Introduction
Esophageal cancer (EC) is the sixth leading cause of
cancer-related mortality and the eighth most frequently diagnosed
cancer, worldwide. There are 481,000 new cases of esophageal cancer
each year, which also indicates a rapidly increasing incidence rate
(1,2). Based on histology, esophageal cancer
can be mainly categorized into esophageal squamous cell carcinoma
(ESCC) and esophageal adenocarcinoma (EAC), which accounts for ~90%
of all esophageal cancers. Compared with EAC, ESCC has a higher
incidence worldwide, particularly in the so-called Asian belt
(Turkey, Northeastern Iran, Kazakhstan and Northern and Central
China), where ESCC has been noted to account for ~90% of the total
EC cases (3). Such as other
carcinomas, ESCC is likewise a complex and heterogeneous
disease.
Molecular markers in EC patients have been widely
studied, and a meta-analysis summarized the results of published
studies regarding the prognostic role (4), including 109 studies for 13 different
markers. It showed that VEGF, cyclin D1, Ki-67 and squamous cell
carcinoma antigen can be used in ESCC, and COX-2, HER-2 in EAC as
prediction markers for overall survival. In the early event of
ESCC, DNA hypermethylation is frequently found in promoter regions,
and some molecular markers such as DAPK, p16, MGMT, MLH1, RARβ2,
HIN1, TFPI-2, DACH1 and SOX17 were found methylated in the
precursor lesions of human esophageal epithelia (5–10).
Others, such as CDH1, RASSF1A, P16 and FHIT were found to be
involved in tumor progression and poor prognosis (11–15).
Even though some molecules may serve as biomarkers, little is known
concerning the carcinogenesis mechanism of ESCC, particularly the
transcriptional regulatory network. Thus, more integrative,
systems-level analyses are necessary in order to better understand
how ESCC develops and progresses, and how it may respond to
different therapeutic interventions.
Gene expression profiling assay has been used to
explore the molecular mechanisms in various types of tumors, which
provides a powerful approach to examine the expression profiles of
virtually all known genes at once (16–19).
However, profiling analyses only detect differentially expressed
genes and provide limited insight into the underlying mechanism.
More information could be obtained only through integrated analysis
combining data from different dimensions.
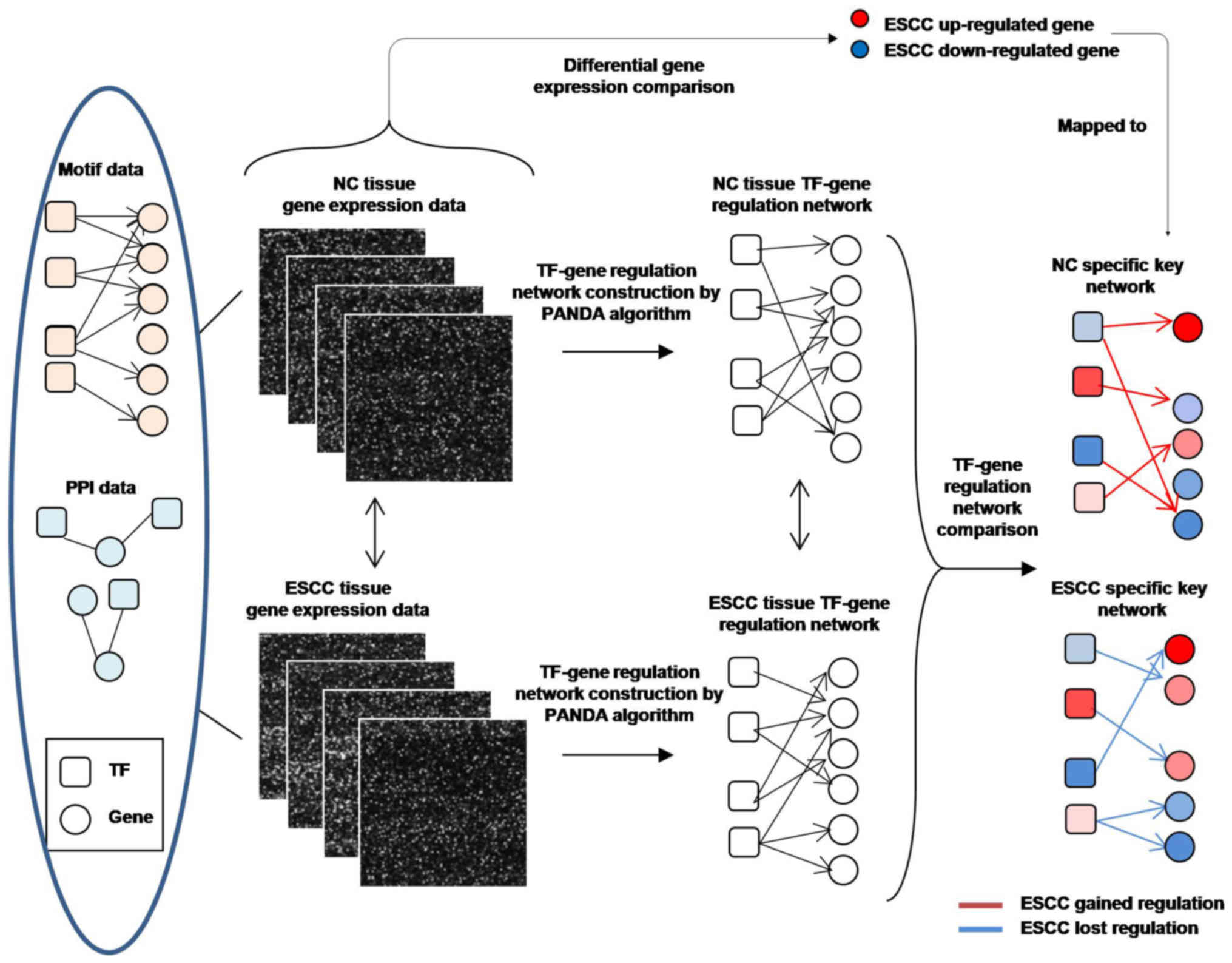
Passing Attributes between Networks for Data
Assimilation (PANDA) is an integrative network inference method
based on a message-passing approach (20,21).
