Introduction
Atherosclerosis, a major underlying factor in stroke
and cardiovascular disease, crucial causes of mortality and
morbidity worldwide (1), is a
chronic arterial disease featuring lipid deposition and
inflammation in the vessel wall (2). Formation of foam cells is the early
event in atherosclerotic pathogenesis. In this stage, exorbitantly
oxidized low-density lipoprotein (ox-LDL) uptake or damaged
cholesterol efflux in macrophages is the leading cause of foam cell
formation (3). Scavenger receptors
(SRs) on macrophage membranes are responsible for ox-LDL uptake,
such as cluster of differentiation 36 (CD36) and class A scavenger
receptor (SR-A) (4). In addition,
to lipid influx, efflux of intracellular lipids occurs mostly via
reverse cholesterol transport (RCT). Several membrane proteins,
such as SR-B type І (SR-BІ) and ATP-binding cassette transporter A1
(ABCA1) and ABCG1, have been reported to play a critical role in
the RCT pathway (5).
The cellular cholesterol efflux from tissues is
dependent on extracellular lipid receptors including lipid-poor
apoproteins and high-density lipoprotein (HDL) (6). ABCA1 promotes free cholesterol efflux
from macrophages into apolipoprotein A1 (ApoA1), whereas ABCG1
plays a key role in mediating cholesterol efflux to HDL (7–9).
Therefore, formation of foam cells is mainly regulated by the RCT
pathway and SRs. Evidence has suggested that modulation of SRs or
the RCT pathway by antioxidants prevents lipid accumulation in foam
cells and then retards the progression of atherosclerosis (10,11).
TMP, the predominant active ingredient in Rhizoma
Ligustici wallichii (Chuanxiong), possesses antiproliferative
and apoptosis-inducing activities in diverse cancer cell types
(12–14). However, it has been reported that
TMP was effective in the treatment of a few cardiovascular
complications, including angina pectoris and cerebrovascular and
thrombotic vascular diseases (15–17),
as a result of its biological activities including vasodilation
(17) and antiplatelet aggregation
(18). Additionally, TMP exerts
anti-atherosclerotic effects via inhibition of endothelial
dysfunction (19), regulation of
lipid levels in the plasma (20),
attenuation of oxidative stress (21) and inflammation (22). However, the effects and molecular
mechanisms involved in TMP-mediated lipid accumulation in
macrophage-derived foam cells have not been well documented. In
addition, a variety of genetic population research has highlighted
the value of the p38 and PI3K/Akt signaling pathways in the
promotion of human atherosclerotic lesions (23,24).
However, whether p38 and PI3K/Akt are connected with
the anti-atherogenic effect of TMP on foam cell formation warranted
further investigation. In a recent study, we investigated the
impact of TMP on atherosclerosis and the potential mechanisms in
RAW264.7 cells (mouse macrophage cell line) and apolipoprotein
E-deficient (ApoE−/−) mice. We observed that TMP
markedly inhibited not only the formation of foam cells in
vitro, but also the atherosclerotic plaque area in aortas from
ApoE−/− mice. The anti-atherosclerotic impact of TMP may
be attributed to the upregulation of ABCA1 and ABCG1 and the
downregulation of CD36 and SR-A by p38 MAPK and PI3K signaling.
Materials and methods
Reagents
TMP (purity, 98.0%), cycloheximide (CHX), LY294002,
SB203580 were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Goat anti-SR-A antibody (sc-166184, 1:2,000) and protein
A/G-Sepharose were obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Mouse anti-ABCA1 (ab7360; 1:800) as well as
anti-p-AKT (ab38449; 1:800), anti-p-p38 (ab47363; 1:800),
anti-ABCG1 (ab52617; 1:5,000), anti-CD36 (ab133625; 1:5,000),
anti-β-actin (ab20272; 1:5,000) and anti-SR-BI (ab52629; 1:2,000)
rabbit antibodies were obtained from Abcam (Cambridge, MA, USA).
Goat anti-mouse IgG (bs-0295GS; 1:2,000) and goat anti-rabbit IgG
(bs-0296G; 1:2,000) were obtained from Bioss Biotechnology
(Beijing, China). DiI-labeled ox-LDL and ox-LDL were purchased from
Guangzhou Yiyuan Biotechnology Co., Ltd. (Guangzhou, China).
Cell culture
RAW264.7 was obtained from the American Type Culture
Collection (ATCC; Manassas, VA, USA), and maintained in RPMI-1640
medium (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum
in a 37°C incubator with 5% CO2. TMP was dissolved in
dimethyl sulfoxide (DMSO), and further diluted with complete
RPMI-1640 medium (DMSO <0.1%) for the treatment of in
vitro cells. The control group was cultured in medium
containing the same volume of DMSO.
Animals
Twenty mice (10 for each group) used in the present
study were 8-week-old male ApoE−/− mice (22–24 g/mouse),
from Jackson Laboratory (Bar Harbor, ME, USA), which were procured
from Tengxin Technology Company (Chongqing, China), and were housed
in barrier facilities on a 12-h light/dark cycle. All experimental
mice were permitted food and water ad libitum. The animal
procedures were approved by the Animal Care and Use Committee of
the Chongqing Medical University (Chongqing, China).
Animal experimental protocols
ApoE−/− mice were orally treated for 8
weeks with TMP (150 mg/kg/day) or vehicle (20 ml/kg/day, 0.5%
sodium carboxyl methyl cellulose) by gastric gavages (n=10, each
group), until being fed a high-fat diet (15.8% fat and 1.25%
cholesterol) for an additional 8 weeks. In the present study, we
adopted TMP doses in accordance with a previous study (25). Mice were euthanized using
CO2 following treatment with TMP (total diet-fed period
was 16 weeks), and then the hearts and aortas were collected for
Oil Red O staining and western blotting.
Cell viability assay with MTT
Macrophages were seeded in 96-well plates at a
density of 7.5×104 cells/well and the cell viability via
methyl thiazolyl tetrazolium (MTT) assay was detected. Before
removing the culture supernatant, the cells were treated with or
without TMP for 24 h. The following steps were performed as
previously described (26).
Assessment of foam cell formation by
Oil Red O staining
Oil Red O staining was performed as previously
described (3). After being washed
3X with phosphate-buffered saline (PBS), the cells were fixed in 4%
paraformaldehyde for 20 min, and then stained with 0.5% Oil Red O
staining for 10 min to visualize cellular lipid accumulation. Light
microscopy with a magnification of ×200 was used to photograph the
stained cells. After Oil Red O staining, alcohol extraction was
used to detect the density of the lipid content. The absorbance at
500 nm was assessed using a microplate reader.
Cholesterol efflux assay
Cholesterol efflux was assessed as previously
described (27). After
pre-incubation with or without TMP for 24 h, the RAW264.7 cells
were labeled with 6 µg/ml BODIPY-cholesterol in medium containing
0.5% CD (methyl-β-cyclodextrin) for 1 h at 37°C. Next, the cells
were washed with PBS, then incubated with serum/phenol red-free
medium containing 10 µg/ml ApoA-1 and 10 µg/ml HDL for 2 h at 37°C
during the total cholesterol efflux experiments. In the ABCA1- or
ABCG1-mediated cholesterol efflux experiments, the cells were
incubated with serum/phenol red-free media containing 10 µg/ml
ApoA-1 (for ABCA1) or 10 µg/ml HDL (for ABCG1) for 2 h at 37°C. The
collected medium was centrifuged at 2,000 × g for 10 min to remove
the unattached cells. BODIPY-cholesterol fluorescence was detected
with an excitation/emission wavelength of 485/515 nm using a Wallac
1420 VICTOR 2TM fluorometer (Perkin-Elmer, Inc., Waltham, MA, USA).
The percentage of fluorescence in the medium relative to the total
fluorescence (cells + medium) was used to calculate the cholesterol
efflux.
DiI-Ox-LDL uptake
The DiI-Ox-LDL uptake assay was performed as
previously described (27).
RAW264.7 cells were treated with or without TMP for 24 h, and then
incubated in RPMI-1640 medium containing 10 µg/ml DiI-Ox-LDL for 4
h at 37°C. The medium containing DiI-Ox-LDL was collected, and the
cells were washed with probe-free medium. The fluorescence
intensities of the medium and the cell lysates were detected with
an excitation/emission wavelength of 514/550 nm using the Wallac
1420 VICTOR 2TM fluorometer.
Western blotting
Cells or tissues were collected and protein extracts
were prepared as previously described (3). The proteins were applied to western
blotting using primary antibodies. Then, a chemiluminescence method
(Pierce Biotechnology, Inc., Rockford, IL, USA) and Quantity One
(Bio-Rad, Hercules, CA, USA) software program were used to
visualize and quantify the proteins.
