Introduction
Camptothecin (CPT) is a cytotoxic quinoline alkaloid
isolated from Camptotheca acuminta, a type of tree natively
growing in China, which was discovered in the 1960s (1). The antitumor activity of CPT depends
on a highly specific inhibition of Topo-1. This activity is
achieved by docking at the enzyme-DNA interface to stabilize the
formation of Topo-1-DNA cleavable complexes, thus prohibiting DNA
strand religation. Once the stable form of the cleavable complex is
broken by some mechanism, such as replication or transcription
caused by some cytotoxic drugs, the breakage of the Topo-1-linked
DNA single-strand can cause DNA double-strand breaks (DSBs)
(2). Subsequently, these DSBs will
trigger a DNA damage response to activate serine-threonine kinases
to drive the ATM-CHK2- and ATR-CHK1-mediated checkpoint pathways as
well as H2AX phosphorylation to arrest the cell cycle at the
G1/S and G2/M phases (3). The clinical application of CPT is
limited due to its low solubility as well as serious and
unfathomable side-effects (4,5). To
overcome these drawbacks, several CPT derivatives have been
developed to date, including topotecan (9-dimethyl amino-10-hydroxy
camptothecin; TPT) and irinotecan
(7-ethyl-10-[4-(1-piperidino)-1-piperidino]
carbonyloxycamptothecin; CPT-11) (6,7). The
US Food and Drug Administration has approved these CPT derivatives
for ovarian and colon cancer treatment (8,9).
Furthermore, their anticancer potential on other tumors also has
been reported, such as lung (7,10),
breast (11), pancreatic cancer
(12), lymphoma (13), glioma (14) and leukemia (15).
SN38, the prodrug of CPT-11, is approximately 100-
to 1,000-fold more cytotoxic than CPT-11 (16). CPT-11 plays an antitumor role in
vivo through the release of its active form SN38 by liver
carboxylesterase. However, there are significant individual
differences among the antitumor activity of CPT-11 due to its low
enzymatic conversion rate in vivo and its uncertain
pharmacokinetic properties among individuals. In addition, the
CPT-11 prodrug group (4-piperidinyl piperidine) causes AchE
inhibition, which easily leads to acetylcholine syndrome, resulting
in early severe diarrhea and other side-effects (10).
In order to overcome the above drawbacks of CPT-11,
scientists have made several attempts to improve it (10,17–19).
In the present study, we designed and synthesized a novel
10-hydroxy CPT prodrug with a high efficiency and low toxicity
(20,21). Its cytotoxic activity, side-effects,
antitumor activity and possible mechanism were analyzed in multiple
assays. The results demonstrated that ZBH-ZM-06 has optimal
antitumor properties and fewer side-effects than those of CPT-11
and SN38.
Materials and methods
Derivative of CPT
The new 10-hydroxy CPT prodrug, ZBH-ZM-06, was
designed and synthesized by the Institute of Pharmacology and
Toxicology Academy of Military Medical Sciences (20). By using a linear amino acid as a
linker, the N-terminal amino acid was ligated with 10-OH of SN38
through a urethane bond. Next, through removal of the benzyl
protecting group by catalytic hydrogenation, a carboxyl group was
liberated. The selected N-methylpiperazine, which has good
biocompatibility, was conjugated with the free carboxyl group of
the amino acid through an amide bond. The basic nitrogen atom of
N-methylpiperazine can form a salt with the carboxyl group
to improve the water solubility of the compound.
CPT-11 and SN38 were also provided by the Institute
of Pharmacology and Toxicology Academy of Military Medical
Sciences. All of the compounds were dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich, St. Louis, MO, USA) at 10 mmol/l as a stock
reagent. Further dilutions were made with Iscoves modified
Dulbeccos medium (IMDM; Life Technologies, Grand Island, NY, USA)
at the appropriate concentrations and stored at −20°C.
AchE inhibition assay and stability
test
The AchE inhibition test by ZBH-ZM-06 was performed
as previously described (20). The
stability of ZBH-ZM-06 was analyzed at 1, 2, 4, 8 and 12 h in
phosphate-buffered saline (PBS; pH 7.4 and pH 5.0) by
high-performance liquid chromatography (HPLC) with a C18 analytical
column, as previously described (20).
Cell culture and cytotoxicity
assay
Twelve human tumor cell lines, including SW1116,
SAOS-2, A549, SGC-7901, 7860, HeLa, SK-OV-3, K562, NCI-H446, A375,
MCF-7, SMMC-7721 and NCM460, a normal human colon mucosal
epithelial cell line, were incubated with IMDM or RPMI-1640 medium
(Gibco, Carlsbad, CA, USA) supplemented with 10% fetal calf serum
(FCS), 100 U/ml penicillin, 2 mmol/l glutamine, and 100 µg/ml
streptomycin at 37°C in a humidified atmosphere incubator
containing 5% CO2. Cells were harvested during the
logarithmic growth phase and plated in 96-well plates at
2.5×103 cells/well in 100 µl of medium. After incubation
for 24 h, 0.0032 µmol/l to 50 µmol/l ZBH-ZM-06, CPT-11 or SN38 was
added to the indicated plate. The cellular viability was determined
by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) assay 72 h later (22). The
absorbance at 490 nm was detected by a microplate reader. The
half-maximal inhibition concentration (IC50) values of
the analyzed drugs were calculated. Each experiment was repeated at
least three times.
DNA relaxation assay
To evaluate the effects of ZBH-ZM-06 and CPT-11 on
DNA relaxation, the TopoGEN Topoisomerase 1 Drug Screening kit
(TopoGEN, Inc., Port Orange, FL, USA) was employed, according to
the manufacturers instructions. Closed loop superhelix plasmid DNA
(pHOT1) was co-incubated with recombinant wild-type human Topo-1 (2
U, TG2005H-RC1; TopoGEN) at 37°C for 30 min in the presence or
absence of the drugs in Topo-1 reaction buffer. Reactions were
quenched by incubation with sodium dodecyl sulfate (1%) and
proteinase K (50 µg/ml) for 15 min at 37°C. The DNA samples were
then analyzed by electrophorese on 1% agarose gel containing 1
µg/ml ethidium bromide. Gels were visualized and photographed under
ultraviolet light. Each experiment was performed in triplicate.
Cell cycle analysis
K562, SK-OV-3 and SW1116 cells were seeded in 6-well
plates at 5×105/well in FCS-free IMDM. After incubation
for 24 h, the cells were exposed to ZBH-ZM-06, CPT-11, or SN38 at
10 µmol/l in IMDM containing 10% FCS and harvested after 24, 48 and
72 h, respectively. The cells were stained with the Coulter DNA
PREP™ reagents kit (Beckman Coulter, Brea, CA, USA), according to
the manufacturers protocol. The cell cycle was analyzed by flow
cytometry using a Coulter EPICS XL-MCL instrument. Data were
analyzed using MultiCycle 32-bit Version Software (Phoenix Flow
Systems, San Diego, CA, USA).
