Introduction
Multiple myeloma (MM) is a malignancy of the bone
marrow characterized by the clonal proliferation of
antibody-secreting plasma cells (1). The incidence of MM has exceeded that
of acute leukemia, ranking as the second most common of all
hematological malignancies. Worldwide, >80,000 new cases of MM
are reported each year, representing 10% of all hematological
malignancies and ~1% of all new cancer cases (2–3).
Abnormal proliferation of plasma cells produces monoclonal
immunoglobulins (paraprotein, M-component, M-protein) and gives
rise to symptoms and conditions including hypercalcemia, renal
impairment, pathological bone fractures, hyperviscosity and bone
marrow deficiency by suppressing the function of normal
immunoglobulins.
The etiology of MM is not fully understood.
Radiation, chemicals, certain viruses, weakened immunity and
chromosomal abnormalities are thought to be involved in MM etiology
(4). An association between
proteasome 26S subunit non-ATPase 10 (PSMD10) and the tumorigenesis
of MM has not been widely investigated; however, aberrant
expression of PSMD10 has been reported to have an important
function in various other malignancies, including gastric cancer,
intrahepatic cholangiocarcinoma, testicular germ cell tumors and
hepatocellular carcinomas (5–9). In a
preliminary study, a Protein Turnover Assay (ProTA) was developed;
the assay insures strict co-translational expression of duplicate
monomeric fluorescent proteins that possess the natural N-terminal
residues of 15,000 human proteins, which therefore allows precise
snapshot profiling of global protein stability at the system level
(10). Subsequently, ProTA was
applied to profile changes in human protein degradation induced by
bortezomib (BTZ), aiming to elucidate the potential molecular basis
of the effects of BTZ and tumor drug resistance. Additionally, it
was ascertained that BTZ stabilized proteasome 26S subunit ATPase 1
(PSMC1) and the proteasome assembly factor PSMD10 (10). PSMD10, also known as gankyrin or p28
(GANK), is one of the non-ATPase subunits that is a regulatory
component of the human 26S proteasome. PSMD10, the mammalian
homolog of yeast proteasome assembly factor NAS6 (11), controls the balance between the cell
cycle and apoptosis by degrading RB1 and TP53 (12,13),
which have important effects on the pathogenesis of cancer.
Dysregulation of PSMD10 expression leads to cancer progression, and
has crucial functions in the processes of cell survival, migration
and proliferation (14–16). Previous studies reported that the
direct downregulation of PSMD10 expression in MM cells induced an
increase of P53 mRNA levels and subsequent upregulation of
cyclin-dependent kinase inhibitor 1A (CDKN1A; also known as
Cip1/p21Waf1) and BAX transcripts, which are direct transcriptional
targets of P53 (17). PSMD10 acts
as an oncoprotein and is involved in the negative regulation of the
tumor suppressors RB1 and P53 (13). The imbalance of P53 in myeloma cells
may play an important role in myeloma pathogenesis. Upon
BTZ-induced proteasome inhibition, myeloma cells appear to acutely
stabilize the proteasome assembly factor, PSMD10, which ultimately
facilitates the assembly of proteasome subunits into functional
proteasomes (10). Thus, we
hypothesized that this may functionally abrogate BTZ-induced
proteasome inhibition and help cells survive proteotoxic stress,
which may provide an escape route for cells, resulting in
tumorigenesis, progression and drug resistance during BTZ-based
chemotherapy. This hypothesis may be further examined by designing
RNA interference (RNAi) tools that specifically target the
pro-survival functions of PSMD10 in tumorigenic cells on the basis
of the tissue distributions and the differential functions of the
various PSMD10 splice variants.
RNAi is a sequence-specific gene silencing process
that occurs at the mRNA level. In mammalian cells, short dsRNAs
(<30 bp) knockdown the expression of specific mRNAs via base
pairing (18). Based on this
physiological process, silencing of gene expression by RNAi is
currently used as a standard tool in cultured mammalian cells and
has had a considerable impact on research into the function of
human genes and gene therapy (19).
Although chemically synthetic siRNAs can be introduced into
cultured cells and induce a transient knockdown effect of target
mRNAs, this does not always result in high efficiency of RNAi
delivery and stable transcript knockdown. Alternatively, in
mammalian cells, expression vectors driven by RNA polymerase III
enable the permanent production of small dsRNAs (20,21).
In addition, a lentiviral system can be employed to reversibly
induce gene silencing in a spatially and temporally restricted
manner.
The relationship between PSMD10 and MM, and the
function of PSMD10 in MM cells, have not been clearly established
yet. In the present study, a recombinant lentivirus-mediated short
hairpin RNA (shRNA) vector targeting human PSMD10 gene was
designed. The vector-produced RNAs were processed by Dicer in a
similar manner to siRNAs. The recombinant lentivirus vector
mediating the RNAi-targeting of the PSMD10 gene was then used to
examine the effect of PSMD10 knockdown on RPMI-8226 MM cells in
vitro.
Materials and methods
Cell culture
RPMI-8226 MM and 293T cells were obtained from the
Cell Institute of the Chinese Academy of Sciences (Shanghai,
China). RPMI-8226 cells were cultured in RPMI-1640 culture-medium
and 293T cells in Dulbecco's modified Eagle's medium (DMEM),
supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/ml
penicillin and 100 mg/ml streptomycin (all from Invitrogen,
Carlsbad, CA, USA). Cells were incubated at 37°C with 5%
CO2, the medium was refreshed every 24 or 48 h, and 293T
cells were passaged using trypsin solution when confluent
monolayers were reached.
Design of human PSMD10-shRNA
The mRNA sequence of human PSMD10 (GenBank;
NM_002814.3) was inputted into the Invitrogen online RNAi Designer
to design siRNAs. We selected three optimal siRNAs that targeted
different sequences, as not every siRNA sequence is equally
effective when incorporated into an shRNA (22). A control scrambled shRNA sequence
was specifically designed. All four sequences were aligned using
the GenBank BLAST program, no homologous sequences matched other
than the PSMD10 gene. In order to obtain the dsRNA configuration
required for shRNA formation from single-stranded RNA, a sense
siRNA sequence was linked to its complementary antisense sequence
via a 12-bp loop region, and combined with two supplementary T
nucleotides flanked by restriction enzyme recognition sequences and
protective bases (23). Two
complementary single-stranded DNA oligonucleotides of the four
shRNAs were chemically synthesized by the Laboratory of
Pharmacology, Second Military Medical University (Shanghai, China).
These oligonucleotides were annealed to produce double-stranded
oligonucleotides (Table I).
 | Table I.Sequences of 4 designed
PSMD10-shRNA. |
Table I.
Sequences of 4 designed
PSMD10-shRNA.
| PSMD10-shRNA
name | Sequences |
|---|
| shRNA1-f |
5′-GATCCGCCGATAAATCCCTGGCTACTTCCTGTCAGATAGCCAGGGATTTATCGGCTTTTTG-3 |
| shRNA1-r |
5′-AATTCAAAAAGCCGATAAATCCCTGGCTATCTGACAGGAAGTAGCCAGGGATTTATCGGCC-3′ |
| shRNA2-f |
5′-GATCCGGCTGTACTCCCTTACATTCTTCCTGTCAGAAATGTAAGGGAGTACAGCCTTTTTG-3′ |
| shRNA2-r |
5′-AATTCAAAAAGGCTGTACTCCCTTACATTTCTGACAGGAAGAATGTAAGGGAGTACAGCCC-3′ |
| shRNA3-f |
5′-GATCCGGCATGAGATCGCTGTCATCTTCCTGTCAGAATGACAGCGATCTCATGCCTTTTTG-3′ |
| shRNA3-r |
5′-AATTCAAAAAGGCATGAGATCGCTGTCATTCTGACAGGAAGATGACAGCGATCTCATGCCC-3′ |
| NC-f |
5′-GATCCGAAGCCAGATCCAGCTTCCCTTCCTGTCAGAGGAAGCTGGATCTGGCTTCTTTTTG-3′ |
| NC-r |
5′-AATTCAAAAAGAAGCCAGATCCAGCTTCCTCTGACAGGAAGGGAAGCTGGATCTGGCTTCC-3′ |
Construction of pSIH-PSMD10-shRNA
vectors
The annealed PSMD10-shRNA oligonucleotides were
cloned into linearized pSIH1-H1-copGFP shRNA empty vectors (catalog
no. SI501A-1; System Biosciences) using the following steps:
pSIH1-H1-GFP shRNA vectors and double-stranded shRNA
oligonucleotides were digested with both BamHI and
EcoRI; and the large fragment of the vector and the digested
shRNA fragment were then ligated using T4 DNA ligase according to
the manufacturer's instructions. The ligated products were
transformed into competent DH5α cells (catalog no. D9057; Takara,
Shiga, Japan) using the heat shock method. The transformed cells
were grown on an LB-agar plate containing ampicillin at 37°C
overnight.
Identification of double digestion and
DNA sequence
To ensure that the shRNAs were inserted into the
vectors, positive clones were collected and identified by PCR
amplification. PCR products (5 µl) were separated using 2% agarose
gel electrophoresis and the lengths of the amplified bands were
examined. The primer sequences used for PCR identification were as
follows: forward, 5′-ATAT TTGC ATGT CGCT ATGT G-3′ and reverse,
5′-CAGG CTAG ATCT GGTC TAAC CA-3′. Positive clones were cultured
overnight at 37°C in 3 ml LB broth plus 100 µg/ml ampicillin with
vigorous agitation at 200 rpm. The plasmid DNA was prepared using
Mini Plasmid Purification kit Ver 2.0 (catalog no. SK1191; Shanghai
ShengGong Biotech, Shanghai, China) according to the manufacturer's
instructions. Extracted recombinant plasmids were used for DNA
sequencing to identify the inserted PSMD10-shRNA fragments. The
pSIH1-PSMD10-shRNA sequencing forward primer was:
5′-GCAGAGCTCGTTTAGTGAACC-3′.
