Introduction
The P53 family of tetrameric transcription factors
is composed of the P53, P63 and P73 proteins (1–3) that
influence many cellular pathways including cell proliferation,
apoptosis, DNA repair, angiogenesis, metabolism and differentiation
(4–6). All three proteins share an N-terminal
transactivation domain, a central sequence-specific DNA-binding
domain and a C-terminal oligomerization domain. Only P63 and P73
proteins contain an additional sterile-α-motif domain within the
C-terminus. Moreover, the P53 family members can be expressed as
different isoforms both at the N-terminus (ΔN and TA) and at the
C-terminus (α-γ for P53; α-ε for P63; α-η for P73) through the
usage of two alternative promoters and alternative splicing
mechanisms, respectively.
Despite protein and gene structure similarities, the
overlap in cellular functions between P53, P63 and P73 is limited
as highlighted by comparing their role in human physiology. P53 is
a well-known tumor suppressor protein encoded by one of the most
frequently mutated genes in sporadic types of cancer. Furthermore,
TP53 germline mutations are associated with the development
of the cancer prone Li-Fraumeni and Li-Fraumeni like syndromes
(7–9). Conversely, P63 is critical for the
correct development of ectodermal-derived tissues, whereas P73
contributes to neural and immune system functions (10,11).
No genetic disorder has been linked to TP73 germinal
mutations (12), whereas
heterozygous mutations in the TP63 gene underlie a subset of
human ectodermal dysplasia syndromes (EDs) (13,14).
Despite the fact that cancer development is rarely
associated with TP63 and TP73 mutations, both genes
play an active role in carcinogenesis. Flores et al
(15) demonstrated the contribution
of P63 and P73 in tumor development by revealing that
TP63+/− and TP73+/− mice
develop malignant tumors at a higher frequency with respect to the
wild-type counterparts. In addition,
TP53+/−TP63+/− and
TP53+/−TP73+/− compound mutant mice
developed a more severe phenotype (e.g., more metastases) compared
to the TP53+/− mice, indicating that P63 and P73
have functions independent of P53.
The tumor-suppressive activities of P63 and P73
appear to be tissue-specific. In fact, the tumor spectrum detected
in mice defective for TP63 and TP73 is quite
different from that of TP53-deficient mice (16). TP63+/− and
TP73+/− mice develop primarily lung adenomas
(TP63+/−, 25%; TP73+/−, 40%)
squamous cell hyperplasia (TP63+/−, 50%;
TP73+/−, 30%), while TP53+/−
mice primarily develop thymic lymphomas and osteosarcomas. These
differences are probably due to the different patterns of
expression of the P53 family members, while P53 is ubiquitous, the
P63 and P73 proteins are highly expressed in epithelial
tissues.
The observation that mutagens are carcinogens and
that different mutagens induce specific spectra of mutations in
genes frequently mutated in cancer (17), suggested that the analysis of the
site and nature of DNA changes in genes mutated in tumors could be
useful at identifying the factors contributing to the development
of a specific tumor type (18,19).
Molecular epidemiology converged to the TP53 gene,
positioning it at the crossroad of molecular carcinogenesis and
risk assessment (18). This was due
to the fact that the TP53 gene: i) is very sensitive to
point mutations; ii) alterations in its coding sequence occur in
>50% of all human types of cancer; and iii) ~80% of these
alterations are missense mutations leading to proteins with a
single amino acid substitution (20). The relationship between the exposure
to a carcinogen and the presence of specific TP53 mutations
in specific tumors has been extensively studied and described
(21). TP53 mutations
identified in tumors and cell lines have been collected in a
database that contains more than 30,000 entries (http://p53.fr/) (20).
With regard to sunlight, the International Agency
for Research on Cancer in 1992 classified UV radiation as a human
carcinogen (22). Upon reaching the
skin, UV photons are absorbed by electrons of DNA, leading to the
formation between adjacent pyrimidines of photoproducts known as
cyclobutane pyrimidine dimers (CPDs) and pyrimidine 6–4 pyrimidone
photoproducts (6–4PPs) (23).
Incorrect DNA repair leads to fixation of specific C→T and CC→TT
transitions at di-pyrimidine sites (24). About 50% of skin cancers exhibit the
signature of UV-induced mutagenesis on TP53 (18). TP53 mutations are frequent in
both normal-appearing sun-exposed skin and premalignant actinic
keratosis lesions, which are considered precursors of squamous cell
carcinoma.
The introduction of the next generation sequencing
(NGS) approaches on cancer cell genomes to determine the whole
landscape of mutations in cancers, profoundly changed the
perspective of mutational fingerprints. While the Sanger sequencing
imposed a technical threshold (a mutation is detectable in a single
sample if it is present in ~15–20% of the alleles), NGS can detect
with accuracy hundreds of mutations in a single sample even if it
is present in 1% (and even less) of the alleles.
The identification of gene mutations that directly
or indirectly confer a selective growth advantage to the cell in
which they occur (‘driver mutations’) among a plethora of other
mutations devoid of a role in the cancer process (‘passenger
mutations’) is a formidable challenge. The situation is even more
complicated, since a driver gene (i.e., that can harbor driver
mutations) may also contain passenger mutations. A review suggests
that, considering all types of tumors, the total number of driver
genes in cancer which was discovered through the NGS approach is
~125, 29% of which was discovered through the NGS approach
(25). Numerous statistical methods
have been described to distinguish between driver versus passenger
mutations (26–28). Furthermore, the number of driver
mutations sufficient for a cell to evolve into an advanced cancer
is thought to be extremely low. In solid tumors of adults,
including melanoma, alterations in as few as three driver genes are
sufficient (29,30). This pointed towards the idea that
pathways, rather than individual genes, appear to govern the
tumorigenic process (25), which
has rendered complex cancer-genome landscapes intelligible and
exploitable.
Cutaneous melanoma represents 4% of skin cancers but
is responsible for 80% of skin cancer related deaths, corresponding
to ~1–2% of all cancer related deaths (31). Cutaneous melanoma, which develops
from epidermal melanocytes of the skin, represents the most common
melanoma. Sun exposure is accepted as a major causative factor of
melanomas (32). Cutaneous
melanomas have been studied by NGS since 2010 (33). This analysis generated the first
comprehensive catalogue of somatic mutations from a malignant
melanoma, revealing that the predominant mutational signature
reflects DNA damage due to UV exposure. Moreover, through analysis
of large-scale melanoma exome data, Hodis et al (34) discovered six novel driver melanoma
genes (PPP6C, RAC1, SNX31, TACC1, STK19 and ARID2), providing
unequivocal genomic evidence for a direct mutagenic role of UV
light in melanoma pathogenesis. Recently, the sequence of genetic
alterations during melanoma progression was defined, revealing
distinct evolutionary paths for different melanoma subtypes
(35,36). While benign lesions harbored BRAF
V600E mutations exclusively, those classified as ‘intermediate’
were enriched for NRAS mutations and additional driver mutations.
TERT promoter mutations were observed at a high incidence in
intermediate lesions and melanomas in situ. Biallelic CDKN2A
inactivation emerged exclusively in invasive melanomas, while PTEN
and TP53 mutations were found only in advanced primary melanomas
and their presence appears to represent the transition to
metastatic tumors (36).