Measurement of tumor cell
apoptosis
The FITC Annexin V Apoptosis Detection kit I (BD
Biosciences, San Diego, CA, USA) and the FITC Active Caspase-3
Apoptosis kit (550480; BD Biosciences) were employed for the
measurement of cell apoptosis. K562, SK-OV-3 and SW1116 cells
(5×105) were seeded on 6-well plates and cultured
overnight. The next day, 10 µmol/l ZBH-ZM-06, CPT-11, or SN38 was
added for the indicated time (24, 48 and 72 h). After treatment,
the cells were harvested, washed and resuspended in 100 µl of
binding buffer containing 5 µl of Annexin V-FITC and 5 µl of
propidium iodide (PI), or stained with FITC-conjugated cleaved
caspase-3 antibody for 15–20 min in the dark at 20°C. Next, viable,
apoptotic, necrotic cells, and activated caspase-3+
cells were analyzed by an Epics XL/MCL flow cytometer.
Western blot analysis
SW1116 cells were incubated with 10 µmol/l
ZBH-ZM-06, CPT-11, or SN38 for 72 h. The cell lysate preparation
and immunoblot analysis were performed as previously described
(21).
Statistical analysis
Experimental data were analyzed by the t-test,
one-way analysis of variance, and chi-squared test with SPSS 23.0
software (version 23.0; SPSS, Inc., Chicago, IL, USA). Data were
expressed as the mean ± standard deviation (SD) using a minimum of
triplicate determinations. P<0.05 was considered to be
statistically significant.
Results
Decreased AchE inhibitory activity of
ZBH-ZM-06
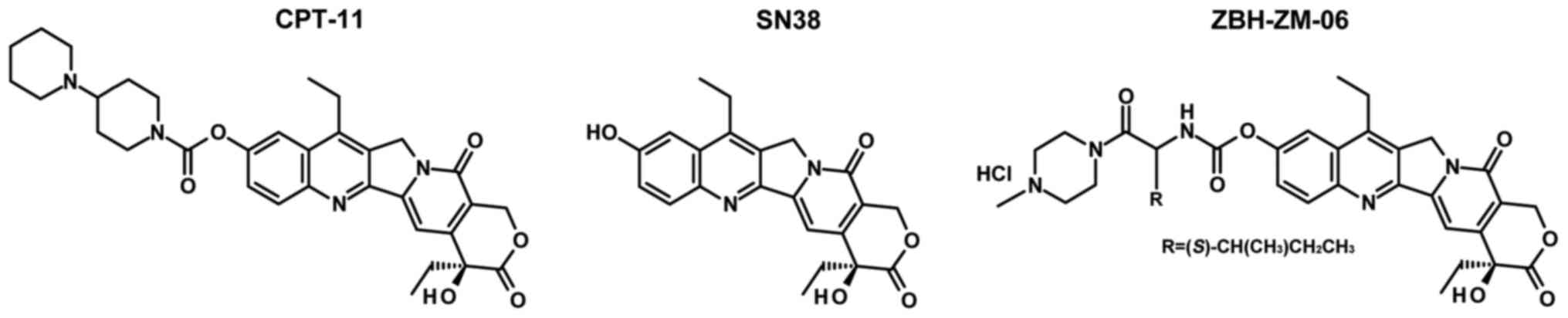
The molecular structures of CPT-11, SN38 and
ZBH-ZM-06 are shown in Fig. 1.
CPT-11 is a potent inhibitor of AchE and can cause acute
cholinergic diarrhea. In order to test whether ZBH-ZM-06 causes
this side-effect, the inhibitory activity of ZBH-ZM-06 on AchE was
detected. Obviously, ZBH-ZM-06 revealed a lower potential to
inhibit AchE compared with CPT-11 at different concentrations
(Table I). This result indicates
that the toxic side-effect of AchE inhibition of ZBH-ZM-06 is less
than that of CPT-11.
 | Table I.The AchE inhibitory activity of
ZBH-ZM-06 and CPT-11. |
Table I.
The AchE inhibitory activity of
ZBH-ZM-06 and CPT-11.
|
| Inhibition (%) |
|---|
|
|
|
|---|
|
| 0.1 µM | 1 µM | 10 µM | 100 µM |
|---|
| ZBH-ZM-06 | 4.9 | 8.4 | 12.5 | 44.2 |
| CPT-11 | 45.7 | 78.9 | 96.5 | – |
ZBH-ZM-06 can fully release the active
ingredient SN38 under physiological pH conditions
The stability of ZBH-ZM-06 was investigated in
phosphate-buffered saline (PBS) by HPLC using a C18 analytical
column. The mobile phase consisted of water and acetonitrile at a
ratio of 70:30, respectively, containing 1% trifluoroacetic acid.
ZBH-ZM-06 and its metabolite SN38 were separated at a flow rate of
0.8 ml/min and detected at a wavelength of 370 nm. ZBH-ZM-06 was
dissolved in PBS (0.2 mg/ml, pH 7.4 and pH 5.0), incubated at 37°C
and analyzed at 0, 1, 2, 4, 8 and 12 h (20). The results are summarized in
Table II. ZBH-ZM-06 showed a
pH-dependent stability. ZBH-ZM-06 was relatively stable, as shown
by the finding that 92.8% still remained after 12 h at pH 5.0,
37°C. However, at pH 7.4, only 4.9% remained after incubation for 4
h. The antitumor activity of CPT-11 is achieved through the release
of its active ingredient, SN38 (23), by liver carboxylesterase.
Nevertheless, there are significant individual differences in the
antitumor activity of CPT-11 due to its low enzymatic conversion
rate in vivo and its uncertain pharmacokinetic properties
among individuals. ZBH-ZM-06 can fully release the active
ingredient, SN38, under physiological pH (pH 7.4); therefore, this
non-liver release-dependent property may improve the drug
efficacy.
| Table II.The chemical stability of ZBH-ZM-06
as indicated by the percentage of remaining compound after
incubation for various times and pH values. |
Table II.
The chemical stability of ZBH-ZM-06
as indicated by the percentage of remaining compound after
incubation for various times and pH values.
|
| 0 h | 1 h | 2 h | 4 h | 8 h | 12 h |
|---|
| pH 7.4 | 98.1 | 38.8 | 17.7 | 4.9 | – | – |
| pH 5.0 | 99.8 | – | – | 97.8 | 95.0 | 92.8 |
Inhibition of tumor cell viability by
ZBH-ZM-06
The inhibitory activity of ZBH-ZM-06 on cellular
viability was detected by MTT assay in 12 tumor cell lines. The
NCM460 cell line was used as a normal cell line control. The
IC50 values are summarized in Table III. The most sensitive cell lines
were SW1116, SAOS-2, HeLa, SK-OV-3, K562 and A375. ZBH-ZM-06 showed
weak inhibitory activity against NCM460 cells.
| Table III.IC50 values of ZBH-ZM-06,
CPT-11 and SN38 in tumor cell lines and NCM460 cells. |
Table III.
