Introduction
Lung cancer is the leading cause of cancer-related
death worldwide. Lung adenocarcinoma is the most common type of
lung cancer and is highly associated with smoking. Nonetheless,
among non-smokers, >60% lung cancers are adenocarcinoma as well
(1). With regard to the processes
of cancer metastasis and acquired drug resistance, genetic
variations and microenvironmental cues are increasingly gaining
attention (2). Tumor hypoxia, a
prominent microenvironmental cue, has long been associated with
increased malignancy, poor prognosis and resistance to chemotherapy
(3,4). During tumor growth, tumor cells
located away from blood vessels become hypoxic, and recent studies
have focused on the mechanisms by which hypoxic tumor cells alter
their transcriptional profiles to modulate glycolysis,
proliferation, survival, migration and invasion to resist this
hypoxic stress.
Hypoxia-inducible factors (HIFs) are
O2-regulated transcriptional factors that play critical
roles in low-oxygen adaptive mechanism. Increased levels of HIFs
have been identified in many solid tumors, such as brain, breast,
cervical, gastrointestinal, lung, oropharyngeal, and ovarian
cancers (3,5,6). As
HIFs induce expression of angiogenic factors and lead to
angiogenesis, tumors can acquire more oxygen and nutrients for
survival and proliferation (7,8). Two
major HIFs, HIF-1α and HIF-2α, have similar structures and regulate
both unique and common target genes. HIF-1α specifically regulates
glycolytic genes, including phosphoglycerate kinase (PGK), lactate
dehydrogenase A (LDHA), pyruvate dehydrogenase kinase 1 (PDK1),
carbonic hydrase-9 (CA IX) and BNIP3. HIF-2α exclusively regulates
the Pou transcription factor Oct-4, cyclin D1, and transforming
growth factor α (TGF-α) (9–12). Overall, hypoxia increases tumor
malignancy and metastasis via activation of multiple
hypoxia-responsive genes to regulate cancer cell proliferation,
cell survival, and spread.
Tumor hypoxia has been associated with Wnt signaling
pathway in the modulation of cancer progression. The Wnt pathway is
one of the fundamental mechanisms that regulate cell proliferation,
polarity, and fate determination during embryonic development and
tissue homeostasis (13,14). In the absence of Wnt ligands,
β-catenin adheres to the plasma membrane and cytoplasmic β-catenin
is kept in a lower level by a β-catenin destruction complex. Upon
Wnt ligand stimulation, β-catenin is stabilized, accumulated, and
then translocated to the nucleus to activate its downstream genes
(15–17). Loss of Wnt pathway regulation is
often linked to birth defects, cancers, and other diseases
(18).
Several studies have reported crosstalk between
hypoxia and Wnt signaling in the modulation of cancer malignancy
and metastasis (14,18). For example, HIF-1α modulates
Wnt/β-catenin signaling in hypoxic embryonic stem cells by
enhancing β-catenin activation and expression of the downstream
effectors LEF-1 and TCF-1 (19).
Moreover, HIF-2α interacts with β-catenin and promotes cell
proliferation in renal cell carcinoma (RCC) (20). It is worth noting that RCC cells are
HIF-1α deficient and constitutively express HIF-2α only, whereas
hypoxic lung cancer cells express both HIF-1α and HIF-2α. Although
deregulation of Wnt signaling pathway and activation of the hypoxia
pathway in lung cancers have been reported, the precise functional
crosstalk between hypoxia and Wnt signaling in lung cancer has not
yet been determined.
In this study, we investigated the involvement of
Wnt signaling pathway and hypoxia in lung cancer progression. Our
results showed that Wnt signaling activity is upregulated under
hypoxia, and β-catenin is stabilized and translocated into the
nucleus to stimulate expression of downstream Wnt target genes. We
further identified that HIF-2α is the major effector and
downregulation of HIF-2α and β-catenin reduces cell migration,
invasion and colony formation upon hypoxia treatment. Our results
also discovered that hypoxia-induced AKT1 phosphorylation is
responsible for Wnt signaling activation upon hypoxia treatment.
Based on these observations, we suggest that HIF-2α and β-catenin
cooperatively play essential roles in resisting hypoxia-induced
stress in lung cancer cells.
Materials and methods
Construction of DNA plasmids
A β-catenin gene (CTNNB1) fragment was PCR-amplified
from a human cDNA library using a PCR primer set (forward,
5′-GGATCCATGGCTACTCAAGCTGATTTGATG-3′; reverse,
5′-GTCGACTCACTTATCGTCGTCATCCTTGTA-3′), and this fragment was then
subcloned into the HR'-puro vector to generate the
HR'-β-catenin-puro plasmid. The plasmids
HR'-HIF-1α-P402A/P564A-puro and HR'-HIF-2α-P405A/P531A-puro, which
carry constitutively expressed HIF-1α and HIF-2α, were subcloned
from HA-HIF-1α-P402A/P564A-pBabe-puro and
HA-HIF-2α-P405A/P531A-pBabe-puro (obtained from Addgene), into the
HR'-puro vector. Lentiviral plasmids pLKO.1-shHIF-1α
(TRCN0000003810; TRCN0000000819), pLKO.1-shHIF-2α (TRCN0000003805;
TRCN0000003806) and pLKO.1-shβ-catenin (TRCN0000314990; TRCN
0000314991) were obtained from the RNAi core facility (Academia
Sinica, Taipei, Taiwan).
Cell culture
A549, H1975, and HEK293T cell lines were obtained
from the American Type Culture Collection (ATCC). Human lung
adenocarcinoma cell lines A549 and H1975 were cultured at 37°C in
5% CO2 in RPMI-1640 supplemented with 10% fetal bovine
serum (FBS) and 1% penicillin/streptomycin. HEK293T cells were
maintained at 37°C in 5% CO2 in DMEM supplemented with
10% FBS and 1% penicillin/streptomycin.
Lentiviral preparation and
infection
Lentiviral preparation and infection were performed
as previously described (21).
Briefly, 10 µg of the HR'-puro-based or pLKO.1-based lentiviral
vector, together with 9 µg of ∆8.9 plasmid and 2.5 µg of vesicular
stomatitis virus G protein (VSVG) plasmid, was co-transfected into
HEK293T cells in a 100-mm culture dish using a standard calcium
phosphate transfection protocol. The culture medium was replaced
with fresh medium 16 h after transfection, and the virus-containing
supernatants were collected 48 h after transfection. Lentiviral
infection was performed by adding virus solution to the cells in
the presence of 8 ng/ml polybrene. Fresh culture medium containing
puromycin (2.0 µg/ml) was added to the cells at 24 h after
infection. The surviving cells were pooled and cultured for further
analysis.
RNA isolation and reverse
transcription-PCR
Total RNA was extracted from cultured cells using
MaestroZol™RNA extraction reagent (OmicsBio, Taipei, Taiwan)
according to the manufacturer's protocol. Extracted RNA was then
reverse-transcribed into cDNA using NCode™ VILO™ miRNA cDNA
Synthesis kit (Invitrogen) and T3 Thermocycler (Biometra).