Recently, using NGS, missense mutations in the
TP63 gene were frequently and specifically found in
cutaneous melanomas (~15%) (37).
This observation prompted us to study the features of TP63
mutations from the molecular epidemiology point of view and to
ascertain whether all three members of the P53 family were mutated
in melanomas with equal frequency.
Materials and methods
Data collection
A total of 605 cases were considered by reviewing
all studies documenting TP53/TP63/TP73
mutations in cutaneous melanomas retrieved from cBioPortal
(http://www.cbioportal.org) (38,39).
Eighty nine of them exhibited a total of 111 TP63 mutations,
91 cases exhibited a total of 105 TP53 mutations and only 9
cases exhibited a total of 10 TP73 mutations (40,41).
TP63 mutations associated to EDs were
retrieved from literature reporting amino acid substitutions and
the causative single base pair substitution deduced from the
genetic code (13,42,43).
Excluding a tandem mutation (CGC>CAA, R318Q), a total of 101
TP63 base pair substitutions found in EDs were
considered.
In order to infer the potential role of the
different mutations in terms of driver or passenger mutations, the
output of two bioinformatic predictors, namely mutation assessor
and functional analysis through hidden Markov models, FATHMM-MKL
(an algorithm which predicts the functional, molecular and
phenotypic consequences of protein missense variants using hidden
Markov models, and classifies as ‘pathogenic’ or ‘neutral’
mutations if the scores are ≥0.7 or ≤0.5, respectively) were
retrieved from cBioPortal.
Statistical analysis
In order to rigorously compare mutational spectra
observed at the same locus, i.e., considering where along the
nucleotide sequence the mutation was observed, and how many
independent mutations were observed in each specific position, the
Adams and Skopek algorithm (44)
and the program from Cariello et al were used (45).
The Adams and Skopek algorithm uses a Monte Carlo
method to simulate a P-value of the standard hypergeometric test
for a contingency table (44).
Unlike the χ2 test, which can also be applied to
contingency tables and requires that all cells contain five or more
events, the hypergeometric test is appropriate when applied to
sparse data sets, often found in mutational spectra analyses. The
Cariello et al program (45)
uses a random number generator to produce a large number of
simulated spectra based on the hypergeometric probability of the
experimentally observed input spectra. The degree to which the
simulated spectra differ from the input spectra is used to estimate
the probability that the two input spectra were derived from the
same population. A P-value <0.05 leads to the rejection of the
null hypothesis and to the conclusion that the input spectra are
different.
Yeast strains and media
The issue of P53 family transactivation specificity
was thoroughly investigated by our group taking advantage of the
previously described yeast functional assay (luciferase
reporter-based assay) (8,9,20,46).
In particular, in order to evaluate the functionality of the P63
R379C allele, it was cloned into a yeast expression vector as
ΔN-P63α and TA-P63α isoforms; their functionality was evaluated in
two yeast reporter strains containing the luciferase gene under the
control of the P63 responsive genes P21 and PERP. yLFM-P21-5′ and
yLFM-PERP yeast strains were used (14); the two strains are isogenic except
for the different Response Element (RE) (20 bp) located upstream
the luciferase reporter gene. Cells were grown in YPDA medium (1%
yeast extract, 2% peptone, 2% dextrose, 200 mg/l adenine) or in a
selective medium containing dextrose or raffinose as a carbon
source and adenine (200 mg/l) but lacking tryptophan
(Sigma-Aldrich, St. Louis, MO, USA; BiokarDiagnostics, Pantin,
France). Galactose was added to the medium to modulate P63
expression under the inducible GAL1-GAL10 promoter.
Yeast vectors
pTSG-based vectors expressing wild-type ΔN-P63α and
TA-P63α isoforms were already available (14). Yeast vectors expressing mutant
isoforms (ΔN-P63α R379C and TA-P63α R379C) were constructed using
the experimental strategy as previously described (9,14).
Briefly, for R379C mutation, a pair of complementary
oligonucleotides (which served as forward and reverse primers) was
synthesized, with the mutated nucleotide (underlined the
corresponding codon) adjacent to the central position (R379C
forward, 5′-aagcgcccgt tttgtcagaacacacatggt-3′ and reverse,
5′-accatgtgtgttctgacaaaacgggcgctt-3′). Forward and reverse primers,
paired respectively with α-Cter and ΔN-Nter (or TA-Nter) primers
(ΔN-Nter,
5′-caagctataccaagcatacaatcaactatctcatatacagttaactcgagatgttgta
cctggaaaacaat-3′; TA-Nter,
5′-caagctataccaagcatacaatcaactatctcatatacagttaactcgagatgtcccagagcacacagaca-3′),
were used in two separate PCR reactions with the pcDNA3.1
TP63α plasmid (ΔN or TA isoform) as template. The following
PCR conditions were used: 1 min of denaturation at 94°C, 1 min of
annealing at 55°C and 2 min of elongation at 72°C (35 cycles)
(Exact polymerase, 5 prime). Unpurified aliquots of PCR products
were then transformed in yIG397 yeast strain together with an
XhoI/NotI (New England Biolabs, Ipswich, MA, USA)
digested pTS-based vector. In yeast, the plasmid is re-sealed
together with the PCR products by recombination, exploiting the
sequence homology at the end of the fragments (gap repair assay)
(8,9). Plasmid DNA was recovered from yeast
colonies, expanded in E. coli, and checked by digestion; the
presence of the specific mutation was verified by DNA sequencing
(BMR Genomics, Padova, Italy).
Yeast vectors expressing R379C mutant protein
(ΔN-P63α and TA-P63α isoforms) under the inducible GAL1-GAL10
promoter (pTSG-based), were constructed by double-digesting the
pTS-based expression vectors with the XhoI and NotI
restriction enzymes. The DNA fragment containing the mutant ΔN- or
the TA-TP63α coding sequence (cDNA) was then cloned in a
double-digested pTSG-based vector (14).
Yeast functional assay
P63 wild-type and mutant isoforms (ΔN-P63α and
TA-P63α) were expressed by a pTSG-based vector (TRP1 selection
marker) (14). Plasmid pRS314
(TRP1) was used as an empty vector. The functional assay was
conducted according to the miniaturized protocol we developed
(14,46). Briefly, yeast strains were
transformed (LiAc method) with the pTSG-based expression vectors
along with the empty vector. Yeast transformants were resuspended
in minimal synthetic medium containing raffinose plus galactose
(0,128%) in a final volume of 200 µl (96-well plates) and were
grown for 8 h with vigorous shaking at 30°C. The OD (600 nm) was
then directly assessed in the transparent 96-well plate. Twenty
microliters of cell suspension were transferred in a white plate
and mixed with an equal volume of PLB buffer 2X (Passive Lysis
Buffer; Promega, Madison, WI, USA) in order to obtain the lysis of
yeast cells. After 15 min of shaking at room temperature, 20 µl of
Firefly luciferase substrate (Bright Glo; Promega) were added. The
luciferase activity was assessed using a multilabel plate reader
(Mithras LB 940; Berthold Technologies, Bad Wildbad, Germany) and
normalized to OD 600 nm. The transactivation ability of the
wild-type and mutant P63 proteins was expressed as fold of
induction over empty vector for each reporter strain; the
percentage of the luciferase activity of the P63 mutants with
respect to the wild-type was also calculated.