It defines transcription factors (TFs) as transmitters and their
target genes as receivers. By integrating mRNA expression data with
the TF motif data, which link TFs with their potential targets, it
generates a Z-score reflecting the confidence level of regulatory
relationship for each TF-target edge. PANDA has been successfully
used to study several diseases including chronic obstructive
pulmonary disease (COPD), emphysema and ovarian cancer
In the present study, we applied PANDA for
constructing and comparing regulatory networks in ESCC and its
paired non-cancerous (NC) tissues, and integrate this information
with differential expression data to obtain a better understanding
of the carcinogenesis mechanism of ESCC, as well as identification
of new biomarkers.
Materials and methods
Tissue sample handling
For GeneChip analysis, ESCC and paired normal
esophageal mucosa samples were obtained from 8 ESCC patients who
underwent esophagectomy at Beijing Friendship Hospital from
September to December 2012. The median age of the patients when
diagnosed was 63 years (range, 46–73 years). Two of the 8 ESCC
patients were men, and 6 were women. Well-differentiated,
moderately differentiated, poorly differentiated ESCC were found in
1, 4 and 3 cases, respectively (more details are shown in Table I). Another 5 ESCC tissues from
patients who underwent esophagectomy at Beijing Friendship Hospital
from November 2016 to January 2017 were recruited as a validation
cohort. Study protocols were approved by the Ethics Committee of
the Affiliated Hospital of Capital Medical University, and all
experiments were performed in accordance with approved guidelines
of the Affiliated Hospital of Capital Medical University. Written
informed consent was obtained from the patients for publication of
the present study and any accompanying images.
 | Table I.Demographic and pathological
characteristics of the 8 ESCC patients. |
Table I.
Demographic and pathological
characteristics of the 8 ESCC patients.
| Pt. no. | Sex | Age | Differentiation | TNM stage | AJCC stage |
|---|
| 1 | M | 63 | Poor | T3N1M0 | III |
| 2 | M | 72 | Moderate | T3N0M0 | IIA |
| 3 | M | 46 | Moderate | T1N0M0 | I |
| 4 | M | 55 | Moderate | T1N1M0 | IIA |
| 5 | M | 59 | Well | T1N0M0 | I |
| 6 | M | 51 | Poor | T2N1M0 | IIB |
| 7 | F | 68 | Moderate | T4N1M0 | III |
| 8 | F | 53 | Poor | T3N1M0 | III |
Isolation and GeneChip analysis
Total RNA extraction, quality detection, GeneChip
assay protocols as well as preliminary analysis reports of
differentially expressed gene were described in our previous study
(22). All CEL raw data were
background corrected by the RMA method. Then, log2 transformation
along with quantile normalization were also subsequently
applied.
TF-target network construction and
comparison
We used PANDA models to evaluate the regulatory
relationship between TFs and their targeted genes in both ESCC and
NC tissues (Fig. 1). A ‘prior’
regulatory network by mapping transcription factor motifs to a
reference genome was derived from JASPAR database. For
protein-protein interactions (PPI), a publicly available dataset
was used as an estimate. Then, subnetworks of edges that are most
distinct between each pair of our reconstructed network models were
identified by AnaPANDA program (details are shown in the Results
section).
Co-activation and co-repression
analysis
ESCC- and NC-specific edges were respectively
extracted for co-activation and co-repression analysis. All
extracted edges linked with a differentially expressed target were
selected for further analysis. For each 2-paired TFs, a Fisher's
exact test was performed and a p-value was calculated to evaluate
ESCC-specific co-activation effects and NC-specific co-activation
effects (ESCC-specific co-repression effects).