IC50 values of ZBH-ZM-06,
CPT-11 and SN38 in tumor cell lines and NCM460 cells.
|
|
| IC50
(µmol/l) | P-value |
|---|
|
|
|
|
|
|---|
| No. | Cell line | ZBH-ZM-06 | CPT-11 | SN38 | vs. CPT-11 | vs. SN38 |
|---|
| 1 | SW1116 | 0.0679±0.0588 | 2.7742±0.7676 |
862.2826±151.3390 | 0.0563 | 0.0045 |
| 2 | SAOS-2 | 0.0000±0.0000 | 0.3781±0.1157 | 2.2989±0.5484 | 0.0217 | 0.0002 |
| 3 | A549 | 0.9331±0.9234 | 3.7853±0.2473 |
683.9418±255.6370 | 0.2747 | 0.0072 |
| 4 | SGC-7901 | 1.3539±0.4708 | 19.5366±7.5020 | 13.2479±6.2612 | 0.0642 | 0.1331 |
| 5 | 7860 | 0.0369±0.1414 | 0.4515±0.1000 | 4.9959±2.5830 | 0.3400 | 0.0087 |
| 6 | HeLa | 0.3025±0.1996 |
68.4969±21.7217 |
7630.3091±6997.9112 | 0.0037 | 0.0054 |
| 7 | SK-OV-3 | 0.0475±0.0404 | 3.3985±1.2949 |
88.4301±61.3723 | 0.0256 | 0.0004 |
| 8 | K562 | 0.0000±0.0000 | 0.7551±0.3578 | 1.2429±0.1687 | 0.0298 | 0.0059 |
| 9 | NCI-H446 | 0.3710±0.3924 | 4.5701±4.6985 | 26.0500±8.1501 | 0.1056 | 0.0053 |
| 10 | A375 | 0.0000±0.0000 | 1.1970±0.7749 | 7.3358±1.3535 | 0.0169 | 0.0008 |
| 11 | MCF-7 | 5.3157±1.3016 | 1.4336±0.3022 | 3.3145±0.0007 | 0.5764 | 0.1083 |
| 12 | SMMC-7721 | 1.7157±2.2853 | 12.2966±6.0580 | 0.0028±0.0016 | 0.7424 | 0.4478 |
| 13 | NCM460 |
189.1652±49.3190 |
31.8162±10.9849 | 22.7732±6.1773 | 0.8134 | 0.9053 |
ZBH-ZM-06 inhibited the relaxation of
supercoiled DNA
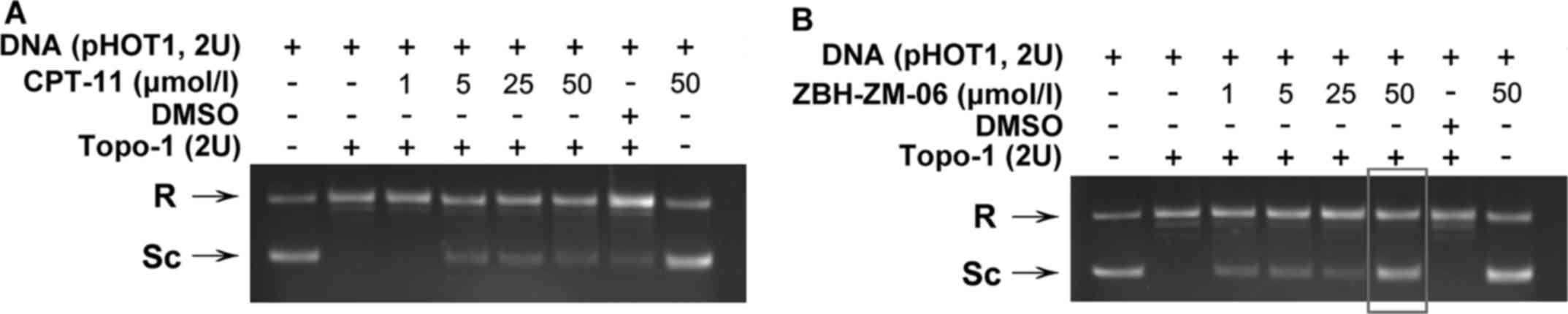
To investigate whether ZBH-ZM-06 inhibits cell
growth through the inhibition of Topo-1 activity, a DNA relaxation
assay was performed in the presence of ZBH-ZM-06 or CPT-11 on
supercoiled plasmid pHOT1 in vitro (Fig. 2). The results demonstrated that
supercoiled DNA was relaxed by Topo-1 in the absence of drug (lane
2 of Fig. 2). In contrast,
incubation with ZBH-ZM-06 or CPT-11 inhibited the relaxation of
supercoiled DNA, as demonstrated by the increased intensity of the
band corresponding to the supercoiled plasmid DNA. Furthermore, the
inhibition of ZBH-ZM-06 occurred at a concentration as low as 1
µmol/l, compared to CPT-11 (at 5 µmol/l), indicating that ZBH-ZM-06
has a stronger inhibitory activity against Topo-1 than CPT-11.
