
|
|
7
|
Karmakar S, Weinberg MS, Banik NL, Patel
SJ and Ray SK: Activation of multiple molecular mechanisms for
apoptosis in human malignant glioblastoma T98G and U87MG cells
treated with sulforaphane. Neuroscience. 141:1265–1280. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Clarke JD, Hsu A, Williams DE, Dashwood
RH, Stevens JF, Yamamoto M and Ho E: Metabolism and tissue
distribution of sulforaphane in Nrf2 knockout and wild-type mice.
Pharm Res. 28:3171–3179. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dinkova-Kostova AT and Kostov RV:
Glucosinolates and isothiocyanates in health and disease. Trends
Mol Med. 18:337–347. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yao A, Shen Y, Wang A, Chen S, Zhang H,
Chen F, Chen Z, Wei H, Zou Z, Shan Y, et al: Sulforaphane induces
apoptosis in adipocytes via Akt/p70s6k1/Bad inhibition and ERK
activation. Biochem Biophys Res Commun. 465:696–701. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang L, Tian Z, Yang Q, Li H, Guan H, Shi
B, Hou P and Ji M: Sulforaphane inhibits thyroid cancer cell growth
and invasiveness through the reactive oxygen species-dependent
pathway. Oncotarget. 6:25917–25931. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li C, Zhou Y, Peng X, Du L, Tian H, Yang
G, Niu J and Wu W: Sulforaphane inhibits invasion via activating
ERK1/2 signaling in human glioblastoma U87MG and U373MG cells. PLoS
One. 9:e905202014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peng X, Zhou Y, Tian H, Yang G, Li C, Geng
Y, Wu S and Wu W: Sulforaphane inhibits invasion by phosphorylating
ERK1/2 to regulate E-cadherin and CD44v6 in human prostate cancer
DU145 cells. Oncol Rep. 34:1565–1572. 2015.PubMed/NCBI
|
|
14
|
Shankar S, Ganapathy S and Srivastava RK:
Sulforaphane enhances the therapeutic potential of TRAIL in
prostate cancer orthotopic model through regulation of apoptosis,
metastasis, and angiogenesis. Clin Cancer Res. 14:6855–6866. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Z, Li C, Shang L, Zhang Y, Zou R,
Zhan Y and Bi B: Sulforaphane induces apoptosis and inhibits
invasion in U251MG glioblastoma cells. Springerplus. 5:2352016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Myzak MC, Karplus PA, Chung FL and
Dashwood RH: A novel mechanism of chemoprotection by sulforaphane:
Inhibition of histone deacetylase. Cancer Res. 64:5767–5774. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deschênes-Simard X, Kottakis F, Meloche S
and Ferbeyre G: ERKs in cancer: Friends or foes? Cancer Res.
74:412–419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tong WG, Ding XZ, Talamonti MS, Bell RH
and Adrian TE: LTB4 stimulates growth of human pancreatic cancer
cells via MAPK and PI-3 kinase pathways. Biochem Biophys Res
Commun. 335:949–956. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee WJ, Hsiao M, Chang JL, Yang SF, Tseng
TH, Cheng CW, Chow JM, Lin KH, Lin YW, Liu CC, et al: Quercetin
induces mitochondrial-derived apoptosis via reactive oxygen
species-mediated ERK activation in HL-60 leukemia cells and
xenograft. Arch Toxicol. 89:1103–1117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tian H, Zhou Y, Yang G, Geng Y, Wu S, Hu
Y, Lin K and Wu W: Sulforaphane-cysteine suppresses invasion via
downregulation of galectin-1 in human prostate cancer DU145 and PC3
cells. Oncol Rep. 36:1361–1368. 2016.PubMed/NCBI
|
|
21
|
Manero F, Gautier F, Gallenne T, Cauquil
N, Grée D, Cartron PF, Geneste O, Grée R, Vallette FM and Juin P:
The small organic compound HA14-1 prevents Bcl-2 interaction with
Bax to sensitize malignant glioma cells to induction of cell death.