RT-qPCR validation of gene
expression
Five ESCC and paired NC tissues of the validation
cohort were handled in the same way as microarray assays, as
previously described (22). cDNA
was synthesized from ~1 µg RNA and qPCR settings were 94°C for 2
min followed by 40 cycles of 94°C for 15 sec, 60°C for 20 sec and
72°C for 30 sec, and then followed by 72°C for 2 min.
Statistical analysis and data
visualization
All statistical tests were performed using R 3.3.1
software (www.r-project.org). All statistical
tests were two-tailed and p<0.05 was considered statistically
significant. Venn diagram, ggplot2 and pheatmap R packages were
used for data visualization.
Results
Building TF-target regulatory networks
of ESCC and NC tissues
ESCC and paired NC samples were obtained from 8 ESCC
patients who underwent esophagectomy. mRNA was extracted and gene
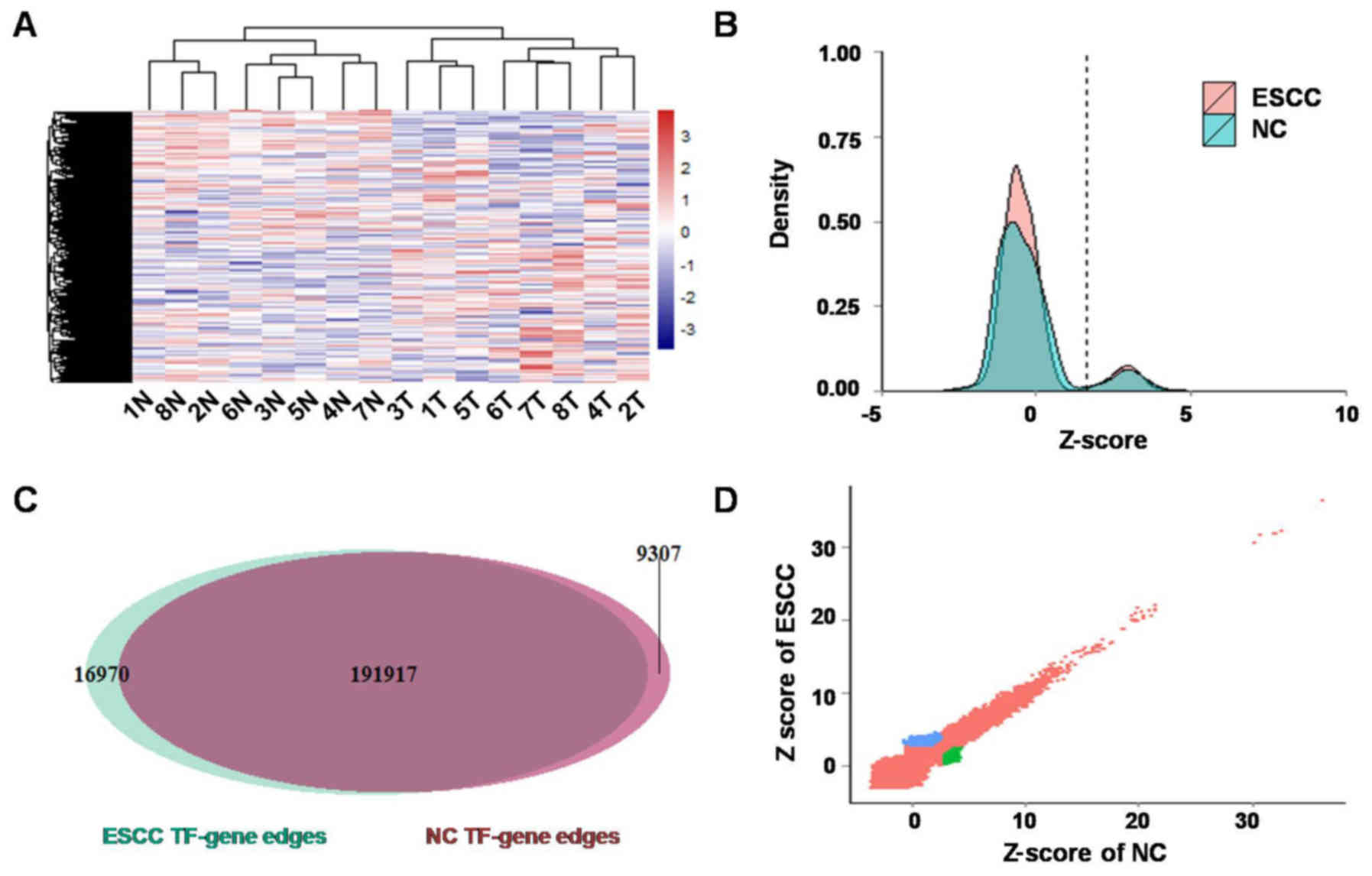
profiling data were obtained. Firstly, gene expression differences
between ESCC and NC were evaluated by paired t-test, and a cut-off
of FDR adjusted p<0.05 was used to determine significant
differentially expressed genes. There were 1,116 upregulated genes
and 1,301 downregulated genes in ESCC compared with NC (Fig. 2A). Then, TF-target regulatory
networks for ESCC and NC were constructed by PANDA integrating gene
profiling data with the TF motif data. TF-target edges (208,887 and
201,224) were identified in ESCC and NC, respectively, among which
16,970 and 9,307 edges were ESCC- and NC-specific (Fig. 2B-D).