Quantitative real-time PCR and primer
sequence
qPCR amplification was performed by the NCode™
Express SYBR® GreenER™ qPCR Reagent System (Invitrogen),
and Universal Probe Library system (Roche). The following primer
sequences are listed below. For UPL system: 18s (Probe: #48)
forward-5′-GCAATTATTCCCCATGAACG-3′,
reverse-5′-GGGACTTAATCAACGCAAGC-3′; CTNNB1 (Probe: #21)
forward-5′-GCTTTCAGTTGAGCTGACCA-3′,
reverse-5′-CAAGTCCAAGATCAGCAGTCTC-3′. For SYBR Green: N-cadherin
forward-5′-CCTTGAGCTCCCTTAATTCC-3′,
reverse-5′-CCACCATACATGTCAGCAAG-3′; c-Jun
forward-5′-CGGAGAGGAAGCGCATGA-3′,
reverse-5′-ACCTGTTCCCTGAGCATGTTG-3′; cyclin D1 (CCND1)
forward-5′-CCGAGAAGCTGTGCATCTACAC-3′,
reverse-5′-CGCCTCTGGCATTTTGGA-3′; GAPDH
forward-5′-CGACCACTTTGTCAAGCTCA-3′;
reverse-5′-AGGGGTCTACATGGCAACTG-3′.
Transient transfection and reporter
assay
A total of 1 µg DNA was added per well of a 24-well
culture dish, and transient transfection was performed using the
jetPEI™ transfection reagent (Polyplus-Transfection Inc.) according
to the manufacturer's protocol. TOPFlash, a common Wnt activity
reporter plasmid expressing firefly luciferase, was co-transfected
with plasmids expressing the desired protein into 24-well plates
pre-seeded with lung cancer cells. In each well, an
SV40-Renilla-luc plasmid expressing Renilla luciferase was
co-transfected as a control for transfection efficiency
normalization. Luciferase activity assays were performed using a
Dual-Luciferase® Reporter Assay System (Promega)
according to the manufacturer's protocol. Transfection of each
construct and the reporter plasmid was performed in triplicate in
each assay. Bars represent the averages of the normalized values,
with error bars indicating the range.
Protein extraction and immunoblot
Immunoblot analysis was performed as previously
described (21). Briefly, equal
amounts of proteins were resolved on an SDS polyacrylamide gel and
transferred onto a nitrocellulose membrane. Membranes were then
probed with the indicated primary antibodies and appropriate
secondary antibodies. The specific signals were visualized by
LAS-3000 CCD-imaging system (Fujifilm). The following primary
antibodies were used: anti-HIF-1α (2015-1, Epitomics); anti-HIF-2α
(ab199, Abcam); anti-AKT1 (1081, Epitomics); anti-Phospho-AKT1
Ser473 (2118-1, Epitomics); anti-β-actin (GTX110564, GeneTex);
γ-tubulin (1878-1, Epitomics); anti-β-catenin (GTX61089, GeneTex);
anti-Lamin A/C (346) (Sc-7293, Santa Cruz Biotechnology). All
western blot signal intensities were quantified by NIH Image J
software and further normalized with β-actin (22).
Wound healing cell migration
assay
A Culture-Insert (Ibidi, Martinsried, Germany) was
placed in a 6-well culture plate, and
3×104-4×104 cells were seeded in each Culture
Insert. After 24 h, the Culture-Insert was removed using sterile
tweezers, and a 500-µm single wound was created in the center of
the cell monolayer. After the desired time of incubation, the cells
that had migrated into the wound area or protruded from the border
of the wound were visualized and photographed under an inverted
microscope. The experiment was performed at least three times
independently. The area of scratch region was calculated by NIH
ImageJ software (23). The relative
ratio of scratch area was represented as the ratio of the remaining
scratch area at the indicated time points to the whole scratch area
at 0 h.
Invasion assay
Matrigel (5 mg/ml) (BD Falcon) was diluted in cold
10% Nuserum (BD Falcon) RPMI and then coated onto a Transwell
device (8 µm pore size, BD Falcon). After incubation at 37°C for 30
min, 300 µl 10% Nuserum RPMI was added into the Transwell. Each
Transwell device was then placed into a well containing 700 µl 10%
Nuserum RPMI in a 24-well plate; 2.5×104 cells were
seeded into each Transwell and incubated at 37°C for 24 h. The
cells were then fixed with methanol at −20°C for 20 min and then
stained with propidium iodide (PI, 50 µg/ml, Sigma-Aldrich). Cells
that had penetrated the filter and attached to the other side were
counted.
Cell cycle analysis
Briefly, cells infected with desired lentivirus were
selected by puromycin. After 48 h, cells were harvested and then
fixed with 75% ethanol overnight at −4°C. The fixed cells were
incubated with PI staining solution containing RNase A (1 mg/ml,
Sigma-Aldrich) and PI (50 µg/ml) for 30 min. Cell cycle status was
then examined by flow cytometry on a BD FACSCanto (BD Bioscience)
and analyzed by ModFit software.
Statistical analysis
The results were presented as the mean ± standard
deviation of at least 3 independent experiments. Student's t-test
was performed to analyze the differences between groups. P<0.05
and P<0.01 were considered to be statistically significant.
