Introduction
Chemotherapy is the systemic administration of drugs
(singly or in combination) to destroy cancer cells. Although
chemotherapy has long been used to treat cancer patients, it is
challenged by drug resistance, which remains a major obstacle to
the successful treatment of cancer. Substantial research efforts
have revealed that drug resistance arises from a broad range of
mechanisms, such as, drug efflux, drug target mutations and
engagement of alternative survival pathways (1). Recently, it was reported that drug
resistance is accompanied by epithelial-mesenchymal transition
(EMT), a process whereby epithelial cells lose polarity, and the
ability to adhere to other cells and acquire mesenchymal properties
(2–4). Sommers et al provided the first
evidences of a link between drug resistance and EMT by revealing
loss of epithelial markers and the acquisition of vimentin
expression in adriamycin-resistant MCF-7 cells and
vinblastine-resistant ZR-75-B human breast cancer cells (5). Since then, several studies have
reported that cancer cells exposed to chemotherapeutic agents
exhibit the EMT phenotype and that after undergoing EMT, cells
possess invasive and metastatic properties in diverse cancers
including breast (5–7), gastric (8,9) and
colon cancer (10).
Since it has been increasingly demonstrated that EMT
plays an important role in drug resistance and metastasis, novel
therapeutic strategies targeting EMT have been explored in order to
overcome drug resistance. Several small molecules including the
antibiotic salinomycin (11), the
antiviral drug zidovudine (12),
and the antidiabetic drug metformin (13,14)
have been shown to reverse the process of EMT. In addition,
curcumin, an active ingredient in curry, has been revealed to
suppress doxorubicin (DOX)-induced EMT by inhibiting transforming
grow factor-β (TGF-β) and PI3K/AKT signaling pathways in breast
cancer cells (15).
Deep-sea water (DSW) is defined as sea water
obtained from a depth of more than 200 meters. DSW is rich in
minerals, including calcium (Ca), magnesium (Mg), potassium (K),
sodium (Na) and zinc (Zn) (16),
but particularly, magnesium and calcium. The concentration of
calcium is ~100 mg/l in 1,500 hardness DSW, while the amount of
magnesium is ~300 mg/l. DSW has been shown to improve blood
cholesterol and prevent obesity and atherosclerosis (17–19).
Previously, we revealed that DSW decreased cancer cell metastatic
potential by decreasing the expression of TGF-β, Wnt3a, Wnt5a,
urokinase plasminogen activator (uPA), and matrix
metalloproteinase-9 (MMP-9) (20,21).
Although the precise mechanisms mediating the antimetastatic
effects of DSW have not been clarified yet, studies revealing that
magnesium and/or calcium deficiencies are associated with cancer
risk and metastasis (22–25) suggest that magnesium and calcium may
play important mediatory roles in the antimetastatic effects of
DSW. In addition, in our previous study, it was also observed that
DSW provides a cardioprotective effect against DOX-induced
cardiotoxicity, which compromised clinical use of DOX (26). Thus, in the present study, we
further evaluated whether DSW has the potential to abolish the
side-effects of DOX by targeting EMT in MCF-7 human breast cancer
cells.
Materials and methods
Cell culture
MCF-7 human breast cancer cells were purchased from
the Korean Cell Line Bank (Seoul, Korea). The cells were cultured
in Dulbeccos modified Eagles medium (DMEM) (Welgene, Daegu, Korea)
containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA,
USA), 1% antibiotic-antimycotic solution (Welgene), and 10 µg/ml
insulin (Welgene) at 37°C in a 5% CO2 incubator.
Preparation of DSW
DSW was supplied by the Marine Deep Ocean Water
Application Research Center at the Korean Institute of Ocean
Science and Technology (Goseong, Korea). DSW was taken 6.7 km off
the Goseong (Gangwon-Do, Korea) coast at a depth of 500 meters.
Samples were microfiltered, subjected to reverse osmosis, and
concentrated by electrodialysis to obtain desalinated water
(hardness 0) and 4,000 hardness DSW. To prepare DSW containing
media, DMEM powder (Sigma, St. Louis, MO, USA) was dissolved in
hardness 4,000 DSW and diluted with desalinated DSW (hardness 0) to
obtain hardness 1,500 DSW media. Further serial dilutions were
performed to achieve hardness of 500 and 1,000 from 1,500 hardness
DSW using desalinated media (hardness 0). The ratio of magnesium to
calcium presented in DSW was 3:1. Hardness was calculated using the
following equation:
Hardness of DSW (mg/l) = Mg (mg/l) × 4.1 + Ca (mg/l)
× 2.5
Cell treatment
MCF-7 cells were treated with 0.5 µM DOX (Sigma) or
DSW of various hardness (500, 1,000 and 1,500) and further cultured
for 72 h. The cells were harvested for RNA isolation or the
preparation of protein lysates. To inhibit the ERK1/2, p38 or
PI3K/AKT signaling pathways, the cells were pretreated with 10 µM
U0126 (a MEK1/2 inhibitor), SB203580 (a p38 inhibitor) or LY294002
(a PI3K/AKT inhibitor) (all from LC Laboratories, Woburn, MA, USA)
for 24 h, and then treated with 0.5 µM DOX for 72 h.
Cell viability assay
MCF-7 cells were seeded into 96-well plates at 37°C
and incubated for 24 h. To assess the antitumor effects of DOX or
DSW, the cells were treated with DOX or DSW and cultured for an
additional 24, 48 or 72 h. To study the combined effects of DOX and
DSW, the cells were treated with DSW (hardness 500–1,500) in the
presence of 0.3 µM DOX. Cell viabilities were assessed using
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) reagent (Sigma). Absorbances were assessed at 570 nm using a
Multi-Detection Microplate Reader (Molecular Devices, Sunnyvale,
CA, USA).
Transwell migration assay
Transwell chambers (Corning Inc., Corning, NY, USA)
with 8-µm pore polycarbonate filters coated with Matrigel matrix
(BD Biosciences, Bedford, MA, USA) were used for the assay.
3×104 cells/well were seeded on the upper chamber of
24-well plates, which were coated with Matrigel 1:20 for 1 h. After
24 h of culture, DMEM containing 0.3 µM DOX and 2% FBS was placed
in the upper chamber and media containing 10% FBS was placed in the
lower chamber and cells were cultured for an additional 72 h. The
cells were then fixed with 100% methanol for 10 min and stained
with hematoxylin and eosin (Sigma). Non-invasive cells were removed
with a cotton swab and the membranes were mounted onto glass slides
using mounting solution (Sigma). Membranes were placed on a
microscope slide to take optical microscopic images.
Wound-healing assay
4×105 MCF-7 cells/well were seeded on
collagen-coated (20 µg/ml) (Corning Inc.) 6-well plates and
incubated at 37°C. After growing for 48 h to reach 100% confluence,
the cell layers were scratched with a 200 µl yellow pipette tip to
create wounds and washed with PBS to remove cell debris. The cells
were then treated with 0.3 µM DOX or DSW of various hardness, and
optical images were captured at 0 and 72 h after wounding.
RNA isolation and reverse
transcriptase-polymerase chain reaction (RT-PCR)
Cells were grown and treated in 6-well plates as
described above. Total RNA was extracted using the easy-BLUE™ Total
RNA Extraction kit (iNtRON Biotechnology Inc., Sungnam, Korea).
RT-PCR was performed using 1 µg of total RNA using the Avian
Myeloblastosis Virus RNA PCR kit version 3.0 (Takara Bio, Inc.,
Shiga, Japan). DNA amplicons were subjected to agarose gel
electrophoresis containing 0.5 µg/ml of ethidium bromide (EtBr),
and visualized using an UV transluminometer (CoreBio System, Seoul,
Korea). Primer sequences of the target genes are presented in
Table I.
 | Table I.Primer sequences for RT-PCR. |
Table I.