Quantitative real-time polymerase
chain reaction (RT-qPCR)
RT-qPCR was performed as previously described
(26). Total RNA was isolated using
TRIzol reagent (Invitrogen). cDNA synthesis was performed using
MuLV reverse transcriptase (Applied Biosystems, Foster City, CA,
USA). Real-time PCR was performed using a SYBR-Green PCR Master Mix
kit (Tiangen Biotech Co., Ltd., Beijing, China). Primer sequences
were as follows: ABCA1 forward, 5′-ggtttggagatggttatacaatagttgt-3′
and reverse, 5′-cccggaaacgcaagtcc-3′; ABCG1 forward,
5′-ttcccctggagatgagtgtc-3′ and reverse, 5′-cagtaggccacagggaacat-3′;
SR-A forward, 5′-tggtccacctggtgctcc-3′ and reverse,
5′-acctccagggaagcaattt-3′; CD36 forward,
5′-cagttggagacctgcttatcc-3′ and reverse,
5′-gcgtcctgggttacattttc-3′; SR-BІ forward,
5′-accctaacccaaaggagcat-3′ and reverse, 5′-cacagcaacggcagaactac-3′;
β-actin forward, 5′-ttgtccctgtatgcctctgg-3′ and reverse,
5′-gaggtctttacggatgtcaacg-3′. The RT-qPCR reaction was performed
under the following conditions: 3 min at 95°C for 1 cycle, 10 sec
at 95°C, 30 sec at 60°C for 39 cycles, and 95°C for 5 sec.
Statistical analysis
Data are presented as the mean ± SEM. Statistical
analysis was carried out using one-way ANOVA followed by Bonferroni
post hoc test or unpaired Student's t-test. Continuous variables
were tested for normal distribution using the Kolmogorov-Smirnov
test. Differences were considered statistically significant when
P<0.05. All calculations were carried out using SPSS 15.0
version (SPSS, Inc., Chicago, IL, USA).
Results
TMP enhances cholesterol efflux and
suppresses lipid accumulation of RAW264.7 macrophages
An MTT assay was used to detect the toxicity of TMP
on macrophages. The cell viability was unchanged with the treatment
of TMP (12.5, 25 and 50 µg/ml) for 24 h (Fig. 1C). Accordingly, we chose a
concentration range of 12.5–50 µg/ml for subsequent research. Lipid
accumulation was examined in macrophages with ox-LDL in the
presence or absence of TMP, which is a sign of foam cell formation.
The image results for foam cell formation are shown in Fig. 1A and B. TMP significantly inhibited
lipid accumulation in macrophages.
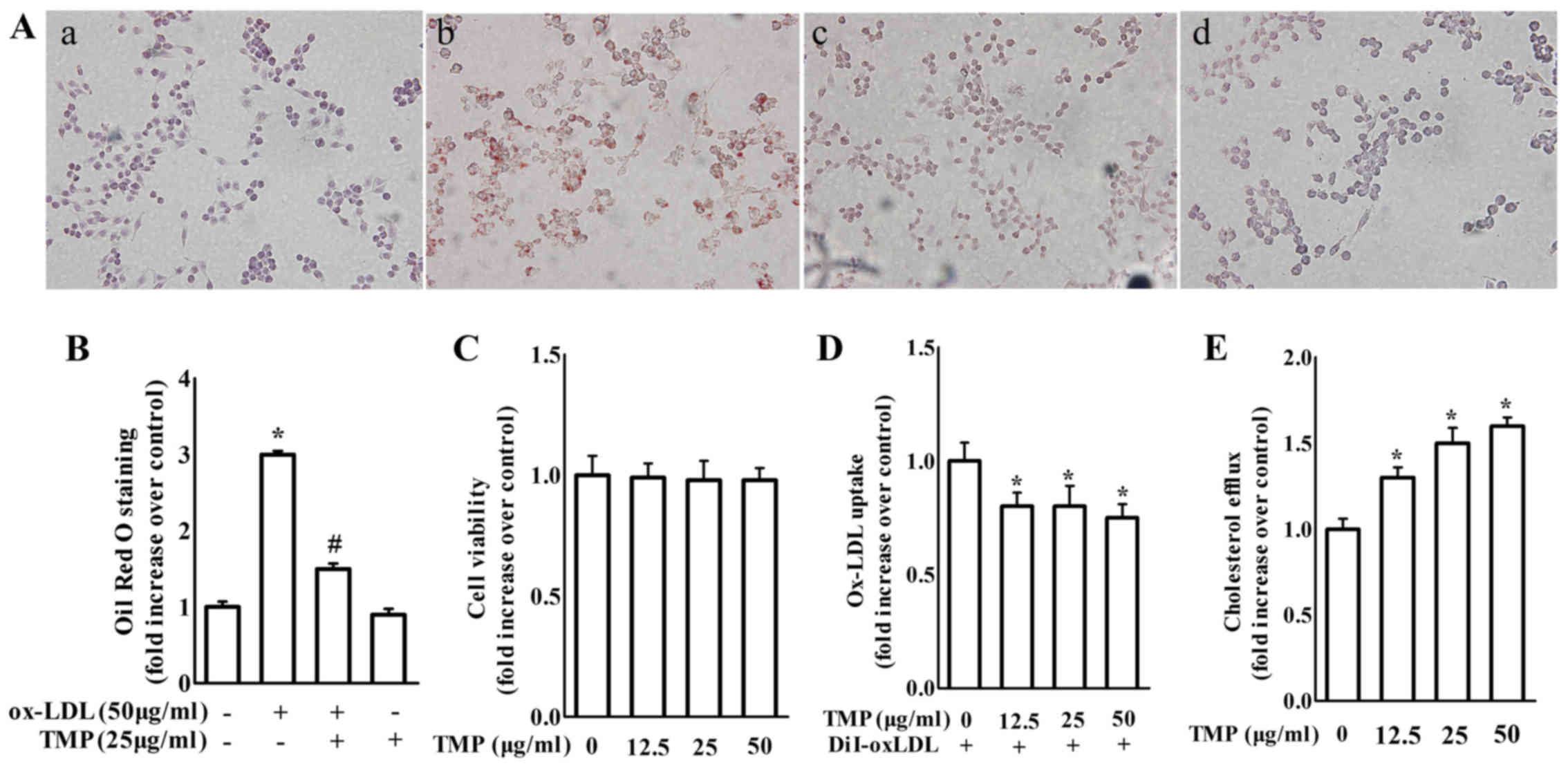 | Figure 1.TMP at non-cytotxic concentrations
without cytotoxicity inhibits lipid accumulation and promotes
cholesterol efflux in RAW264.7 macrophages. (A) Macrophages were
treated with ox-LDL (50 µg/ml) in the presence or absence of TMP
(25 µg/ml) for 24 h. a, control group; b, the ox-LDL-treated group;
c, the ox-LDL plus TMP group; d, the TMP-treated group. The cells
with various treatments were fixed, and then stained with Oil Red O
(magnification, ×400). (B) After Oil Red O staining, alcohol
extraction was used to detect the density of the lipid content. The
absorbance at 500 nm was assessed with a microplate reader. (C)
Macrophages were treated with TMP (0, 12.5, 25 and 50 µg/ml) for 24
h and cell viability was assessed by MTT assay. (D) Macrophages
were treated with TMP (0, 12.5, 25 and 50 µg/ml) for 24 h, and then
incubated with 10 µg/ml DiI-Ox-LDL for 4 h at 37°C. The cell lysate
and medium were collected to assess the absorbance at 540 nm. (E) A
cholesterol efflux assay was performed as described in the
Materials and methods. The data are representative of 3 independent
experiments (mean ± SEM); *P<0.05 compared with control group;
#P<0.05 compared with ox-LDL-treated group. TMP,
tetramethylpyrazine; ox-LDL, oxidized low-density lipoprotein; MTT,
methyl thiazolyl tetrazolium. |
Accordingly, TMP treatment caused a significant
decrease in ox-LDL uptake and an increase in cholesterol efflux
compared with the control group (Fig.
1D and E). These findings revealed that TMP inhibited foam cell
formation by increasing cholesterol efflux and decreasing ox-LDL
uptake in macrophages.
TMP decreases the expression of
scavenger receptors and increases the expression of ABC
transporters in RAW264.7 macrophages
We next examined the effects of TMP on the
expression of scavenger receptors and ABC transporters, such as
SR-A, CD36, SR-BІ, ABCG1 and ABCA1, which play integral roles in
intracellular lipid accumulation in RAW264.7 as a previous study
demonstrated (27). Our findings
revealed that treatment with 12.5–50 µg/ml TMP dose-dependently
increased the mRNA and protein expression of ABCA1 and ABCG1
(Fig. 2). Additionally, the protein
and mRNA levels of CD36 and SR-A in macrophages were significantly
inhibited by TMP (Fig. 2). However,
both the mRNA and protein levels of SR-BI were unchanged with
treatment of TMP in macrophages (Fig.
2). Based on the results in Fig.
2, TMP may inhibit the formation of foam cells by regulating
ABC transporters and scavenger receptors in RAW264.7
macrophages.