Results
Wnt signaling activity is upregulated
by hypoxia
As recent studies indicated that interaction between
hypoxia and Wnt signaling promotes cell proliferation in RCC
(20), we examined whether the
effect of hypoxia (1% O2) on the Wnt pathway exists in
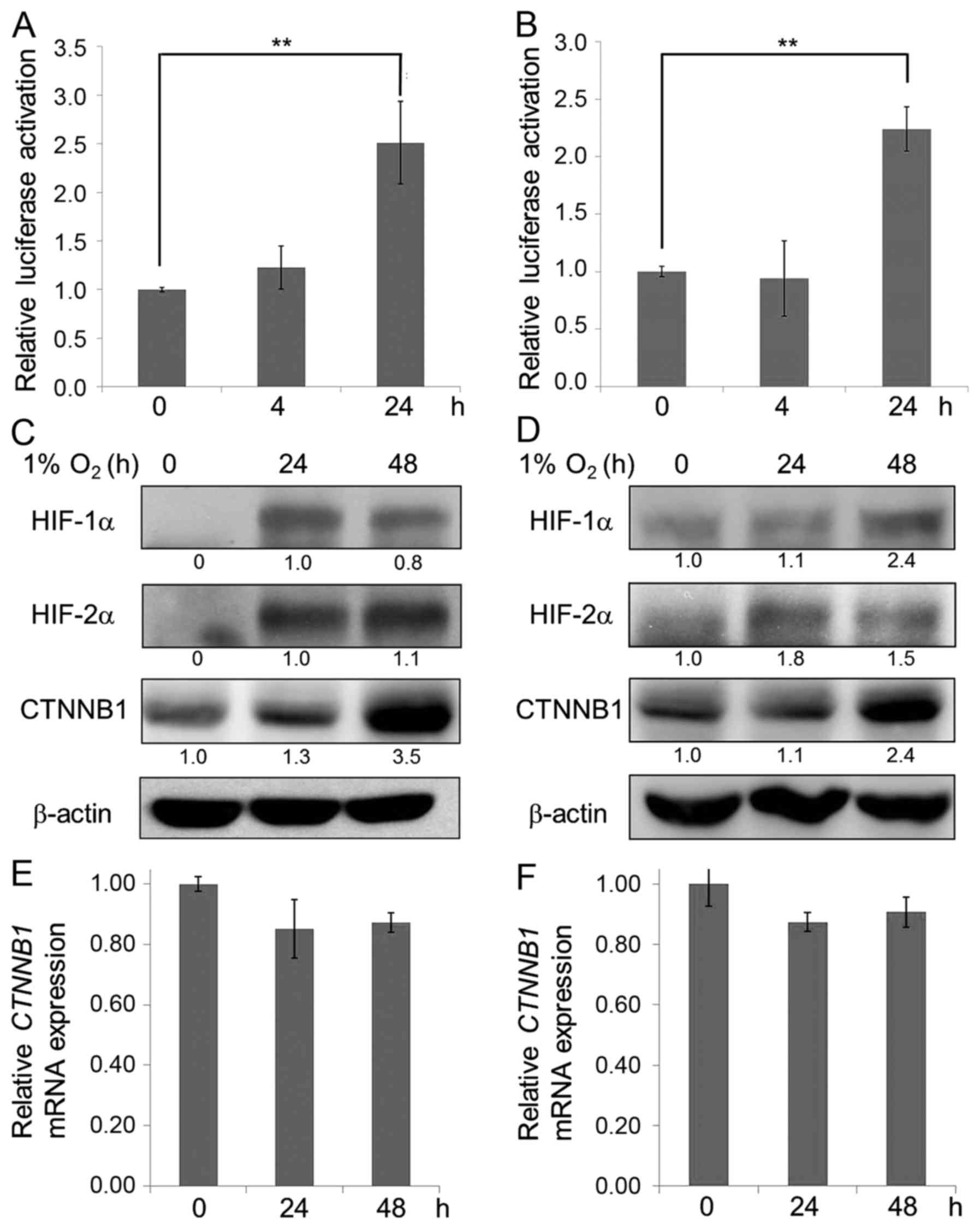
lung adenocarcinoma cells. Reporter assays showed that hypoxia
upregulated Wnt reporter (TOPFlash) luciferase activity in both
A549 and H1975 lung adenocarcinoma cells (Fig. 1A and B). β-catenin is the major
transcriptional co-activator in the Wnt signaling pathway, and
therefore we examined whether hypoxia upregulated expression of
β-catenin. The results showed that hypoxia stabilized HIF-1α and
HIF-2α and upregulated the protein level of β-catenin in both A549
and H1975 cells (Fig. 1C and D). We
further identified that the increase of β-catenin expression under
hypoxia is due to a post-translational regulation since no
significant difference in the mRNA level of β-catenin under hypoxia
compared to normoxia was observed in A549 or H1975 cells (Fig. 1E and F). Together, our data suggest
that hypoxia activates Wnt signaling by stabilizing the β-catenin
protein level via a post-translational modification rather than by
de novo protein synthesis.
β-catenin translocates into the
nucleus upon hypoxia treatment
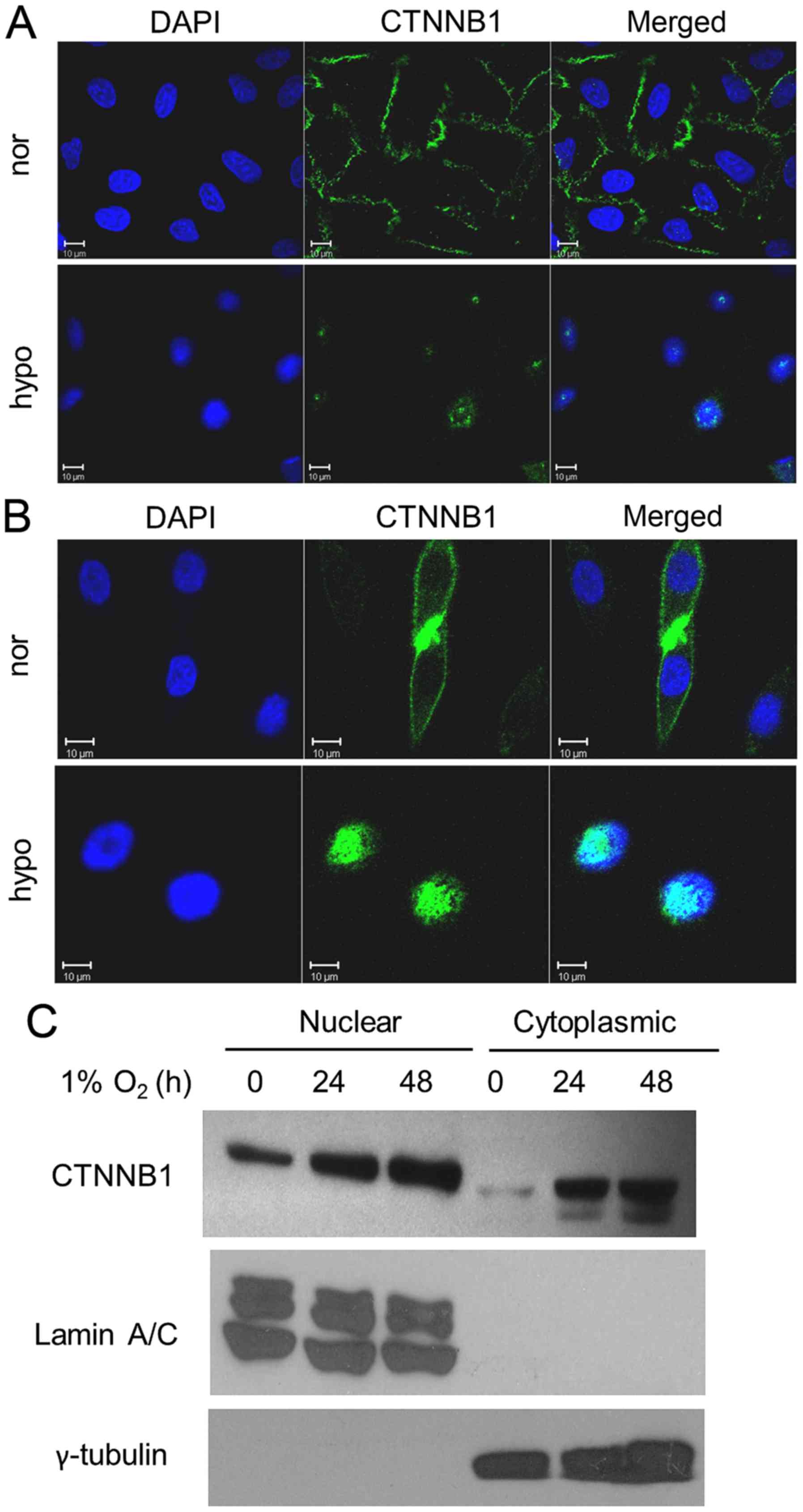
Next, an immunofluorescence assay was performed to
assess the distribution of β-catenin upon hypoxia treatment. We
detected β-catenin at the cell membrane and cell-cell junction in
normoxic A549 and H1975 cells (Fig. 2A
and B, upper panels), whereas β-catenin was found in the
nucleus in hypoxic cells (Fig. 2A and
B, low panels). Nuclear translocation of β-catenin was also
detected in the nuclear fraction (Fig.