Primer sequences for RT-PCR.
| Primers | Forward | Reverse |
|---|
| Snail-1 |
5′-CCGGACCCACACTGGCGAGA-3′ |
5′-CTCGAGGGTCAGCGGGGACA-3′ |
| Slug |
5′-GCTGCTCCATTCCACGCCCA-3′ |
5′-AGGCTTCTCCCCCGTGTGAGTT-3′ |
| Vimentin |
5′-TCGCCAACTACATCGACAAG-3′ |
5′-GTTCTTGGCAGCCACACTTT-3′ |
| Fibronectin |
5′-CAGTGGGAGACCTCGAGAAG-3′ |
5′-TCCCTCGGAACATCAGAAAC-3′ |
| MDR1 |
5′-GCCTGGCAGCTGGAAGACAAATACACAAAATT-3′ |
5′-CAGACAGCAGCTGACAGTCCAAGAACAGGACT-3′ |
| TGF-β |
5′-CGTCTGCTGAGGAGGCTCAAGTTA-3′ |
5′-CAGCCGAGGTCCTTGCGGAA-3′ |
| Wnt3a |
5′-TGAACAAGCACAACAACGAG-3′ |
5′-CAGTGGCATTTTTCCTTCC-3′ |
| Axin2 |
5′-TTATGCTTTGCACTACGTCCCTCCA-3′ |
5′-CGCAACATGGTCAACCCTCAAGAC-3′ |
| Wnt5a |
5′-GGGAGGTTGGCTTGAACATG-3′ |
5′-GAATGGCACGCAATTACCTT-3′ |
| GAPDH |
5′-ATCCCATCACCATCTTCCAG-3′ |
5′-TTCTAGACGGCAGGTCAGGT-3′ |
Western blotting
Cells were grown and treated in 6-well plates as
aforementioned. The cells were lysed with RIPA buffer (150 mM NaCl,
1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl
pH 7.5 and 2 mM EDTA) including a phosphatase and protease
inhibitor cocktail (GenDEPOT, Barker, TX, USA). Lysates were
centrifuged at 13,000 rpm for 20 min to clear debris and protein
concentrations were determined using bicinchoninic acid reagent
(Sigma). Proteins (20 µg) were separated by SDS-PAGE (8–12% gels)
and transferred to polyvinylidene fluoride (PVDF) membranes at 100
V for 40 min. The membranes were blocked in 5% skim milk in
TBS-Tween (50 mM Tris-HCl, 150 mM NaCl and 0.1% Tween-20) for 1 h
at room temperature and incubated with the following primary
antibodies: anti-rabbit phospho-extracellular signal-regulated
kinase 1/2 (p-ERK1/2), total-ERK1/2, phospho-p38 (p-p38),
total-p38, phospho-protein kinase B (p-AKT), total-AKT,
phospho-GSK3β, glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
(Cell Signaling Technology, Inc., Beverly, MA, USA) and β-actin
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), overnight at
4°C. Blots were then incubated with HRP-conjugated secondary
anti-rabbit and -mouse antibodies (Santa Cruz Biotechnology Inc.),
diluted 1:3,000 for 1 h at room temperature and developed by
Luminescent Image Analyzer LAS-4000 (Fujifilm, Tokyo, Japan).
Statistical analysis
Statistical analysis was conducted using the
Student's t-test. All experiments were conducted in triplicate and
the results are presented as the means ± SDs. P-values of <0.05
were considered as statistically significant.
Results
DOX induces EMT, and thus enhances
human breast cancer cell motility
To assess the antitumor effects of DOX, we first
evaluated the viabilities of MCF-7 human breast cancer cells
treated with different concentrations of DOX for 24, 48 or 72 h. As
shown in Fig. 1A, treatment with
DOX significantly and dose-dependently decreased cell viability.
Despite, its therapeutic effects, we observed that DOX-treatment
caused cells to become spindle-shaped and to detach from each other
(Fig. 1B). Since these
morphological changes were reminiscent of the EMT phenotype, we
evaluated the mRNA expression of EMT markers in DOX-treated cells.
DOX increased the expression of mesenchymal markers, vimentin and
fibronectin. Furthermore, the expression of mesenchymal
transcription factors, Snail-1 and Slug, were also enhanced by DOX
(Fig. 1C), suggesting that DOX
induced EMT. Since EMT is associated with cell motility, we
assessed the effects of DOX on the migration and invasion of MCF-7
cells. The migratory ability was examined using in vitro
wound-healing assay. After creating uniform wounds with a 200 µl
pipette tip, the ability of the cells to migrate and close the
wound gap were assessed. MCF-7 cells are weakly invasive in
vitro, but cells treated with 0.3 µM DOX for 72 h were found to
rapidly close wounds (Fig. 1D). We
also performed Transwell migration assay. Treatment of MCF-7 cells
with 0.3 µM DOX for 72 h in the upper chamber stimulated cells to
migrate through the microporous membrane (Fig. 1E). Collectively, our data revealed
that DOX induced EMT, and thus, enhanced the migratory ability of
the cells.
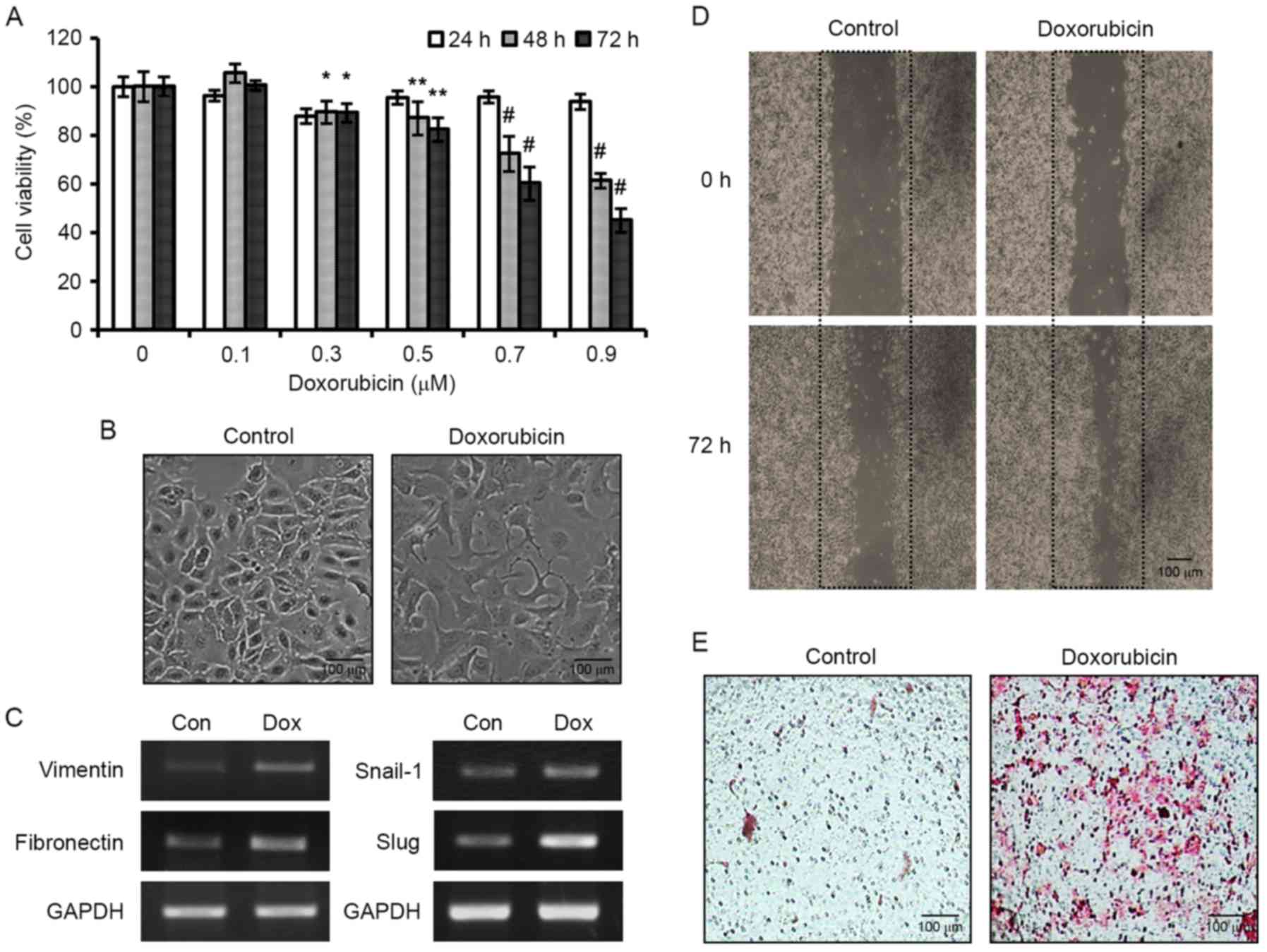 | Figure 1.Stimulation of EMT and enhanced
motility of MCF-7 human breast cancer cells by DOX. (A) Cells were
treated with various concentrations of DOX for 24, 48 or 72 h, and
cell viabilities were assessed by MTT. Results are expressed as the
means ± SDs of three independent experiments performed in
triplicate; *P<0.05, **P<0.01 and #P<0.001
compared to the control. (B) Morphological images of MCF-7 cells
exposed to DOX for 72 h. Scale bar, 100 µm. (C) Effects of DOX on
the mRNA expression of the mesenchymal markers, vimentin and
fibronectin, and on the transcription factors, Snail-1 and Slug.