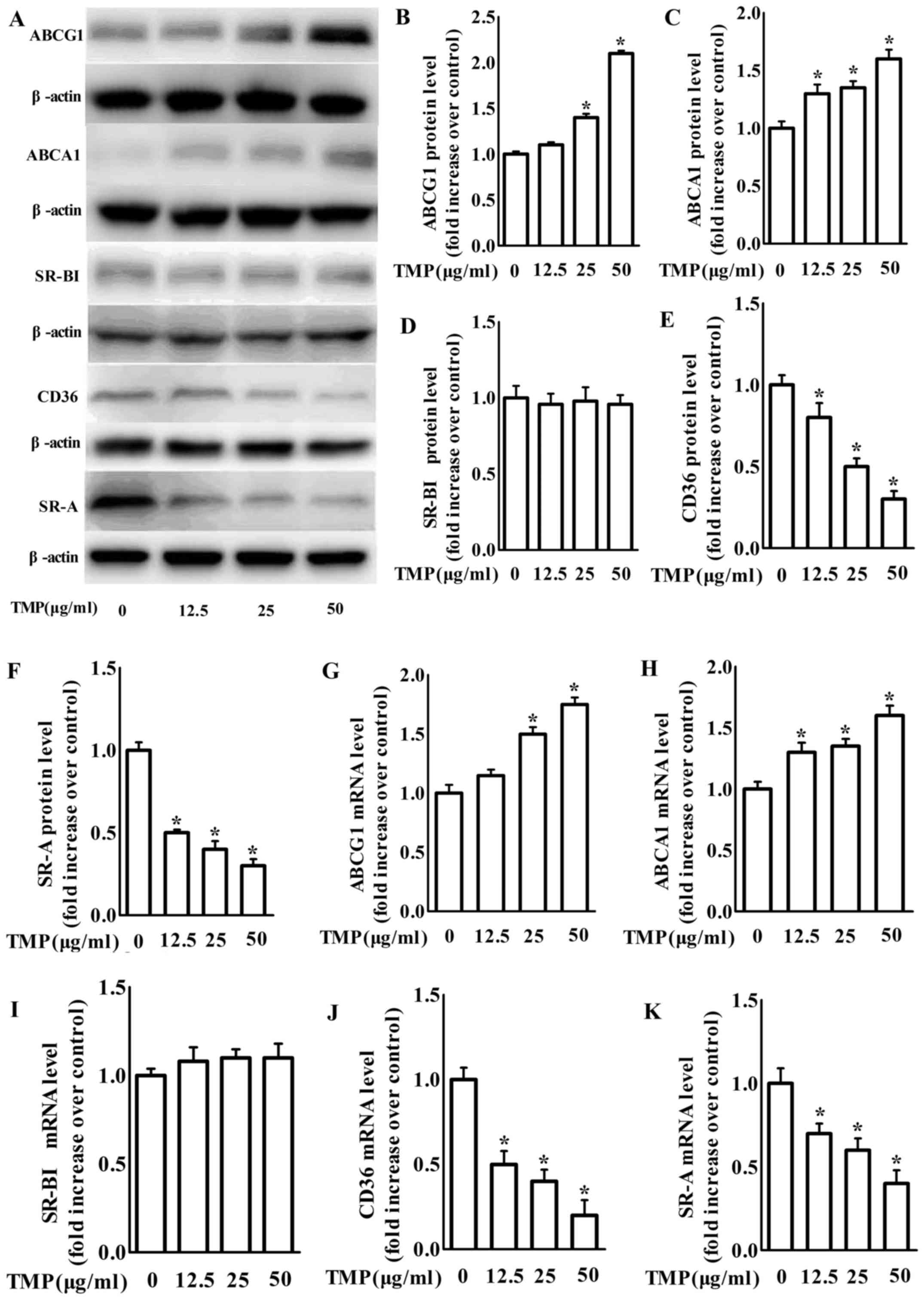 | Figure 2.TMP decreases protein and mRNA
expression of CD36 and SR-A, but increases protein and mRNA
expression of the ABCA1 and ABCG1 in RAW264.7 macrophages. (A)
Macrophages were treated with TMP (0, 12.5, 25 and 50 µg/ml) for 24
h and the protein level of SR-A, CD36, SR-BІ, ABCA1, ABCG1 or
β-actin was determined by western blotting. (B-F) The relative
protein levels of CD36, SR-A, SR-BІ, ABCA1 and ABCG1 are presented
as the mean ± SEM of the optical density from 3 separate
experiments. (G-K) After treatment, total RNA was extracted and
then subjected to RT-qPCR to detect the mRNA expression of SR-A,
CD36, ABCA1, ABCG1 and SR-BІ. The data are representative of 3
independent experiments (mean ± SEM); *P<0.05 compared with the
control group. TMP, tetramethylpyrazine; CD36, cluster of
differentiation 36; SR-A scavenger receptor class A; ABCA1,
ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette
transporter G1; SR-BI, SR-B type I; RT-qPCR, quantitative real-time
polymerase chain reaction. |
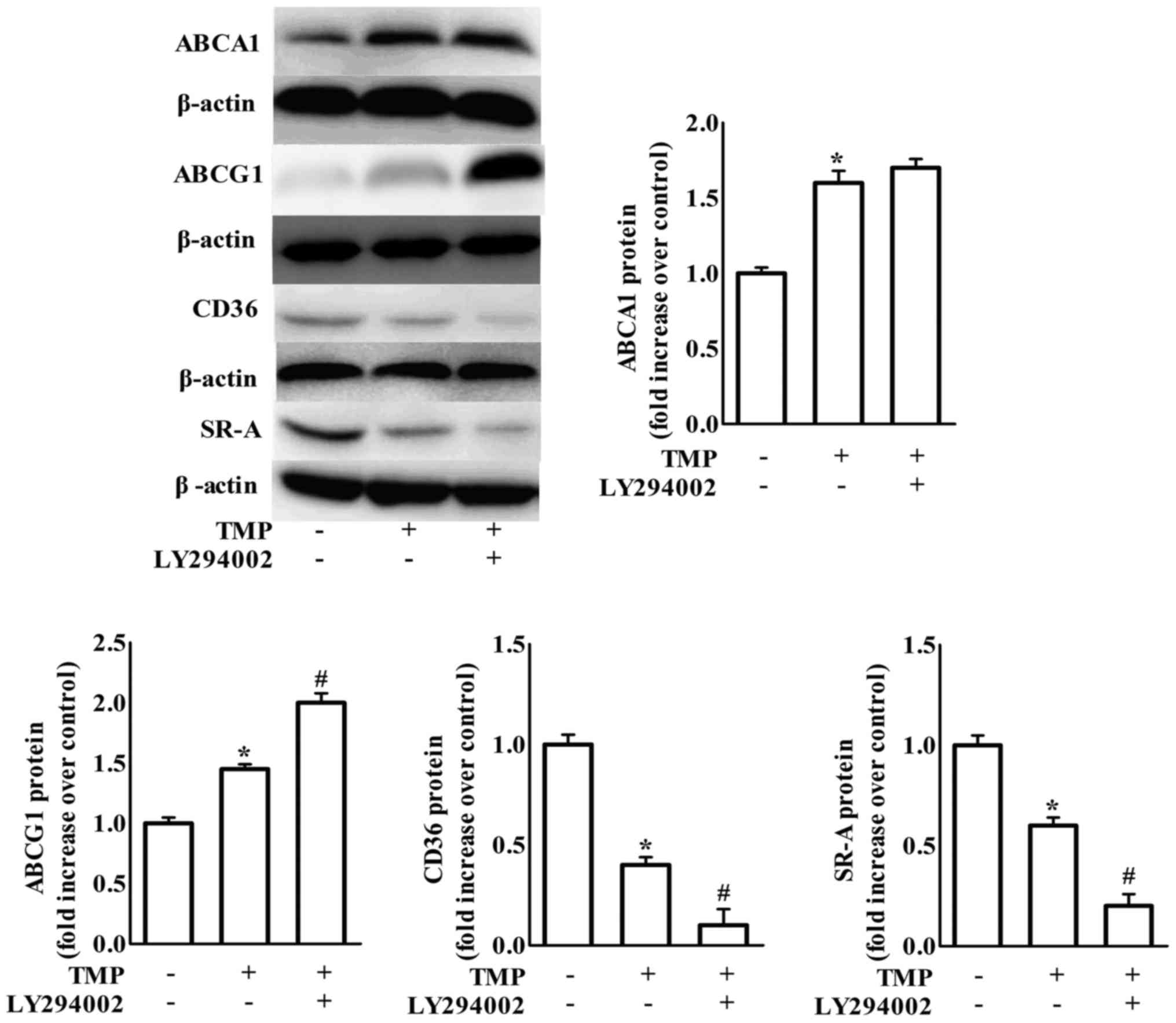
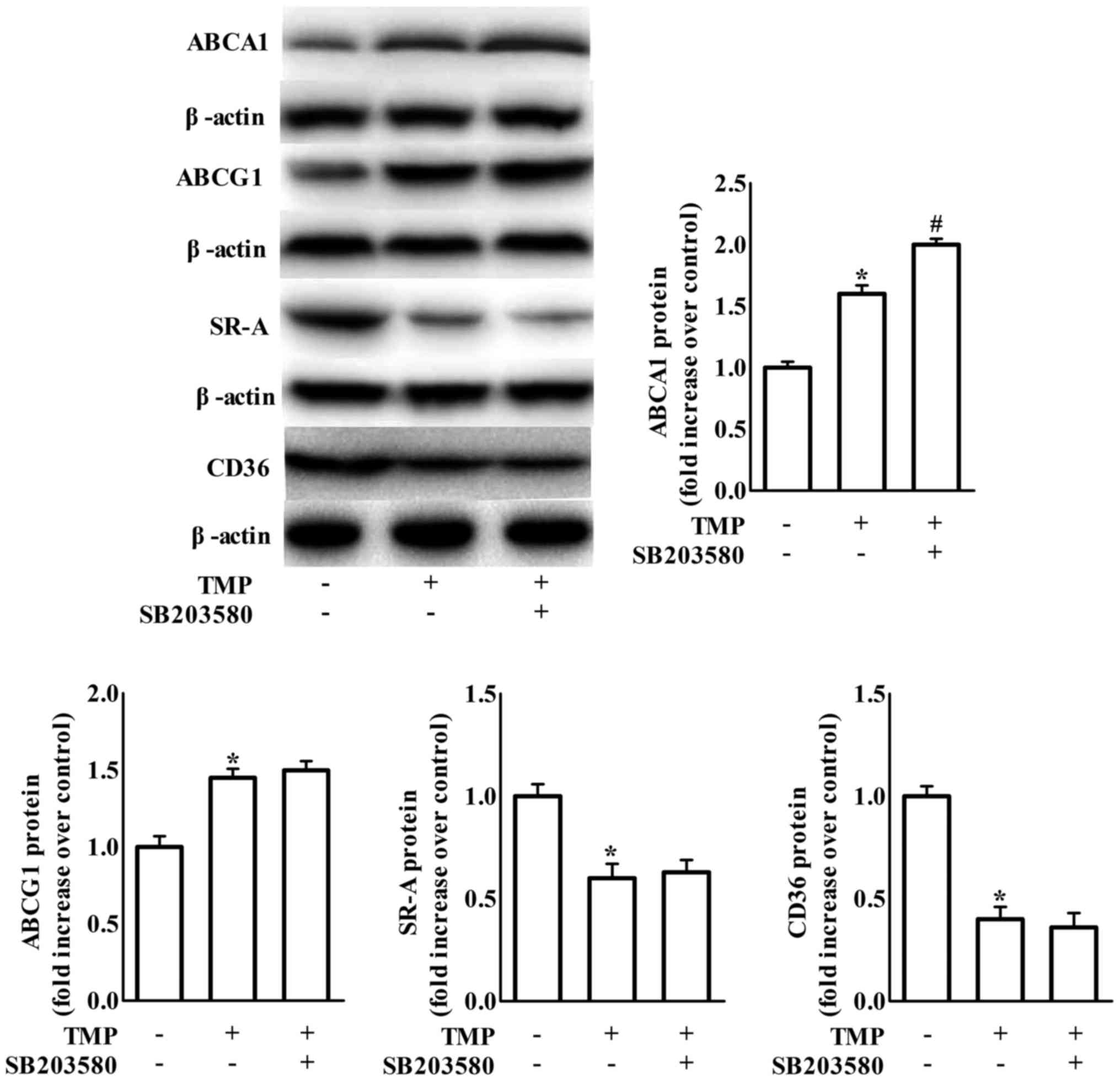