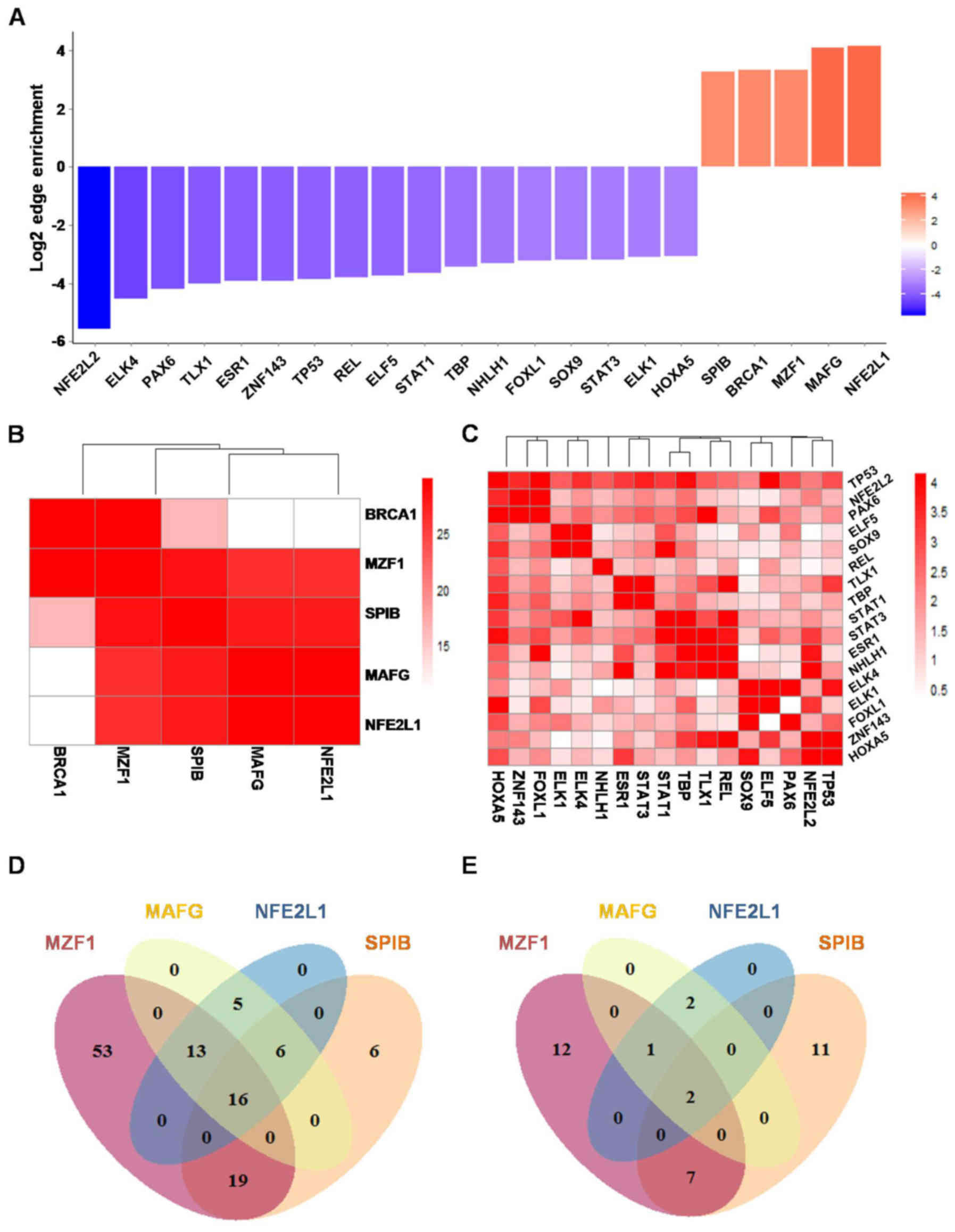
Edge enrichment of TFs in ESCC
In order to find driver TFs which are specifically
activated in ESCC, an edge enrichment score and an associated
p-value derived from AnaPANDA were generated for each TF, to assess
its activity change in ESCC compared to NC. Twenty-two TFs with
absolute value of edge enrichment score >8 and p-value <0.05
were considered activity changed TFs; 17 of them (NFE2L2, ELK4,
PAX6, TLX1, ESR1, ZNF143, TP53, REL, ELF5, STAT1, TBP, NHLH1,
FOXL1, SOX9, STAT3, ELK1, and HOXA5) were repressed in ESCC and 5
(SPIB, BRCA1, MZF1, MAFG and NFE2L1) were activated in ESCC
(Fig. 3A).