(D) Photomicrographs of DOX-induced wound-healing in MCF-7 cells at
0 and 72 h after wounding. (E) Photomicrographs of Transwell
migration assay. Cells were treated with 0.3 µM DOX for 72 h and
the cells that migrated through the membrane were stained with
H&E. Scale bar, 100 µm. EMT, epithelial-mesenchymal transition;
DOX, doxorubicin. |
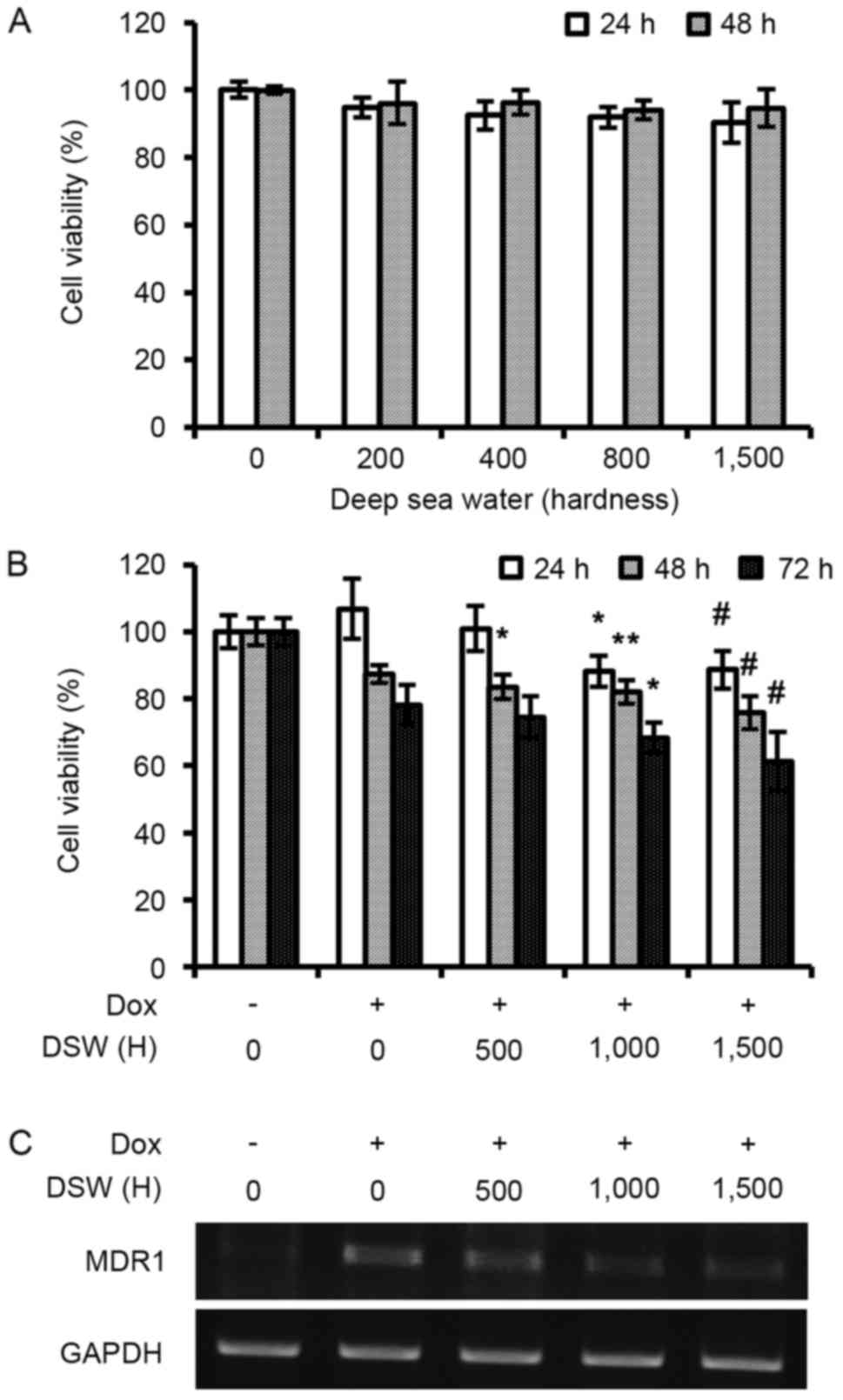
DSW increases the antitumor effects of
DOX by inhibiting MDR1
We first assessed whether DSW itself has anticancer
effects on MCF-7 human breast cancer cells. After MCF-7 cells were
treated with DSW of varying hardness for 24 or 48 h, we assessed
cell viabilities using an MTT assay. As shown in Fig. 2A, cell proliferation was not
affected by DSW treatment even when cells were treated with 1,500
hardness DSW for 48 h (Fig. 2A).
However, we found that DSW enhanced the antitumor effect of DOX
(Fig. 2B). The cells were treated
with DSW of varying hardness in the presence of 0.3 µM DOX and
further incubated for another 24, 48 or 72 h before assessing cell
viability by MTT assay. Notably, mildly enhanced antitumor effects
of DOX were observed in a hardness-dependent manner; a decrease in
cell viabilities of ~20% was found after treatment with DSW of
hardness 1,500 for 72 h (Fig. 2B).
To understand the underlying mechanism of this enhanced antitumor
effect of DOX by DSW, we examined the mRNA expression of the
ATP-binding cassette (ABC) drug transporter, multidrug resistance 1
(MDR1). As shown in Fig. 2C, the
expression of MDR1 was significantly induced by DOX, however
co-treatment with DSW suppressed this DOX-induced MDR1 expression,
suggesting that DSW enhanced the efficacy of DOX by inhibiting the
expression of MDR1.