Effective siRNA sequence screening and
verification
The recombinant expression vector was verified by
sequencing extracted plasmid DNA. Endotoxins were removed using a
plasmid mini kit (catalog no. 12145; Qiagen, Inc., Hilden, Germany)
according to the manufacturer's instructions. The purity and
concentration of plasmid DNA were determined by UV
spectrophotometer after observing the integrity of plasmid DNA on
agarose gel electrophoresis.
When 293T cells cultured in a 10-cm cell culture
dish had reached 70% confluence, a cell suspension was prepared
using trypsin. The cells were then seeded into a 6-well plate in 2
ml complete DMEM (catalog no. 11995073; Invitrogen) plus 10% FBS,
and cultured at 37°C in 5% CO2 overnight. After 24 h,
adherent cells reached ~80% confluence and were in a healthy
condition with spindle-like shape. For transfection, 4 µg plasmid
DNA diluted in 250 µl Opti-MEM (catalog no. 11058021) and 10 µl
Lipofectamine 2000 (catalog no. 1668027) (both from Invitrogen)
diluted in 250 µl Opti-MEM were used/well according to this
protocol. Transfected cells were grouped as follows: 293T, 293T +
pshRNA vector, 293T + pshRNA1-PSMD10, 293T + pshRNA2-PSMD10, 293T +
pshRNA3-PSMD10, 293T + pshRNA-NC. After transfection for 48 h,
fluorescence microscopy indicated that the transfection efficiency
was ~90%.
Total RNA was extracted from the transfected cells
using TRIzol (catalog no. 15596-018; Invitrogen). Extracted RNA was
treated with DNase I to remove any DNA contamination, and
single-stranded cDNA was prepared from RNA using Reverse
Transcriptase M-MLV (catalog no. D2640A; Takara). Quantitative PCR
(qPCR) primers were designed using Primer Premier 5.0 and oligo
7.0. A reverse transcription-qPCR reaction was performed to screen
the most effective recombinant plasmids. qPCR was performed using
Takara SYBR Premix Ex Taq (catalog no. DRR041A; Takara). A standard
PCR program was used as follows: 95°C for 3 min; 40 cycles of 95°C
for 10 sec and 60°C for 20 sec; followed by 95°C for 10 sec and
72°C for 20 sec. A melting curve analysis was performed to verify
the identities of PCR products. Each sample was tested three times
to obtain an average. Relative expression levels of the PSMD10 gene
were normalized to β-actin expression levels by calculating the Cq
value using Thermal Cycler DICE Real-Time System analysis software.
Primer sequences for the amplification of PSMD10 and β-actin are
listed in Table II.
| Table II.Primers of PSMD10 and β-actin. |
Table II.
Primers of PSMD10 and β-actin.
| Name | Primer | Product length
(bp) |
|---|
| Human PSMD10 |
|
|
|
Sense |
5′-CTGGCCGGGATGAGATTGTAAAAG-3′ | 187 |
|
Antisense |
5′-CGGTGCATTGCTGTAGCCTCATAA-3′ |
|
| Human β-actin |
|
|
|
Sense |
5′-CCCAAGGCCAACCGCGAGAAGATG-3′ | 219 |
|
Antisense |
5′-GTCCCGGCCAGCCAGGTCCAGA-3′ |
|
Lv-shRNA2-PSMD10 recombinant virus
packaging and production
pPACKTM Lentivector Packaging System was used to
perform the packaging and production of lentiviral particles. At 24
h before transfection, 293TN packaging cells were seeded in a 10-cm
cell culture dish to achieve ~80% confluence for the transfection
experiment. Per 10-cm culture dish, 4 µl of the most effective
recombinant plasmid pshRNA2-PSMD10 (500 ng/µl) and 20 µl Lentivirus
Package plasmid mix (500 ng/µl) (catalog no. LV500A-1; System
Biosciences) were co-transfected into cells using 40 µl
transfection reagent according to the manufacturer's instructions.
At 24 h after transfection, the medium was refreshed using freshly
prepared DMEM plus 2% FBS. At 48 h after transfection, the cells
were adherent and ~90% confluent with normal growth conditions and
a flat shape. Viral particle-containing supernatants were harvested
by removing the medium from the cells and transferred to a 15-ml
sterile, capped, conical tube. Non-adherent cells and debris were
pelleted by centrifugation at 5,000 × g at 4°C for 5 min. Each
viral titer was calculated as ×104 by counting the
number of GFP+ 293T cells by the gradient dilution
method 2 days after transduction with serial dilutions of the viral
stocks. The virus particle concentration was adjusted to
1×104 ifu/µl using dPBS (pH=7.8) with 1 ml of each tube
packaged and stored at −80°C.
Recombinant lentiviral infection of
target cells
The optimum MOI value of the lentivirus infection of
RPMI-8226 cells was determined according to the results of
preliminary experiments. An MOI of 20 was used. RPMI-8226 cells
were seeded in a 6-well plate and cultured in RPMI-1640 plus 10%
FBS. Infected cells were grouped as follows: RPMI-8226 cells,
RPMI-8226 cells + Lv-NC, RPMI-8226 + Lv-shRNA2-PSMD10. The virus
solution was added to the cells at the optimum MOI of 20.
Fluorescence was enhanced and the number of infected cells (~90%)
was increased at 96 h post-infection compared with that at 72 h.
Some cells were collected for RNA and protein extraction to
evaluate RNAi efficiency, and the remaining cells were stored in
liquid nitrogen for further experiments.
Detection of gene and protein levels
in target cells
Total RNA was extracted from the MM RPMI-8226 cells
using experimental methods and reagents as previously described. MM
cells were lysed in 1 ml M-PER Mammalian Protein Extraction Reagent
(catalog no. 78501; Pierce, Rockford, IL, USA) plus protease
inhibitor, then incubated on ice for 40 min. The lysates were
cleared by centrifugation and the protein concentration was
determined using the BCA method. Proteins were separated by
SDS-PAGE (4% stacking gel, 10% separating gel) and transferred to
polyvinylidene difluoride (PVDF) membranes (catalog no. IPVH00010;
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
skimmed milk in Tris-buffered saline/Tween-20 (TBST) buffer, then
probed with anti-PSMD10 (catalog no. ab154676; Abcam, Cambridge,
MA, USA) and anti-β-actin (catalog no. sc-81178; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) antibodies at room
temperature, followed by incubation with horseradish
peroxidase-conjugated anti-rabbit (for PSMD10; catalog no. 7074)
and anti-mouse (for β-actin; catalog no. 7076) secondary
antibodies. After several washes, the membranes were developed with
an enhanced chemiluminescence system (catalog no. 32106; Pierce ECL
Western Blotting Substrate) and exposed to Kodak BioMax light film.
β-actin protein levels were used as a control to verify equal
protein loading.
Detection of apoptosis by flow
cytometry and fluorescence microscopy
After transduction with the lentivirus, MM cell
lines were seeded in 6-well plates at a density of 1×105
cells/well. After 96 h post-infection, MM cells were harvested and
washed in phosphate-buffered saline (PBS). A cell suspension with a
density of 1×106 cells/ml was prepared for each assay.
Cells were simultaneously stained with phycoerythrin (PE)-labeled
Annexin V and with 7-aminoactinmycin D (7-AAD) to discriminate
viable cells (Annexin−/7-AAD−) from early
apoptotic cells (Annexin+/7-AAD−), and late
apoptotic and necrotic cells
(Annexin+/7-AAD+). Flow cytometric analysis
was performed to detect Annexin V and 7-AAD staining. All
experiments were performed in triplicate.
Statistical analysis
SPSS version 21.0 was used for statistical analysis.