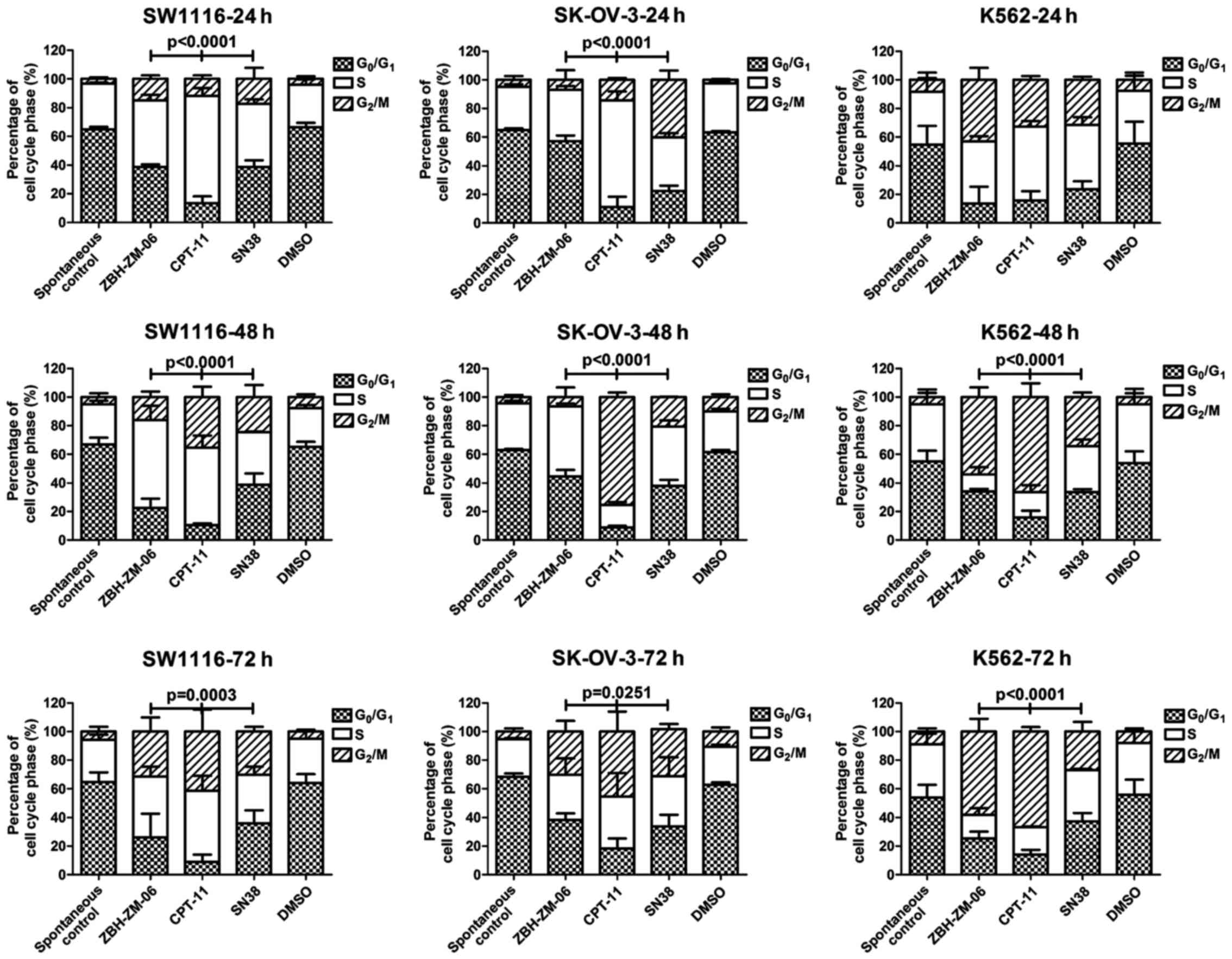
Different patterns of tumor cell cycle
arrest induced by ZBH-ZM-06, CPT-11 or SN38
The cell cycle arrest by ZBH-ZM-06, CPT-11 or SN38
at 24, 48 and 72 h was analyzed (Fig.
3). The cell cycle arresting patterns at the S and
G2/M phases induced by ZBH-ZM-06 were significantly
different from those by CPT-11 or SN38 (P<0.0251–0.0001), except
for K562 cells treated for 24 h, which did not show a significant
difference among the three groups.
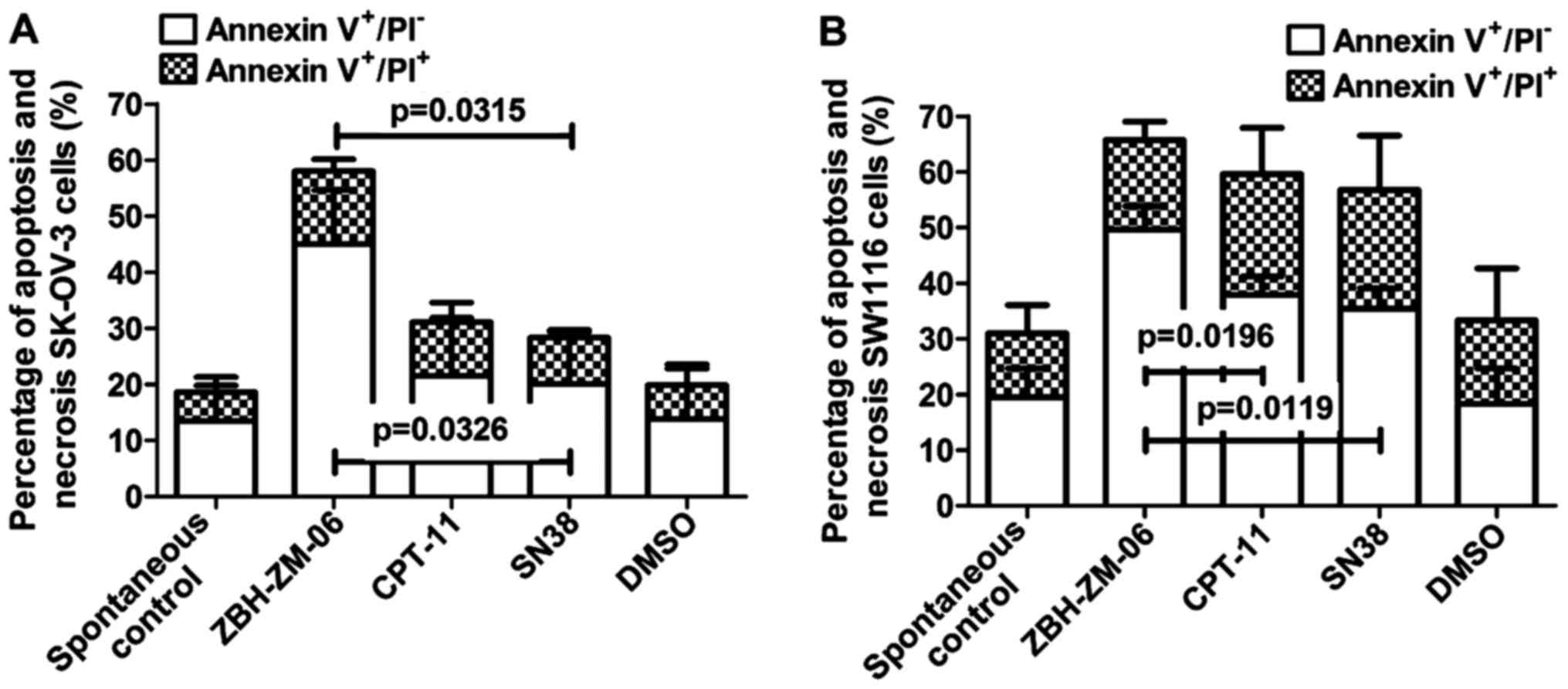
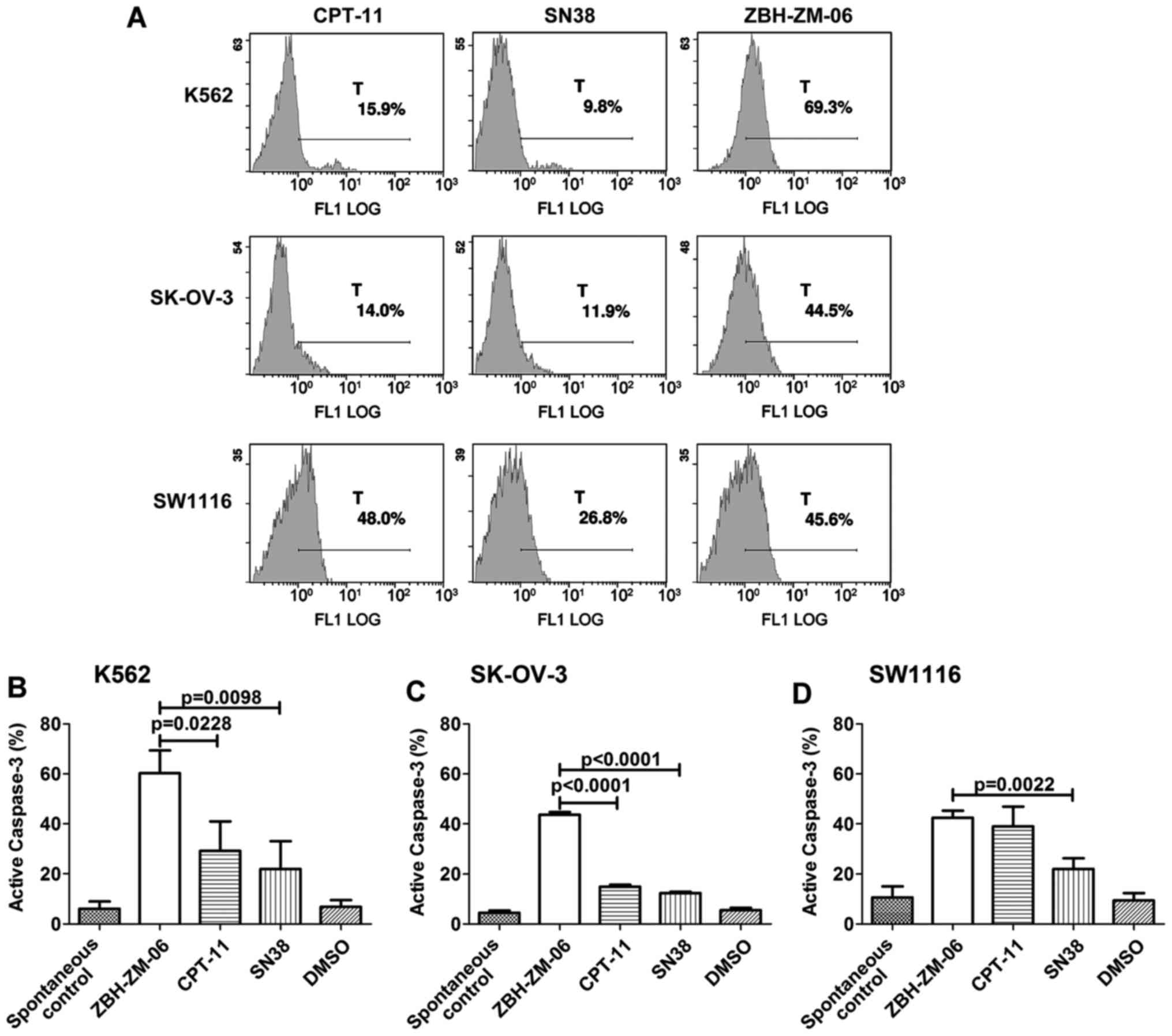
Tumor cell apoptosis induced by
ZBH-ZM-06
Tumor cell apoptosis induction by ZBH-ZM-06, CPT-11
and SN38 was analyzed by an Epics XL-MCL flow cytometer with
FITC-Annexin V and PI staining. Fig.
4 shows the percentages of apoptotic SW1116 and SK-OV-3 cells
induced by 10 µmol/l ZBH-ZM-06, CPT-11 and SN38 after 72 h. The
results indicated that ZBH-ZM-06 more efficiently induced apoptosis
in SK-OV-3 and SW1116 cells either early (Annexin
V+/PI−) or late (Annexin
V+/PI+) than CPT-11 and SN38 (P<0.05). To
test whether the operation process influences the results, we
applied the non-adherent K562 cells to detect the number of
apoptotic cells after 10 µmol/l ZBH-ZM-06, CPT-11 and SN38
treatment for 24, 48 and 72 h (Fig.
5). As shown in Fig. 5, after
24 h of treatment, the number of apoptotic K562 cells in the
ZBH-ZM-06 group was significantly higher than that in the CPT-11
and SN38 groups (P<0.05–0.0001). At this time, although cell
cycle arrest had occurred (Fig. 3),
there was no significant difference among the ZBH-ZM-06, CPT-11 and
SN38 groups, indicating that cell apoptosis occurred earlier after
drug treatment. This result is consistent with our previous
conclusion (21). The number of
apoptotic K562 cells treated with ZBH-ZM-06 for 48 and 72 h was
significantly higher than that of cells treated with CPT-11 or SN38
(P<0.0001).