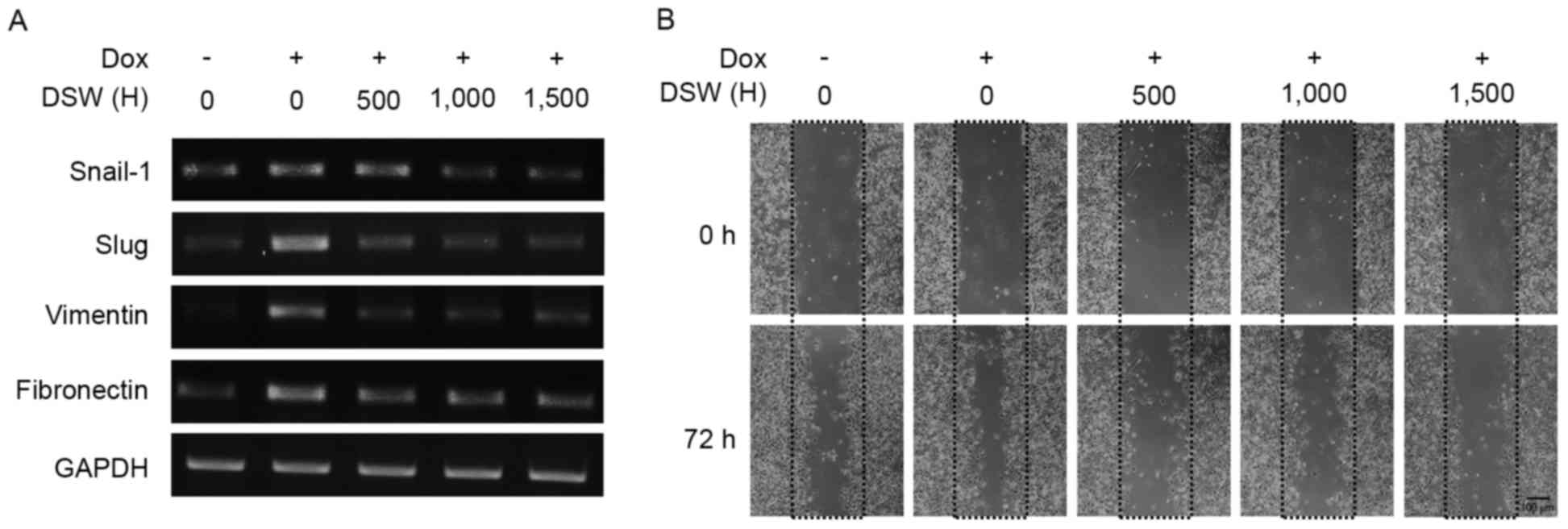
DSW suppresses DOX-induced EMT and in
vitro cancer cell migration
To investigate the effect of DSW on DOX-induced EMT,
we evaluated the expression of EMT markers in mRNA expression after
cells were treated with 0.5 µM DOX or in combination with varying
DSW hardness for three days. As shown in Fig. 3A, Snail-1 and Slug were induced by
DOX however treatment with DSW significantly decreased their
expression. Similar inhibitory effects of DSW were observed in
DOX-induced expression of vimentin and fibronectin (Fig. 3A), suggesting that DSW suppresses
DOX-induced EMT. Consequently, co-treatment of the cells with
varying hardness of DSW for up to 72 h significantly and
hardness-dependently decreased DOX-induced migration vs.
DOX-treated control cells in the wound-healing assay (Fig. 3B).
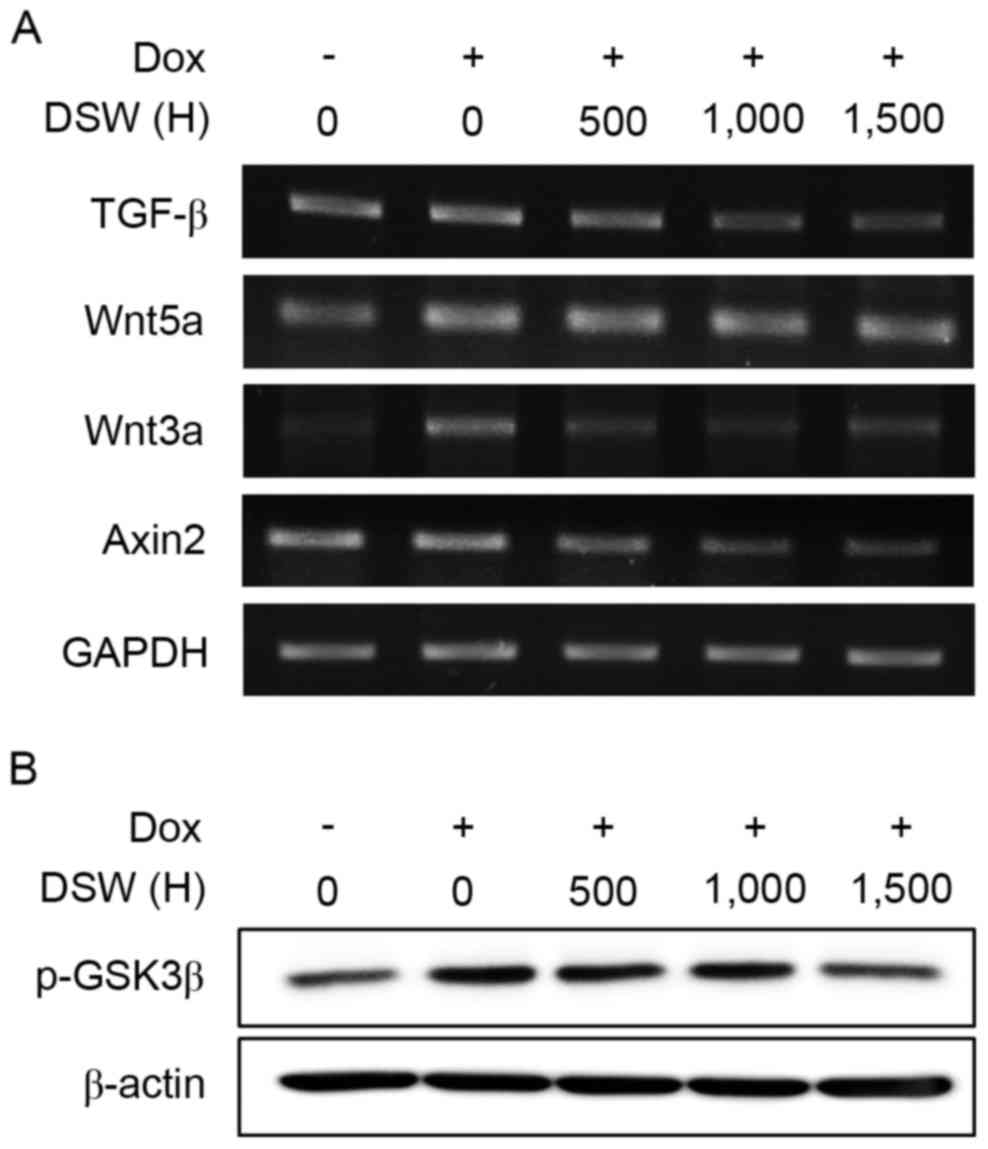
DSW suppresses TGF-β and Wnt signaling
pathways
The TGF-β signaling pathway is well known to induce
EMT (27). Bandyopadhyay et
al revealed that DOX activated the Smad-dependent TGF-β
signaling pathway to induce EMT, consequently promoting invasion in
murine 4T1 breast cancer cells (6).
In addition, DOX resistance in HCT116 human colon cancer cells
promoted EMT by upregulating TGF-β (10). Thus, we explored the effect of DSW
on TGF-β signaling pathways. After cells were treated with 0.5 µM
DOX or in combination with varying hardness of DSW for three days,
we assessed the mRNA expression of TGF-β. As shown in Fig. 4A, treatment with DSW significantly
decreased the mRNA expression of TGF-β, though DOX mildly
stimulated its induction. Since the canonical TGF-β signaling
pathway depends on activation of the Smad family (27), particularly Smad2 and Smad 3, we
tried to assess the level of phosphorylated Smad2/3 in cells
exposed to DOX or DSW. However, we were not able to detect the
activated Smad2/3 expression in MCF-7 cells exposed to DOX, as well
as the endogenous level of Smad2/3 expression in untreated cells
(data not shown). Thus, we were not able to clarify the role of the
Smad-dependent TGF-β signaling pathway in mediating the inhibitory
effects of DSW on DOX-induced EMT in the present study. Since
recent studies revealed that TGF-β, directly upregulated
non-canonical Wnt ligand, Wnt5a expression through the Smad complex
(28), we also tried to evaluate
the mRNA expression of Wnt5a. Although the expression of Wnt5a was
induced by stimulation of the cells with DOX, this DOX-induced
Wnt5a expression was not decreased by treatment with DSW (Fig. 4A), implying that the inhibitory
effects of DSW on DOX-induced EMT are independent of the
non-canonical Wnt signaling pathway and possibly independent of the
Smad-dependent TGF-β signaling pathway.