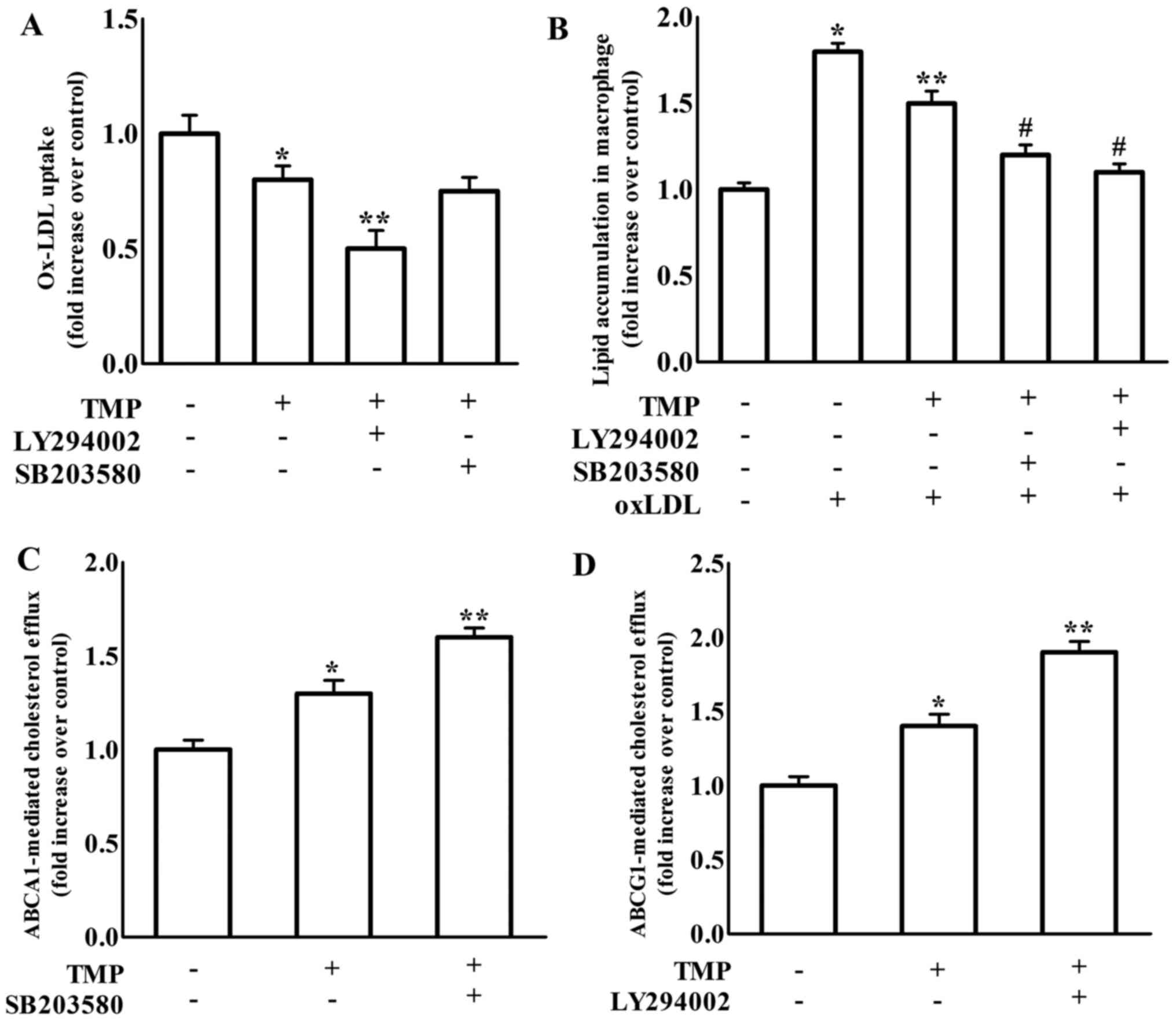
TMP regulates ABCA1, ABCG1, CD36 and
SR-A expression and inhibits foam cell formation via PI3K/Akt- and
p38-dependent pathways
Previous studies demonstrated that PI3K/Akt plays a
crucial role in the uptake of modified lipoproteins and cholesterol
efflux in various cells (28,29).
The present study detected whether PI3K/Akt was involved in the
expression of scavenger receptors and ABC transporters in
TMP-induced inhibition of formation of foam cells. As shown in
Fig. 3, treatment with LY294002, a
PI3K/Akt inhibitor, increased TMP-mediated downregulation of CD36
and SR-A and upregulation of ABCG1 protein expression, but not
ABCA1 expression. It has been suggested that blockage of p38 MAPK
signaling is involved in TMP-invoked inhibition of inflammation in
endothelial cells (22). To clarify
whether p38 is involved in TMP-regulated expression of ABC
transporters and scavenger receptors, we examined the effects of
SB203580 (a bicyclic imidazole compound, a specific inhibitor of
p38 MAPK). We revealed that the inhibition of p38 only further
increased TMP-induced ABCA1 protein expression in RAW264.7
macrophages (Fig. 4).