2C). These data show that upon hypoxia treatment, β-catenin
translocates into the nucleus to stimulate expression of downstream
Wnt target genes.
Hypoxia induces Wnt signaling in a
HIF-2α dependent manner
As HIF-1α and HIF-2α are two major effectors of
hypoxia, we sought to define which factor is more significant in
activation of hypoxia-induced Wnt signaling. HIF-1α, HIF-2α and
β-catenin, together with a Wnt reporter plasmid TOPFlash, were
transiently co-overexpressed in A549 cells, and luciferase activity
was then determined. Our results showed that overexpression of
HIF-2α and β-catenin each activated Wnt reporter activity and that
co-overexpression of HIF-2α and β-catenin further enhanced this
activity (Fig. 3A). Western blot
analysis also showed significant upregulation of the expression
level of β-catenin in HIF-2α-overexpressing A549 cells (Fig. 3B). Real-time qPCR analysis showed
that the mRNA level of Wnt downstream genes, such as c-Jun and
N-cadherin, were significantly enhanced in HIF-2α-overexpressing
A549 cells (Fig. 3C). In addition,
ectopic overexpression of HIF-2α and β-catenin using a lentiviral
system induced morphological changes in A549 cells, whereas
overexpression of HR' control and HIF-1α had no significant effect
(Fig. 3D). After performing
lentiviral-based RNA silencing of HIF-1α and HIF-2α in A549 cells,
upregulation of β-catenin upon hypoxia (1% O2) was
inhibited in the cells knocked down for HIF-2α but not in those
knocked down for HIF-1α (Fig. 3E).
These results indicate that HIF-2α might play an even more
essential role in hypoxia-induced morphological changes and Wnt
activation in lung cancer cells.
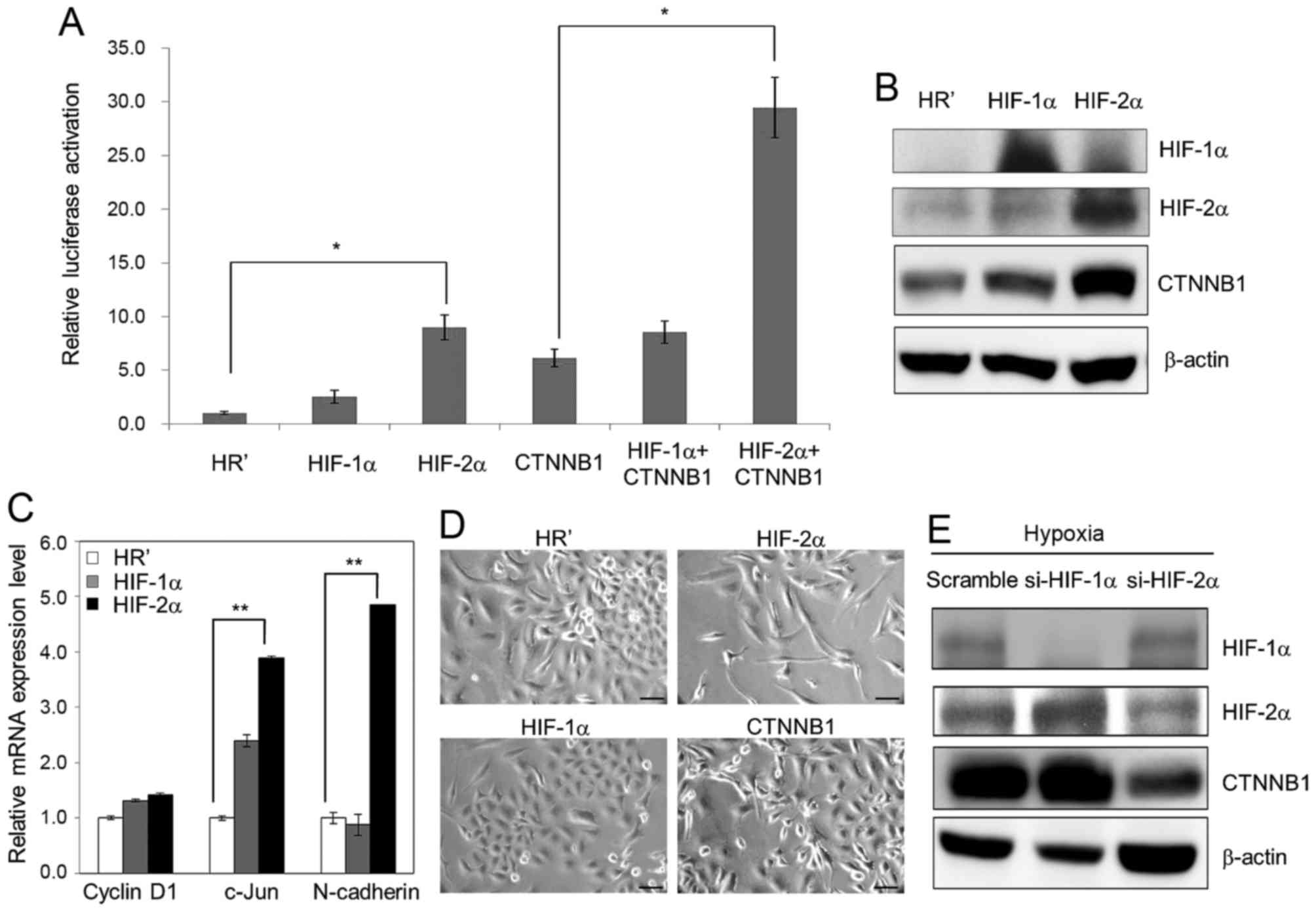 | Figure 3.HIF-2α is the major effector of Wnt
signaling activation upon hypoxia treatment. (A) TOPFlash (100 ng)
and SV40-Renilla-luc (50 ng) plasmids were transiently
co-transfected with plasmids carrying HIF-1α, HIF-2α and β-catenin
(CTNNB1) into A549 cells. Reporter luciferase activity assay was
examined at 48 h after transfection. (B) Lentiviral-based ectopic
expression of HR' control, HIF-1α and HIF-2α was performed using
A549 cells. Protein expression of HIF-1α, HIF-2α and β-catenin was
then examined by western blotting. (C) The mRNA level of cyclin D1,
c-Jun and N-cadherin was examined in HR' control-, HIF-1α- and
HIF-2α-overexpressing A549 cells. (D) Lentiviral-based ectopic
expression of HR' control, HIF-1α, HIF-2α and β-catenin was
performed in A549 cells. The morphology of all stable cell lines
was examined by microscopy. Scale bar, 50 µm. (E) Lentiviral-based
knockdown of scramble control, si-HIF-1α and si-HIF-2α was
performed using A549 cells, and the protein expression of HIF-1α,
HIF-2α and β-catenin under hypoxia was then examined by western
blot analysis. In all cases, 1% O2 was the hypoxic
condition. β-actin was used as a loading control. Data represent as
the means ± SD from three independent experiments. *P<0.05.