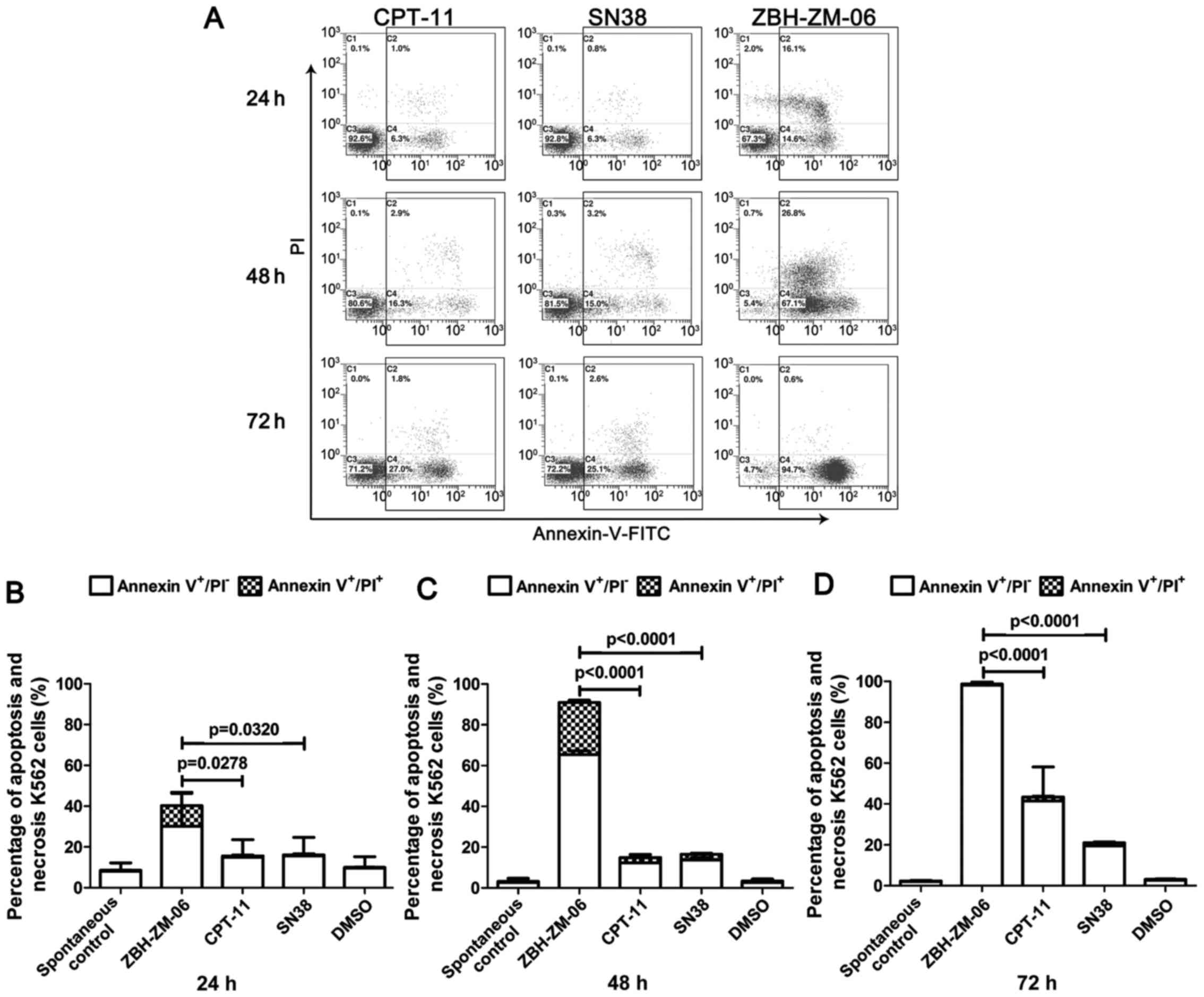 | Figure 5.ZBH-ZM-06, CPT-11 and SN38 induce
K562 cell apoptosis. K562 cells were treated with 10 µmol/l
ZBH-ZM-06, CPT-11 and SN38, respectively, for 24, 48 and 72 h. The
percentages of apoptotic cells were detected by an Epics XL/MCL
flow cytometer with FITC-Annexin V and PI staining. The Annexin
V+/PI− and Annexin
V+/PI+ cells indicated early (dotted) and
late (open) apoptosis, respectively. DMSO-treated cells were used
as negative controls. The typical biparametric images provided by
the flow cytometer are presented in A. The summarized data (mean ±
standard deviation from three independent experiments) are
presented in B, 24 h, C, 48 h, D, 72 h. |
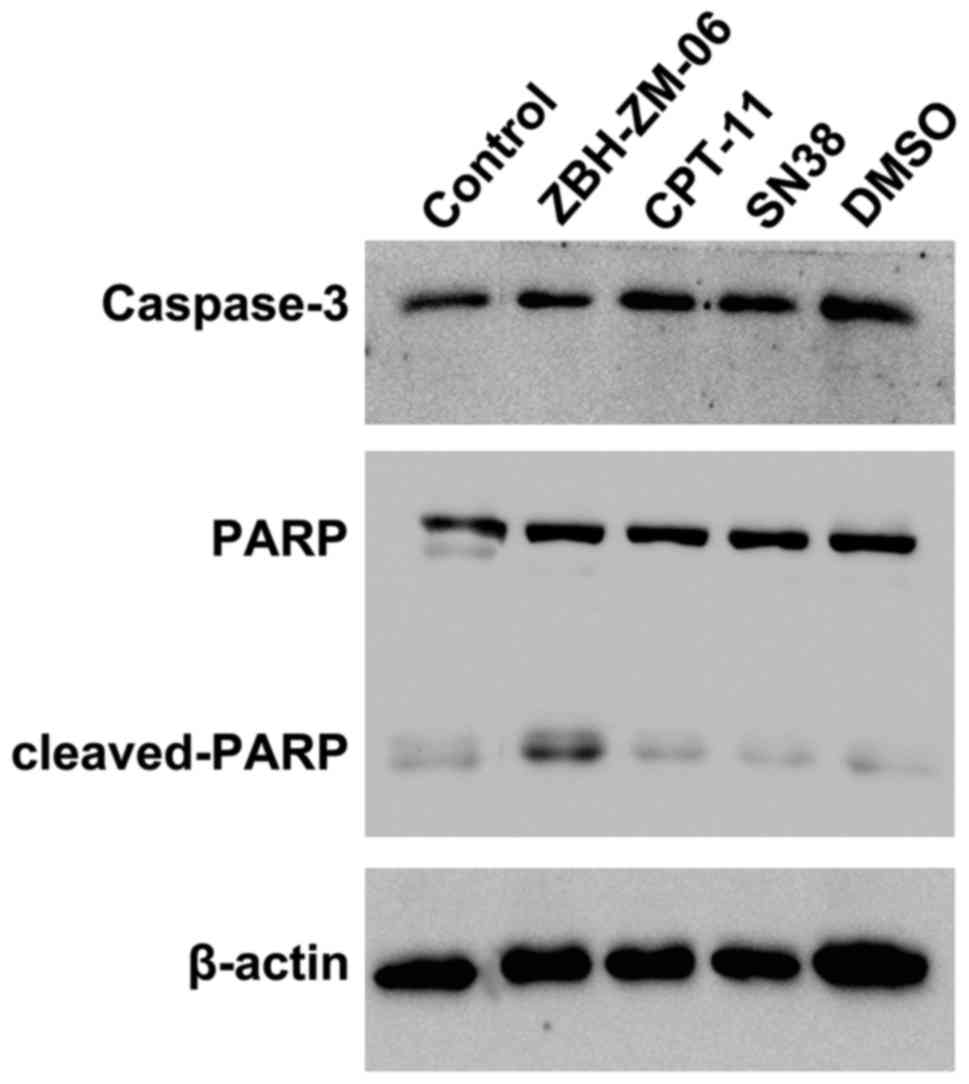
ZBH-ZM-06 promotes tumor cell
apoptosis by activating caspase-3 and poly(ADP-ribose)polymerase
(PARP)
Caspase-3 is a key player in the execution of the
apoptotic cascade by cleaving PARP. To determine whether ZBH-ZM-06
induced apoptosis through this pathway, the expression levels of
cleaved capase-3 and PARP were examined by western blot analysis.
As shown in Fig. 6, ZBH-ZM-06
efficiently activated caspase-3 to cleave PARP proteins.
The levels of the active form of caspase-3 were
determined by flow cytometry in the similarly treated groups as
well. Consistent with the western blot results, the percentage of
activated caspase-3+ K562, SK-OV-3 and SW1116 cells in
the ZBH-ZM-06 group was higher than that of the CPT-11 and SN38
groups (Fig. 7).