Values presented are representative of triplicate determinations in
no less than three experiments. Data are expressed as the mean ±
standard deviation and compared using a two-sided Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Sequencing profile of the recombinant
PSMD10-shRNA expression vectors
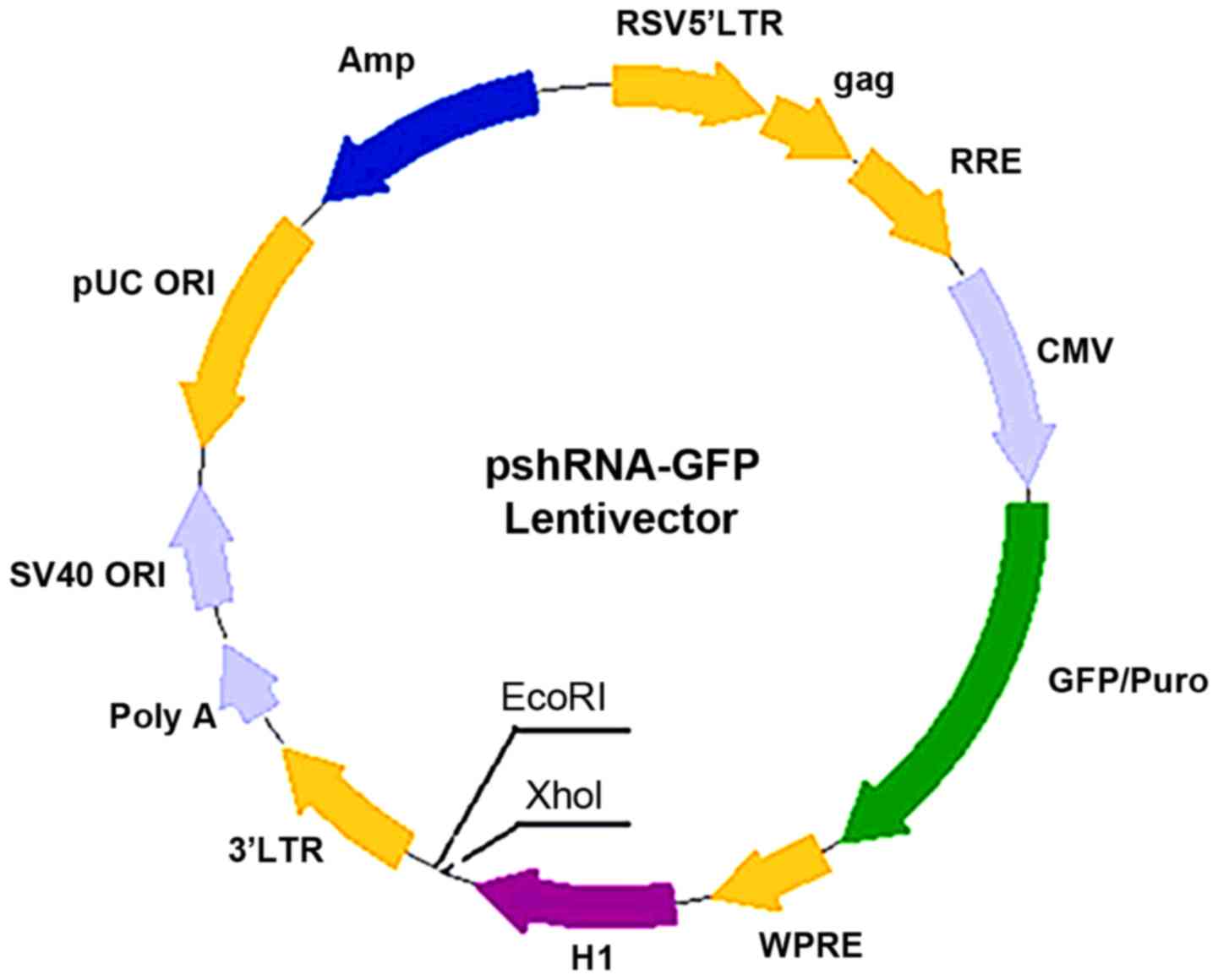
A schematic diagram of the construction of the
pSIH1-H1-shRNA expression vector to silence PSMD10 gene expression
is presented in Fig. 1. Recombinant
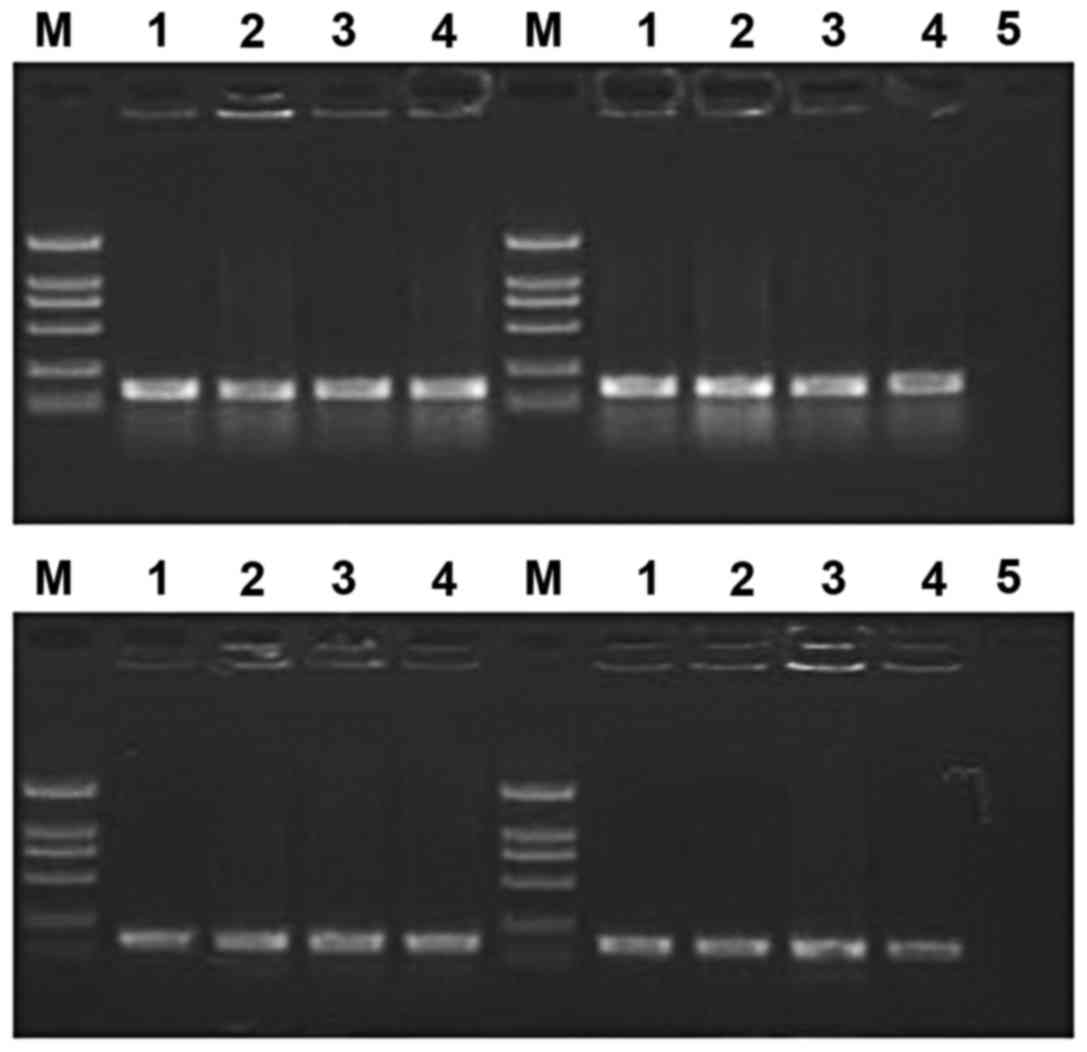
plasmids were digested with BamHI and EcoRI, and
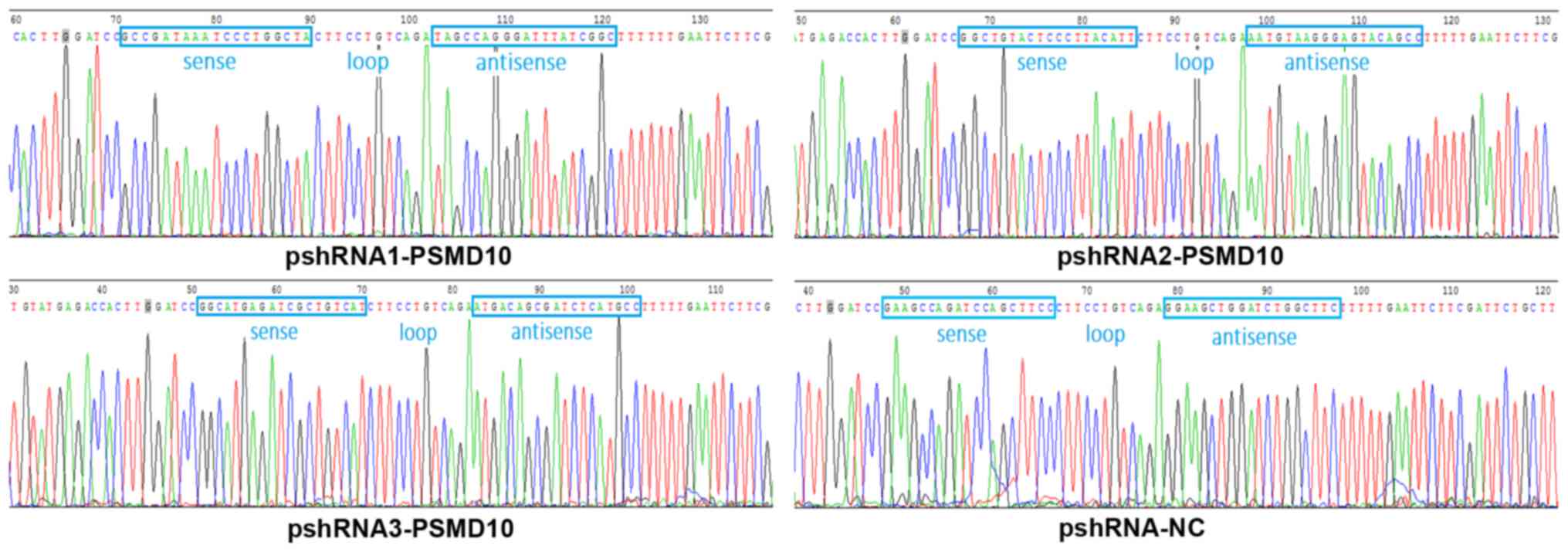
fragments were identified on 2% agarose gels (Fig. 2). DNA sequencing results provided
further verification of the presence of the recombinant plasmids,
indicating that the correct sequences were carried by all the shRNA
expression plasmids (Fig. 3).