Co-activation of 4 TFs were identified
in ESCC
To evaluate co-activation and co-repression effect
among those TFs, hypergeometric distribution model-based target
profile similarity analysis was performed. Only ESCC- and
NC-specific edges and significantly differentially expressed
targets were included in this model. SPIB, MZF1, MAFG and NFE2L1
exhibited a strong co-activation effect with each other (Fig. 3B; p-value <1×10−25).
TP53, NFE2L2 and PAXB were found to be moderately co-repressed in
ESCC (Fig. 3C; p-value
<1×10−4). STAT1, STAT3, ESR1 and NHLH1 also were
found to be moderately co-repressed in ESCC (Fig. 3C; p-value
<1×10−4).
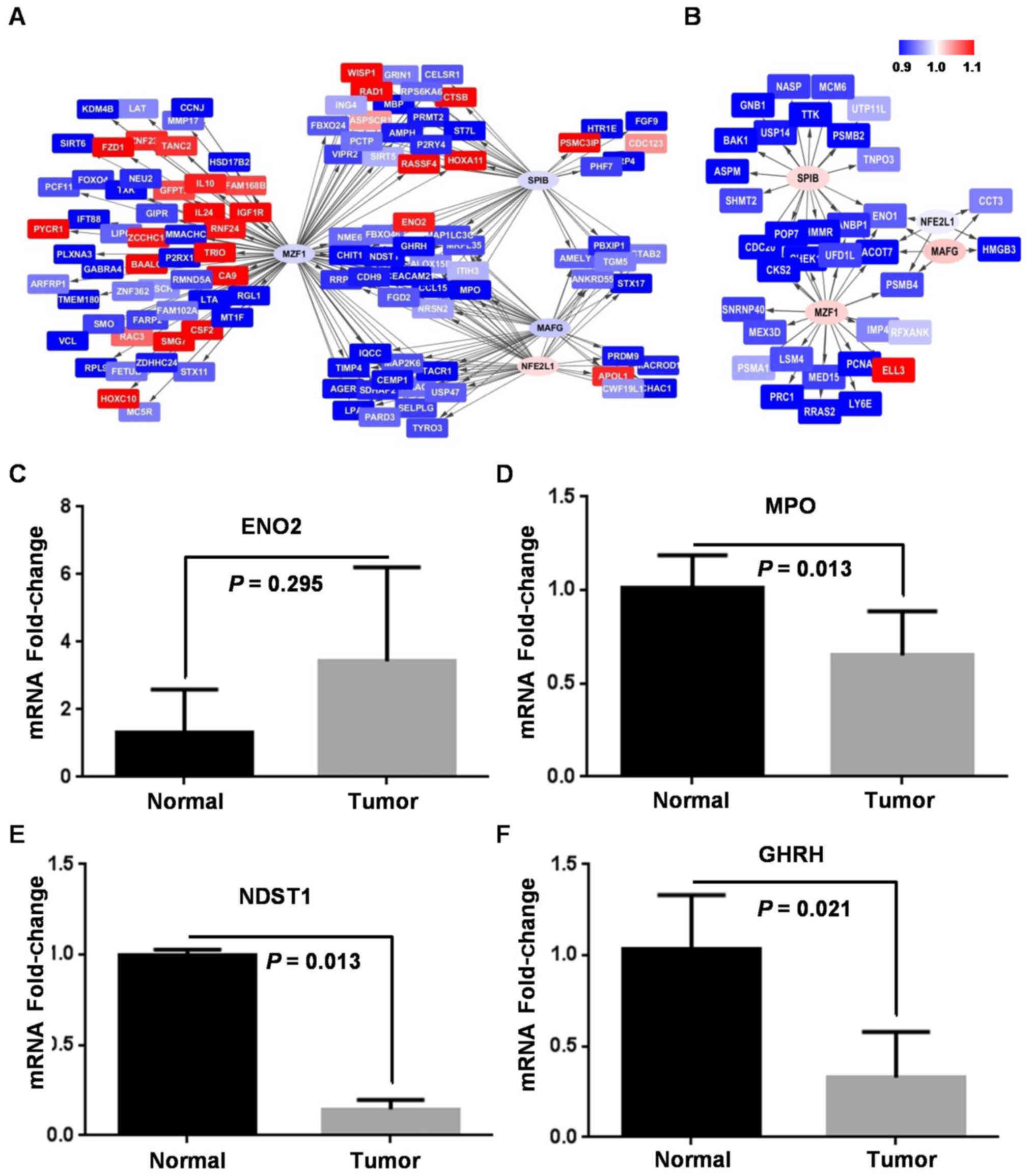
Specific TF-target regulatory network
was identified in ESCC
To elucidate core ESCC TF-target regulatory network,
the overlap of targets among the 4 co-activated TFs in ESCC was
further investigated. Among all 118 genes targeted at least by one
of SPIB, MZF1, MAFG and NFE2L1, there were 16 genes co-targeted by
all the 4 TFs, 19 co-targeted by three, 24 co-targeted by 2 of them
in ESCC (Fig. 3D). However, there
were only 2 genes co-targeted by all the 4 TFs, 1 co-targeted by
three, 9 co-targeted by 2 of them in NC (Fig. 3E). Finally, a network diagram was
shown to provide intuitive insight into this ESCC-specific
TF-target regulatory network (Fig. 4A
and B).