**P<0.01. |
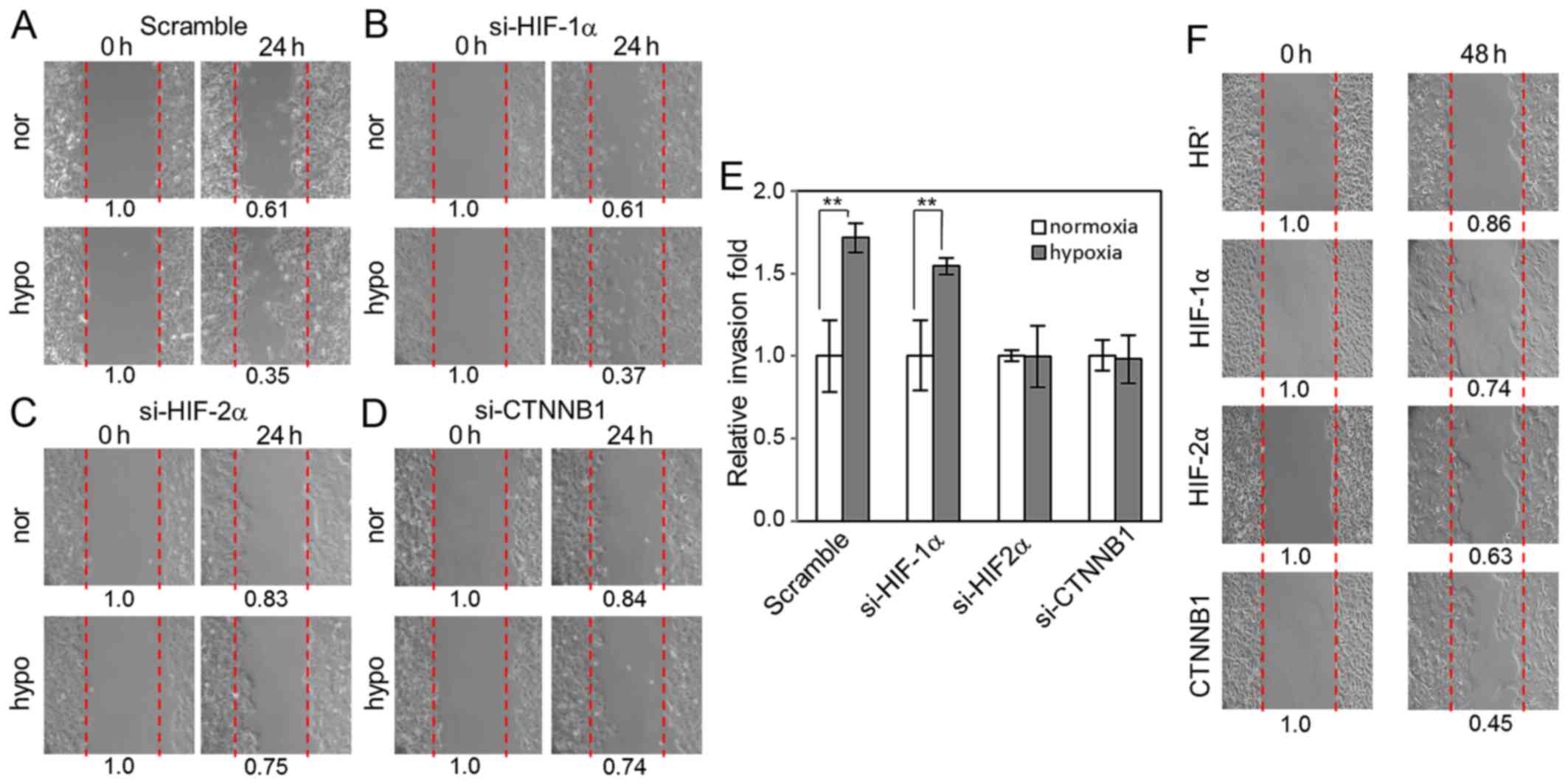
HIF-2α and β-catenin are essential for
hypoxia-induced cell migration and invasion
Next we attempted to define whether Wnt signaling,
as well as HIF-1α and HIF-2α, has an important function in
hypoxia-induced migration of lung cancer cells by knocking down
HIF-1α, HIF-2α and β-catenin in A549 cells using lentiviral-based
small interfering RNAs. Our data showed that knockdown of HIF-1α
did not affect hypoxia-induced cell migration into the scratched
area compared to scramble control cells (Fig. 4A and B). In contrast, knockdown of
HIF-2α and β-catenin both significantly disrupted hypoxia-induced
cell migration into the scratched area (Fig. 4C and D). Invasion assays revealed
that cells with HIF-1α knockdown exhibited similar invasion ability
compared to scramble control cells upon hypoxia. However, knockdown
of HIF-2α and β-catenin significantly suppressed hypoxia-induced
invasion (Fig. 4E). Stable A549
cell lines carrying HR' control-, HIF-1α-, HIF-2α-, and
β-catenin-overexpression were used to examine their cell migration
ability. We did not observe significant differences in migration
ability between these cells under normal culture condition, but
under low-serum condition (0.5% FBS), we observed that
overexpression of HIF-2α and β-catenin increased cell migration
compared to HR' control- and HIF-1α-overexpressing cells (Fig. 4F). These results indicate that Wnt
signaling may regulate hypoxia-induced cell migration and invasion
via β-catenin activation in a HIF-2α-dependent manner.