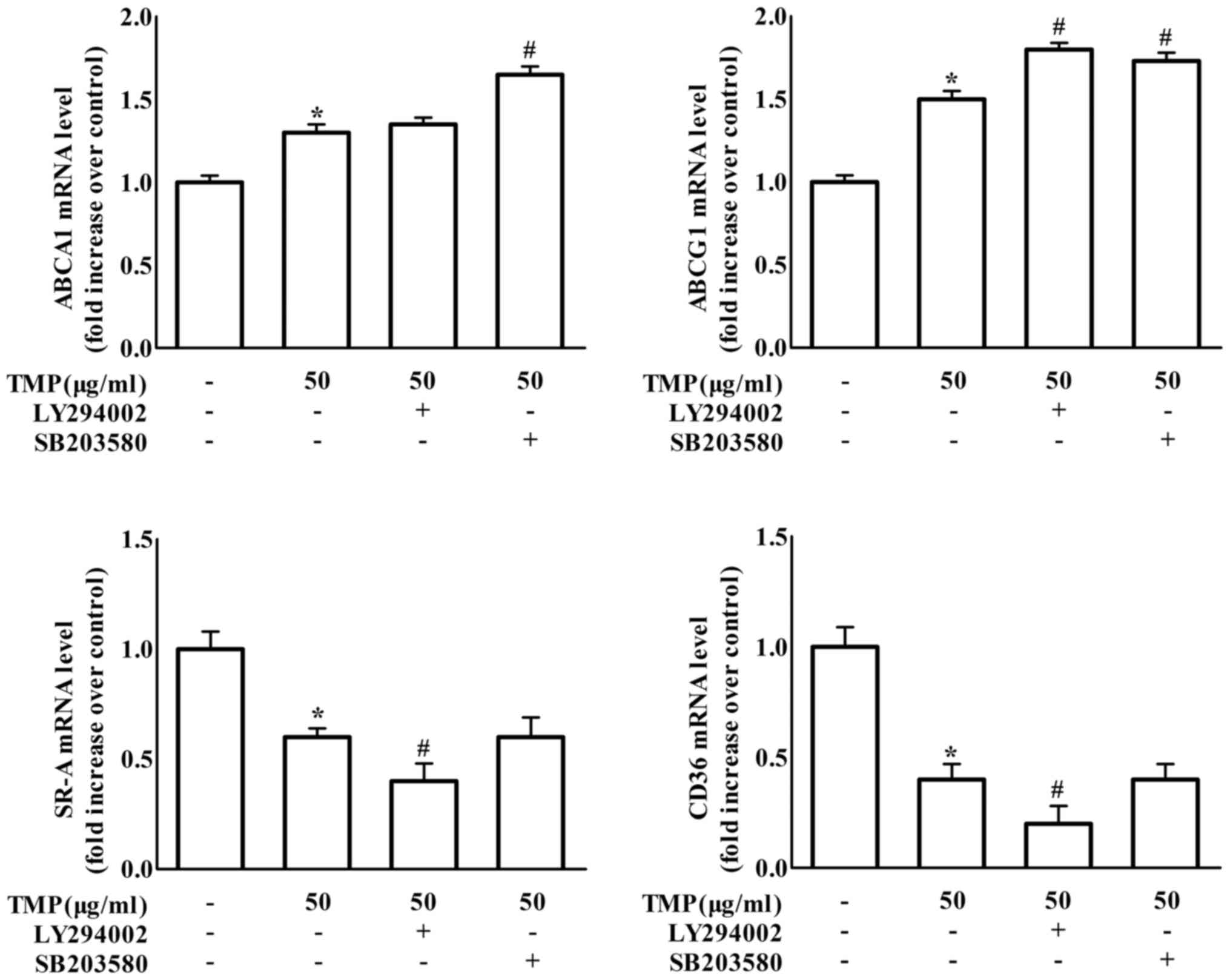
To further confirm whether TMP results in changes in
foam cell formation-related proteins by affecting transcriptional
levels, the mRNA levels of these proteins were also examined by
real-time PCR in RAW264.7 macrophages. The findings in Fig. 5 revealed that treatment with the
PI3K inhibitor LY294002 significantly enhanced the effects of TMP
on the decrease of CD36 and SR-A gene expression. Meanwhile, the
p38 inhibitor SB203580 treatment caused an increase in TMP-induced
ABCA1 gene expression. Moreover, treatment with either the PI3K
inhibitor LY294002 or the p38 inhibitor SB203580 increased
TMP-induced ABCG1 gene expression. We further detected the effect
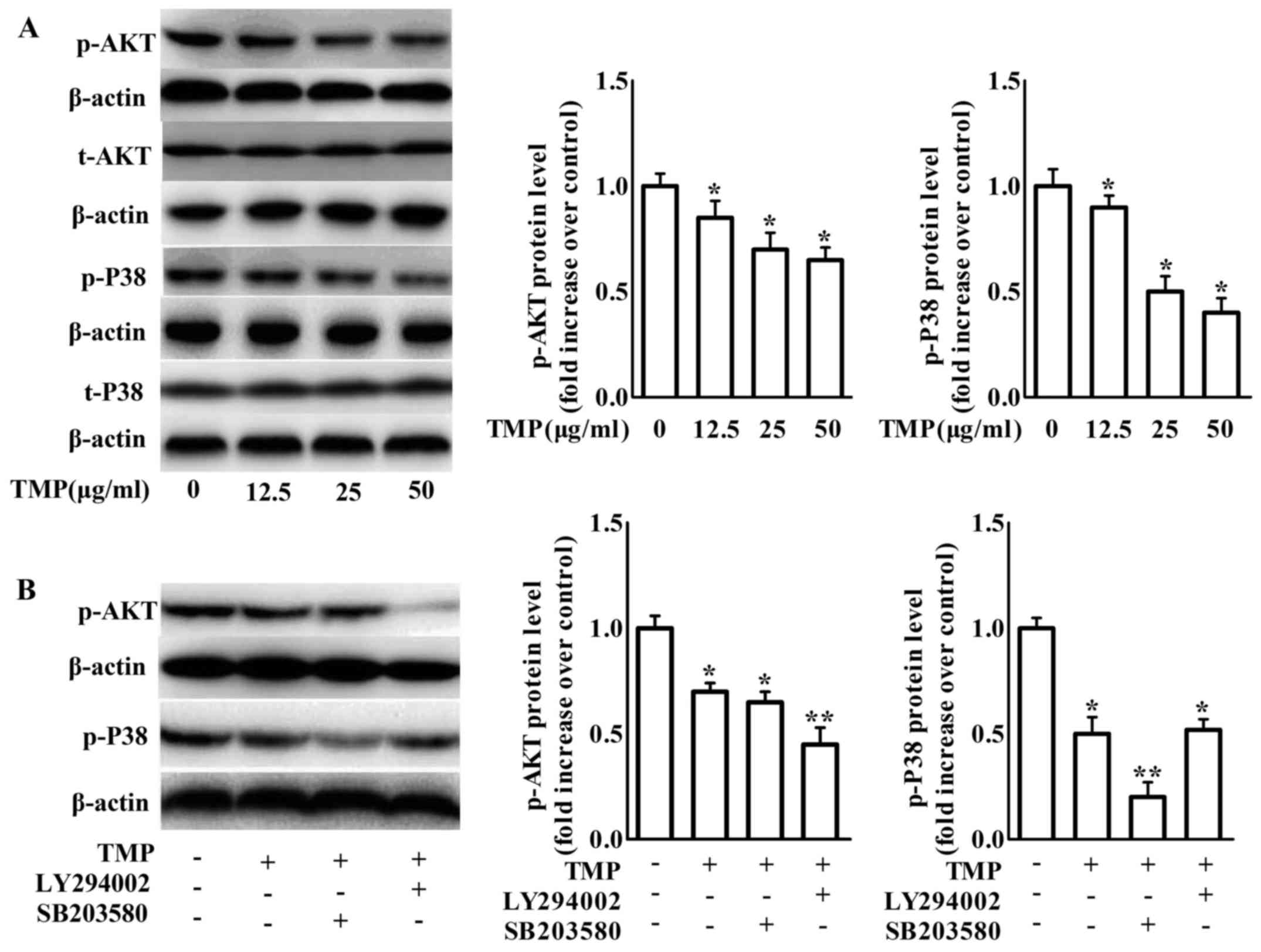
of TMP on PI3K/Akt and p38 phosphorylation in RAW264.7 macrophages.
The phosphorylation of PI3K/Akt and p38 was inhibited after 24 h of
TMP treatment (Fig. 6A).
Pretreatment with the p38 inhibitor SB203580 further increased the
effects of TMP on the inhibition of p38 phosphorylation without
affecting PI3K/Akt phosphorylation. In addition, pretreatment with
the PI3K inhibitor LY294002 increased TMP-decreased PI3K/Akt
phosphorylation, but not TMP-decreased p38 phosphorylation
(Fig. 6B). These results revealed
that TMP can inhibit PI3K/Akt and p38 phosphorylation independently
in RAW264.7 macrophages. Combined with the data of Figs. 3–5,
these results indicated that the effects of TMP on the upregulation
of ABCA1 and ABCG1 expression and the downregulation of CD36 and
SR-A expression were mediated by the inactivation of PI3K/Akt and
p38.