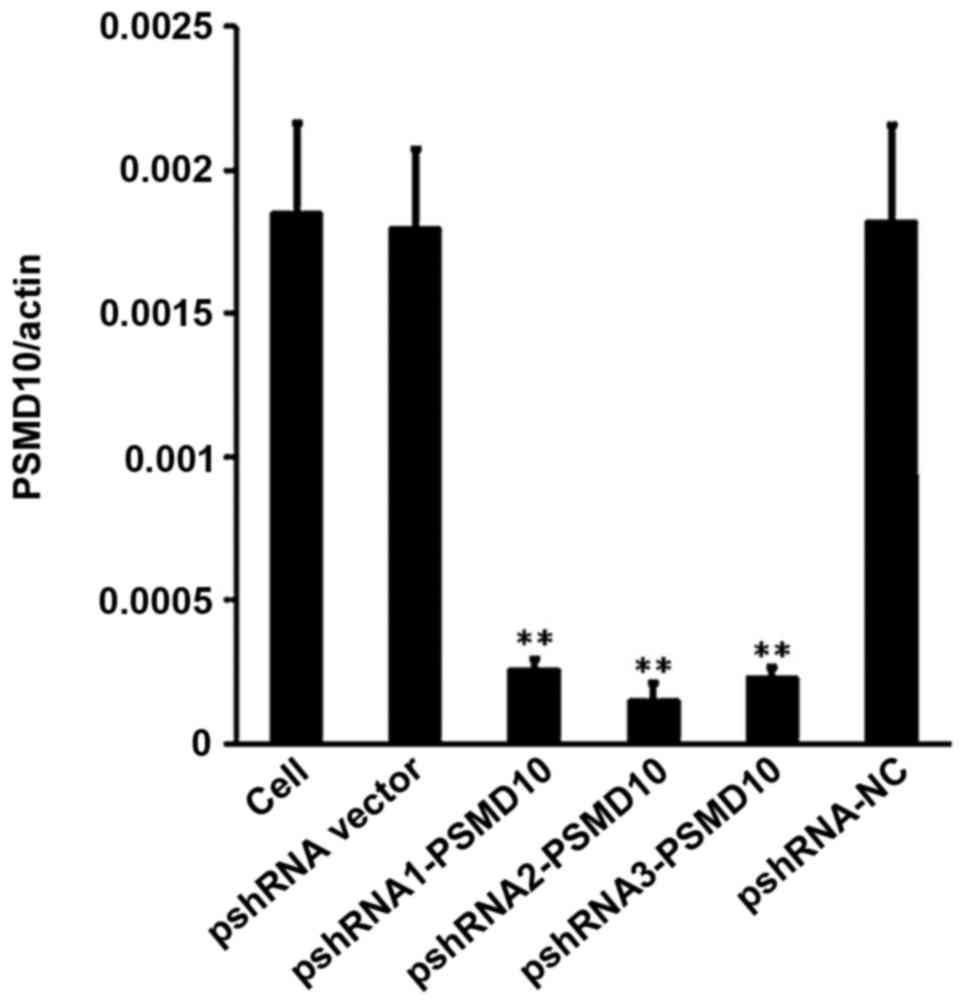
Detection of 293T cell transfection
efficiency and RT-qPCR
After transfection for 48 h, a fluorescence
microscope was used to observe the transfection efficiency of 293T
cells transfected with the pSIH1-H1-shRNA expression vector; the
transfection efficiency was ~90% (Fig.
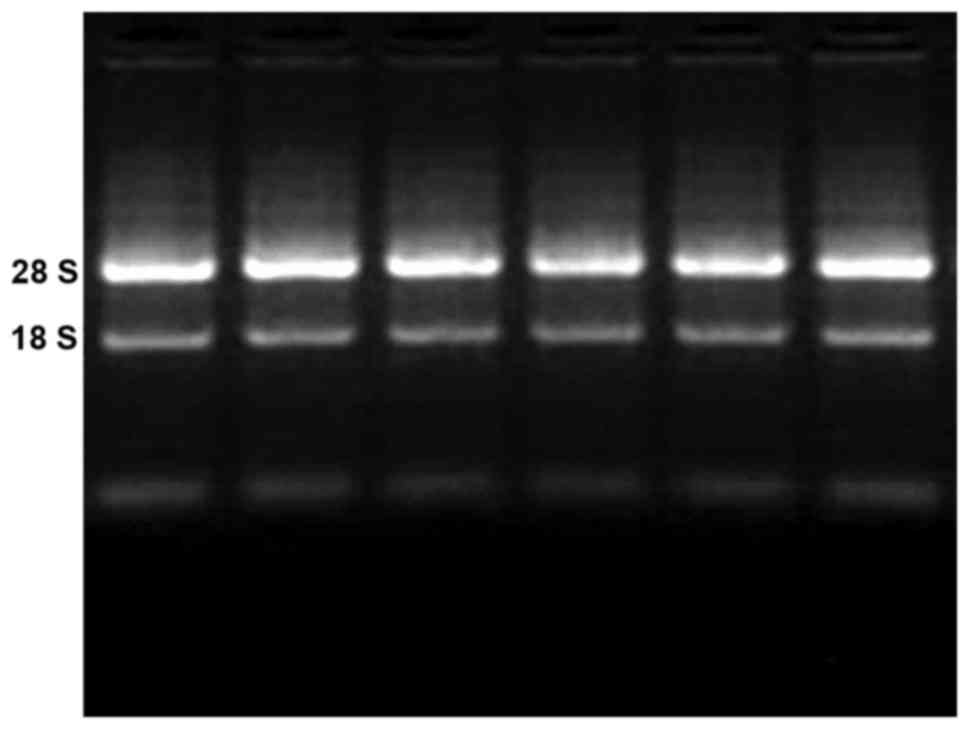
4). RNA electrophoresis was performed to demonstrate the
integrity of extracted RNA (Fig.
5). RT-qPCR was performed to analyze the relative expression
levels of PSMD10 normalized to β-actin by calculating the Ct value
using Thermal Cycler DICE Real-Time System analysis software
(Fig. 6).
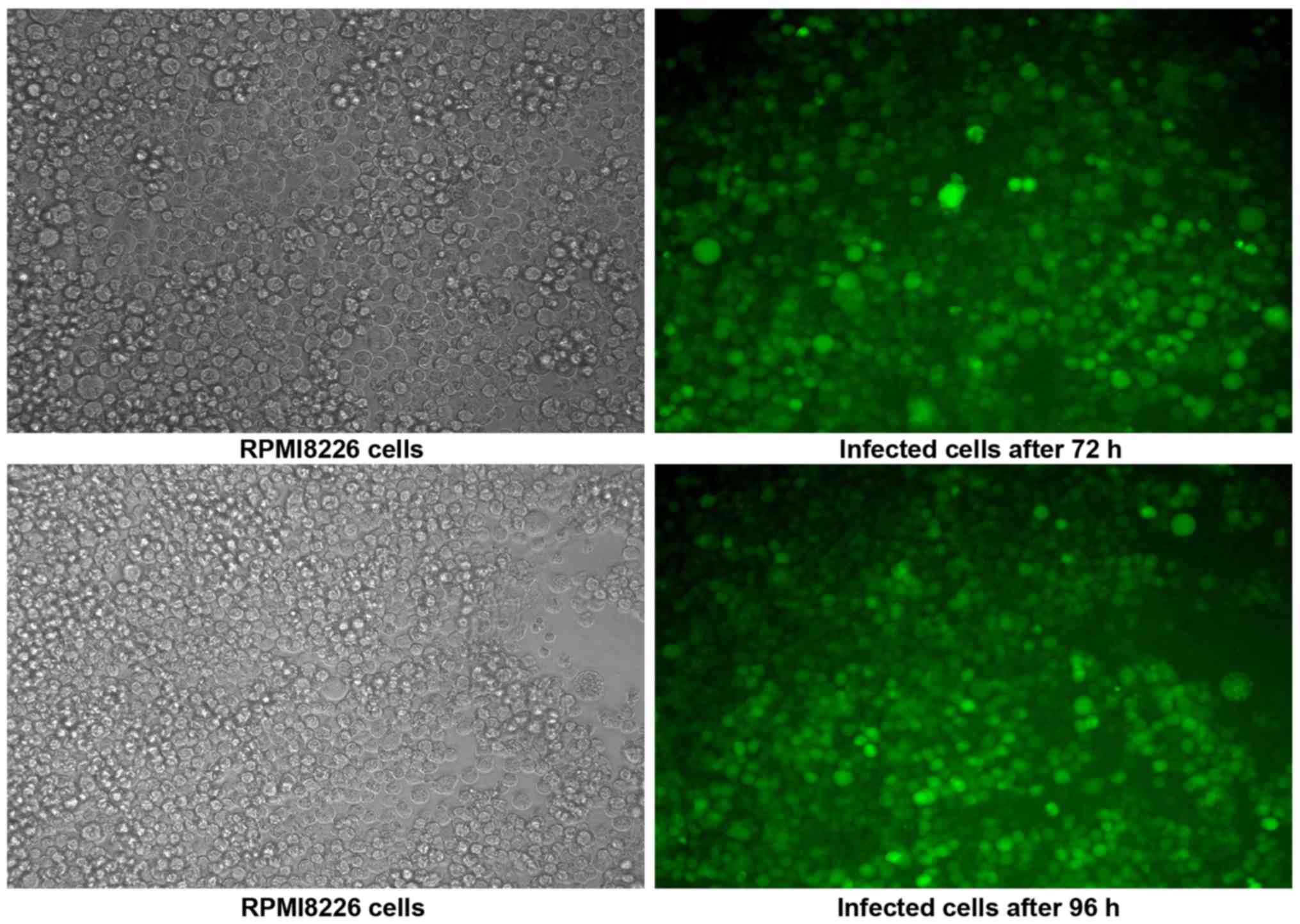
Infection of RPMI-8226 cells with
Lv-shRNA2-PSMD10
RPMI-8226 cells infected with Lv-shRNA2-PSMD10 were
observed to exhibit green fluorescence under a fluorescence
microscope (Fig. 7).