Wnt5a is a non-canonical signaling member of the Wnt
family, and its activation is independent of β-catenin. In
contrast, activation of the canonical Wnt signaling pathway depends
on β-catenin (29). Upon binding of
canonical Wnt signaling members, such as Wnt1 and Wnt3a, to
frizzled (FZD) and low-density lipoprotein receptor-related protein
5/6 (LRP 5/6), GSK3β is phosphorylated to its inactive form, and
thus functional β-catenin accumulates in the cytosol and is further
transported into the nucleus to activate target genes, such as,
Axin2 and Snail-1. Since the Wnt signaling pathway is another
signaling pathway that promotes EMT in coordination with the TGF-β
signaling pathway (30,31), we evaluated the mRNA expression of
Wnt3a and its target gene, Axin2. Non-treated control MCF-7 cells
exhibited barely detectable levels of Wnt3a mRNA, while DOX-treated
cells exhibited significant Wnt3a expression. However, co-treatment
with DSW efficiently suppressed the expression of Wnt3a, and its
target gene, Axin2 (Fig. 4A).
Moreover, we found that the inactive form of GSK3β was decreased by
DSW co-treatment (Fig. 4B), which
suggested that the inhibitory effects of DSW on the canonical Wnt
signaling pathway are involved in targeting EMT.
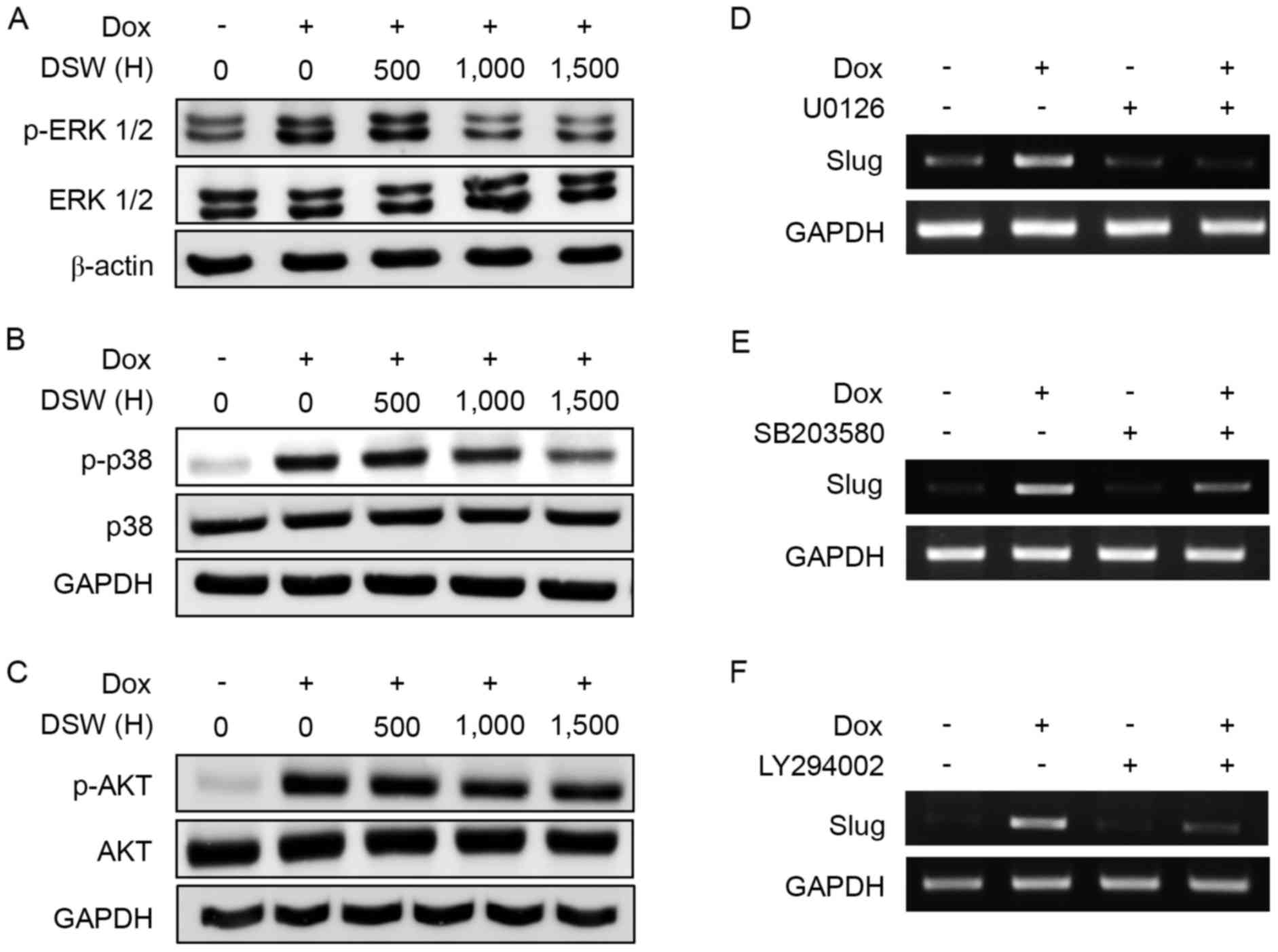
The inhibitory effects of DSW on
DOX-induced EMT are mediated by the inhibition of the ERK1/2, p38
MAPK and PI3K/AKT signaling pathways
TGF-β activates various kinase pathways, including
extracellular signal-regulated kinase 1/2 (ERK1/2), PI3K/AKT and
p38 mitogen-activated protein kinase (MAPK) through a
Smad-independent signaling pathway (27), and these pathways are implicated in
the activation of EMT. Since DSW efficiently suppressed the
expression of TGF-β, we further analyzed whether the inhibitory
effect of DSW on TGF-β expression subsequently affected the ERK1/2,
PI3K/AKT and p38 MAP kinase signaling pathways. After cells were
treated with 0.5 µM DOX or in combination with varying hardness of
DSW for three days, western blot analysis was performed. DOX
significantly induced the phosphorylation of ERK1/2, but
co-treatment with DSW efficiently decreased this phosphorylation
(Fig. 5A). Similarly, the
phosphorylation of p38 and AKT were also found to be induced in
DOX-treated MCF-7 cells, but DSW co-treatment significantly
inhibited the phosphorylation of p38 and AKT (Fig. 5B and C). To further confirm the role
of the PI3K/AKT and MAP kinase signaling pathways in DOX-induced
EMT, we blocked the ERK1/2, p38 and PI3K/AKT signaling pathways
using U0126, SB203580 and LY294002, respectively. Cells were
treated with 10 µM U0126, 10 µM SB203580 or 10 µM LY294002 for 24 h
prior to the addition of 0.5 µM DOX, and then incubated for another
72 h. Notably, Slug upregulation in DOX-treated cells was
efficiently prevented by blocking the ERK1/2 (Fig. 5D), p38 (Fig. 5E) or PI3K/AKT (Fig. 5F) signaling pathways, which
suggested that the MAPK and PI3K/AKT signaling pathways played a
critical role in DOX-induced EMT. Collectively, our data revealed
that the inhibitory effects of DSW on DOX-induced EMT are mediated
through the inhibition of the ERK1/2, p38 and AKT signaling
pathways.