Furthermore, only 2 out of 16 genes co-targeted by
the 4 TFs were significantly differentially expressed between stage
I/II (n=5) and stage III (n=3) ESCC, and only 1 gene co-targeted by
the 4 TFs was significantly differentially expressed between
well/moderately (n=5) and poorly (n=3) differentiated ESCC. These
results indicated that co-activation of the 4 TFs is not associated
with ESCC stage or grade of differentiation.
To further validate the ESCC-specific network,
expression of 4 co-targeted genes of the 4 TFs was quantified by
qPCR assays in an independent ESCC cohort. ENO2 was ~3-fold
upregulated in ESCC (paired t=1.203, p=0.295), while MPO (paired
t=8.639, p=0.013), NDST1 (paired t=48.851, p=0.013), and CDH9
(paired t=4.457, p=0.021) were significantly downregulated in ESCC
(Fig. 4C-F). Expression of all the
co-targeted genes was in accordance with their expression in our
predicated network.
Discussion
There have been numerous studies of high-throughput
omics analysis in cancer. However, studies focusing on regulatory
spectrums are not common. For cancers with a low incidence in
developed areas, such as ESCC, transcriptional regulatory network
analysis is lacking.
By combining multiple sources of data to model
TF-target regulatory networks, integrative analyses showed strong
power in the investigation of pathologic mechanisms (23,24).
PANDA is a well-established integrative analysis tool which is
successfully used to study chronic disease (25) and cancer (20). PANDA predicts TF-gene regulatory
relationships based on information from gene expression and
TF-sequence-motif data with a message-passing approach. In the
present study, we used PANDA and its appended program AnaPANDA to
investigate transcriptional regulatory networks in ESCC and NC
tissues. We found 1,116 upregulated genes and 1,301 downregulated
genes in ESCC and identified 16,970 ESCC-specific TF-target edges
and 9,307 NC-specific TF-target edges.
Further edge enrichment analysis found 17 TFs
repressed in ESCC and 5 TFs activated in ESCC. Co-activation and
co-repression effects were also identified in these TFs, which
indicated that the change in transcriptional regulatory networks in
ESCC was coupled and not isolated. Four TFs, i.e. SPIB, MZF1, MAFG
and NFE2L1, exhibited a strong and significant co-activation effect
in ESCC, which was suggested to be an important TF regulatory
module in carcinogenesis of ESCC.
MZF-1, a Kruppel family protein, functions as a
crucial TF in hematopoietic development (26), which exerts oncogenic or
tumor-suppressive functions in various types of cancer. In
colorectal cancer (CRC) and gastric cancer (GC), MZF1 was found to
increase migration and invasion ability by facilitating
transcription of MMP-14 (27,28).
However, in cervical cancers, MZF1 repressed expression of MMP2 to
exert tumor-suppressive functions (29).
NFE2L1 is a member of the CNC-bZIP family, which has
been reported to regulate the mTORC1 signaling pathway and increase
cellular proteasome levels. Various studies have shown that NFE2L1
is involved in hypoxia and oxidative stress response of cancer
cells (30,31). In ESCC, the role of NFE2L1 is poorly
understood, and our results suggest a similar role as that in
gastroenterological tumors.
MAFG is a transcription factor of the Maf family
proteins (32), which have been
reported to regulate bile acid homeostasis (33). MAFG was also found to exert an
oncogenic function in hepatocellular carcinoma (HCC) and colorectal
cancer, enhance tumor invasiveness by upregulating β-catenin in HCC
and promote CpG island methylation in BRAF-mutant colorectal cancer
(34,35).
SPIB, known as a member of the Ets family,
participates in the differentiation of mature B-cells and
plasmacytoid dendritic cells (36–39),
which have been mostly reported in hematologic-associated cancers
with a role of mediating apoptosis via the PI3K-AKT pathway
(40). A gene profiling assay
showed that SPIB promotes the progression of gastric cancers
(41). Our results also suggested
that SPIB is oncogenic in ESCC.
These 4 TFs were found to be oncogenic in tumors of
the digestive system, but their co-activation effect was not
reported, nor were their effects in ESCC. Our results not only
indicate that they have a potential carcinogenic role in ESCC, but
also indicate that further investigation may contribute to the
further understanding of their co-regulation and co-targeting
profile in ESCC.
In conclusion, in the present study, we constructed
transcriptional regulatory networks of ESCC and NC tissues.