Cancer Res. 66:2757–2764. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cartron PF, Oliver L, Martin S, Moreau C,
LeCabellec MT, Jezequel P, Meflah K and Vallette FM: The expression
of a new variant of the pro-apoptotic molecule Bax, Baxpsi, is
correlated with an increased survival of glioblastoma multiforme
patients. Hum Mol Genet. 11:675–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
George J, Banik NL and Ray SK: Combination
of taxol and Bcl-2 siRNA induces apoptosis in human glioblastoma
cells and inhibits invasion, angiogenesis and tumour growth. J Cell
Mol Med. 13:4205–4218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wick W, Grimmel C, Wild-Bode C, Platten M,
Arpin M and Weller M: Ezrin-dependent promotion of glioma cell
clonogenicity, motility, and invasion mediated by BCL-2 and
transforming growth factor-beta2. J Neurosci. 21:3360–3368.
2001.PubMed/NCBI
|
|
25
|
Thomas S, Quinn BA, Das SK, Dash R, Emdad
L, Dasgupta S, Wang XY, Dent P, Reed JC, Pellecchia M, et al:
Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther
Targets. 17:61–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van Delft MF and Huang DC: How the Bcl-2
family of proteins interact to regulate apoptosis. Cell Res.
16:203–213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yin XM, Oltvai ZN and Korsmeyer SJ: BH1
and BH2 domains of Bcl-2 are required for inhibition of apoptosis
and heterodimerization with Bax. Nature. 369:321–323. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi L, Chen J, Yang J, Pan T, Zhang S and
Wang Z: MiR-21 protected human glioblastoma U87MG cells from
chemotherapeutic drug temozolomide induced apoptosis by decreasing
Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 1352:255–264.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Skała E, Sitarek P, Toma M, Szemraj J,
Radek M, Nieborowska-Skorska M, Skorski T, Wysokińska H and
Śliwiński T: Inhibition of human glioma cell proliferation by
altered Bax/Bcl-2-p53 expression and apoptosis induction by
Rhaponticum carthamoides extracts from transformed and
normal roots. J Pharm Pharmacol. 68:1454–1464. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yating Q, Yuan Y, Wei Z, Qing G, Xingwei
W, Qiu Q and Lili Y: Oxidized LDL induces apoptosis of human
retinal pigment epithelium through activation of ERK-Bax/Bcl-2
signaling pathways. Curr Eye Res. 40:415–422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nuñez G, Benedict MA, Hu Y and Inohara N:
Caspases: The proteases of the apoptotic pathway. Oncogene.
17:3237–3245. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Walters J, Pop C, Scott FL, Drag M, Swartz
P, Mattos C, Salvesen GS and Clark AC: A constitutively active and
uninhibitable caspase-3 zymogen efficiently induces apoptosis.
Biochem J. 424:335–345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu SW, Wang H, Poitras MF, Coombs C,
Bowers WJ, Federoff HJ, Poirier GG, Dawson TM and Dawson VL:
Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by
apoptosis-inducing factor. Science. 297:259–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang TY, Chang WC, Wang MY, Yang YR and
Hsu YC: Effect of sulforaphane on growth inhibition in human brain
malignant glioma GBM 8401 cells by means of mitochondrial- and
MEK/ERK-mediated apoptosis pathway. Cell Biochem Biophys.
63:247–259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang H, Shang X, Wu H, Huang G, Wang Y,
Al-Holou S, Gautam SC and Chopp M: Combination treatment with
resveratrol and sulforaphane induces apoptosis in human U251 glioma
cells. Neurochem Res. 35:152–161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shapiro TA, Fahey JW, Dinkova-Kostova AT,
Holtzclaw WD, Stephenson KK, Wade KL, Ye L and Talalay P: Safety,
tolerance, and metabolism of broccoli sprout glucosinolates and
isothiocyanates: A clinical phase I study. Nutr Cancer. 55:53–62.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Alejandro EU and Johnson JD: Inhibition of
Raf-1 alters multiple downstream pathways to induce pancreatic
beta-cell apoptosis. J Biol Chem. 283:2407–2417. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yuan S, Wen J, Cheng J, Shen W, Zhou S,
Yan W, Shen L, Luo A and Wang S: Age-associated up-regulation of
EGR1 promotes granulosa cell apoptosis during follicle atresia in
mice through the NF-κB pathway. Cell Cycle. 15:2895–2905. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou Z, Lu X, Wang J, Xiao J, Liu J and
Xing F: microRNA let-7c is essential for the anisomycin-elicited
apoptosis in Jurkat T cells by linking JNK1/2 to AP-1/STAT1/STAT3
signaling. Sci Rep. 6:244342016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Al-Sarraj A and Thiel G: Substance P
induced biosynthesis of the zinc finger transcription factor Egr-1
in human glioma cells requires activation of the epidermal growth
factor receptor and of extracellular signal-regulated protein
kinase. Neurosci Lett. 332:111–114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li L, Zhao LM, Dai SL, Cui WX, Lv HL, Chen
L and Shan BE: Periplocin extracted from cortex periplocae
induced apoptosis of gastric cancer cells via the ERK1/2-EGR1
pathway. Cell Physiol Biochem. 38:1939–1951. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang M, Teng W, Qu Y, Wang H and Yuan Q:
Sulforaphene inhibits triple negative breast cancer through
activating tumor suppressor Egr1. Breast Cancer Res Treat.