Discussion
In the present study, we have evaluated the
potential health benefit of DSW on DOX-induced EMT in MCF-7 human
breast cancer cells. Our data revealed that MCF-7 cells treated
with DOX displayed characteristics of EMT by exhibiting the
acquisition of mesenchymal markers, vimentin and fibronectin, and
the EMT-related transcription factors, Slug and Snail-1 in their
mRNA expression. In addition, we observed that DOX stimulated the
migratory ability of MCF-7 cells in an in vitro
wound-healing assay. It was consistent with other studies which
revealed that chemotherapy agents, such as, doxorubicin and
paclitaxel, promoted EMT of treated cancer cells, and cells that
underwent EMT exhibited the enhanced properties of migration and
invasion (5–10).
Cooperation of signaling pathways, such as, TGF-β,
Sonic Hedgehog (SHH) and Wnt signaling pathways, is known to
promote EMT (30,31). Particularly, the TGF-β signaling
pathway is the most well-established pathway that induces EMT and
tumor metastasis (27). The
canonical TGF-β signaling pathway is mediated by activation of the
Smad family. Upon ligand binding, phosphorylation of TGF-β receptor
I (TGF-βRI) by TGF-βRII results in the recruitment of Smad2 and
Smad3, which are then transported from the cytosol to the nucleus
after complex formation with the coactivator Smad4. In the nucleus,
the Smad complex binds to regulatory elements to induce the
transcription of target genes associated with EMT, such as, Snail
and Slug (27). Since we were not
able to detect activated Smad2 or Smad3 expression in MCF-7 cells
exposed to DOX, or endogenous Smad2/3 in untreated cells (data not
shown), the role of the Smad-dependent TGF-β signaling pathway in
DOX-induced EMT was not clarified in the present study. However,
Chen et al suggested that the Smad-dependent TGF-β signaling
pathway may play a minor role in DOX-induced EMT since a specific
inhibitor of TGF-β receptor kinase, SB431542, was not able to
abrogate DOX-induced E-cadherin downregulation and altered Snail
expression in BT-20 breast cancer cells (15).
In addition to Smad-dependent mechanisms,
TGFβ-induced EMT is also mediated through Smad-independent
mechanisms (27,31). Particularly, the ERK MAPK, p38 MAPK
and PI3K/AKT signaling pathways have been linked to TGFβ-induced
EMT (27,32). Activation of ERK1/2 in response of
TGF-β is initiated by Ras, leading to the activation of Raf and
MEK1/2 kinases. Increased Ras-ERK MAP kinase signaling then results
in the downregulation of E-cadherin expression, whereas blocking
the kinase function of MEK1/2 inhibits TGFβ-induced EMT (33–35).
TGF-β also induces the activation of p38 MAP kinase (36,37).
The interaction between TNF receptor-associated factor 6 (TRAF6)
and TGF-βRI causes the activation of TGFβ-activated kinase 1 (TAK1)
and leads to the activation of p38 MAP kinase. Furthermore,
blocking the activation of p38 MAP kinase prevents TGFβ-induced
EMT, which suggests that p38 MAP kinase plays a role in
TGFβ-induced EMT (36,37). TGF-β activates PI3 kinase directly
through its own receptors (38,39).
Upon binding of the regulatory subunit of PI3 kinase to TGF-βRI and
TGF-βRII, AKT is activated, resulting in the activation of mTOR/S6
kinase. Notably, a dominant negative form of AKT was found to
inhibit TGFβ-induced EMT (38,40).
In the present study we found that the Smad-independent TGF-β
signaling pathways were involved in DOX-induced EMT in MCF-7 cells.
Induction of the phosphorylation of ERK1/2, p38 and AKT were
observed in cells treated with DOX. Since the treatment of cells
with selective inhibitors of each pathway markedly prevented the
process of EMT, our data revealed that the ERK1/2, p38 and AKT
signaling pathways are involved in DOX-induced EMT. However, DSW
efficiently inhibited the phosphorylation of ERK1/2, p38 and AKT.
Consequently, the expressions of mesenchymal markers, vimentin and
fibronectin, and EMT-related transcription factors, Slug and
Snail-1 were decreased by DSW. Moreover, DSW significantly
suppressed the DOX-induced increase in the migratory ability of
MCF-7 cells as determined by the wound-healing assay. Collectively,
our data revealed that DSW targeted these Smad-independent TGF-β
signaling pathways to prevent DOX-induced EMT in MCF-7 cells.
The Wnt signaling pathway is another signaling
pathway that promotes EMT in coordination with the TGF-β signaling
pathway (29–31). Without Wnt signals, the tumor
suppressor, GSK3β exists in its active form, which phosphorylates
β-catenin for degradation. When the canonical Wnt signaling pathway
is activated by binding canonical Wnt signaling members, such as,
Wnt1 and Wnt3a, to FZD and LRP 5/6, GSK3β is phosphorylated, and
thus inactivated. Functional β-catenin is then accumulated in the
cytosol and is further transported into the nucleus to activate
target genes, such as, Axin2 and Snail-1. Since it is established
that TGF-β can also inactivate GSK3β by activating Smad-independent
pathways, including the ERK MAPK (41), p38 MAPK (42) and PI3K/AKT (43) signaling pathways, GSK3β is a major
converging element between the Wnt and TGF-β signaling pathways.
Furthermore, inactivation of GSK3β can increase Snail-1 expression
directly by transporting Snail-1 from the nucleus to the cytosol
for degradation (44). We
previously reported that DSW inhibits the Wnt signaling pathway,
which mediates cell migration and invasion in coordination with the
TGF-β signaling pathway (20). In
the present study, we confirmed that DSW efficiently suppressed the
expression of canonical Wnt signaling ligand, Wnt3a, and its target
gene, Axin2. Furthermore, we found that the inactive form of GSK3β
was decreased by DSW, which implies that increasing the active form
of GSK3β by DSW may inhibit the expression of Slug or Snail-1
directly or through the Smad-independent signaling pathways
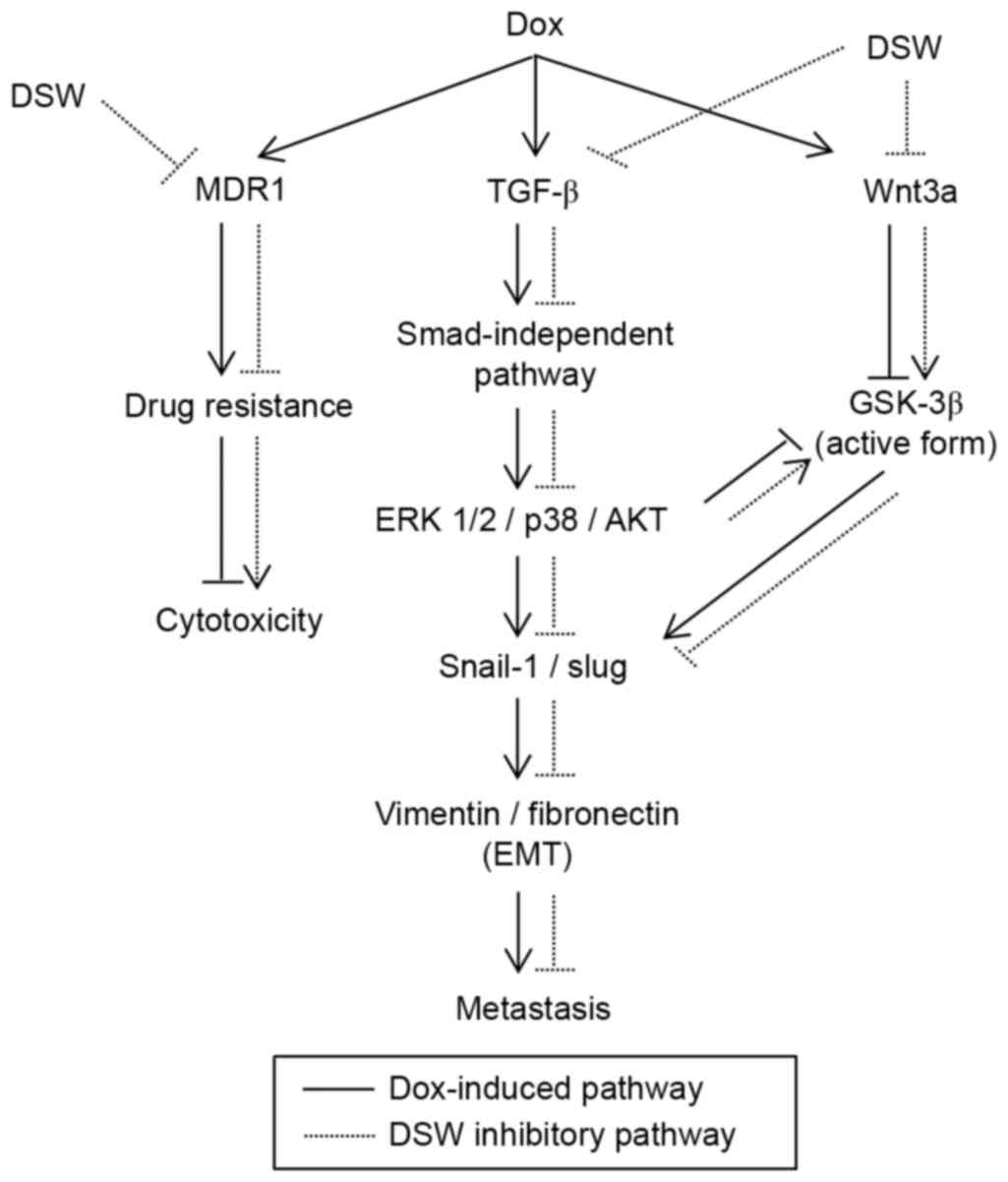
(Fig. 6).