Western blot analysis
Yeast transformants in the yLFM-P21-5′ and yLFM-PERP
strains were grown 8 h in selective medium containing 0.128%
galactose and collected by centrifugation. Cells were resuspended
in 100 µl distilled water, and 100 µl 0.2 M NaOH were then added.
The mixture was then incubated for 5 min at room temperature,
pelleted, resuspended in 50 µl SDS sample buffer (0.06 M Tris-HCl,
pH 6.8, 5% glycerol, 2% SDS, 4% β-mercaptoethanol, 0.0025%
bromophenol blue), boiled for 3 min and pelleted again (47). Ten microliters of yeast supernatant
were resolved on 4–15% Mini-Protean TGX gels and transferred to
nitrocellulose membranes by Trans-Blot Turbo Blotting System using
the Trans-Blot Transfer Pack, (Bio-Rad, Hercules, CA, USA). A
specific antibody directed against P63 (clone 4A4, sc-8431; Santa
Cruz Biotechnology, Santa Cruz, CA, USA) was diluted in 1% non-fat
dry milk dissolved in PBS-T. Phosphoglycerate kinase 1 (PGK1) was
used as a loading control (clone 22C5D8; Life Technologies; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). After washing, the
blots were incubated with the appropriate IgG horseradish
peroxidase-conjugated secondary antibody (anti-mouse IgG HRP,
A9044; Sigma-Aldrich, St. Louis, MO, USA), and immune complexes
were visualized with ECL Fast Pico (Immunological Sciences, Rome,
Italy). Chemiluminescence was analyzed by Alliance LD (Uvitec,
Cambridge, UK).
Results
TP53 and TP63, but not TP73, are
frequently mutated in cutaneous melanomas
All TP53, TP63 and TP73
mutations in cutaneous melanomas were retrieved from the cBioPortal
(http://www.cbioportal.org) (38,39).
Overall, the incidence of TP53 and TP63 mutations was
similar [15.0% (91/605) and 14.7% (89/605) for TP53 and
TP63, respectively] while the frequency of TP73
mutations [1.5% (9/605)] was significantly lower (p<0.0001).
Since some melanomas exhibited more than a single mutation, a total
of 105 TP53, 111 TP63 and 10 TP73 mutations
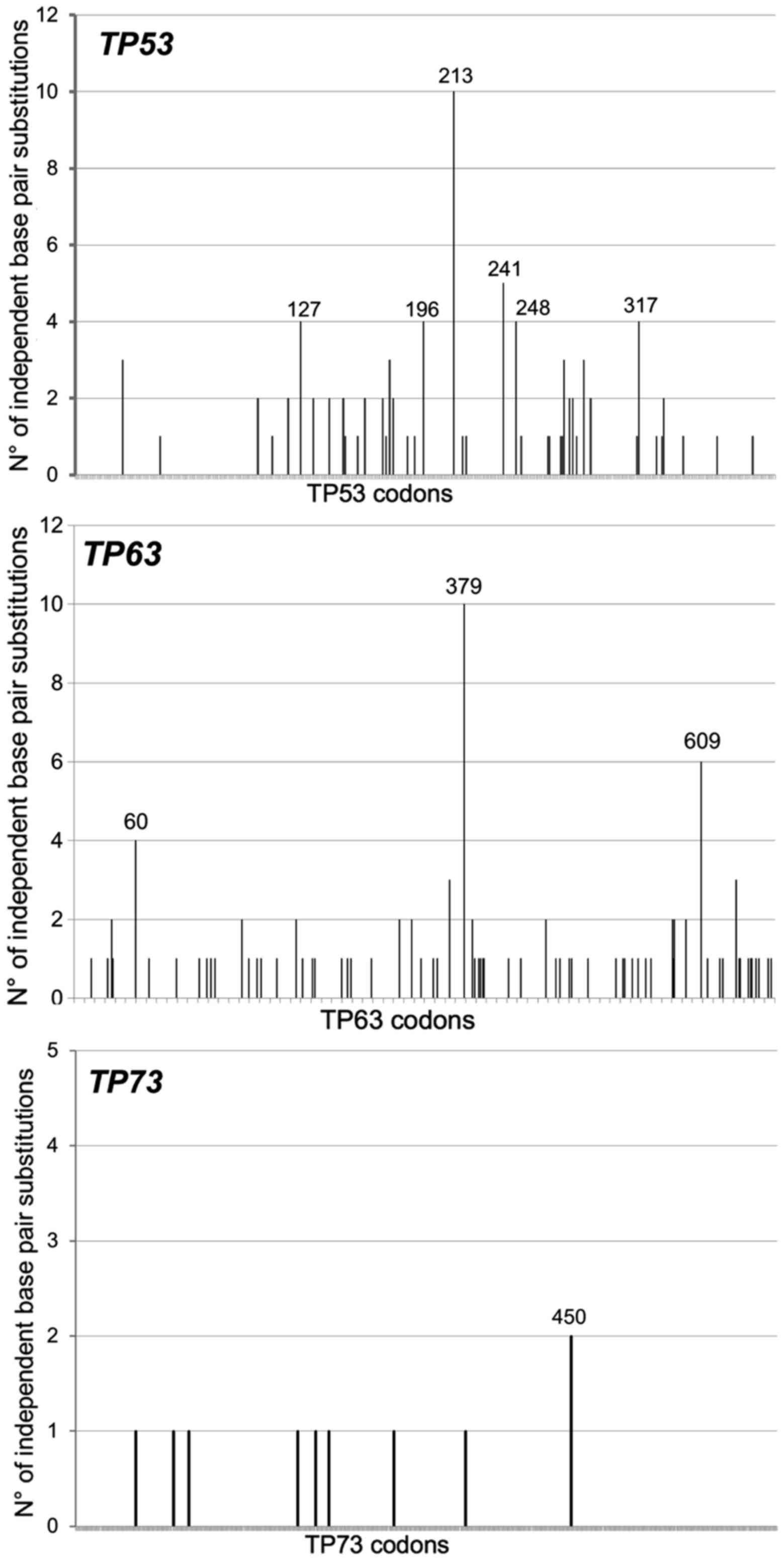
were retrieved and their molecular features analyzed (Table I). Clearly, for all three genes the
majority (>83%) of mutations were single base pair substitutions
(TP53, 88/105=83.8%; TP63, 101/111=91%; TP73,
10/10=100%), at least 80% of which were localized at adjacent
pyrimidines (PyPy). At least 50% of all base pairs substitutions
for all three genes were GC>AT transitions. Their codon
localization along each cDNA sequence is represented in Fig. 1.
 | Table I.Comparison of TP53, TP63 and
TP73 mutations in cutaneous melanomas (n=605). |
Table I.