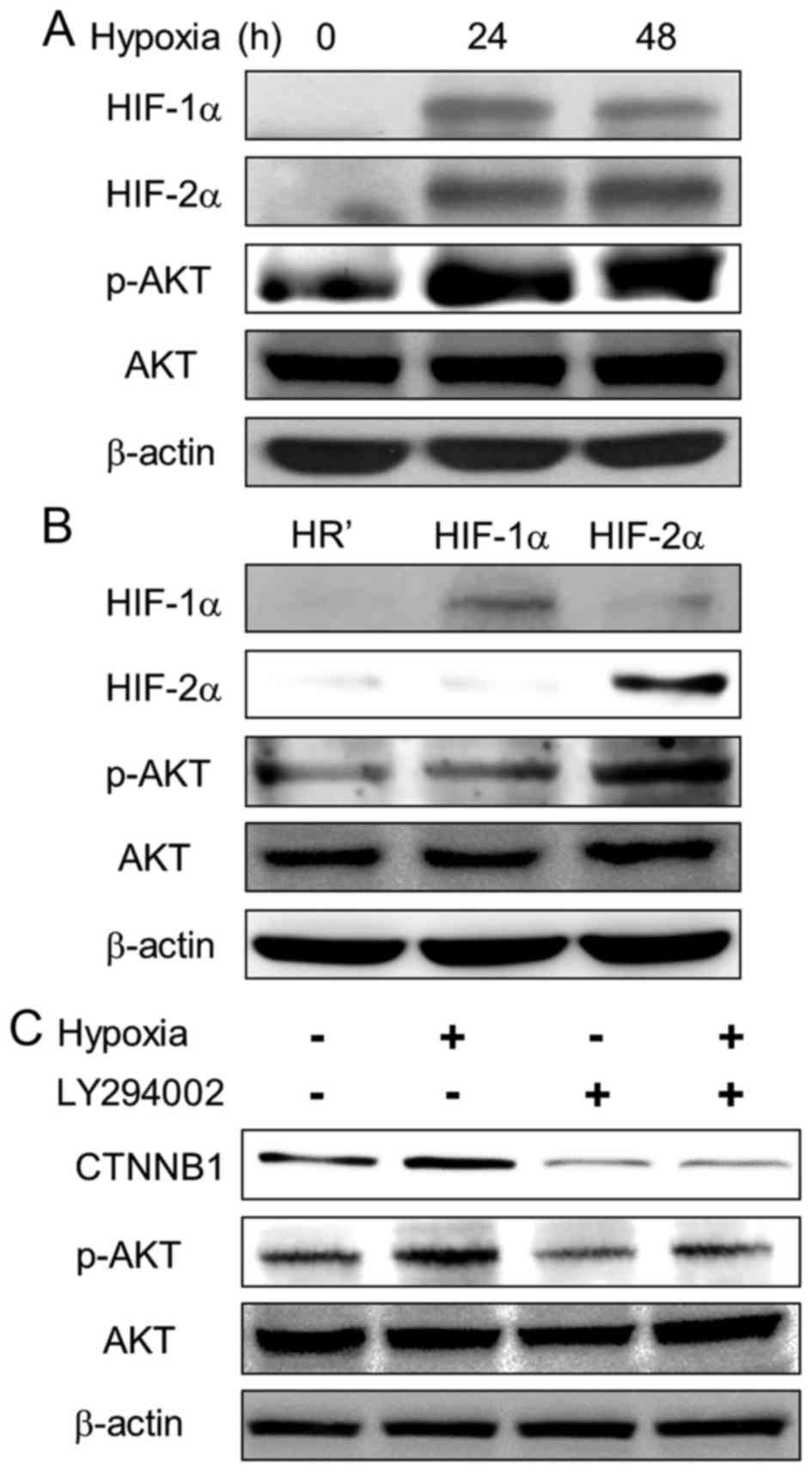
Hypoxia increases β-catenin expression
through the PI3K/AKT pathway
Previous studies suggested that hypoxia induces the
PI3K/AKT pathway in many cell types (24–26).
We further examined the effect of hypoxia on the PI3K/AKT pathway
in lung cancer cells. Upon hypoxia, phosphorylation of AKT1
(Ser473) was upregulated in A549 cells (Fig. 5A). Furthermore, phospho-AKT1
expression was increased in HIF-2α-overexpressing cells compared to
HR' control and HIF-1α-overexpressing cells (Fig. 5B). To define whether the PI3K/AKT
pathway is involved in hypoxia-induced β-catenin upregulation, a
PI3K inhibitor, LY294002 (Sigma-Aldrich), was employed to inhibit
AKT1 phosphorylation. The results showed decreased expression of
β-catenin and AKT1 phosphorylation upon LY294002 treatment, even
under hypoxia (Fig. 5C). Our
results suggest that PI3K/AKT pathway is activated by HIF-2α and is
essential in hypoxia-induced Wnt signaling activation.
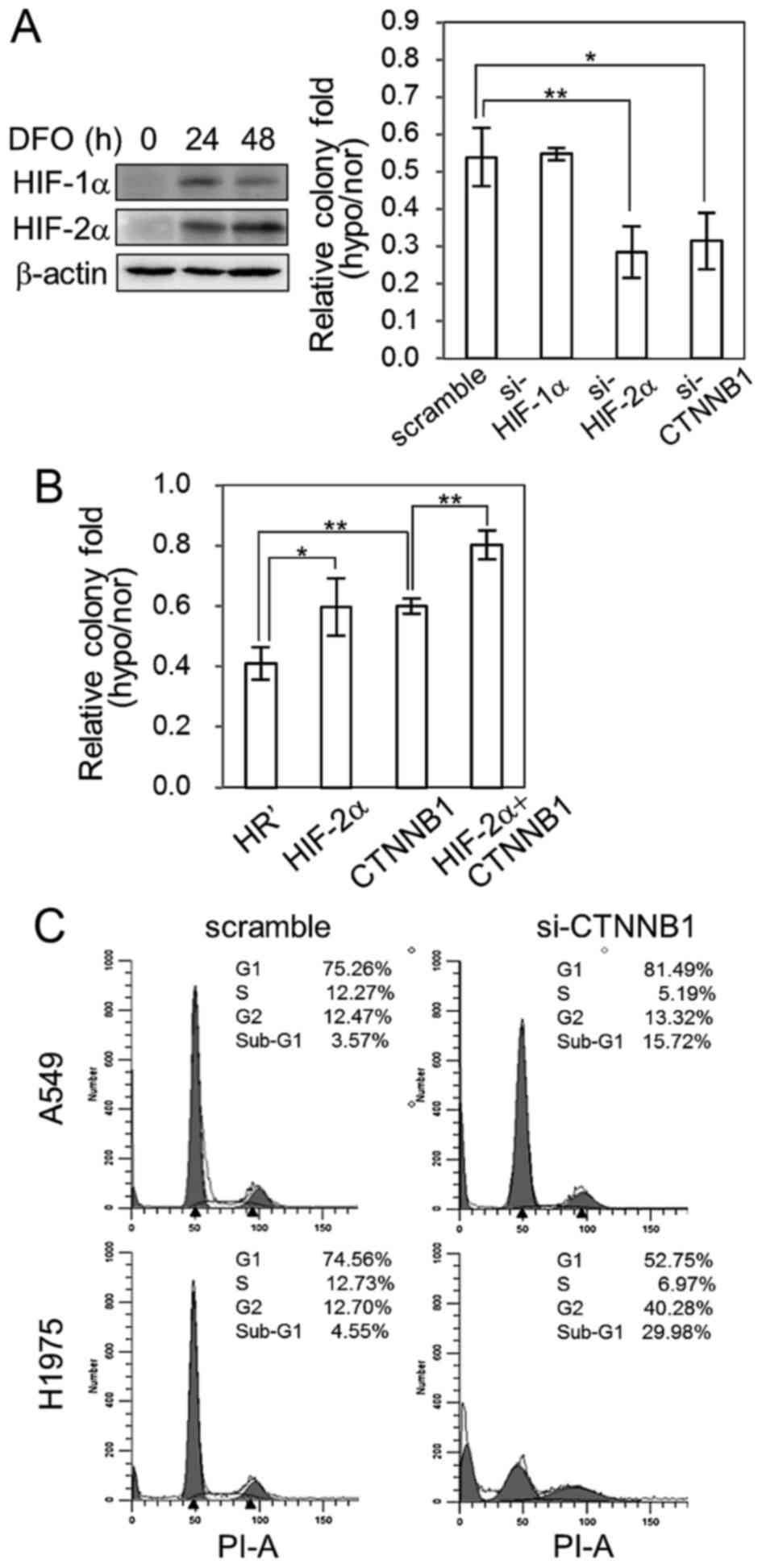
HIF-2α and β-catenin protect lung
cancer cells from long-term hypoxia-induced stress
To further investigate the significant role of
HIF-2α and β-catenin under hypoxia, we incubated cells with
desferrioxamine (DFO, Sigma-Aldrich) to mimic long-term exposure to
hypoxia. When incubated in 100 µM DFO for 24 and 48 h, HIF-1α and
HIF-2α were successfully activated in A549 cells (Fig. 6A, left panel). Our results showed
that after 21 days of incubation with 100 µM DFO in culture,
knockdown of HIF-2α and β-catenin in A549 cells significantly
reduced their colony numbers compared to scramble control and
HIF-1α-silenced cells in colony formation assays (Fig. 6A, right panel); additionally, upon
chronic hypoxic stress, HIF-2α- and β-catenin-overexpressing A549
cells possessed higher colony numbers compared to HR' control
cells, and co-overexpression of HIF-2α/β-catenin can enhance the
growth advantage even higher than overexpression of HIF-1α or
β-catenin alone (Fig. 6B). As our
results suggested that β-catenin plays an essential role in
controlling cell growth under hypoxic stress, we further examined
the effect of β-catenin in lung cancer cell growth by examining
cell cycle progression in β-catenin-silenced A549 and H1975 cells.