We further investigated the effects of PI3K and p38
inhibition on TMP-mediated ox-LDL uptake, cholesterol efflux and
intracellular lipid accumulation, which are necessary for
macrophage-derived foam cell formation. As shown in Fig. 7A, TMP-decreased ox-LDL uptake in
macrophages was exacerbated by LY294002, which was consistent with
the enhanced effect of LY294002 on TMP-mediated scavenger
receptors. However, SB203580 did not affect TMP-regulated ox-LDL
uptake. ABCA1 promotes free cholesterol efflux from macrophages to
ApoA1 and ABCG1 is responsible for cholesterol efflux to HDL
(7–9). Therefore, we used separate experiments
to investigate ABCA1- and ABCG1-regulated cholesterol efflux. The
results in Fig. 7C revealed that
TMP-increased ABCA1-regulated cholesterol efflux was increased by
SB203580, which was consistent with the promotive effect of
SB203580 on TMP-mediated upregulation of ABCA1. TMP-increased
ABCG1-mediated cholesterol efflux was enhanced by LY294002, which
was consistent with the reinforced effect of LY294002 on
TMP-mediated ABCG1 expression (Fig.
7D). Additionally, the TMP-invoked inhibition of lipid
accumulation in RAW264.7 macrophages was significantly enhanced by
both LY294002 and SB203580 (Fig.
7B). These results revealed that the TMP-mediated changes in
foam cell formation-related proteins in RAW264.7 macrophages were
involved in the PI3K- and p38-signaling pathways. Hence, the
inactivation of PI3K and p38 is necessary for the suppressive
effects of TMP on macrophage-derived foam cell formation.
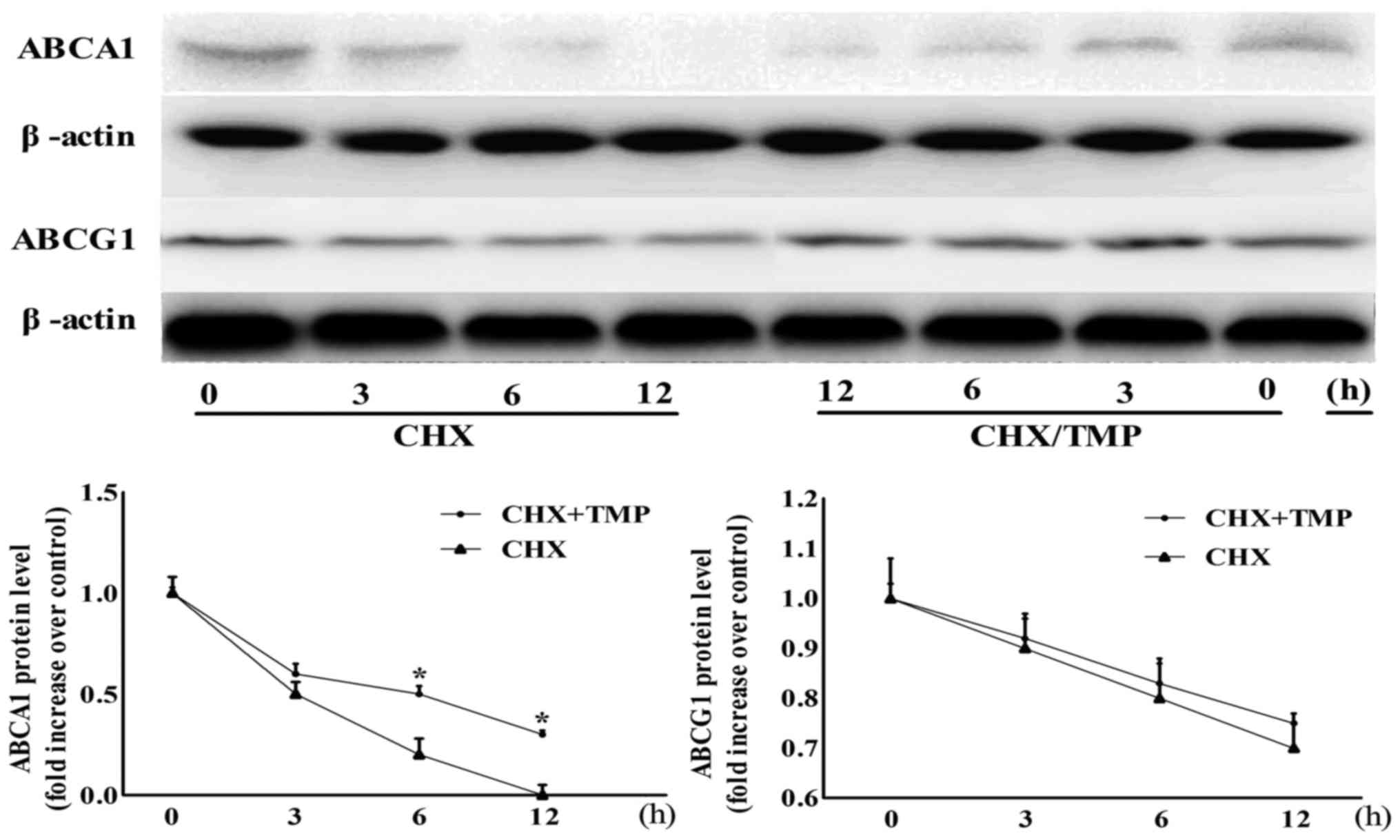
TMP decreases ABCA1 protein stability,
but does not affect ABCG1 protein stability
The present study demonstrated that TMP upregulated
the protein expression and mRNA levels of ABCG1 and ABCA1, however,
whether the protein stabilities of these proteins were affected by
TMP remained to be examined. As shown in Fig. 8, no difference was found in the
ABCG1 degradation between TMP-treated and control groups in the
presence of CHX (an inhibitor of de novo protein synthesis)
during a 12-h period, indicating that TMP-increased ABCG1
expression was independent of protein stability. However, in the
presence of CHX, the degradation rate of the ABCA1 protein was
time-dependently suppressed by TMP.
TMP retards atherosclerotic lesion
formation in ApoE−/− mice
We further examined the anti-atherogenic action of
TMP in vivo. Sixteen week-old ApoE−/− mice
treatment with TMP (150 mg/kg/day) for 8 weeks markedly retarded
lesion formation in atherosclerosis compared to the vehicle-fed
ApoE−/− mice (Fig. 9A and
B). Meanwhile, TMP decreased the protein expression of SR-A and
CD36, but increased the protein expression of ABCG1 and ABCA1 in
aortas (Fig. 9C). Our findings are
in agreement with the results of the in vitro
experiments.