Comparison of TP53, TP63 and
TP73 mutations in cutaneous melanomas (n=605).
| Types of
mutations | TP53 | TP63 | TP73 |
|---|
| No. of cases with
mutations | 91 | 89 | 9 |
| No. of cases with
mutations/total (%) | 91/605
(15.0)a | 89/605
(14.7)b | 9/605
(1.5)a,b |
| No. of
mutations | 105 | 111 | 10 |
| Type of
mutations |
|
|
|
|
Frameshift Del/Ins | 8 | 1 | 0 |
| Tandem
mutations | 2 | 1 | 0 |
| Splice
mutations | 7 | 8 | 0 |
| Single
bp | 88 | 101 | 10 |
|
Classes of bp, n
(%) |
|
|
|
|
AT>CG | 5 (6) | 0 (0) | 1 (10) |
|
AT>GC | 6 (7) | 4 (4) | 1 (10) |
|
AT>TA | 5 (6) | 3 (3) | 1 (10) |
|
GC>AT | 65 (73) | 92 (91) | 5 (50) |
|
GC>CG | 5 (6) | 1 (1) | 2 (20) |
|
GC>TA | 2 (2) | 1 (1) | 0 (0) |
| Localization at
CpGs, n/total (%) | 23/88 (26) | 37/101 (37) | 1/10 (10) |
| Localization at
PyPy, n/total (%) | 75/88 (85) | 98/101 (97) | 8/10 (80) |
Thus, TP53 and TP63, but not
TP73, mutations are frequently found in cutaneous melanomas
and their molecular features are reminiscent of a UV-exposure,
suggesting that TP53 and TP63 genes behave like two
‘recorders of UV-exposure’. This conclusion is indirectly supported
by the fact that among 80 patients with uveal melanomas, a form of
melanoma that should be less UV-dependent since the UV radiation
should not have the power to hit the target tissue, none of them
displayed TP53 and TP63 mutations (TCGA study)
(http://www.cbioportal.org).
The TP63 mutation fingerprint in
cutaneous melanomas is distinguishable from the one observed in
P63-associated EDs
To verify the assumption of molecular epidemiology,
namely that carcinogens leave distinguishable fingerprints, 101
germinal TP63 mutations found in patients affected by a
specific subset of EDs were retrieved from the literature (13,42,43).
Excluding a tandem mutation (1 CGC>CAA, R318Q) with no feature
of UV-induced tandem mutation, a total of 100 TP63 base pair
substitutions were analyzed and compared with the TP63
mutations found in cutaneous melanomas (Table II). The genesis of the former
mutations is most likely unrelated to UV-exposure. If the classes
of mutations were considered, the only significant difference
between the two spectra is a higher percentage of GC>AT
transitions in melanomas than in EDs (92/101; 91% vs. 76/101; 76%;
p<0.0005). However, significant and interesting differences
emerged when the localization of mutations at CpGs or PyPy sites
was considered. Indeed, significantly more TP63 mutations
were located at CpGs in EDs than in melanomas (74 vs. 37%;
p<0.0001), and vice versa, significantly more TP63
mutations were located at PyPy in melanomas than in EDs (97 vs.
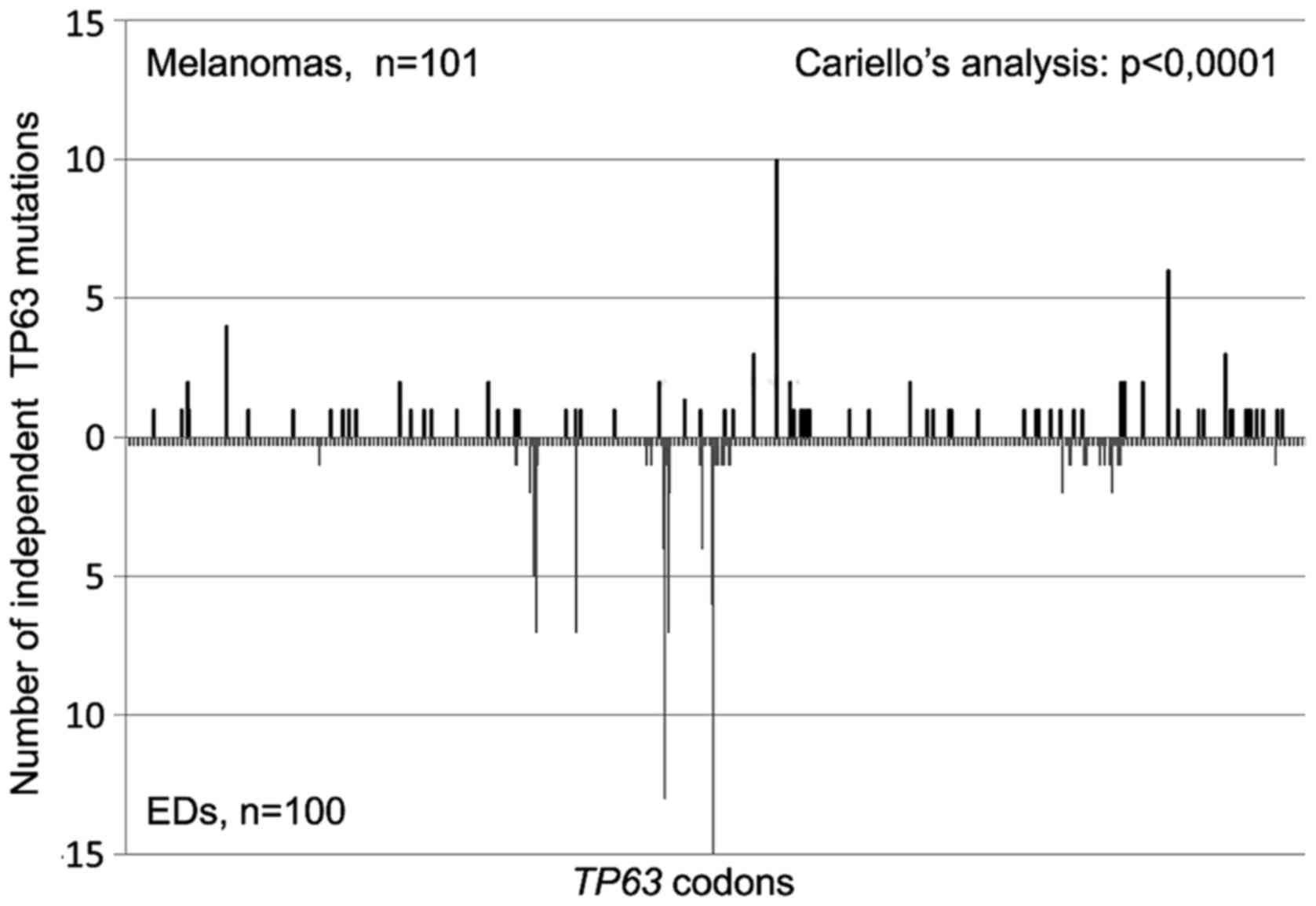
70%; p<0.0001). Finally, the two TP63 mutation spectra
appeared significantly different (p<0.0001; Fig. 2) also when the sequence distribution
of independent base pair substitutions along the TP63
nucleotide sequence was compared using the Cariello et al
program (45), which provides a
rigorous statistical test of the relatedness of two spectra at the
same locus.
| Table II.Comparison of TP63 mutations
in cutaneous melanomas and in a subset of human EDs. |
Table II.