Minerals are essential for all living organisms and
recent studies revealed that the intake of certain minerals
decreases the incidence of cancer. Wark et al reported that
a higher intake of dietary magnesium was associated with lower risk
of colorectal tumors (45).
Furthermore, decreased serum levels of magnesium are frequently
observed in patients with tumors (22,46).
Zinc is an essential mineral that acts as a co-factor for diverse
enzymes. Notably, zinc differentially modulated DNA damage in
normal and cancer cells. Zinc exhibited a protective action against
DNA damage induced by hydrogen peroxide in normal cells, whereas
zinc inhibited the DNA repair for hydrogen peroxide-induced DNA
damage in cancer cells (47). Fong
et al also reported the role of zinc, demonstrating that
dietary zinc deficiency led to an increased tumor incidence in mice
(48). Epidemiologic studies
suggest that intake of calcium appears to decrease the risk of
colon cancer (25). In addition,
individuals with improved calcium and vitamin D nutritional status
substantially decreased all cancer risk in postmenopausal women
(23). Since DSW is rich in
minerals, such as, calcium, magnesium and zinc, we presumed that
the combined ionic action of several minerals may play important
roles in inhibiting DOX-induced EMT.
In summary, our results revealed that DSW inhibits
DOX-induced EMT without interfering with the anticancer effects of
DOX. We found that DSW inhibited the Smad-independent signaling
pathway and the Wnt canonical signaling pathway to prevent the
process of EMT. The mechanism responsible for the DSW-mediated
suppression of DOX-induced EMT was not fully elucidated in the
present study and further investigation is warranted on this topic.
However, this is the first study to reveal that DSW has a
substantial inhibitory effect on DOX-induced EMT. Collectively, our
findings suggest that DSW may improve the clinical use of DOX by
suppressing its side-effects by targeting EMT in MCF-7 human breast
cancer cells.
Acknowledgements
The present study was supported financially by the
National R&D Project ‘Development of New Application Technology
for the Deep Seawater Industry’ of the Korean Ministry of Oceans
and Fisheries.
Glossary
Abbreviations
Abbreviations:
|
DSW
|
deep-sea water
|
|
DOX
|
doxorubicin
|
|
EMT
|
epithelial-mesenchymal transition
|
|
TGF-β
|
transforming growth factor-β
|
|
TGF-βR
|
TGF-β receptor
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
AKT
|
protein kinase B
|
|
FZD
|
frizzled
|
|
LRP 5/6
|
low-density lipoprotein
receptor-related protein 5/6
|
|
MAPK
|
mitogen-activated protein kinase
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
MDR1
|
multidrug resistance protein 1
|
References
|
1
|
Garraway LA and Jänne PA: Circumventing
cancer drug resistance in the era of personalized medicine. Cancer
Discov. 2:214–226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat. 154:8–20. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Y and Zhou BP: Epithelial-mesenchymal
transition in breast cancer progression and metastasis. Chin J
Cancer. 30:603–611. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sommers CL, Heckford SE, Skerker JM,
Worland P, Torri JA, Thompson EW, Byers SW and Gelmann EP: Loss of
epithelial markers and acquisition of vimentin expression in
adriamycin- and vinblastine-resistant human breast cancer cell
lines. Cancer Res. 52:5190–5197. 1992.PubMed/NCBI
|
|
6
|
Bandyopadhyay A, Wang L, Agyin J, Tang Y,
Lin S, Yeh IT, De K and Sun LZ: Doxorubicin in combination with a
small TGFβ inhibitor: A potential novel therapy for metastatic
breast cancer in mouse models. PLoS One. 5:e103652010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Işeri OD, Kars MD, Arpaci F, Atalay C, Pak
I and Gündüz U: Drug resistant MCF-7 cells exhibit
epithelial-mesenchymal transition gene expression pattern. Biomed
Pharmacother. 65:40–45. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han R, Xiong J, Xiao R, Altaf E, Wang J,
Liu Y, Xu H, Ding Q and Zhang Q: Activation of β-catenin signaling
is critical for doxorubicin-induced epithelial-mesenchymal
transition in BGC-823 gastric cancer cell line. Tumour Biol.
34:277–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han RF, Ji X, Dong XG, Xiao RJ, Liu YP,
Xiong J and Zhang QP: An epigenetic mechanism underlying
doxorubicin induced EMT in the human BGC-823 gastric cancer cell.
Asian Pac J Cancer Prev. 15:4271–4274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Liu H, Yu J and Yu H:
Chemoresistance to doxorubicin induces epithelial-mesenchymal
transition via upregulation of transforming growth factor β
signaling in HCT116 colon cancer cells. Mol Med Rep. 12:192–198.
2015.PubMed/NCBI
|
|
11
|
Zhou Y, Liang C, Xue F, Chen W, Zhi X,
Feng X, Bai X and Liang T: Salinomycin decreases doxorubicin
resistance in hepatocellular carcinoma cells by inhibiting the
β-catenin/TCF complex association via FOXO3a activation.
Oncotarget. 6:10350–10365. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Namba T, Kodama R, Moritomo S, Hoshino T
and Mizushima T: Zidovudine, an anti-viral drug, resensitizes
gemcitabine-resistant pancreatic cancer cells to gemcitabine by
inhibition of the Akt-GSK3β-Snail pathway. Cell Death Dis.
6:e17952015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vazquez-Martin A, Oliveras-Ferraros C,
Cufí S, Del Barco S, Martin-Castillo B and Menendez JA: Metformin
regulates breast cancer stem cell ontogeny by transcriptional
regulation of the epithelial-mesenchymal transition (EMT) status.
Cell Cycle. 9:3807–3814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao Z, Cheng X, Wang Y, Han R, Li L,
Xiang T, He L, Long H, Zhu B and He Y: Metformin inhibits the
IL-6-induced epithelial-mesenchymal transition and lung
adenocarcinoma growth and metastasis. PLoS One. 9:e958842014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen WC, Lai YA, Lin YC, Ma JW, Huang LF,
Yang NS, Ho CT, Kuo SC and Way TD: Curcumin suppresses
doxorubicin-induced epithelial-mesenchymal transition via the
inhibition of TGF-β and PI3K/AKT signaling pathways in
triple-negative breast cancer cells. J Agric Food Chem.