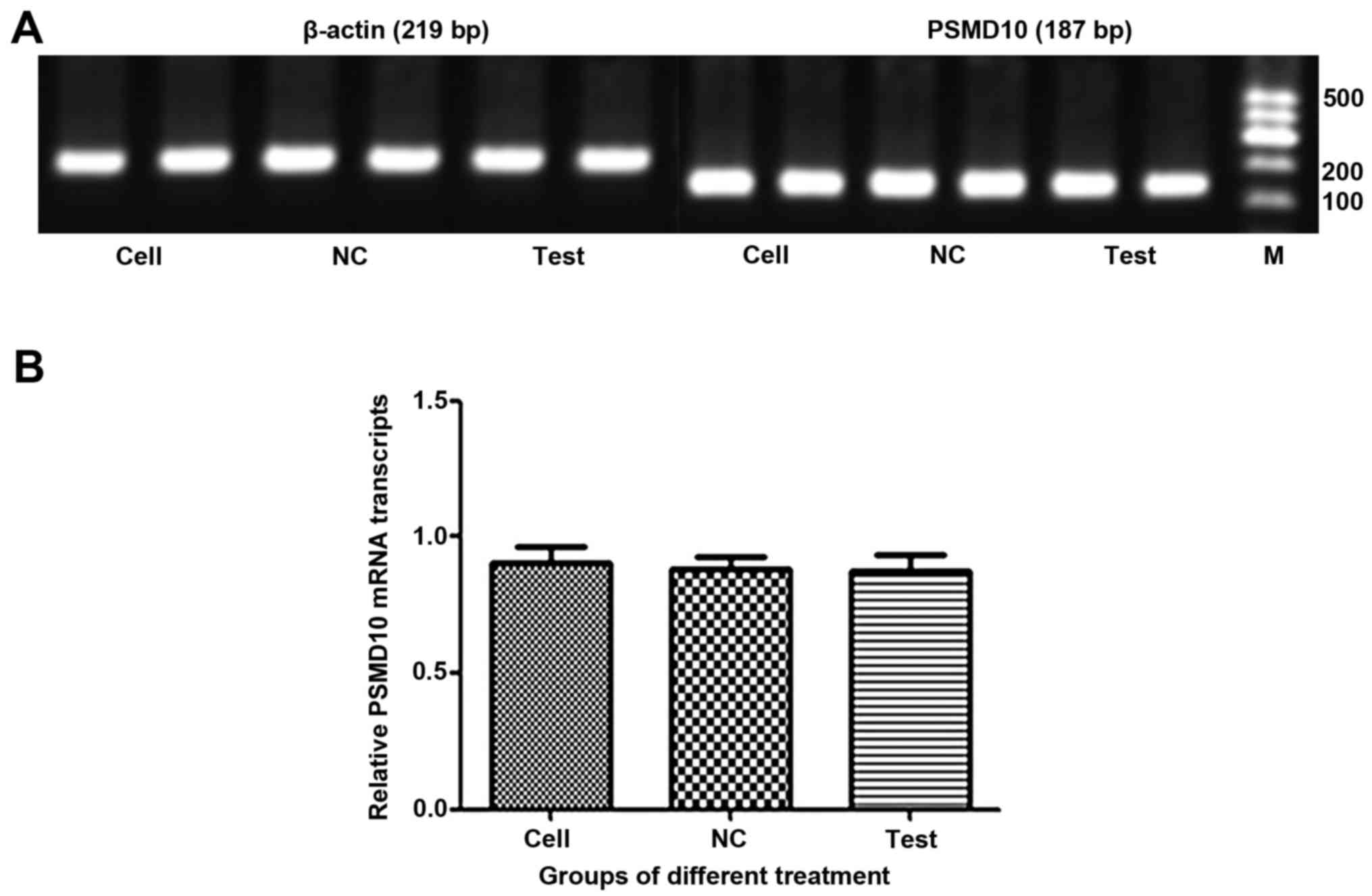
Detection of PSMD10 mRNA levels after
infection
The mRNA levels of PSMD10 were determined by RT-PCR
(Fig. 8A) and RT-qPCR (Fig. 8B). Surprisingly, there was no
significant difference in expression levels of PSMD10 mRNA between
cells infected with Lv-shRNA2-PSMD10 and those infected with
Lv-NC.
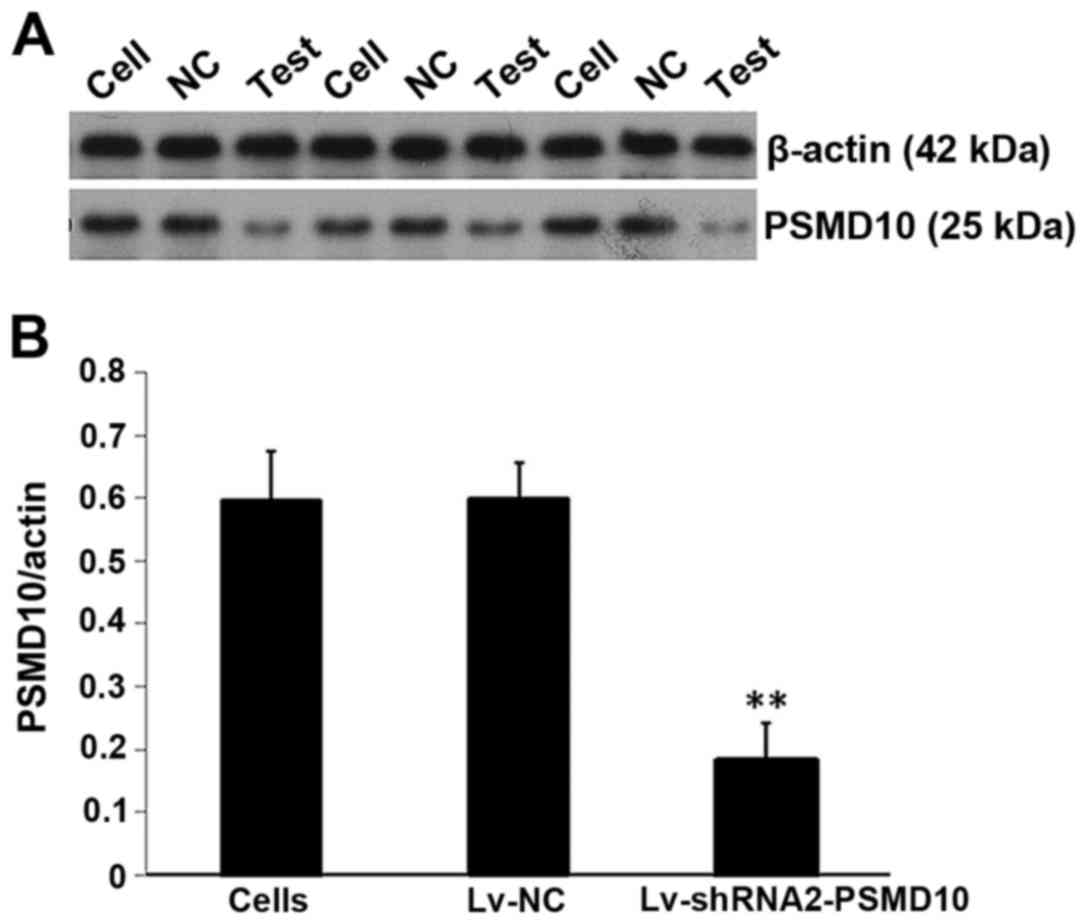
Silencing of PSMD10 protein expression
after infection
Protein levels of PSMD10 gene expression were
determined by western blotting. As demonstrated in Fig. 9A, compared with the non-infected
cells and the Lv-NC group, the PSMD10 protein levels in the
Lv-shRNA2-PSMD10 group were significantly decreased. Furthermore,
there was a significant decrease in the protein expression of
PSMD10 as demonstrated by densitometric analysis (P<0.01;
Fig. 9B).