Discussion
The present study reports the capacity of the novel
CPT derivative ZBH-ZM-06 as a potential antitumor agent in
vitro. ZBH-ZM-06 was created by reconstruction of SN38 to
reduce several of the disadvantages of CPT-11, for example, poor
solubility and causing severe diarrhea. SN38 is the active
metabolite of CPT-11, which is 100–1000 times more potent than
CPT-11 (24). However, only a small
portion (2–8%) of it is finally converted to SN38. The variability
of the conversion from CPT-11 to SN38 causes substantial dangerous
toxicity risks and difficulties for the clinical management of the
corresponding complications. Therefore, the direct administration
of SN38 might be of benefit for cancer patients. However, the poor
solubility in aqueous and other pharmaceutical solvents (including
ethanol, cremophor and polysorbate 80) limits its application
(25), which has been proven
previously by our group and others (26,27).
Generally, CPT derivatives are expected to have
improved solubility and stability. Yu et al (18) synthesized a series of 6-substituted
indolizinoquinolinedione derivatives and evaluated them for their
biochemical and biological activities. Zhou et al (28) evaluated the cytotoxicity of MXN-004
(a small-molecule compound of PEGylated SN38) in vitro and
demonstrated that it has good water solubility. They further
investigated the pharmacokinetics and tissue distribution of
MXN-004 and its active metabolite SN38 in rats. We designed and
synthesized the novel compound ZBH-ZM-06 by using a linear amino
acid as a prodrug carrier. The amine of the amino acid was linked
to the 10-OH of SN38 via a carbamate linkage.
N-methylpiperazine, which has good physiological
compatibility, was linked to the C-terminus of the amino acid by an
amide bond. The basic nitrogen atom of N-methylpiperazine
can form a salt with the carboxyl group to improve the water
solubility of the compound. One of the disadvantages of CPT-11 is
that it is too stable under physiological conditions to be
converted into the active form SN38, which results in a low
efficiency. ZBH-ZM-06 overcame this disadvantage. It released SN38
rapidly and completely under physiological conditions (pH 7.4),
while remaining stable under acidic conditions (pH 5.0). Therefore,
it could be used as a lead compound for further drug
development.
The anticancer activity of ZBH-ZM-06 was evaluated
in 12 cancer cell lines representing diverse histologies. The
results revealed that ZBH-ZM-06 had a stronger cytotoxic activity
on nine cell lines (Table III).
Such effects are comparable with those of previous studies.
Demarquay et al (29)
reported the characterization of BN80927, a novel CPT analog, and
demonstrated that it was a very potent antiproliferative agent, as
shown by the fact that the IC50 values were consistently
lower than those of SN38 in tumor cell lines as well as in their
related drug-resistant lines. Lansiaux et al (26) determined whether an E-ring ketone
derivative such as S36272 functions as a typical Topo-1 inhibitor,
in a manner similar to TPT and SN38. The cytotoxicity of S38809 was
studied on a panel of 34 cell lines, including 31 human tumor cell
lines, P388 and P388CPT5 sublines, and normal pulmonary artery
endothelial cells. S38809 proved to be a potent cytotoxic agent
against the 31 human tumor cell lines, with a mean IC50
value of 5.4 vs. 11.6 nM for TPT and 3.3 nM for SN38; the colon,
leukemia and ovary cell lines were relatively more sensitive.
Consistent with our previous report, the correlation of the drug
effect with the drug concentration was not considerable in
ZBH-ZM-06-treated NCM460 cells, compared with CPT-11- or
SN38-treated cells (21).
The 4-piperidinyl piperidine moiety of CPT-11 is
responsible for inhibiting AchE activity, causing acute cholinergic
diarrhea. This fact hinted to design a novel CPT derivative without
AchE inhibition (30). ZBH-ZM-06
exhibited only weak inhibitory activity against AchE, compared to
CPT-11, and showed that it can reduce acute cholinergic diarrhea
associated with CPT-11. This result was comparable to those of
previous studies (31,32).
During DNA replication, Topo-1 primarily produces
single-stranded breaks in DNA, which causes DNA relaxation to allow
DNA replication. Once DNA replication is complete, Topo-1 will
religate the single-stranded breaks to restore the double-stranded
DNA structure. In order to explore the cytotoxic mechanism on DNA,
we performed a conversional DNA relaxation assay to evaluate the
inhibitory effect of ZBH-ZM-06 on Topo-1 activity.
Topo-1-regulating drugs, such as CPT-11, can stabilize the covalent
enzyme-DNA complex to prevent DNA religation, thereby triggering a
series of unstoppable DNA replications to induce cell death
eventually (33). Compared to our
previously reported structure (21)
of ZBH-1205, the effective inhibitory concentration of ZHB-ZM-06
for DNA relaxation is 1 µmol/l. The effective inhibitory
concentration for DNA relaxation for ZBH-1205 is 50 µmol/l. This
DNA relaxation activity of ZHB-ZM-06 is even higher than that of
CPT-11 (at 5 µmol/l).
According to the cell cycle arrest analysis, our
previously constructed compounds did not show an obvious effect on
tumor cell cycle arrest, even though they induced apoptosis. In the
present study, we modified the structure of ZBH-ZM-06 to improve
the solubility and the stability, thus providing good effects. We
treated SW1116, SK-OV-3 and K562 cells with ZBH-ZM-06 at a lower
concentration (10 µmol/l) and analyzed the cell cycle at 24, 48 and
72 h after treatment. The results suggested that ZBH-ZM-06 arrested
the cell cycle of SW1116, SK-OV-3 and K562 cells at the S and
G2/M phases (Fig. 3) at
24 h after treatment. With the extension of treatment, the S-phase
arrest ratio was obviously increased and accompanied with a
decrease of the G2/M-phase arrest ratio. During the cell
cycle in eukaryotes, the G2/M phase is a critical highly
complex multi-stage process before cell division. Cell cycle arrest
at the G2/M phase is a common cellular response to a
variety of DNA-damaging agents. Topo-1 poisons, including CPT-11,
induce replication-mediated double-stranded DNA breaks by a
replication-fork collision mechanism and induce cell cycle arrest
of cancer cells at the G2/M phase. Treatment of human
colon cancer cells with SN38 also resulted in G2/M cell
cycle arrest (34). Our results are
consistent with previous reports, suggesting that the mechanism of
ZBH-ZM-06 is similar to that of CPT-11 and SN38.
Cell cycle arrest allows time for the repair of DNA
lesions. Cells re-enter the cell cycle if the damage can be
adequately repaired, or they die by apoptosis if the damage is too
severe. The apoptotic signal is transferred through a series of
signaling cascades. Activation of cell surface receptors triggers
the extrinsic signaling pathway to activate caspase-8, while
cytochrome c release from the mitochondria can initiate the
intrinsic signaling pathway to subsequently induce caspase-9 and
caspase-3 activation (35,36). Caspase-3 is a cysteine protease and
a well-characterized effector of programmed cell death signaling.