Combining these networks with a gene expressing analysis, we found
SPIB, MZF1, MAFG and NFE2L1 co-activated in ESCC, which could be an
important co-regulatory mechanism underlying the carcinogenesis of
ESCC.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81302160 and
81272447), and the Beijing Natural Science Foundation Program and
Scientific Research Key Program of Beijing Municipal Commission of
Education (grant no. KZ201410025024).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu HC, Yang X, Xu LP, Zhao LJ, Tao GZ,
Zhang C, Qin Q, Cai J, Ma JX, Mao WD, et al: Meat consumption is
associated with esophageal cancer risk in a meat- and
cancer-histological-type dependent manner. Dig Dis Sci. 59:664–673.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: Defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen M, Huang J, Zhu Z, Zhang J and Li K:
Systematic review and meta-analysis of tumor biomarkers in
predicting prognosis in esophageal cancer. BMC Cancer. 13:5392013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo M, Ren J, House MG, Qi Y, Brock MV and
Herman JG: Accumulation of promoter methylation suggests epigenetic
progression in squamous cell carcinoma of the esophagus. Clin
Cancer Res. 12:4515–4522. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jia Y, Yang Y, Zhan Q, Brock MV, Zheng X,
Yu Y, Herman JG and Guo M: Inhibition of SOX17 by microRNA 141 and
methylation activates the WNT signaling pathway in esophageal
cancer. J Mol Diagn. 14:577–585. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu L, Herman JG, Brock MV, Wu K, Mao G,
Yan W, Nie Y, Liang H, Zhan Q, Li W, et al: Silencing DACH1
promotes esophageal cancer growth by inhibiting TGF-β signaling.
PLoS One. 9:e955092014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo M, Ren J, Brock MV, Herman JG and
Carraway HE: Promoter methylation of HIN-1 in the progression to
esophageal squamous cancer. Epigenetics. 3:336–341. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jia Y, Yang Y, Brock MV, Cao B, Zhan Q, Li
Y, Yu Y, Herman JG and Guo M: Methylation of TFPI-2 is an early
event of esophageal carcinogenesis. Epigenomics. 4:135–146. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang JS, Guo M, Montgomery EA, Thompson
RE, Cosby H, Hicks L, Wang S, Herman JG and Canto MI: DNA promoter
hypermethylation of p16 and APC predicts neoplastic progression in
Barrett's esophagus. Am J Gastroenterol. 104:2153–2160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yun T, Liu Y, Gao D, Linghu E, Brock MV,
Yin D, Zhan Q, Herman JG and Guo M: Methylation of CHFR sensitizes
esophageal squamous cell cancer to docetaxel and paclitaxel. genes.
Cancer. 6:38–48. 2015.
|
|
12
|
Rong R, Jiang LY, Sheikh MS and Huang Y:
Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates
RASSF1A-mediated microtubule interaction and M-phase cell cycle
regulation. Oncogene. 26:7700–7708. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuroki T, Trapasso F, Yendamuri S,
Matsuyama A, Alder H, Mori M and Croce CM: Promoter
hypermethylation of RASSF1A in esophageal squamous cell carcinoma.
Clin Cancer Res. 9:1441–1445. 2003.PubMed/NCBI
|
|
14
|
Maesawa C, Tamura G, Nishizuka S,
Ogasawara S, Ishida K, Terashima M, Sakata K, Sato N, Saito K and
Satodate R: Inactivation of the CDKN2 gene by homozygous deletion
and de novo methylation is associated with advanced stage
esophageal squamous cell carcinoma. Cancer Res. 56:3875–3878.
1996.PubMed/NCBI
|
|
15
|
Lee EJ, Lee BB, Kim JW, Shim YM, Hoseok I,
Han J, Cho EY, Park J and Kim DH: Aberrant methylation of Fragile
Histidine Triad gene is associated with poor prognosis in early
stage esophageal squamous cell carcinoma. Eur J Cancer. 42:972–980.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barabási AL: Network medicine - from
obesity to the ‘diseasome’. N Engl J Med. 357:404–407. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Silverman EK and Loscalzo J: Developing
new drug treatments in the era of network medicine. Clin Pharmacol
Ther. 93:26–28. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Silverman EK and Loscalzo J: Network
medicine approaches to the genetics of complex diseases. Discov
Med. 14:143–152. 2012.PubMed/NCBI
|
|
19
|
Papin JA, Reed JL and Palsson BO:
Hierarchical thinking in network biology: The unbiased
modularization of biochemical networks. Trends Biochem Sci.
29:641–647. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Glass K, Quackenbush J, Spentzos D,
Haibe-Kains B and Yuan GC: A network model for angiogenesis in
ovarian cancer. BMC Bioinformatics. 16:1152015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lao T, Glass K, Qiu W, Polverino F, Gupta
K, Morrow J, Mancini JD, Vuong L, Perrella MA, Hersh CP, et al:
Haploinsufficiency of Hedgehog interacting protein causes increased
emphysema induced by cigarette smoke through network rewiring.