Comparison of TP63 mutations
in cutaneous melanomas and in a subset of human EDs.
| Type of
mutations |
Melanomasa | EDsb | Melanoma vs. EDs
(P-value) |
|---|
| No. of
mutations | 111 | 101 |
|
| Type of
mutations |
|
|
|
|
Frameshift Del/Ins | 1 | 0 |
|
| Tandem
mutations | 1 | 1 |
|
| Splice
mutations | 8 | 0 |
|
| Single
bp substitutions | 101 | 100 |
|
| Classes of bp
substitutions, n (%) |
|
|
|
|
AT>CG | 0 (0) | 4 (4) | NS |
|
AT>GC | 4 (4) | 9 (9) | NS |
|
AT>TA | 3 (3) | 3 (3) | NS |
|
GC>AT | 92 (91) | 76 (76) | <0.0005 |
|
GC>CG | 1 (1) | 3 (3) | NS |
|
GC>TA | 1 (1) | 5 (5) | NS |
| Localization at
CpGs, n/total (%) | 37/101 (37) | 74/100 (74) | <0.0001 |
| Localization at
PyPy, n/total (%) | 98/101 (97) | 70/100 (70) | <0.0001 |
In conclusion, using a mutational spectrometry
approach we demonstrated that a UV fingerprint could be recognized
in the TP63 mutations found in cutaneous melanomas and that
such a mutation spectrum is distinguishable from the one observed
in EDs.
TP53 and TP63 mutations in cutaneous
melanomas: drivers, or passengers? A tentative answer from
bioinformatics prediction of the functional consequences of TP53
and TP63 mutations
Since TP53 and TP63 mutations are
relatively frequent in cutaneous melanomas a question arises. Do
these mutations play a role as potential drivers or passengers in
the tumorigenic process? To answer this question, for each mutation
we first registered the classifications (Mutation Assessor and
FATHMM-MKL) reported at the cBioPortal (http://www.cbioportal.org) (41,42).
To act as a driver, the mutations should have consequences on the
functionality of the P53/P63 proteins (i.e., amino acid
substitution should cause a change in transactivation capacity of
the mutant with respect to the wild-type protein). When the
functional consequences of mutations were evaluated using the
Mutation Assessor bioinformatics tool a significantly larger
proportion of TP53 mutations (TP53: 60/64, 93.7%;
TP63: 32/95, 33.7%; p<0.0001, Fisher's exact test) were
classified to have a medium impact on protein functionality, while
a significantly larger proportion of TP63 mutations
(TP63: 42/95, 44.2%; TP53: 2/64, 3.1%; p<0.0001
Fisher's exact test), were classified to have a neutral impact on
protein functionality (Table
III). When prediction was performed with the FATHMM-MKL
bioinformatics tool each and every single amino acid substitution
in TP63 was predicted to be pathogenic (score>0.89)
(Table III). Furthermore, no
significant difference emerged among specific mutations since
almost all TP53 and TP63 mutations were classified as
pathogenic. Therefore, independent bioinformatics tools reveal a
role of TP53 as a driver gene in cutaneous melanomas, while
for TP63 mutations the situation is less clear. However, the
abundance of TP63 mutations (similar to the one of
TP53) along with the FATHMM-MKL prediction reveal that
TP63 mutations may play some role in cutaneous
melanomas.
| Table III.Functional evaluation of amino acid
substitutions using bioinformatics tools. |
Table III.
Functional evaluation of amino acid
substitutions using bioinformatics tools.
| Type of
mutations | TP53 | TP63 | TP73 | TP53 vs.
TP63 (P-value) |
|---|
| No. of
mutations | 105 | 111 | 10 |
|
| Single bps | 88 | 101 | 10 |
|
| Missense mutations
with predicted functional consequences according to: |
|
|
|
|
|
Mutation Assessor | 64 | 95 | 9 |
|
|
Neutral | 2 | 42 | 6 | <0.0001 |
|
Low | 2 | 21 | 1 | <0.009 |
|
Medium | 60 | 32 | 2 | <0.0001 |
|
High | 0 | 0 | 0 |
|
| FATHMM
prediction | 88 | 79 | 7 |
|
|
Neutral | 2 | 0 | 3 | NS |
|
Pathogenic | 86 | 79 | 4 | NS |
From bioinformatics prediction to
experimental determination of P63 functionality: the P63 R379C
hotspot mutant protein in melanomas retains residual wild-type
function
In order to shed more light on the functional
effects of amino acid substitutions, we experimentally determined
the outcome of the substitution of the arginine at codon 379 with a
cysteine (R379C) on the transactivation ability of the P63 protein.
This mutant protein represents the most frequent amino acid
substitution observed in cutaneous melanomas (mutation hotspot).
Furthermore, while Mutation Assessor predicts that this amino acid
substitution will have a low impact on functionality, conversely
FATHMM predicts it as pathogenic. For this purpose the P63 R379C
allele was cloned into a yeast expression vector as ΔN-P63α and
TA-P63α isoforms, and their functionality was evaluated in two
yeast reporter strains containing the luciferase gene under the
control of the P63 responsive genes P21 and PERP (see materials and
methods for details).
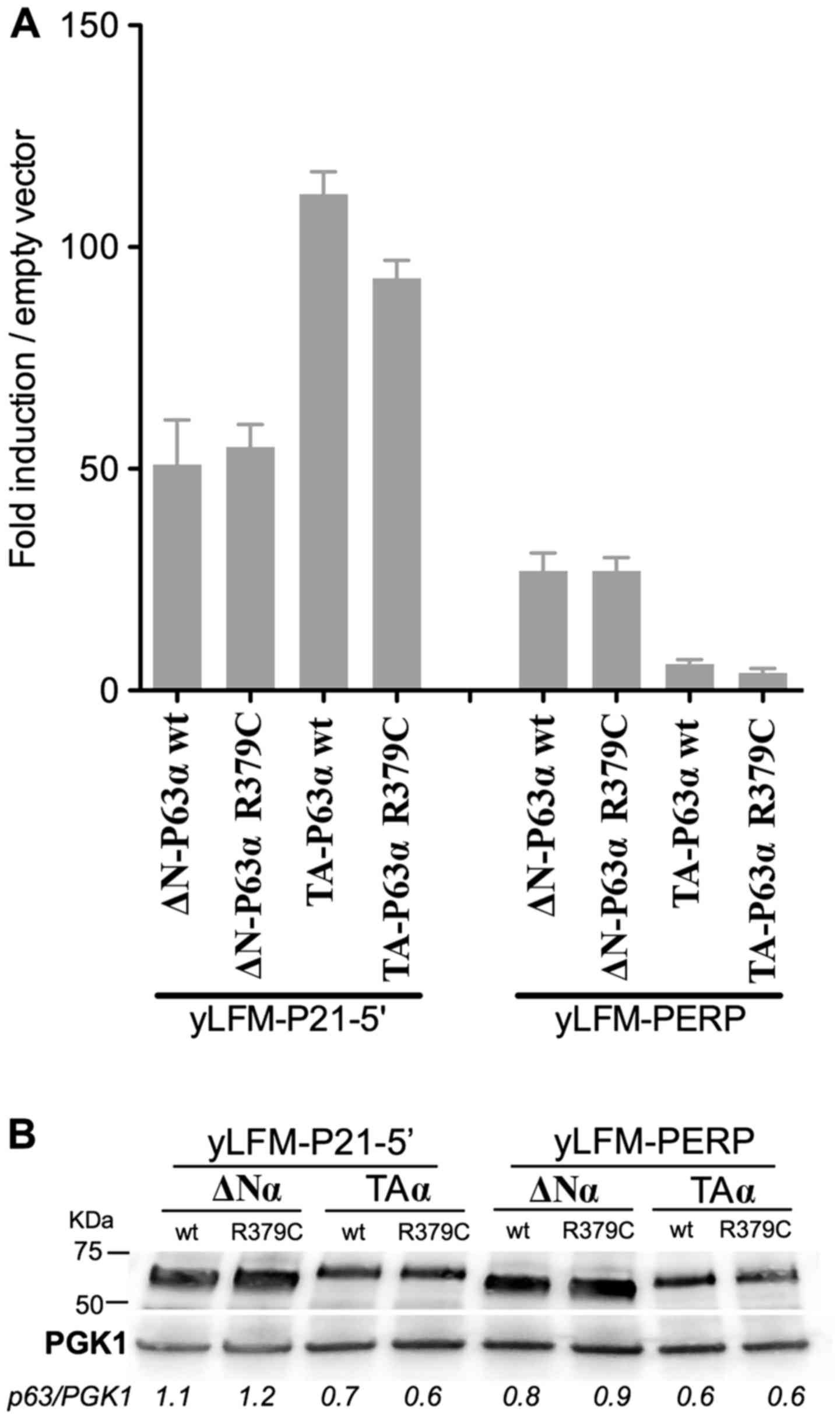
The results revealed that the R379C amino acid
substitution did not markedly affect the transactivation ability of
ΔN-P63α and TA-P63α proteins with respect to the wild-type
(Fig. 3A). Specifically, when
expressed as the ΔN-P63α isoform the R379C protein exhibited a
transactivation activity comparable to that of the wild-type P63.