The results showed increase in the sub-G1 population in
β-catenin-silenced cells, indicating that β-catenin has an
essential role in aiding the survival of lung cancer cells
(Fig. 6C). Together, these data
suggest that HIF-2α and β-catenin are essential factors in
resisting chronic hypoxia-induced cell death and crosstalk between
hypoxia and Wnt pathway is involved in lung cancer progression.
Discussion
Intratumoral hypoxia has long been associated with
increased malignancy and cancer metastasis. Both HIF-1α and HIF-2α
are overexpressed in many cancer types and are associated with poor
prognosis in cancers of the breast, brain, cervix, ovary, and
uterus (27–30). Crosstalk between hypoxia and Wnt
signaling has also been noted. However, most studies focus on
either HIF-1α or HIF-2α, respectively. In this study, we determined
that HIF-2α is the major effector of lung cancer cells in Wnt
signaling activation under hypoxia and crosstalk between Wnt
pathway and hypoxia participates in lung cancer progression
processes, such as metastasis and cell survival. Overexpression of
HIF-2α in lung cancer cells increases β-catenin expression and
induces morphological changes similar to EMT. Knockdown of HIF-2α
decreases β-catenin expression and inhibits hypoxia-induced cell
migration and invasion, whereas no significant effect was observed
with knockdown of HIF-1α (Fig.
4A-E). HIF-1α is only responsible for a minor effect of
hypoxia-induced cell invasion (Fig.
4E), and therefore it is reasonible that si-HIF-2α can abolish
the majority of hypoxia-induced cell invasion. Our results also
showed that si-CTNNB1 abolished hypoxia-induced invasion. It is not
surprising since β-catenin is an important factor in WNT signaling
pathway as well as, in this study, hypoxia pathway. We also proved
that hypoxia increases β-catenin expression, resulting in β-catenin
translocation to the nucleus to stimulate Wnt downstream target
genes. Thus, we demonstrate that hypoxia increases Wnt signaling
activity and tumor cell malignancy by stabilizing β-catenin protein
levels in a HIF-2α-dependent manner.
The roles of hypoxia in regulating hypoxic cell
death or promoting cell survival remain controversial. For example,
hypoxia-induced cell death has been observed in E1A and Ha-ras
transformed cell lines, several nonglioma tumor cells, glioblastoma
cells, and nontransformed cells (31–33);
in addition, HIF-1α not only activates genes involving in metabolic
adaption to hypoxic environment to promote cell survival, but also
induces several pro-apoptotic factors, including P53, BNIP3, and
BNIP3L, to promote cell death (34). In another perspective,
hypoxia-induced autophagy has been reported to play protective
roles in cell survival under hypoxic stress (35). The present study provides an
alternative survival way in showing that ectopic expression of
HIF-2α upregulates AKT1 phosphorylation and inhibition of PI3K/AKT
activity by LY294002 reduced hypoxia-induced β-catenin expression,
indicating that PI3K/AKT upregulation is essential for
hypoxia-induced Wnt activation. These data suggest that
hypoxia-induced β-catenin upregulation may be important for
resistance to hypoxic stress. In colony formation assays using DFO
to mimic long-term hypoxia exposure, knockdown of HIF-2α and
β-catenin significantly reduces colony numbers, whereas
overexpression of either HIF-2α, β-catenin or HIF-2α/β-catenin
increases colony numbers. These data further emphasize the
significant role of Wnt signaling in resisting hypoxic stress.
In this study, we also examined the expression level
of Wnt target genes under HIF-1α- and HIF-2α-overexpression. While
our results showed that overexpression of HIF-2α upregulates Wnt
target genes to a greater extent, it is rather remarkable that not
all of Wnt target genes examined were upregulated accordingly,
i.e., for example, we were unable to observe a significant
elevation of cyclin D1 expression in HIF-1α- and
HIF-2α-overexpressing cells. Cyclin D1 associates with CDK4 to form
a protein kinase complex that phosphorylates and inactivates
retinoblastoma protein pRb, playing a critical role in regulating
cell cycle progression (36).
Cyclin D1 is frequently amplified and overexpressed in cancer
cells, implying its important role in malignant development
(37). The lack of elevated cyclin
D1 expression can partially explain why we did not observe a
significant growth advantage in HIF-2α- and
β-catenin-overexpressing cells under normal culture condition.
However, when cells were cultured in a high stringent condition
such as chronic hypoxia or low serum, overexpression of HIF-2α and
β-catenin showed elevated migration and colony formation
ability.
In summary, our results discriminate the functional
differences between HIF-1α and HIF-2α in the hypoxic environment
and also suggest the involvement of HIF-2α and the PI3K/AKT pathway
in activating Wnt signaling under hypoxia, thus promoting the
survival and malignancy of lung cancer cells.
Acknowledgements
This work was supported by the Ministry of Science
and Technology Grant MOST 105-2325-B-010-003, Ministry of Health
and Welfare Grant MOHW106-TDU-B-211-144-003, and National Yang Ming
University Grant 106AC-P902, Taiwan.