Genome Med. 7:122015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu CQ, Zhu ST, Wang M, Guo SL, Sun XJ,
Cheng R, Xing J, Wang WH, Shao LL and Zhang ST: Pathway analysis of
differentially expressed genes in human esophageal squamous cell
carcinoma. Eur Rev Med Pharmacol Sci. 19:1652–1661. 2015.PubMed/NCBI
|
|
23
|
Banks CA, Lee ZT, Boanca G,
Lakshminarasimhan M, Groppe BD, Wen Z, Hattem GL, Seidel CW,
Florens L and Washburn MP: Controlling for gene expression changes
in transcription factor protein networks. Mol Cell Proteomics.
13:1510–1522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Buckingham M and Rigby PW: Gene regulatory
networks and transcriptional mechanisms that control myogenesis.
Dev Cell. 28:225–238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Glass K, Quackenbush J, Silverman EK,
Celli B, Rennard SI, Yuan GC and DeMeo DL: Sexually-dimorphic
targeting of functionally-related genes in COPD. BMC Syst Biol.
8:1182014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hromas R, Collins SJ, Hickstein D, Raskind
W, Deaven LL, O'Hara P, Hagen FS and Kaushansky K: A retinoic
acid-responsive human zinc finger gene, MZF-1, preferentially
expressed in myeloid cells. J Biol Chem. 266:14183–14187.
1991.PubMed/NCBI
|
|
27
|
Deng Y, Wang J, Wang G, Jin Y, Luo X, Xia
X, Gong J and Hu J: p55PIK transcriptionally activated by MZF1
promotes colorectal cancer cell proliferation. Biomed Res Int.
2013:8681312013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng L, Jiao W, Mei H, Song H, Li D,
Xiang X, Chen Y, Yang F, Li H, Huang K, et al: miRNA-337-3p
inhibits gastric cancer progression through repressing myeloid zinc
finger 1-facilitated expression of matrix metalloproteinase 14.
Oncotarget. 7:40314–40328. 2016.PubMed/NCBI
|
|
29
|
Tsai SJ, Hwang JM, Hsieh SC, Ying TH and
Hsieh YH: Overexpression of myeloid zinc finger 1 suppresses matrix
metalloproteinase-2 expression and reduces invasiveness of SiHa
human cervical cancer cells. Biochem Biophys Res Commun.
425:462–467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Katsuoka F and Yamamoto M: Small Maf
proteins (MafF, MafG, MafK): History, structure and function. Gene.
586:197–205. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang YM, Roh YS, Seki E and Maf G: MafG, a
novel target of FXR that regulates bile acid homeostasis.
Gastroenterology. 149:1981–1983. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ding X, Yang Y, Han B, Du C, Xu N, Huang
H, Cai T, Zhang A, Han ZG, Zhou W, et al: Transcriptomic
characterization of hepatocellular carcinoma with CTNNB1 mutation.
PLoS One. 9:e953072014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
No authors listed: MAFG mediates CIMP in
BRAF-mutant colorectal cancer. Cancer Discov. 4:OF112014.
View Article : Google Scholar
|
|
34
|
Rui L, Schmitz R, Ceribelli M and Staudt
LM: Malignant pirates of the immune system. Nat Immunol.
12:933–940. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lenz G, Wright GW, Emre NC, Kohlhammer H,
Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al:
Molecular subtypes of diffuse large B-cell lymphoma arise by
distinct genetic pathways. Proc Natl Acad Sci USA. 105:13520–13525.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sasaki I, Hoshino K, Sugiyama T, Yamazaki
C, Yano T, Iizuka A, Hemmi H, Tanaka T, Saito M, Sugiyama M, et al:
Spi-B is critical for plasmacytoid dendritic cell function and
development. Blood. 120:4733–4743. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schmidlin H, Diehl SA, Nagasawa M,
Scheeren FA, Schotte R, Uittenbogaart CH, Spits H and Blom B: Spi-B
inhibits human plasma cell differentiation by repressing BLIMP1 and
XBP-1 expression. Blood. 112:1804–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schotte R, Nagasawa M, Weijer K, Spits H
and Blom B: The ETS transcription factor Spi-B is required for
human plasmacytoid dendritic cell development. J Exp Med.
200:1503–1509. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Takagi Y, Shimada K, Shimada S, Sakamoto
A, Naoe T, Nakamura S, Hayakawa F, Tomita A and Kiyoi H: SPIB is a
novel prognostic factor in diffuse large B-cell lymphoma that
mediates apoptosis via the PI3K-AKT pathway. Cancer Sci.
107:1270–1280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Y and Manning BD: mTORC1 signaling
activates NRF1 to increase cellular proteasome levels. Cell Cycle.
14:2011–2017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim HM, Han JW and Chan JY: Nuclear factor
erythroid-2 like 1 (NFE2L1): Structure, function and regulation.
Gene. 584:17–25. 2016. View Article : Google Scholar : PubMed/NCBI
|