Apoptosis is increased in MM cells by
knockdown of endogenous PSMD10
To determine whether apoptosis was increased in MM
RPMI-8226 cells by silencing PSMD10 gene expression, Annexin V-PE
and 7-AAD staining was performed. Flow cytometric analysis was used
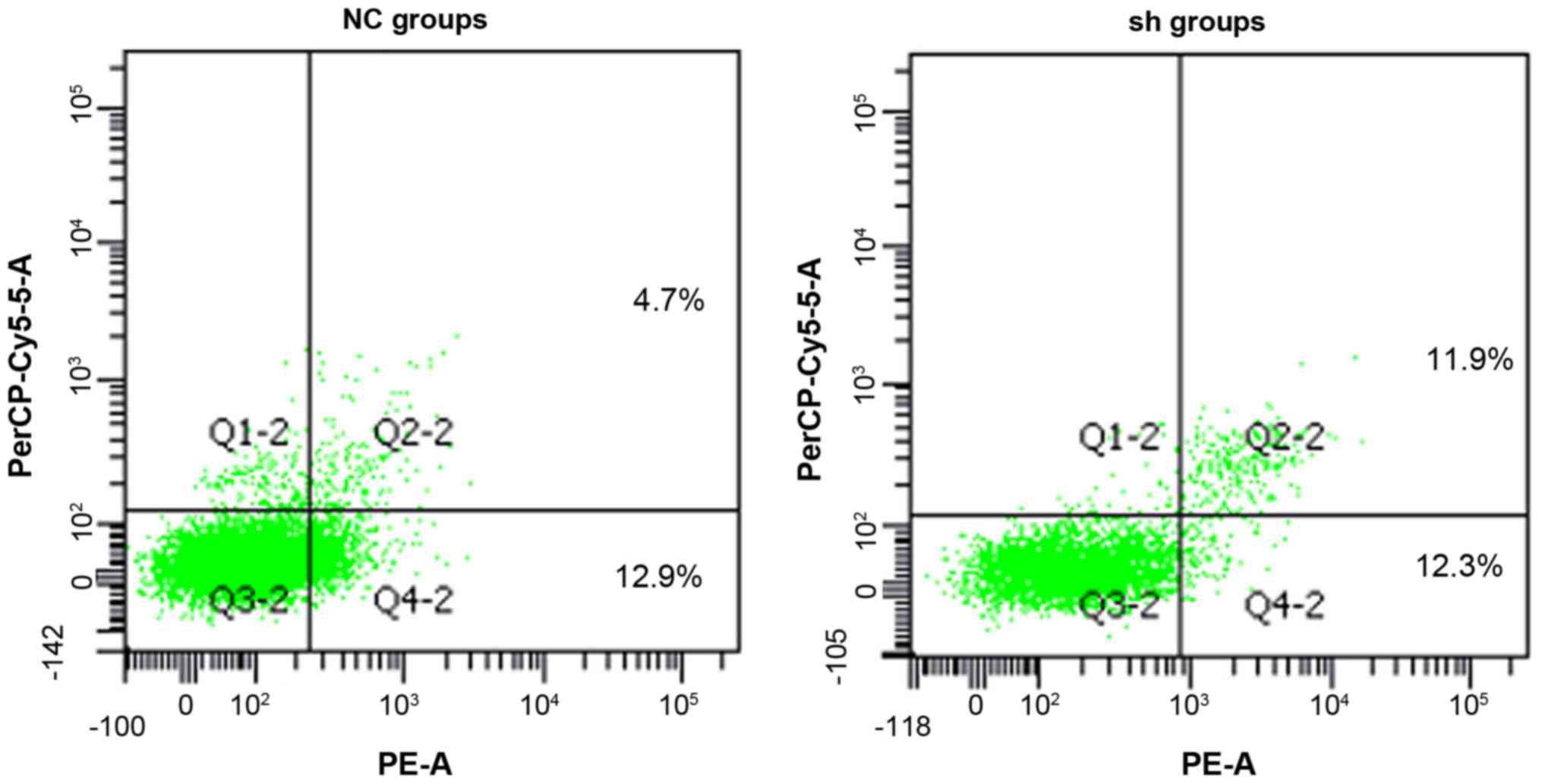
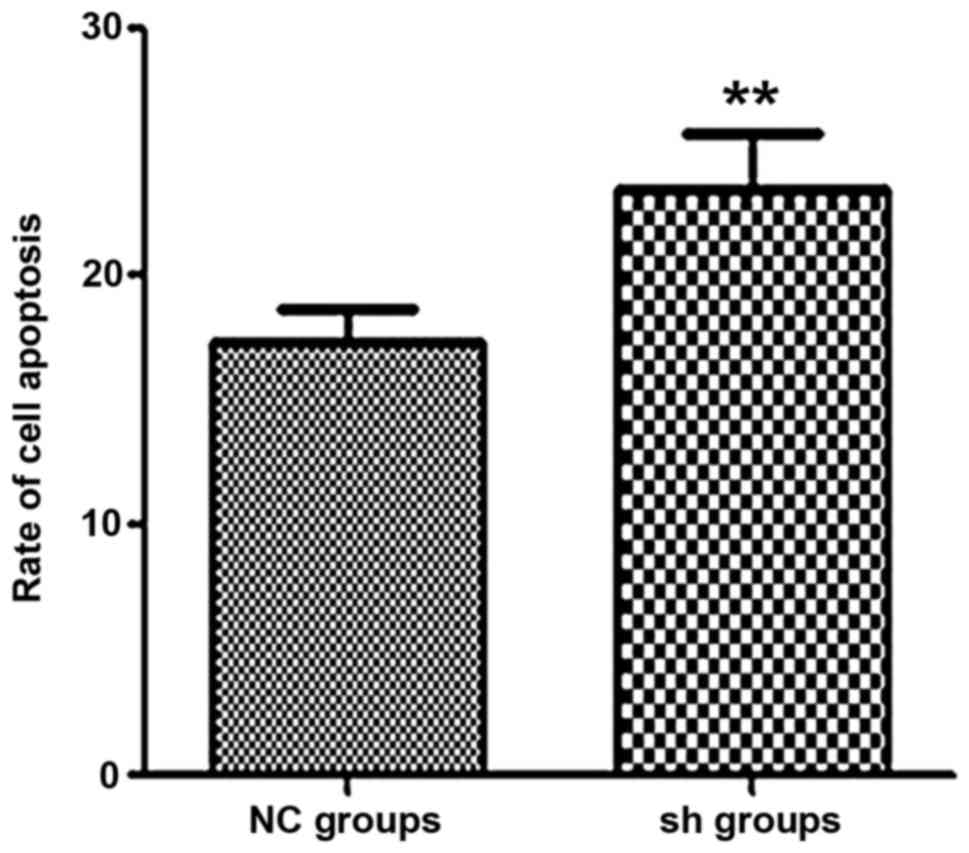
to evaluate the number of apoptotic cells (Fig. 10). Silencing of PSMD10 increased
the percentage of apoptotic cells (quadrant 2 + quadrant 4) in the
Lv-shRNA2-PSMD10 group compared with the NC groups (Fig. 11).
Discussion
Using conventional treatments, MM remains an
essentially incurable disease. The overall median survival time
after an MM diagnosis is 5–6 years (24), yet disease outcomes are highly
influenced by the characteristics of the cancer (for example,
high-risk cytogenetics) or the patients (for example, age).
Treatment of MM remains highly individualized, with multiple
factors used to determine the most appropriate course of therapy.
In younger patients, progression-free survival (PFS) and overall
survival (OS) have been greatly improved by treatment with
autologous stem cell transplantation (25,26).
Unfortunately, the decision-making process of treatment choices for
myeloma patients at each phase of the disease has become complex;
this is in large part due to the deeper understanding of plasma
cell biology, resulting in more therapeutic options at each stage.
MM is an incurable plasma cell malignancy characterized by initial
responsiveness to treatment, followed by the appearance of
increasingly refractory disease and ultimately, death due to
infection, renal failure and cytopenia (27). Thus, the development of new
therapeutic strategies for MM remains important.
In recent years, the proteasome inhibitors (PIs) BTZ
and carfilzomib have emerged as frontline treatments for relapsed
and refractory MM; however, resistance to these drugs occurs
through poorly defined mechanisms. Numerous studies have identified
different acquired-resistance models, such as β5 proteasome subunit
mutations, and stabilization of tumor suppressors and pro-apoptosis
proteins. In MM cells, sensitivity to proteasome inhibition arises
since these malignant proliferative B cells are highly reliant on
protein synthesis and turnover, and thus, highly dependent on the
ubiquitin proteasome system for the processing of defective
proteins (28–30). Various E1, E2 and E3 ligases tag
targeted proteins with multiple ubiquitin residues, and the tagged
proteins are subsequently degraded by the 26S proteasome. A 19S lid
complex and a 20S core complex form the 26S proteasome. The 20S
core carries out the cleavage of unfolded proteins. Upon immune
stimulation, for instance, by interferon-γ, the 20S constitutive
catalytic subunits, β1 (PSMB6, caspase-like activity), β2 (PSMB7,
trypsin-like activity), and β5 (PSMB5, chymotrypsin-like activity),
are replaced by their immune cognate forms; β1i (PSMB9/LMP2), β2i
(PSMB10/MECL-1) and β5i (PSMB8/LMP7) (31,32).
BTZ is a reversible inhibitor that targets primarily the β5-subunit
and also targets the caspase-like activity of the β1 proteasome
subunit to a lesser extent. At higher concentrations, BTZ inhibits
the trypsin-like proteolytic activity facilitated by the β2
proteasome subunits.
Previous research indicated that the molecular
mechanisms involved in acquired BTZ resistance may be associated
with the upregulation of β5 subunits, the point mutation of β5
genes and downregulation of β5i subunits (33). Some data suggested that β5 subunits
may be the key regulatory factors linked to the PI resistance of
tumor cells. However, mutations of β5 subunits may not be uniquely
responsible for the resistance of tumor cells to PI drugs. An
aberration in the PSMB8 gene was demonstrated to breakdown the
tertiary structure of β5i and decrease chymotrypsin-like activity
but did not affect β5i protein expression (34). Defects in immunoproteasome assembly
in PSMB8 mutants can be expected, as β5i is required for the
maturation of immunoproteasomes by processing and incorporation of
the β1i and β2i subunits (35).