158:277–286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang DX, Ma DY, Yao ZQ, Fu CY, Shi YX,
Wang QL and Tang QQ: ERK1/2/p53 and NF-κB dependent-PUMA activation
involves in doxorubicin-induced cardiomyocyte apoptosis. Eur Rev
Med Pharmacol Sci. 20:2435–2442. 2016.PubMed/NCBI
|
|
46
|
Wang Q, Liu S, Tang Y, Liu Q and Yao Y:
MPT64 protein from Mycobacterium tuberculosis inhibits apoptosis of
macrophages through NF-κB-miRNA21-Bcl-2 pathway. PLoS One.
9:e1009492014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sitarek P, Skała E, Toma M, Wielanek M,
Szemraj J, Nieborowska-Skorska M, Kolasa M, Skorski T, Wysokińska H
and Śliwiński T: A preliminary study of apoptosis induction in
glioma cells via alteration of the Bax/Bcl-2-p53 axis by
transformed and non-transformed root extracts of Leonurus
sibiricus L. Tumour Biol. 37:8753–8764. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fulda S and Vucic D: Targeting IAP
proteins for therapeutic intervention in cancer. Nat Rev Drug
Discov. 11:109–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ashkenazi A and Dixit VM: Death receptors:
Signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Indran IR, Tufo G, Pervaiz S and Brenner
C: Recent advances in apoptosis, mitochondria and drug resistance
in cancer cells. Biochim Biophys Acta. 1807:735–745. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death - apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Abdi A, Sadraie H, Dargahi L, Khalaj L and
Ahmadiani A: Apoptosis inhibition can be threatening in Aβ-induced
neuroinflammation, through promoting cell proliferation. Neurochem
Res. 36:39–48. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Virág L and Szabó C: The therapeutic
potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev.
54:375–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang XH and Mitch WE: Muscle wasting from
kidney failure - a model for catabolic conditions. Int J Biochem
Cell Biol. 45:2230–2238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sung M and Giannakakou P: BRCA1 regulates
microtubule dynamics and taxane-induced apoptotic cell signaling.
Oncogene. 33:1418–1428. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Giussani P, Bassi R, Anelli V, Brioschi L,
De Zen F, Riccitelli E, Caroli M, Campanella R, Gaini SM, Viani P,
et al: Glucosylceramide synthase protects glioblastoma cells
against autophagic and apoptotic death induced by temozolomide and
Paclitaxel. Cancer Invest. 30:27–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chen H, Landen CN, Li Y, Alvarez RD and
Tollefsbol TO: Epigallocatechin gallate and sulforaphane
combination treatment induce apoptosis in paclitaxel-resistant
ovarian cancer cells through hTERT and Bcl-2 down-regulation. Exp
Cell Res. 319:697–706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Balasubramanian S, Chew YC and Eckert RL:
Sulforaphane suppresses polycomb group protein level via a
proteasome-dependent mechanism in skin cancer cells. Mol Pharmacol.
80:870–878. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yakovlev A, Khafizova M, Abdullaev Z,
Loukinov D and Kondratyev A: Epigenetic regulation of caspase-3
gene expression in rat brain development. Gene. 450:103–108. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lee P, Murphy B, Miller R, Menon V, Banik
NL, Giglio P, Lindhorst SM, Varma AK, Vandergrift WA III, Patel SJ,
et al: Mechanisms and clinical significance of histone deacetylase
inhibitors: Epigenetic glioblastoma therapy. Anticancer Res.
35:615–625. 2015.PubMed/NCBI
|