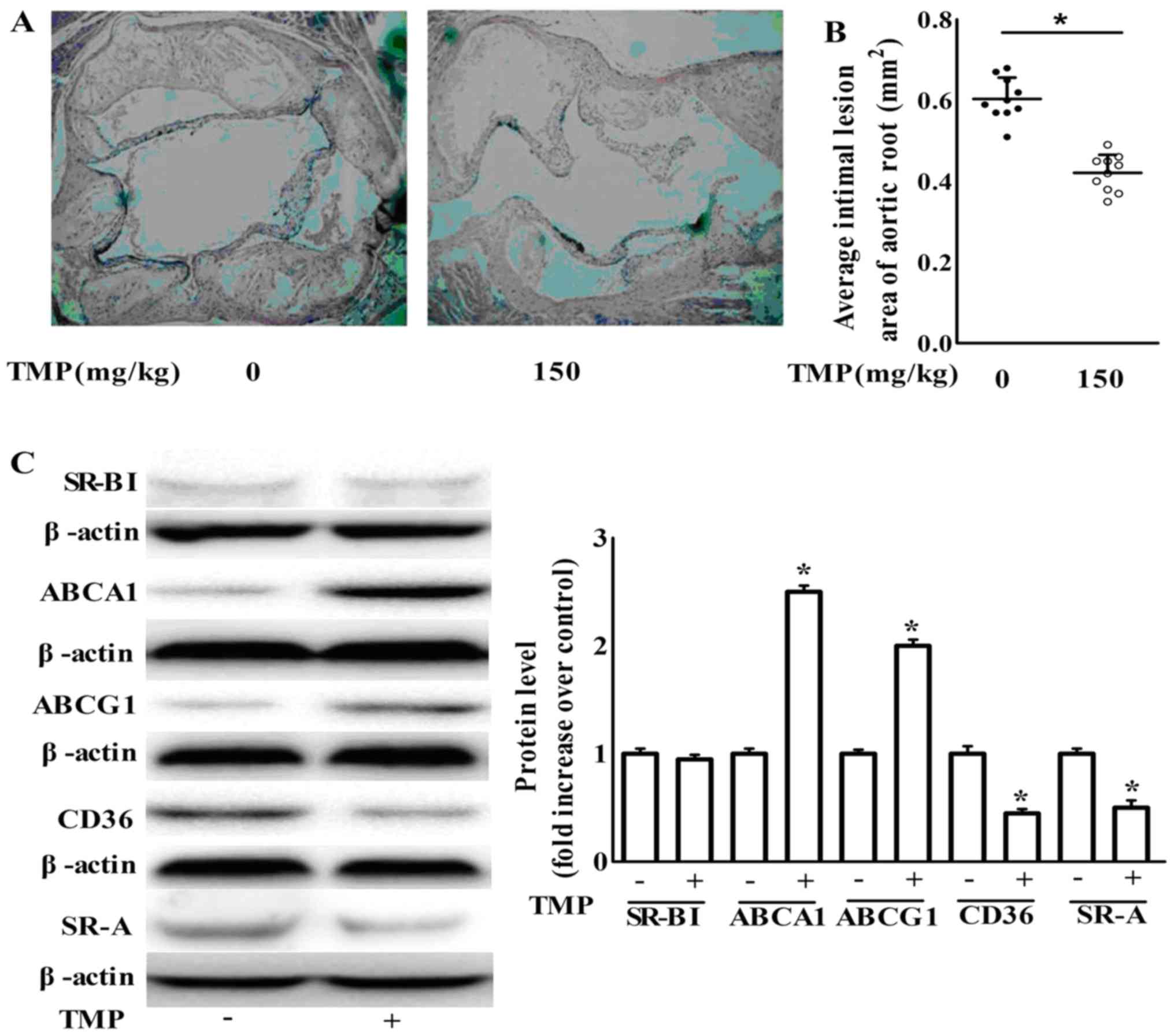 | Figure 9.TMP inhibits atherosclerotic lesion
formation in ApoE−/− mice via an increase in the
expression of ABCA1 and ABCG1 and a decrease in the expression of
CD36 and SR-A. (A) Representative H&E stained aortic sections
(magnification, ×40). (B) Average size of the atherosclerotic
plaques in the aortic root. Data are expressed as the mean ± SEM
(n=10 for each group). (C) After the treatment of
ApoE−/− mice as described in the Materials and methods
section, aortas were collected and subjected to western blotting to
detect the protein expression of ABCA1, ABCG1, SR-BІ, SR-A and
CD36. One representative blot is shown. The relative protein levels
of CD36, SR-A, SR-BІ, ABCA1 and ABCG1 are presented as the mean ±
SEM of the optical density from 3 separate experiments; *P<0.05
compared with the ApoE−/− mice. TMP,
tetramethylpyrazine; ApoE−/−, apolipoprotein
E-deficient; ABCA1, ATP-binding cassette transporter A1; ABCG1,
ATP-binding cassette transporter G1; CD36, cluster of
differentiation 36; SR-A scavenger receptor class A; SR-BI, SR-B
type I. |
Discussion
Tetramethylpyrazine (TMP) has been recognized as a
protective agent against atherosclerosis (19–22).
However, the effect and underlying mechanism by which TMP regulated
lipid accumulation in macrophage-derived foam cells remained to be
investigated. The present study, supplied novel insights into the
molecular mechanisms of the anti-atherogenic characteristic of TMP
in ApoE−/− mouse aortas and in the macrophage-derived
foam cell formation. TMP treatment suppressed ox-LDL uptake and
induced cholesterol efflux, which are co-responsible for subsequent
inhibition of lipid accumulation in macrophages. These effects can
be attributed to decreased CD36 and SR-A expression, and to
increased ABCG1 and ABCA1 in macrophages via inactivation of PI3K-
and p38-dependent pathways. Moreover, TMP increased the expression
of ABCA1 by increasing the protein stability of ABCA1. These
findings revealed that TMP has a beneficial effect on sustaining
lipid levels during the conversion of macrophages into foam cells
in atherosclerosis lesion formation and this potential mechanism
was described.
CD36 and SR-A are the main receptors for ox-LDL
uptake, SR-BI along with ABCG1 and ABCA1 are amenable to
cholesterol efflux. These actions play crucial roles in the
regulation of intracellular lipid levels during foam cell formation
(26). The present study may be the
first to demonstrate that TMP inhibits intracellular lipid
accumulation in macrophage-derived foam cells potentially through
the decrease of ox-LDL uptake and the increase of cholesterol
efflux. Moreover, TMP decreased both the mRNA and protein
expression of CD36 and SR-A. CD36 and SR-A are two predominant
types of scavenger receptors responsible for ox-LDL uptake in
macrophages (26). Research
utilizing LDL receptor knockout-mice (LDLR−/−)
transplanted with SR-A-deficient fetal liver cells indicated that
SR-A in macrophages is conducive to lesion formation of
atherosclerosis. ApoE−/− recipient mice injected with
CD36−/− macrophages and fed with a high-fat diet
revealed that CD36-dependent signaling cascades are essential for
foam cell formation. Extensive studies also revealed that
anti-atherogenic antioxidants can decrease the expression of SR-A
and CD36, which indicate the crucial role of SR-A and CD36 in
atherosclerotic pathogenesis (8).
In the present study, we demonstrated that TMP suppresses ox-LDL
uptake in macrophages via downregulation of the expression of SR-A
and CD36, which could have resulted from the inhibition of the
activity of the PI3K/Akt pathway. Notably, PI3K/Akt signaling
inhibition enhanced the influence of TMP on SR-A and CD36
expression. These findings demonstrate that inhibition of PI3K/Akt
signaling is necessary for the function of TMP athero-protection in
macrophages. In view of the action of SR-A and CD36, the effect of
TMP attenuation of SR-A and CD36 may facilitate the decrease of
ox-LDL uptake and subsequent suppression of foam cell
formation.
Except for its suppressive effect on the expression
of scavenger receptors, TMP increased the expression of ABCA1 and
ABCA1-mediated cholesterol efflux via the inactivation of p38
signaling, and increased the expression of ABCG1 and ABCG1-mediated
cholesterol efflux via the inactivation of PI3K/Akt signaling. The
expression of ABCA1/ABCG1 was upregulated by the activation of
Liver X receptor (LXR) signaling, thus promoting macrophage RCT
(30) and decreasing
atherosclerosis in mouse models (31). In addition, various antioxidants
with anti-atherogenic properties are capable of upregulating the
expression of ABCG1 and ABCA1 (32,33).
In view of the function of ABCG1 and ABCA1, the increasing effect
of TMP on the expression of ABCG1 and ABCA1 observed in the present
study may conduce to the inhibition of ox-LDL uptake and subsequent
suppression of foam cell formation.
More crucially, ABCA1 protein was stabilized by TMP.
This is in agreement with previous findings that revealed that
stabilization of the ABCA1 protein increased cholesterol efflux and
decreased lipid accumulation in foam cells (34). Conversely, destabilization of the
ABCA1 protein induced cholesterol accumulation in macrophages and
foam cell formation (35).
Collectively, our results indicate that the increase of ABCA1
stability by TMP may facilitate the suppression of the formation of
foam cells.
In addition to TMP-invoked ABCA1 protein stability,
we also found that TMP increased the mRNA expression of ABCA1.
These data revealed the TMP-induced increase of the protein
expression of ABCA1 via post-translational and transcriptional
regulation.
These findings demonstrate that TMP inhibits foam
cell formation but does not restrict the cell culture system. The
in vivo study revealed that atherosclerotic progression in
ApoE−/− mice was retarded by TMP, which is conformable
to a previous study (20). Indeed,
TMP exhibited some beneficial effects on atherosclerosis such as
preventing endothelial dysfunction (19), regulating lipid levels in the plasma
(20), attenuating oxidative stress
(21), and inhibiting inflammation
(22). In the present study, we
further found that TMP decreased the expression of CD36 and SR-A
and increased the expression of ABCA1 and ABCG1 in TMP-treated
ApoE−/− mice. Therefore, it appears that these
therapeutic actions are pervasive. Accordingly, TMP may play
beneficial roles in different organs through disparate
mechanisms.
In brief, the present study revealed that TMP
decreased CD36 and SR-A expression and ox-LDL uptake via the
inactivation of PI3K signaling. Furthermore, TMP increased the
expression of ABCA1 and ABCA1-mediated cholesterol efflux via the
inactivation of p38 signaling, and increased ABCG1 expression and
ABCG1-mediated cholesterol efflux via the inactivation of PI3K
signaling. This TMP-invoked regulation of the expression of CD36,
SR-A, ABCA1 and ABCG1 resulted in the inhibition of foam cell
formation induced by ox-LDL. The present study offered novel
insights for better understanding the underlying mechanisms
involved in TMP-invoked suppression of foam cell formation in
atherosclerosis. The only limitation of this study is that we did
not perform an LY294002 or SB203580 control without TMP, and the
effects between TMP and LY294002 or SB203580 were not compared.
Therefore, whether other signaling pathways are involved in the
protection of TMP against macrophage-derived foam cell formation
remains to be investigated.
Acknowledgements
The present study was supported by grants from the
Medical Association of Sichuan Province (S15040), the National
Natural Science Fund (81500357), the Affiliated Hospital of
Southwest Medical University, and the Beijing Medical Award
Foundation.