Certain studies have reported that there may be an association
between PSMB8 polymorphisms and BTZ resistance, but the direct link
remains elusive. Overall, PSMB8 polymorphisms can result in altered
proteasome assembly; however, a direct link between PSMB8
polymorphisms and BTZ-resistance remains elusive. Notably,
mechanistic studies have demonstrated that overexpression of β1 or
β5 subunits led to increased proteasome activity, and enhanced cell
survival following exposure to proteasome inhibitors (36,37).
Further studies revealed that in BTZ-resistant RPMI-8226 cells,
upregulation of β5 expression was clearly opposed by a parallel
decrease in β5i expression levels, presumably to balance the total
proteasome levels in drug-resistant cells compared with parental
cells (38). Furthermore, it should
be noted that chymotrypsin-like activity probes were used to detect
β5i and β1i subunit activities, which are often downregulated in
BTZ-resistant cells, and thus, can influence the overall activity
measurements (39). In summary,
several studies (38,40,41)
have demonstrated that overexpression of β5 subunits is an initial
cellular response mechanism to BTZ treatment, which may precede the
acquisition of mutations following prolonged exposure to BTZ.
Better targeted, subunit-specific probes that can discriminate
between β5, β5i and β1i-related activities are required to
understand these mechanisms in more detail, and may provide an
insight into the concentrations of various proteasome inhibitors
that lead to the development of drug resistance (39).
It has been previously reported that the direct
downregulation of the expression of PSMD10 in MM cells induced an
increase in P53 mRNA levels and subsequent upregulation of CDKN1A
(p21Waf1/Cip1) and BAX transcripts, which are direct
transcriptional targets of P53 (17). PSMD10 acts as an oncoprotein and is
involved in the negative regulation of RB1 and P53 tumor
suppressors (13). The imbalance of
P53 in myeloma cells may play an important role in myeloma
pathogenesis, which can be induced through post-transcriptional
mechanisms. In our preliminary study, many proteins were identified
to be markedly stabilized by BTZ treatment. Similarly, BTZ also
stabilized proteasome subunit PSMC1 and proteasome assembly factor
PSMD10 (10). Thus, we hypothesize
that PSMD10 may be associated with the occurrence and progression
of MM, and drug resistance. Thus, a lentivirus-mediated RNAi
experiment in MM was designed to validate the growth effects of
PSMD10 downregulation on MM cells.
RNAi is a process in which double-stranded RNA is
employed to enhance the degradation of endogenous mRNA. siRNA can
induce transient and efficient RNAi effects (21). siRNA-producing plasmid vectors allow
transient expression of siRNA, and are widely used in gene
silencing, but tend to have low transfection efficiency (42). Lentivirus-mediated shRNA systems can
produce highly efficient, whole organism expression of shRNAs and
allow the induced gene knockdown to be reversed at a given time.
This peculiarity of the shRNA system offers specific applications
to study gene function in animal experiments, which cannot be
achieved with other gene knockdown technologies (43,44).
Although conditional lentivirus vectors have been used for several
years, the employment of lentiviral vectors expressing shRNA as a
therapeutic tool for MM has not been thoroughly explored.
In the present study, lentivirus-mediated shRNA was
used to silence endogenous PSMD10 expression and the effects of
PSMD10 downregulation on the phenotypes of MM RPMI-8226 cells were
examined. Three shRNAs were designed to target PSMD10 mRNA and were
successfully transfected into 293T cells with the lentivirus
plasmids. DNA sequencing and agarose gel electrophoresis further
confirmed that three PSMD10 shRNAs were correctly inserted into the
multiple cloning sites of the pSIH1-H1-copGFP expression plasmid.
Lv-PSMD10-shRNA2 was previously identified as the most effective
one, as the 293T transfectants exhibited a significantly decreased
level of PSMD10 mRNA. In the present study, LvPSMD10-shRNA2
effectively decreased the level of PSMD10 protein, which indicated
that our lentivirus-mediated PSMD10-specific shRNA was able to
specifically silence the expression of the PSMD10 protein in
RPMI-8226 cells. The efficiency of protein silencing was estimated
to be 70% in the present study. However, notably, there was no
obvious change in PSMD10 expression at the mRNA level. Therefore,
presumably, the knockdown effect on the expression of PSMD10 may
take place at the translational level. In addition, the depletion
of PSMD10 resulted in increased apoptosis in RPMI-8226 cells in
vitro. This effect may be mediated by interference with the P53
signal transduction pathway. The potential oncogenic mechanism of
PSMD10 may be to increase the proteasomal degradation of P53 via
the MDM2/P53 complex and boost the turnover of P53 in an
MDM-dependent manner. In this process, PSMD10 protein binds to MDM2
protein and enhances the ability of MDM2 to ubiquitinate P53
(45). Thus, overexpression of
PSMD10 may block P53-dependent apoptosis in human RPMI-8226 MM
cells. However, this hypothesis remains to be elucidated in detail.
It has been previously demonstrated that when PSMD10 is silenced in
hepatocellular carcinoma cell lines with wild-type and mutated P53,
a different induced phenotype can be observed in the cells with
wild-type P53, with upregulation of P53 levels leading to
activation of caspase-9 and the induction of apoptosis (46). BTZ increases P53 protein levels by
inhibiting the proteasomal degradation of P53, and subsequently
activates the P53 signal transduction pathway, which emphasizes the
importance of P53 regulation in the pathogenesis of MM (47). Collectively, the fact that
downregulation of PSMD10 expression in myeloma cells induces
apoptosis supports the potentially important role of the P53/MDM2
regulatory mechanism in MM.
In the event that PSMD10 shRNA can decrease the
resistance of MM to BTZ, it may act as a potential anticancer
therapy that could be administered along with BTZ for MM treatment.
Therefore, in follow-up experiments, a cell line stably expressing
PSMD10 shRNA should be used to review the role of PSMD10 in
regulating BTZ sensitivity in MM. Subsequently, different
concentrations of BTZ treatment could be used to observe the drug
sensitivity in MM cells, and the effect on cell proliferation and
apoptosis. In general, the chemosensitivity of MM to BTZ should be
examined further, and whether chemosensitivity can be induced or
restored by decreasing the expression of PSMD10 should be
investigated.
In conclusion, the present study established an
efficient method for screening the most effective shRNA the
suppression of PSMD10 expression in MM RPMI-8226 cells, and
provided a novel strategy for further investigation of the role of
PSMD10 in MM. In the exploration of biochemical mechanisms of RNAi
pathways, this method is likely to be useful and has the potential
to provide more rational strategies for MM treatment. Collectively,
the results of the present study provide further evidence that
PSMD10 may have potential as a pharmacological target. The results
provide a new perspective for the study of the function of PSMD10
in tumors, and it may be beneficial for future gene therapy
strategies in the treatment of MM.
Acknowledgements
The present study received the support of the
National Natural Science Fund of China (no. 81670195). We are
sincerely grateful to Professor Tao Zhang and Professor Zi Chen for
providing technical assistance and experimental supplies. We are
particularly indebted to the help offered by the pharmacological
laboratory of the Second Military Medical University and the
Central Laboratory of Huashan Hospital.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
National Cancer Institute SEER database:
Surveillance, Epidemiology, and End Results Program. 2014.(cited
June 30). http://seer.cancer.gov/
|
|
3
|
Becker N: Epidemiology of multiple
myeloma. Multiple Myeloma: Recent Results in Cancer Research.
Moehler T and Goldschmid H: Berlin, Heidelberg, Germany:
Springer-Verlag; pp. 25–35. 2011, View Article : Google Scholar
|
|
4
|
Bergsagel PL: Epidemiology, etiology, and
molecularpathogenesis. Multiple Myeloma. Richardson PG and Anderson
KC: London, Chicago: Remedica Publi-shing; pp. 1–24. 2004
|
|
5
|
Liu Y, Xu J, Jiang M, Ni L, Chen Y and
Ling Y: Association between functional PSMD10 Rs111638916 variant
regulated by MiR-505 and gastric cancer risk in a Chinese
population. Cell Physiol Biochem. 37:1010–1017. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Uddin MH, Choi MH, Kim WH, Jang JJ and
Hong ST: Involvement of PSMD10, CDK4, and tumor suppressors in
development of intrahepatic cholangiocarcinoma of Syrian Golden
Hamsters induced by Clonorchis sinensis and
N-nitrosodimethylamine. PLoS Negl Trop Dis. 9:e00040082015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Tian F, Li D, Chen J, Jiang P, Zheng
S, Li X and Wang S: MiR-605 represses PSMD10/Gankyrin and inhibits
intrahepatic cholangiocarcinoma cell progression. FEBS Lett.
588:3491–3500. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen BF, Suen YK, Gu S, Li L and Chan WY:
A miR-199a/miR-214 self-regulatory network via PSMD10, TP53 and
DNMT1 in testicular germ cell tumor. Sci Rep. 4:64132014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo T, Fu J, Xu A, Su B, Ren Y, Li N, Zhu
J, Zhao X, Dai R, Cao J, et al: PSMD10/gankyrin induces autophagy
to promote tumor progression through cytoplasmic interaction with
ATG7 and nuclear transactivation of ATG7 expression. Autophagy.