61:11817–11824. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakasone T and Akeda S: The application of
deep sea water in japan. UJNR Technical Report. 28:69–75. 1999.
|
|
17
|
Hwang HS, Kim SH, Yoo YG, Chu YS, Shon YH,
Nam KS and Yun JW: Inhibitory effect of deep-sea water on
differentiation of 3T3-L1 adipocytes. Mar Biotechnol. 11:161–168.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee KS, Kwon YS, Kim S, Moon DS, Kim HJ
and Nam KS: Regulatory mechanism of mineral-balanced deep sea water
on hypocholesterolemic effects in HepG2 hepatic cells. Biomed
Pharmacother. 86:405–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Radhakrishnan G, Yamamoto M, Maeda H,
Nakagawa A, KatareGopalrao R, Okada H, Nishimori H, Wariishi S,
Toda E, Ogawa H, et al: Intake of dissolved organic matter from
deep seawater inhibits atherosclerosis progression. Biochem Biophys
Res Commun. 387:25–30. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim S, Chun SY, Lee DH, Lee KS and Nam KS:
Mineral-enriched deep-sea water inhibits the metastatic potential
of human breast cancer cell lines. Int J Oncol. 43:1691–1700.
2013.PubMed/NCBI
|
|
21
|
Lee KS, Lee DH, Kwon YS, Chun SY and Nam
KS: Deep-sea water inhibits metastatic potential in HT-29 human
colorectal adenocarcinomas via MAPK/NF-kB signaling pathway.
Biotechnol Bioproc Eng. 19:733–739. 2014. View Article : Google Scholar
|
|
22
|
Kohli GS, Bhargava A, Goel H, Yadav SP,
Saini AS, Singh GP and Lal H: Serum magnesium levels in patients
with head and neck cancer. Magnesium. 8:77–86. 1989.PubMed/NCBI
|
|
23
|
Lappe JM, Travers-Gustafson D, Davies KM,
Recker RR and Heaney RP: Vitamin D and calcium supplementation
reduces cancer risk: Results of a randomized trial. Am J Clin Nutr.
85:1586–1591. 2007.PubMed/NCBI
|
|
24
|
Nasulewicz A, Wietrzyk J, Wolf FI, Dzimira
S, Madej J, Maier JA, Rayssiguier Y, Mazur A and Opolski A:
Magnesium deficiency inhibits primary tumor growth but favors
metastasis in mice. Biochim Biophys Acta. 1739:26–32. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu K, Willett WC, Fuchs CS, Colditz GA and
Giovannucci EL: Calcium intake and risk of colon cancer in women
and men. J Natl Cancer Inst. 94:437–446. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee DH, Kim S and Nam KS: Protective
effects of deep sea water against doxorubicin-induced
cardiotoxicity in H9c2 cardiac muscle cells. Int J Oncol.
45:2569–2575. 2014.PubMed/NCBI
|
|
27
|
Xu J, Lamouille S and Derynck R:
TGF-β-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Katoh M and Katoh M: Transcriptional
mechanisms of WNT5A based on NF-κB, Hedgehog, TGFβ, and
Notch signaling cascades. Int J Mol Med. 23:763–769. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Howe LR and Brown AM: Wnt signaling and
breast cancer. Cancer Biol Ther. 3:36–41. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith BN and Bhowmick NA: Role of EMT in
metastasis and therapy resistance. J Clin Med. 5:172016. View Article : Google Scholar :
|
|
31
|
Zhang J, Tian XJ and Xing J: Signal
transduction pathways of EMT induced by TGF-β, SHH, and WNT and
their crosstalks. J Clin Med. 5:412016. View Article : Google Scholar :
|
|
32
|
Chen XF, Zhang HJ, Wang HB, Zhu J, Zhou
WY, Zhang H, Zhao MC, Su JM, Gao W, Zhang L, et al: Transforming
growth factor-β1 induces epithelial-to-mesenchymal transition in
human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling
pathways. Mol Biol Rep. 39:3549–3556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Grände M, Franzen A, Karlsson JO, Ericson
LE, Heldin NE and Nilsson M: Transforming growth factor-β and
epidermal growth factor synergistically stimulate epithelial to
mesenchymal transition (EMT) through a MEK-dependent mechanism in
primary cultured pig thyrocytes. J Cell Sci. 115:4227–4236. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Janda E, Lehmann K, Killisch I, Jechlinger
M, Herzig M, Downward J, Beug H and Grünert S: Ras and TGFβ
cooperatively regulate epithelial cell plasticity and metastasis:
Dissection of Ras signaling pathways. J Cell Biol. 156:299–313.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lehmann K, Janda E, Pierreux CE, Rytömaa
M, Schulze A, McMahon M, Hill CS, Beug H and Downward J: Raf
induces TGFβ production while blocking its apoptotic but not
invasive responses: A mechanism leading to increased malignancy in
epithelial cells. Genes Dev. 14:2610–2622. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFβ-mediated fibroblastic transdifferentiation and cell migration.
J Cell Sci. 115:3193–3206. 2002.PubMed/NCBI
|
|
37
|
Yu L, Hébert MC and Zhang YE: TGF-β
receptor-activated p38 MAP kinase mediates Smad-independent TGF-β
responses. EMBO J. 21:3749–3759. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor β-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:36803–36810. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lien SC, Usami S, Chien S and Chiu JJ:
Phosphatidylinositol 3-kinase/Akt pathway is involved in
transforming growth factor-β1-induced phenotypic modulation of
10T1/2 cells to smooth muscle cells. Cell Signal. 18:1270–1278.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kattla JJ, Carew RM, Heljic M, Godson C
and Brazil DP: Protein kinase B/Akt activity is involved in renal
TGF-β1-driven epithelial-mesenchymal transition in vitro and in
vivo. Am J Physiol Renal Physiol. 295:F215–F225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Caraci F, Gili E, Calafiore M, Failla M,
La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A and Vancheri
C: TGF-β1 targets the GSK-3β/β-catenin pathway via ERK activation
in the transition of human lung fibroblasts into myofibroblasts.
Pharmacol Res. 57:274–282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bikkavilli RK, Feigin ME and Malbon CC:
p38 mitogen-activated protein kinase regulates canonical
Wnt-β-catenin signaling by inactivation of GSK3β. J Cell Sci.
121:3598–3607. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by GSK-3β-mediated
phosphorylation in control of epithelial-mesenchymal transition.
Nat Cell Biol. 6:931–940. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim
NH, Cha SY, Ryu JK, Choi YJ, Kim J, et al: A Wnt-Axin2-GSK3β
cascade regulates Snail1 activity in breast cancer cells. Nat Cell
Biol. 8:1398–1406. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wark PA, Lau R, Norat T and Kampman E:
Magnesium intake and colorectal tumor risk: A case-control study
and meta-analysis. Am J Clin Nutr. 96:622–631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sartori S, Nielsen I, Tassinari D,
Mazzotta D, Vecchiatti G, Sero A and Abbasciano V: Serum and
erythrocyte magnesium concentrations in solid tumours: Relationship
with stage of malignancy. Magnes Res. 5:189–192. 1992.PubMed/NCBI
|
|
47
|
Sliwinski T, Czechowska A, Kolodziejczak
M, Jajte J, Wisniewska-Jarosinska M and Blasiak J: Zinc salts
differentially modulate DNA damage in normal and cancer cells. Cell
Biol Int. 33:542–547. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fong LY and Magee PN: Dietary zinc
deficiency enhances esophageal cell proliferation and
N-nitrosomethylbenzylamine (NMBA)-induced esophageal tumor
incidence in C57BL/6 mouse. Cancer Lett. 143:63–69. 1999.
View Article : Google Scholar : PubMed/NCBI
|