References
|
1
|
Trigatti BL and Fuller M: HDL signaling
and protection against coronary artery atherosclerosis in mice. J
Biomed Res. 30:94–100. 2016.PubMed/NCBI
|
|
2
|
Cai Y, Li JD and Yan C: Vinpocetine
attenuates lipid accumulation and atherosclerosis formation.
Biochem Biophys Res Commun. 434:439–443. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsai JY, Su KH, Shyue SK, Kou YR, Yu YB,
Hsiao SH, Chiang AN, Wu YL, Ching LC and Lee TS: EGb761 ameliorates
the formation of foam cells by regulating the expression of SR-A
and ABCA1: Role of haem oxygenase-1. Cardiovasc Res. 88:415–423.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rahaman SO, Swat W, Febbraio M and
Silverstein RL: Vav family Rho guanine nucleotide exchange factors
regulate CD36-mediated macrophage foam cell formation. J Biol Chem.
286:7010–7017. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chistiakov DA, Bobryshev YV and Orekhov
AN: Macrophage-mediated cholesterol handling in atherosclerosis. J
Cell Mol Med. 20:17–28. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fielding CJ and Fielding PE: Cellular
cholesterol efflux. Biochim Biophys Acta. 1533:175–189. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McNeish J, Aiello RJ, Guyot D, Turi T,
Gabel C, Aldinger C, Hoppe KL, Roach ML, Royer LJ, de Wet J, et al:
High density lipoprotein deficiency and foam cell accumulation in
mice with targeted disruption of ATP-binding cassette
transporter-1. Proc Natl Acad Sci USA. 97:4245–4250. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kennedy MA, Barrera GC, Nakamura K, Baldán
A, Tarr P, Fishbein MC, Frank J, Francone OL and Edwards PA: ABCG1
has a critical role in mediating cholesterol efflux to HDL and
preventing cellular lipid accumulation. Cell Metab. 1:121–131.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haghpassand M, Bourassa PA, Francone OL
and Aiello RJ: Monocyte/macrophage expression of ABCA1 has minimal
contribution to plasma HDL levels. J Clin Invest. 108:1315–1320.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao JF, Ching LC, Huang YC, Chen CY,
Chiang AN, Kou YR, Shyue SK and Lee TS: Molecular mechanism of
curcumin on the suppression of cholesterol accumulation in
macrophage foam cells and atherosclerosis. Mol Nutr Food Res.
56:691–701. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park SH, Paek JH, Shin D, Lee JY, Lim SS
and Kang YH: Purple perilla extracts with α-asarone enhance
cholesterol efflux from oxidized LDL-exposed macrophages. Int J Mol
Med. 35:957–965. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang C, Yan W, Li B, Xu B, Gong Y, Chu F,
Zhang Y, Yao Q, Wang P and Lei H: A new ligustrazine
derivative-selective cytotoxicity by suppression of NF-κB/p65 and
COX-2 expression on human hepatoma cells. Part 3. Int J Mol Sci.
16:16401–16413. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pan J, Shang JF, Jiang GQ and Yang ZX:
Ligustrazine induces apoptosis of breast cancer cells in vitro and
in vivo. J Cancer Res Ther. 11:454–458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ji AJ, Liu SL, Ju WZ and Huang XE:
Anti-proliferation effects and molecular mechanisms of action of
tetramethypyrazine on human SGC-7901 gastric carcinoma cells. Asian
Pac J Cancer Prev. 15:3581–3586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zeng Z, Zhu W, Zhou X, Jin Z, Liu H, Chen
X, Pan J, Demura H, Naruse M and Shi Y: Tetramethylpyrazine, a
Chinese drug, blocks coronary vasoconstriction by endothelin-1 and
decreases plasma endothelin-1 levels in experimental animals. J
Cardiovasc Pharmacol. 31 Suppl 1:S313–S316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo SK, Chen KJ, Qian ZH, Weng WL and Qian
MY: Tetramethylpyrazine in the treatment of cardiovascular and
cerebrovascular diseases. Planta Med. 47:891983. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dai XZ and Bache RJ: Coronary and systemic
hemodynamic effects of tetramethylpyrazine in the dog. J Cardiovasc
Pharmacol. 7:841–849. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu SY and Sylvester DM:
Antithrombotic/antiplatelet activity of tetramethylpyrazine. Thromb
Res. 58:129–140. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ni X, Wong SL, Wong CM, Lau CW, Shi X, Cai
Y and Huang Y: Tetramethylpyrazine protects against hydrogen
peroxide-provoked endothelial dysfunction in isolated rat aortic
rings: Implications for antioxidant therapy of vascular diseases.
Evid Based Complement Alternat Med. 2014:6271812014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang GF, Shi CG, Sun MZ, Wang L, Wu SX,
Wang HF, Xu ZQ and Chen DM: Tetramethylpyrazine attenuates
atherosclerosis development and protects endothelial cells from
ox-LDL. Cardiovasc Drugs Ther. 27:199–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jiang F, Qian J, Chen S, Zhang W and Liu
C: Ligustrazine improves atherosclerosis in rat via attenuation of
oxidative stress. Pharm Biol. 49:856–863. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li XY, He JL, Liu HT, Li WM and Yu C:
Tetramethylpyrazine suppresses interleukin-8 expression in
LPS-stimulated human umbilical vein endothelial cell by blocking
ERK, p38 and nulear factor-kappaB signaling pathways. J
Ethnopharmacol. 125:83–89. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang Q, Liu W, Liu H and Zhou M: Effect of
compound Chuanxiong capsule on inflammatory reaction and
PI3K/Akt/NF-κB signaling pathway in atherosclerosis. Evid Based
Complement Alternat Med. 2015:5845962015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu Y, Sun B, Liu K, Yan M, Zhang Y, Miao C
and Ren L: Icariin attenuates high-cholesterol diet induced
atherosclerosis in rats by inhibition of inflammatory response and
p38 MAPK signaling pathway. Inflammation. 39:228–236. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen Z, Huo JR, Yang L and Zhu HY: Effect
of ligustrazine on mice model of hepatic veno-occlusive disease
induced by Gynura segetum. J Gastroenterol Hepatol. 26:1016–1021.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li XY, Kong LX, Li J, He HX and Zhou YD:
Kaempferol suppresses lipid accumulation in macrophages through the
downregulation of cluster of differentiation 36 and the
upregulation of scavenger receptor class B type I and ATP-binding
cassette transporters A1 and G1. Int J Mol Med. 31:331–338. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin YT, Jian DY, Kwok CF, Ho LT and Juan
CC: Visfatin promotes foam cell formation by dysregulating CD36,
SRA, ABCA1, and ABCG1 expression in RAW264.7 macrophages. Shock.
45:460–468. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin CS, Lin FY, Ho LJ, Tsai CS, Cheng SM,
Wu WL, Huang CY, Lian CH, Yang SP and Lai JH: PKCδ signalling
regulates SR-A and CD36 expression and foam cell formation.
Cardiovasc Res. 95:346–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang CX, Zhang YL, Wang JF, Jiang JY and
Bao JL: MCP-1 impacts RCT by repressing ABCA1, ABCG1, and SR-BI
through PI3K/Akt posttranslational regulation in HepG2 cells. J
Lipid Res. 54:1231–1240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Naik SU, Wang X, Da Silva JS, Jaye M,
Macphee CH, Reilly MP, Billheimer JT, Rothblat GH and Rader DJ:
Pharmacological activation of liver X receptors promotes reverse
cholesterol transport in vivo. Circulation. 113:90–97. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Terasaka N, Hiroshima A, Koieyama T,
Ubukata N, Morikawa Y, Nakai D and Inaba T: T-0901317, a synthetic
liver X receptor ligand, inhibits development of atherosclerosis in
LDL receptor-deficient mice. FEBS Lett. 536:6–11. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rana M, Kumar A, Tiwari RL, Singh V,
Chandra T, Dikshit M and Barthwal MK: IRAK regulates macrophage
foam cell formation by modulating genes involved in cholesterol
uptake and efflux. BioEssays. 38:591–604. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gui YZ, Yan H, Gao F, Xi C, Li HH and Wang
YP: Betulin attenuates atherosclerosis in apoE−/− mice
by up-regulating ABCA1 and ABCG1. Acta Pharmacol Sin. 37:1337–1348.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Zhou Y, Yu C, Yang H, Zhang C, Ye Y
and Xiao S: Paeonol suppresses lipid accumulation in macrophages
via upregulation of the ATP-binding cassette transporter A1 and
downregulation of the cluster of differentiation 36. Int J Oncol.
46:764–774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fu Y, Mukhamedova N, Ip S, D'Souza W,
Henley KJ, DiTommaso T, Kesani R, Ditiatkovski M, Jones L, Lane RM,
et al: ABCA12 regulates ABCA1-dependent cholesterol efflux from
macrophages and the development of atherosclerosis. Cell Metab.
18:225–238. 2013. View Article : Google Scholar : PubMed/NCBI
|