12:1355–1371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu T, Tao Y, Yang M, Chen P, Gao X, Zhang
Y, Zhang T, Chen Z, Hou J, Zhang Y, et al: Profiling human protein
degradome delineates cellular responses to proteasomal inhibition
and reveals a feedback mechanism in regulating proteasome
homeostasis. Cell Res. 24:1214–1230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tomko RJ Jr, Funakoshi M, Schneider K,
Wang J and Hochstrasser M: Heterohexameric ring arrangement of the
eukaryotic proteasomal ATPases: Implications for proteasome
structure and assembly. Mol Cell. 38:393–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Higashitsuji H, Itoh K, Nagao T, Dawson S,
Nonoguchi K, Kido T, Mayer RJ, Arii S and Fujita J: Reduced
stability of retinoblastoma protein by gankyrin, an oncogenic
ankyrin-repeat protein overexpressed in hepatomas. Nat Med.
6:96–99. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Higashitsuji H, Higashitsuji H, Itoh K,
Sakurai T, Nagao T, Sumitomo Y, Masuda T, Dawson S, Shimada Y,
Mayer RJ, et al: The oncoprotein gankyrin binds to MDM2/HDM2,
enhancing ubiquitylation and degradation of p53. Cancer Cell.
8:75–87. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bai Z, Tai Y, Li W, Zhen C, Gu W, Jian Z,
Wang Q, Lin JE, Zhao Q, Gong W, et al: Gankyrin activates IL-8 to
promote hepatic metastasis of colorectal cancer. Cancer Res.
73:4548–4558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhen C, Chen L, Zhao Q, Liang B, Gu YX,
Bai ZF, Wang K, Xu X, Han QY, Fang DF, et al: Gankyrin promotes
breast cancer cell metastasis by regulating Rac1 activity.
Oncogene. 32:3452–3460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meng Y, He L, Guo X, Tang S, Zhao X, Du R,
Jin J, Bi Q, Li H, Nie Y, et al: Gankyrin promotes the
proliferation of human pancreatic cancer. Cancer Lett. 297:9–17.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Misiewicz-Krzeminska I, Sarasquete ME,
Quwaider D, Krzeminski P, Ticona FV, Paíno T, Delgado M, Aires A,
Ocio EM, García-Sanz R, et al: Restoration of microRNA-214
expression reduces growth of myeloma cells through positive
regulation of P53 and inhibition of DNA replication. Haematologica.
98:640–648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Karagiannis TC and El-Osta A: siRNAs:
Mechanism of RNA interference, in vivo and potential clinical
applications. Cancer Biol Ther. 3:1069–1074. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brummelkamp TR, Bernards R and Agami R: A
system for stable expression of short interfering RNAs in mammalian
cells. Science. 296:550–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Paddison PJ, Caudy AA, Bernstein E, Hannon
GJ and Conklin DS: Short hairpin RNAs (shRNAs) induce
sequence-specific silencing in mammalian cells. Genes Dev.
16:948–958. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reynolds A, Leake D, Boese Q, Scaringe S,
Marshall WS and Khvorova A: Rational siRNA design for RNA
interference. Nat Biotechnol. 22:326–330. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barøy T, Sørensen K, Lindeberg MM and
Frengen E: shRNA expression constructs designed directly from siRNA
oligonucleotide sequences. Mol Biotechnol. 45:116–120. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy
MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust
JA, et al: Improved survival in multiple myeloma and the impact of
novel therapies. Blood. 111:2516–2520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rovira M, Rosinol L, Martinez C,
Fernández-Avilés F and Blade J: Is there a curative potential of
autologous stem cell transplantation in multiple myeloma? Long-term
results from a single-centre series. Bone Marrow Transplant.
42:S147(abstract P592). 2009.
|
|
26
|
Gahrton G, Svensson H, Cavo M, Apperly J,
Bacigalupo A, Björkstrand B, Bladé J, Cornelissen J, de Laurenzi A,
Facon T, et al: European Group for Blood and Marrow
Transplantation: Progress in allogenic bone marrow and peripheral
blood stem cell transplantation for multiple myeloma: A comparison
between transplants performed 1983–93 and 1994–98 at European Group
for Blood and Marrow Transplantation centres. Br J Haematol.
113:209–216. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kyle RA and Rajkumar SV: Multiple myeloma.
Blood. 111:2962–2972. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adams J: The proteasome: A suitable
antineoplastic target. Nat Rev Cancer. 4:349–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bianchi G, Oliva L, Cascio P, Pengo N,
Fontana F, Cerruti F, Orsi A, Pasqualetto E, Mezghrani A, Calbi V,
et al: The proteasome load versus capacity balance determines
apoptotic sensitivity of multiple myeloma cells to proteasome
inhibition. Blood. 113:3040–3049. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meister S, Schubert U, Neubert K, Herrmann
K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann
M, et al: Extensive immunoglobulin production sensitizes myeloma
cells for proteasome inhibition. Cancer Res. 67:1783–1792. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ortiz-Navarrete V, Seelig A, Gernold M,
Frentzel S, Kloetzel PM and Hämmerling GJ: Subunit of the 20S
proteasome (multicatalytic proteinase) encoded by the major
histocompatibility complex. Nature. 353:662–664. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rivett AJ, Bose S, Brooks P and Broadfoot
KI: Regulation of proteasome complexes by gamma-interferon and
phosphorylation. Biochimie. 83:363–366. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Niewerth D, Jansen G, Assaraf YG, Zweegman
S, Kaspers GJ and Cloos J: Molecular basis of resistance to
proteasome inhibitors in hematological malignancies. Drug Resist
Updat. 18:18–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Agarwal AK, Xing C, DeMartino GN, Mizrachi
D, Hernandez MD, Sousa AB, de Martínez Villarreal L, dos Santos HG
and Garg A: PSMB8 encoding the β5i proteasome subunit is
mutated in joint contractures, muscle atrophy, microcytic anemia,
and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet.
87:866–872. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kincaid EZ, Che JW, York I, Escobar H,
Reyes-Vargas E, Delgado JC, Welsh RM, Karow ML, Murphy AJ,
Valenzuela DM, et al: Mice completely lacking immunoproteasomes
show major changes in antigen presentation. Nat Immunol.
13:129–135. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chondrogianni N, Stratford FLL, Trougakos
IP, Friguet B, Rivett AJ and Gonos ES: Central role of the
proteasome in senescence and survival of human fibroblasts:
Induction of a senescence-like phenotype upon its inhibition and
resistance to stress upon its activation. J Biol Chem.
278:28026–28037. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chondrogianni N, Tzavelas C, Pemberton AJ,
Nezis IP, Rivett AJ and Gonos ES: Overexpression of proteasome
beta5 assembled subunit increases the amount of proteasome and
confers ameliorated response to oxidative stress and higher
survival rates. J Biol Chem. 280:11840–11850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Franke NE, Niewerth D, Assaraf YG, van
Meerloo J, Vojtekova K, van Zantwijk CH, Zweegman S, Chan ET, Kirk
CJ, Geerke DP, et al: Impaired bortezomib binding to mutant β5
subunit of the proteasome is the underlying basis for bortezomib
resistance in leukemia cells. Leukemia. 26:757–768. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Blackburn C, Gigstad KM, Hales P, Garcia
K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS, et
al: Characterization of a new series of non-covalent proteasome
inhibitors with exquisite potency and selectivity for the 20S
beta5-subunit. Biochem J. 430:461–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rückrich T, Kraus M, Gogel J, Beck A, Ovaa
H, Verdoes M, Overkleeft HS, Kalbacher H and Driessen C:
Characterization of the ubiquitin-proteasome system in
bortezomib-adapted cells. Leukemia. 23:1098–1105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Niewerth D, Franke NE, Jansen G, Assaraf
YG, van Meerloo J, Kirk CJ, Degenhardt J, Anderl J, Schimmer AD,
Zweegman S, et al: Higher ratio immune versus constitutive
proteasome level as novel indicator of sensitivity of pediatric
acute leukemia cells to proteasome inhibitors. Haematologica.
98:1896–1904. 2013.b. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Heckmann D, Laufs S, Maier P, Zucknick M,
Giordano FA, Veldwijk MR, Eckstein V, Wenz F, Zeller WJ, Fruehauf
S, et al: A lentiviral CXCR4 overexpression and knockdown model in
colorectal cancer cell lines reveals plerixafor-dependent
suppression of SDF-1α-induced migration and invasion. Onkologie.
34:502–508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Christoph T, Bahrenberg G, De Vry J,
Englberger W, Erdmann VA, Frech M, Kögel B, Röhl T, Schiene K,
Schröder W, et al: Investigation of TRPV1 loss-of-function
phenotypes in transgenic shRNA expressing and knockout mice. Mol
Cell Neurosci. 37:579–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sacca R, Engle SJ, Qin W, Stock JL and
McNeish JD: Genetically engineered mouse models in drug discovery
research. Methods Mol Biol. 602:37–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lozano G and Zambetti GP: Gankyrin: An
intriguing name for a novel regulator of p53 and RB. Cancer Cell.
8:3–4. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Umemura A, Itoh Y, Itoh K, Yamaguchi K,
Nakajima T, Higashitsuji H, Onoue H, Fukumoto M, Okanoue T and
Fujita J: Association of gankyrin protein expression with early
clinical stages and insulin-like growth factor-binding protein 5
expression in human hepatocellular carcinoma. Hepatology.
47:493–502. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ooi MG, Hayden PJ, Kotoula V, McMillin DW,
Charalambous E, Daskalaki E, Raje NS, Munshi NC, Chauhan D,
Hideshima T, et al: Interactions of the Hdm2/p53 and proteasome
pathways may enhance the antitumor activity of bortezomib. Clin
Cancer Res. 15:7153–7160. 2009. View Article : Google Scholar : PubMed/NCBI
|