When expressed as the TA-P63α isoform, the same amino acid
substitution exhibited, in both yeast strains, a decrease of
transactivation ability of ~30% with respect to the wild-type. Such
a difference in transactivation ability cannot be due to a lower
amount of mutant TA-P63α protein compared to the wild-type
counterpart, since both proteins were equally expressed (Fig. 3B). These results suggest that,
according to our yeast-based P63 functional assay (14), the R379C mutant P63 cannot be
immediately classified as a potential pathogenic. In this line it
is worth noting that this amino acid is not evolutionarily
conserved within P53 family members, suggesting the lack of a key
role in the P63 protein functionality; consequently, it is possible
to hypothesize that amino acidic substitutions at this position can
be well tolerated without markedly altering the transactivation
ability of the protein.
Discussion
NGS approaches of cancer genomes introduce a new
scenario also for cutaneous melanomas. Cutaneous melanomas were
submitted to NGS for the first time in 2010 (33). Two years later, Hodis et al
(34) discovered novel melanoma
genes and revealed that the spectrum of mutations provided evidence
for a direct mutagenic role of UV light in melanomagenesis. More
recently, a distinct sequence of genetic alterations during
melanoma progression for different melanoma subtypes was defined
(35,36). Benign lesions exclusively harbored
BRAF V600E mutation, whereas those categorized as ‘intermediate’
were enriched for NRAS mutations and additional driver mutations.
Biallelic CDKN2A inactivation emerged exclusively in invasive
melanomas, while the presence of PTEN and TP53
mutations appears to represent the transition to metastatic tumors
(36).
Only recently missense mutations in the TP63
gene were noted to be frequently and specifically found in
cutaneous melanoma (~15%) (37) in
contrast to the previous prevailing notion that TP63
mutations are seldom found in cancer. This observation prompted us
to ascertain whether all three members of the P53 family of
transcription factors were found mutated in cutaneous melanomas
(http://www.cbioportal.org) (38,39).
Notably, TP53 and TP63 were frequently mutated in
cutaneous melanomas (15.0 and 14.7%, respectively) while the
incidence of TP73 mutations (1.5%) was significantly lower
(p<0.0001).
The TP63 and TP53 mutations found in
cutaneous melanomas revealed a UV-recognizable mutation
fingerprint, high percentage of GC>AT transitions at PyPy sites,
along with a few tandem mutations (Table I). These observations strongly
suggest that TP63 and TP53 mutations in cutaneous
melanomas are UV-induced. To reinforce this suggestion the
TP63 mutations found in cutaneous melanomas were compared
with those observed at the germinal level in patients affected by a
specific subset of P63-associated disorders (EDs). Since the two
sets of mutations should have been induced by different mutagenic
events, based on the mutation spectrometry assumption, we
anticipated that the two TP63 mutation spectra would be
distinguishable. Indeed, they were significantly different. The
TP63 mutations in EDs were significantly more frequent at
CpGs sites (p<0.0001; Table
II), while those in skin melanomas were significantly more
frequent at PyPy sites (p<0.0001; Table II). Furthermore, the sequence
distribution of independent TP63 base pair substitutions
along the nucleotide sequence differed significantly in the two
spectra (p<0.0001; Fig. 2).
Altogether these results demonstrated that the TP63 mutations
observed in cutaneous melanomas are UV-specific, while those
observed in EDs are likely of spontaneous origin, with deamination
of 5′-mC at CpG sites being a likely candidate responsible of such
events.
Altogether these results suggest that, besides
TP53, TP63 mutations, but not TP73, may also
play a role in melanomagenesis. However, if this is so, it is
expected that the corresponding amino acid substitutions induced by
those mutations would show significant functional consequences on
the transcriptional activity of the P63 protein. The predictions
based on two bioinformatics tools (Mutation Assessor and FATHMM)
were rather different. While the former discriminated between the
effects of specific amino acid substitutions on the P63 protein
functionality, the latter predicted that all the mutations were
pathogenic (Table III).
Therefore, we experimentally determined the functional consequences
of the TP63 R379C mutation on the transactivation ability
with respect to the wild-type counterpart, since this mutant P63
protein was the most frequent amino acid substitution observed in
cutaneous melanomas and the two bioinformatics tools reported
different prediction for the same mutation. Using our yeast-based
P63 functional assay the R379C protein exhibited a substantial
residual activity compared to the wild-type (70–100%, depending on
the yeast reporter strain) (Fig.
3). This result is in keeping with the prediction of the
Mutation Assessor and appears to not support a major role of this
mutant P63 protein in melanomagenesis, while it is still compatible
with the TP63 gene being a recorder of UV exposure. With
regard to the TP53 hot spot mutation in melanomas, i.e.,
R213Stop, it is obligatorily classified as a loss-of-function
mutant because the mutation results in a truncated P53 protein that
is not able to oligomerize. This observation is in agreement with
the predictions of the bioinformatics tools suggesting that
TP53 mutations may play a role in melanomagenesis.