Glossary
Abbreviations
Abbreviations:
|
HIF
|
hypoxia-induced factor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
VSVG
|
vesicular stomatitis virus G
protein
|
|
PI
|
propidium iodide
|
|
DFO
|
desferrioxamine
|
References
|
1
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rohwer N and Cramer T: Hypoxia-mediated
drug resistance: Novel insights on the functional interaction of
HIFs and cell death pathways. Drug Resist Updat. 14:191–201. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bertout JA, Patel SA and Simon MC: The
impact of O2 availability on human cancer. Nat Rev
Cancer. 8:967–975. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Semenza GL: Evaluation of HIF-1 inhibitors
as anticancer agents. Drug Discov Today. 12:853–859. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Semenza GL: Oxygen homeostasis. Wiley
Interdiscip Rev Syst Biol Med. 2:336–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jain RK: Normalization of tumor
vasculature: An emerging concept in antiangiogenic therapy.
Science. 307:58–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Du R, Lu KV, Petritsch C, Liu P, Ganss R,
Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z, et al: HIF1α
induces the recruitment of bone marrow-derived vascular modulatory
cells to regulate tumor angiogenesis and invasion. Cancer Cell.
13:206–220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu CJ, Wang LY, Chodosh LA, Keith B and
Simon MC: Differential roles of hypoxia-inducible factor 1α
(HIF-1α) and HIF-2α in hypoxic gene regulation. Mol Cell Biol.
23:9361–9374. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Warnecke C, Zaborowska Z, Kurreck J,
Erdmann VA, Frei U, Wiesener M and Eckardt KU: Differentiating the
functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α
(EPAS-1) by the use of RNA interference: Erythropoietin is a HIF-2α
target gene in Hep3B and Kelly cells. FASEB J. 18:1462–1464.
2004.PubMed/NCBI
|
|
11
|
Rankin EB, Higgins DF, Walisser JA,
Johnson RS, Bradfield CA and Haase VH: Inactivation of the
arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von
Hippel-Lindau disease-associated vascular tumors in mice. Mol Cell
Biol. 25:3163–3172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu CJ, Iyer S, Sataur A, Covello KL,
Chodosh LA and Simon MC: Differential regulation of the
transcriptional activities of hypoxia-inducible factor 1 alpha
(HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol.
26:3514–3526. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
MacDonald BT, Tamai K and He X:
Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev
Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hecht A, Vleminckx K, Stemmler MP, van Roy
F and Kemler R: The p300/CBP acetyltransferases function as
transcriptional coactivators of beta-catenin in vertebrates. EMBO
J. 19:1839–1850. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takemaru KI and Moon RT: The
transcriptional coactivator CBP interacts with beta-catenin to
activate gene expression. J Cell Biol. 149:249–254. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barker N, Hurlstone A, Musisi H, Miles A,
Bienz M and Clevers H: The chromatin remodelling factor Brg-1
interacts with β-catenin to promote target gene activation. EMBO J.
20:4935–4943. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clevers H: Wnt/β-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mazumdar J, O'Brien WT, Johnson RS,
LaManna JC, Chavez JC, Klein PS and Simon MC: O2
regulates stem cells through Wnt/β-catenin signalling. Nat Cell
Biol. 12:1007–1013. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi H, Chun YS, Kim TY and Park JW:
HIF-2α enhances β-catenin/TCF-driven transcription by interacting
with β-catenin. Cancer Res. 70:10101–10111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hong C-F, Lin S-Y, Chou Y-T and Wu C-W:
MicroRNA-7 compromises p53 protein-dependent apoptosis by
controlling the expression of the chromatin remodeling factor
SMARCD1. J Biol Chem. 291:1877–1889. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li S, Yang B, Teguh D, Zhou L, Xu J and
Rong L: Amyloid β peptide enhances RANKL-induced osteoclast
activation through NF-κB, ERK, and calcium oscillation signaling.
Int J Mol Sci. 17:16832016. View Article : Google Scholar :
|
|
23
|
Cormier N, Yeo A, Fiorentino E and Paxson
J: Optimization of the wound scratch assay to detect changes in
murine mesenchymal stromal cell migration after damage by soluble
cigarette smoke extract. J Vis Exp. 106:e534142015.
|
|
24
|
Alvarez-Tejado M, Naranjo-Suarez S,
Jiménez C, Carrera AC, Landázuri MO and del Peso L: Hypoxia induces
the activation of the phosphatidylinositol 3-kinase/Akt cell
survival pathway in PC12 cells: Protective role in apoptosis. J
Biol Chem. 276:22368–22374. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deguchi JO, Yamazaki H, Aikawa E and
Aikawa M: Chronic hypoxia activates the Akt and β-catenin pathways
in human macrophages. Arterioscler Thromb Vasc Biol. 29:1664–1670.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beitner-Johnson D, Rust RT, Hsieh TC and
Millhorn DE: Hypoxia activates Akt and induces phosphorylation of
GSK-3 in PC12 cells. Cell Signal. 13:23–27. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhong H, De Marzo AM, Laughner E, Lim M,
Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL and Simons
JW: Overexpression of hypoxia-inducible factor 1alpha in common
human cancers and their metastases. Cancer Res. 59:5830–5835.
1999.PubMed/NCBI
|
|
28
|
Lu X and Kang Y: Hypoxia and
hypoxia-inducible factors: Master regulators of metastasis. Clin
Cancer Res. 16:5928–5935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by HIF-1α
promotes metastasis. Nat Cell Biol. 10:295–305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ouyang G, Liu M, Ruan K, Song G, Mao Y and
Bao S: Upregulated expression of periostin by hypoxia in
non-small-cell lung cancer cells promotes cell survival via the
Akt/PKB pathway. Cancer Lett. 281:213–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Graeber TG, Osmanian C, Jacks T, Housman
DE, Koch CJ, Lowe SW and Giaccia AJ: Hypoxia-mediated selection of
cells with diminished apoptotic potential in solid tumours. Nature.
379:88–91. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Araya R, Uehara T and Nomura Y: Hypoxia
induces apoptosis in human neuroblastoma SK-N-MC cells by caspase
activation accompanying cytochrome c release from mitochondria.
FEBS Lett. 439:168–172. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Steinbach JP, Wolburg H, Klumpp A, Probst
H and Weller M: Hypoxia-induced cell death in human malignant
glioma cells: Energy deprivation promotes decoupling of
mitochondrial cytochrome c release from caspase processing and
necrotic cell death. Cell Death Differ. 10:823–832. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pouysségur J, Dayan F and Mazure NM:
Hypoxia signalling in cancer and approaches to enforce tumour
regression. Nature. 441:437–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang N, Ji N, Jiang W-M, Li ZY, Wang M,
Wen JM, Li Y, Chen X and Chen JM: Hypoxia-induced autophagy
promotes human prostate stromal cells survival and ER-stress.
Biochem Biophys Res Commun. 464:1107–1112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Musgrove EA, Lee CS, Buckley MF and
Sutherland RL: Cyclin D1 induction in breast cancer cells shortens
G1 and is sufficient for cells arrested in G1 to complete the cell
cycle. Proc Natl Acad Sci USA. 91:8022–8026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Santarius T, Shipley J, Brewer D, Stratton
MR and Cooper CS: A census of amplified and overexpressed human
cancer genes. Nat Rev Cancer. 10:59–64. 2010. View Article : Google Scholar : PubMed/NCBI
|