While TP53 mutations have been recognized to
play a role in the transition to metastatic tumors (36), the indications for the role of
TP63 mutations have yet to be elucidated. The fact that
TP63+/− mice developed malignant tumors at a
higher frequency with respect to the wild-type counterpart, and
that the TP53+/−TP63+/− compound
mutant mice developed a more severe phenotype (i.e., higher tumor
burden and more metastases) compared to the
TP53+/− mice, reveals that TP63 is a
tumor/metastasis suppressor gene independent of P53 (15). Thus, TP63 mutations that
inactivate the protein function likely contribute to a carcinogenic
process. However, besides their relative frequency, TP63
mutations in cutaneous melanomas possibly represent a record of
exposure (passenger mutations) rather than driver mutations. This
conclusion is supported by additional arguments. Firstly, through
an extensive study that considers all type of tumors (48) TP53 is present among the 125
drivers genes recognized, while TP63 is not. Secondly,
TP63 and TP53 mutations have a significant tendency
towards co-occurrence (p<0.001) (http://www.cbioportal.org) in melanomas, a situation
that does not support the hypothesis that both mutations may play a
major role during melanomagenesis. Thirdly, a possible mechanism
through which the mutant P53 protein may facilitate the transition
towards metastatic melanomas may be via interference with the
antimetastatic function of the wild-type P63. Adorno et al
(49) proposed such a mechanism in
breast cancer cells, revealing that TGF-β-dependent cell migration,
invasion and metastasis were empowered by the mutant P53 protein
and opposed by P63: a mutant P53/P63 protein complex appeared
assembled with SMAD proteins which served as an essential platform.
Notably, the TGF-β/SMAD pathway appears to be active even in
melanomas (50). Thus, the mere
presence of a mutant P53 protein may be sufficient to antagonize
the P63 function without the need to mutate TP63.
It should also be considered that
TP53/TP63 mutations may also play different roles
according to the phase of cancer development in which they occur.
UV-induced TP53/TP63 mutations at PyPy sites are an
early event in UV-exposed skin that it is still devoid of any
cancer-like phenotype. However, the presence of these mutations
with their effects may allow mutated cells to escape apoptosis,
thus progressing towards more advanced stages of the carcinogenesis
process. In this view, TP53/TP63 may be considered
driver mutations of the early carcinogenesis steps. In more
advanced cancer stages or in full blown cancer the burden of other
mutations prevails, thus giving cancer an aggressive behavior
resulting in chemoresistance, metastatisation and decreased
survival. Accordingly, TP53/TP63 behave as passenger
mutations in these advanced cancer stages.
Two other aspects contribute to maintain the role
of TP63 mutations in cutaneous melanomas uncertain. The
first is related to a recent study by Matin et al (51) who demonstrated that the two
wild-type isoforms of P63, namely TA-P63 and ΔN-P63, play an
anti-apoptotic role in melanoma and confer chemoresistance, even in
the absence of TP53 mutations. In response to DNA damage,
P63 is stabilized in the nuclei and translocates to the
mitochondria where it supresses apoptosis through a direct and
indirect inhibitory function on P53-dependent pro-apoptotic
effectors. In this scenario, the selection of TP63 mutations
during melanomagenesis appears to be unnecessary or even
deleterious for tumor growth, once again pointing toward a
passenger role rather than a driver role for such mutations.
Moreover, this observation highlights functions of the P63 protein
that are not associated to the key role as nuclear transcription
factor. The second aspect is related to the power of the NGS; in
fact, while the Sanger sequencing imposed a technical threshold of
detection (i.e., a mutation could be identified if present in ~20%
of the alleles), NGS can accurately detect mutations at an
abundance in the order of 1% (and even lower). The presence of
every mutation identified with NGS facilitate the identification of
mutational signatures. However, the biological impact of a mutation
that is 1 or 20% abundant may be different on tumor behavior. In
view of understanding which mutations are really important for the
carcinogenic process, an interesting way would be to submit to NGS
consecutive samples, spaced in time, and to verify what happens to
the abundance of specific molecular events in the progression of
the tumor. This is relatively easy for some hematological
malignancies (e.g., chronic lymphocytic leukemia, CLL); however
this appears to be very difficult, or even impossible, for others
kind of tumors. In CLL, Rossi et al (52) revealed that TP53 mutations,
even in <1% of the CLL clone, can be selected upon treatment and
therefore should be taken into account when defining therapy
protocols.
We conclude that TP63 mutations are frequent
in cutaneous melanoma and support UV etiology, however they do not
have a clear role in melanomagenesis. The role of TP63
mutations changes according to the stage of melanoma carcinogenesis
when they occur being drivers in early stages and passengers in
late stages. In the absence of experimental results on gene
mutation functionality, bioinformatics tools are often used to
predict the effect of a single amino acid substitution on the
function of the protein under study. Our results clearly revealed
that the experimental data obtained with our yeast-based functional
assay may be useful to elucidate the role of mutations on P63
function.
Acknowledgements
This study was partially supported by the Italian
Association for Cancer Research, AIRC (IG no. 5506 to G.F., IG no.
15460 to P.G.), by Compagnia S. Paolo, Turin, Italy (Project
2012.1590), and by the Ministero della Salute, Fondi 5×1000 2013,
Fondi Ricerca Corrente 2016.
References
|
1
|
Lane DP and Crawford LV: T antigen is
bound to a host protein in SV40-transformed cells. Nature.
278:261–263. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaghad M, Bonnet H, Yang A, Creancier L,
Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, et al:
Monoallelically expressed gene related to p53 at 1p36, a region
frequently deleted in neuroblastoma and other human cancers. Cell.
90:809–819. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang A, Kaghad M, Wang Y, Gillett E,
Fleming MD, Dötsch V, Andrews NC, Caput D and McKeon F: p63, a p53
homolog at 3q27-29, encodes multiple products with transactivating,
death-inducing, and dominant-negative activities. Mol Cell.
2:305–316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bisio A, Ciribilli Y, Fronza G, Inga A and
Monti P: TP53 mutants in the tower of babel of cancer progression.
Hum Mutat. 35:689–701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Collavin L, Lunardi A and Del Sal G:
p53-family proteins and their regulators: Hubs and spokes in tumor
suppression. Cell Death Differ. 17:901–911. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wei J, Zaika E and Zaika A: p53 family:
Role of protein isoforms in human cancer. J Nucleic Acids.
2012:6873592012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malkin D: Li-fraumeni syndrome. Genes
Cancer. 2:475–484. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Monti P, Ciribilli Y, Jordan J, Menichini
P, Umbach DM, Resnick MA, Luzzatto L, Inga A and Fronza G:
Transcriptional functionality of germ line p53 mutants influences
cancer phenotype. Clin Cancer Res. 13:3789–3795. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Monti P, Perfumo C, Bisio A, Ciribilli Y,
Menichini P, Russo D, Umbach DM, Resnick MA, Inga A and Fronza G:
Dominant-negative features of mutant TP53 in germline carriers have
limited impact on cancer outcomes. Mol Cancer Res. 9:271–279. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang A, Walker N, Bronson R, Kaghad M,
Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, et
al: p73-deficient mice have neurological, pheromonal and
inflammatory defects but lack spontaneous tumours. Nature.
404:99–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mills AA, Zheng B, Wang XJ, Vogel H, Roop
DR and Bradley A: p63 is a p53 homologue required for limb and
epidermal morphogenesis. Nature. 398:708–713. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rufini A, Agostini M, Grespi F, Tomasini
R, Sayan BS, Niklison-Chirou MV, Conforti F, Velletri T, Mastino A,
Mak TW, et al: p73 in Cancer. Genes Cancer. 2:491–502. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rinne T, Brunner HG and van Bokhoven H:
p63-associated disorders. Cell Cycle. 6:262–268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Monti P, Russo D, Bocciardi R, Foggetti G,
Menichini P, Divizia MT, Lerone M, Graziano C, Wischmeijer A,
Viadiu H, et al: EEC- and ADULT-associated TP63 mutations exhibit
functional heterogeneity toward P63 responsive sequences. Hum
Mutat. 34:894–904. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Flores ER, Sengupta S, Miller JB, Newman
JJ, Bronson R, Crowley D, Yang A, McKeon F and Jacks T: Tumor
predisposition in mice mutant for p63 and p73: Evidence for broader
tumor suppressor functions for the p53 family. Cancer Cell.
7:363–373. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jacks T, Remington L, Williams BO, Schmitt
EM, Halachmi S, Bronson RT and Weinberg RA: Tumor spectrum analysis
in p53-mutant mice. Curr Biol. 4:1–7. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vogelstein B and Kinzler KW: Carcinogens
leave fingerprints. Nature. 355:209–210. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harris CC: p53: At the crossroads of
molecular carcinogenesis and risk assessment. Science.
262:1980–1981. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fronza G, Inga A, Monti P, Scott G,
Campomenosi P, Menichini P, Ottaggio L, Viaggi S, Burns PA, Gold B,
et al: The yeast p53 functional assay: A new tool for molecular
epidemiology. Hopes and facts. Mutat Res. 462:293–301. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leroy B, Fournier JL, Ishioka C, Monti P,
Inga A, Fronza G and Soussi T: The TP53 website: An integrative
resource centre for the TP53 mutation database and TP53 mutant
analysis. Nucleic Acids Res. 41(D1): D962–D969. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Greenblatt MS, Bennett WP, Hollstein M and
Harris CC: Mutations in the p53 tumor suppressor gene: Clues to
cancer etiology and molecular pathogenesis. Cancer Res.
54:4855–4878. 1994.PubMed/NCBI
|
|
22
|
International Agency for Research on
Cancer (IARC): IARC Monographs on the Evaluation of Carcinogenic
Risk of Chemicals to Humans: Solar and Ultraviolet Radiation. 55.
IARC; Lyon, France: 1992, http://monographs.iarc.fr/ENG/Monographs/vol55
|
|
23
|
Clingen PH, Arlett CF, Roza L, Mori T,
Nikaido O and Green MH: Induction of cyclobutane pyrimidine dimers,
pyrimidine(6–4)pyrimidone photoproducts, and Dewar valence isomers
by natural sunlight in normal human mononuclear cells. Cancer Res.
55:2245–2248. 1995.PubMed/NCBI
|
|
24
|
Cadet J, Sage E and Douki T: Ultraviolet
radiation-mediated damage to cellular DNA. Mutat Res. 571:3–17.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parmigiani G, Boca S, Lin J, Kinzler KW,
Velculescu V and Vogelstein B: Design and analysis issues in
genome-wide somatic mutation studies of cancer. Genomics. 93:17–21.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carter H, Chen S, Isik L, Tyekucheva S,
Velculescu VE, Kinzler KW, Vogelstein B and Karchin R:
Cancer-specific high-throughput annotation of somatic mutations:
Computational prediction of driver missense mutations. Cancer Res.
69:6660–6667. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Youn A and Simon R: Identifying cancer
driver genes in tumor genome sequencing studies. Bioinformatics.
27:175–181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vogelstein B and Kinzler KW: The path to
cancer - three strikes and you're out. N Engl J Med. 373:1895–1898.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tomasetti C, Marchionni L, Nowak MA,
Parmigiani G and Vogelstein B: Only three driver gene mutations are
required for the development of lung and colorectal cancers. Proc
Natl Acad Sci USA. 112:118–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miller AJ and Mihm MC Jr: Melanoma. N Engl
J Med. 355:51–65. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Marks R: Epidemiology of melanoma. Clin
Exp Dermatol. 25:459–463. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pleasance ED, Cheetham RK, Stephens PJ,
McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR,
Bignell GR, et al: A comprehensive catalogue of somatic mutations
from a human cancer genome. Nature. 463:191–196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hodis E, Watson IR, Kryukov GV, Arold ST,
Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C,
et al: A landscape of driver mutations in melanoma. Cell.
150:251–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shain AH, Yeh I, Kovalyshyn I, Sriharan A,
Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al:
The genetic evolution of melanoma from precursor lesions. N Engl J
Med. 373:1926–1936. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shain AH and Bastian BC: From melanocytes
to melanomas. Nat Rev Cancer. 16:345–358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Romano RA and Sinha S: Family matters:
Sibling rivalry and bonding between p53 and p63 in cancer. Exp
Dermatol. 23:238–239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Berger MF, Hodis E, Heffernan TP, Deribe
YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E,
Ghosh P, et al: Melanoma genome sequencing reveals frequent PREX2
mutations. Nature. 485:502–506. 2012.PubMed/NCBI
|
|
41
|
Krauthammer M, Kong Y, Ha BH, Evans P,
Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et
al: Exome sequencing identifies recurrent somatic RAC1 mutations in
melanoma. Nat Genet. 44:1006–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rinne T, Bolat E, Meijer R, Scheffer H and
van Bokhoven H: Spectrum of p63 mutations in a selected patient
cohort affected with ankyloblepharon-ectodermal defects-cleft
lip/palate syndrome (AEC). Am J Med Genet A. 149A:1948–1951. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Clements SE, Techanukul T, Coman D,
Mellerio JE and McGrath JA: Molecular basis of EEC (ectrodactyly,
ectodermal dysplasia, clefting) syndrome: Five new mutations in the
DNA-binding domain of the TP63 gene and genotype-phenotype
correlation. Br J Dermatol. 162:201–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Adams WT and Skopek TR: Statistical test
for the comparison of samples from mutational spectra. J Mol Biol.
194:391–396. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cariello NF, Piegorsch WW, Adams WT and
Skopek TR: Computer program for the analysis of mutational spectra:
Application to p53 mutations. Carcinogenesis. 15:2281–2285. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Andreotti V, Ciribilli Y, Monti P, Bisio
A, Lion M, Jordan J, Fronza G, Menichini P, Resnick MA and Inga A:
p53 transactivation and the impact of mutations, cofactors and
small molecules using a simplified yeast-based screening system.
PLoS One. 6:e206432011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kushnirov VV: Rapid and reliable protein
extraction from yeast. Yeast. 16:857–860. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Adorno M, Cordenonsi M, Montagner M,
Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo
V, et al: A Mutant-p53/Smad complex opposes p63 to empower
TGFbeta-induced metastasis. Cell. 137:87–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Perrot CY, Javelaud D and Mauviel A:
Insights into the transforming growth factor-β signaling pathway in
cutaneous melanoma. Ann Dermatol. 25:135–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Matin RN, Chikh A, Chong SL, Mesher D,
Graf M, Sanza' P, Senatore V, Scatolini M, Moretti F, Leigh IM, et
al: p63 is an alternative p53 repressor in melanoma that confers
chemoresistance and a poor prognosis. J Exp Med. 210:581–603. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rossi D, Khiabanian H, Spina V, Ciardullo
C, Bruscaggin A, Famà R, Rasi S, Monti S, Deambrogi C, De Paoli L,
et al: Clinical impact of small TP53 mutated subclones in chronic
lymphocytic leukemia. Blood. 123:2139–2147. 2014. View Article : Google Scholar : PubMed/NCBI
|