It is synthesized in normal cells as an inactive proenzyme that can
be rapidly activated and transformed into cleaved caspase-3 (active
form) by autoproteolytic cleavage or cleavage by other caspases.
Cleaved caspase-3 allows caspase-activated DNase, also known as DNA
fragmentation factor, to translocate to the nucleus where it is
responsible for internucleosomal DNA cleavage, generating
oligo-nucleosomal DNA fragments. Therefore, cleaved caspase-3 is
the executor in apoptosis signaling. During the process of
apoptosis, caspase-3 cleaves the death substrate PARP to a specific
85-kDa form.
A previous study has revealed that the pro-apoptotic
activity of CPT-11 is via an intrinsic apoptotic signaling pathway
(37). To validate whether
ZBH-ZM-06 can trigger the intrinsic apoptotic signaling pathway, we
detected activated caspase-3 and cleaved PARP. Our investigation
indicated that ZBH-ZM-06 increased the protein expression level of
activated caspase-3 and cleaved PARP, suggesting that similar to
CPT-11, the pro-apoptotic activity of ZBH-ZM-06 is via an intrinsic
signaling pathway.
According to proapoptotic activity analysis, the
apoptosis of SW1116, SK-OV-3 and K562 cells induced by 10 µmol/l
ZBH-ZM-06 for 72 h showed similar results as we reported
previously. But the early apoptosis rate induced by ZBH-ZM-06 is
higher than that induced by CPT-11 and SN38. Although there were
some differences among these three tumor cell lines (Fig. 5A). Since the experimental procedures
used for adherent cells may injure the cells, we analyzed K562
tumor cells in suspension at 24 and 48 h. The results revealed that
the rates of apoptosis and necrosis, especially the early apoptotic
rate, induced by ZBH-ZM-06 for 24 and 48 h were significantly
higher than those by CPT-11 and SN38 (Fig. 5B). These data suggested that the
novel constructed CPT-11 derivative has a stronger proapoptotic
capacity (38). Our results are in
agreement with those reported by other research groups. Cao et
al (39) explored apoptosis
induced by SN38 and anti-Fas antibody (CH11) in WR/Fas-SMS1 cells
and its possible mechanisms. The results indicated that the
combination of SN38 and CH11 made the cells undergo greater
apoptosis by finally activating caspase-3, whereas SN38 alone was
ineffective at inducing these responses at any time-point. Di
Francesco et al (40) also
reported that Gimatecan (ST1481, LBQ707;
7-t-butoxyiminomethylcamptothecin), a novel lipophilic CPT
derivative, was more cytotoxic than SN38 and TPT in a panel of
neuroblastoma cell lines. Gimatecans superior cytotoxicity is
likely characterized by a marked arrest at the G2/M
phase and induction of caspase 3-dependent apoptosis.
In conclusion, in this study, the CPT derivative
ZBH-ZM-06 was designed and synthesized. It revealed higher water
solubility and fewer side-effects than those of CPT-11 and SN38.
Our results suggested that ZBH-ZM-06 had a greater ability to
inhibit the viability of a broad spectrum of human tumor cells than
CPT-11 and SN38, but it had less of an effect on normal cells. This
antitumor activity may be obtained through Topo-1 inhibition and
intrinsic apoptosis activation. This study provides experimental
evidence that the novel CPT derivative is more effective and has
fewer side-effects than other CPT derivatives. Therefore, it may be
applied in the clinic for cancer treatment.
Acknowledgements
The present study was supported by funding provided
by the Basic Research Project of Science and Technology Development
in Jilin Province (to H.Y., grant no. 20130102097JC) and the Health
Department Research Project of Jilin Province (to D.-W.Z., grant
no. 2012Z037).
References
|
1
|
Wall ME, Wani MC, Cook CE, Palmer KH,
McPhail AT and Sim GA: Plant antitumor agents. I. The isolation and
structure of camptothecin, a novel alkaloidal leukemia and tumor
inhibitor from camptotheca acuminata1,2. J Am Chem Soc.
88:3888–3890. 1966. View Article : Google Scholar
|
|
2
|
Lin RW, Yang CN, Ku S, Ho CJ, Huang SB,
Yang MC, Chang HW, Lin CM, Hwang J, Chen YL, et al: CFS-1686 causes
cell cycle arrest at intra-S phase by interference of interaction
of topoisomerase 1 with DNA. PLoS One. 9:e1138322014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tomicic MT and Kaina B: Topoisomerase
degradation, DSB repair, p53 and IAPs in cancer cell resistance to
camptothecin-like topoisomerase I inhibitors. Biochim Biophys Acta.
1835:11–27. 2013.PubMed/NCBI
|
|
4
|
Joerger M, Hess D, Delmonte A, Gallerani
E, Fasolo A, Gianni L, Cresta S, Barbieri P, Pace S and Sessa C:
Integrative population pharmacokinetic and pharmacodynamic dose
finding approach of the new camptothecin compound namitecan
(ST1968). Br J Clin Pharmacol. 80:128–138. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Joerger M, Hess D, Delmonte A, Gallerani
E, Barbieri P, Pace S and Sessa C: Phase-I dose finding and
pharmacokinetic study of the novel hydrophilic camptothecin ST-1968
(namitecan) in patients with solid tumors. Invest New Drugs.
33:472–479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Naumczuk B, Kawęcki R, Bocian W, Bednarek
E, Sitkowski J and Kozerski L: Preliminary study of mechanism of
action of SN38 derivatives. Physicochemical data, evidence of
interaction and alkylation of DNA octamer d(GCGATCGC)2. Magn Reson
Chem. 55:128–136. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hamilton G, Klameth L, Rath B and
Thalhammer T: Synergism of cyclin-dependent kinase inhibitors with
camptothecin derivatives in small cell lung cancer cell lines.
Molecules. 19:2077–2088. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vladu B, Woynarowski JM, Manikumar G, Wani
MC, Wall ME, Von Hoff DD and Wadkins RM: 7- and 10-substituted
campto-thecins: Dependence of topoisomerase I-DNA cleavable complex
formation and stability on the 7- and 10-substituents. Mol
Pharmacol. 57:243–251. 2000.PubMed/NCBI
|
|
9
|
Chazin EL, Reis RR, Junior WT, Moor LF and
Vasconcelos TR: An overview on the development of new potentially
active camptothecin analogs against cancer. Mini Rev Med Chem.
14:953–962. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zubovych IO, Sethi A, Kulkarni A, Tagal V
and Roth MG: A novel inhibitor of topoisomerase I is selectively
toxic for a subset of non-small cell lung cancer cell lines. Mol
Cancer Ther. 15:23–36. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu YP, Chen HL, Tzeng CC, Lu PJ, Lo CW,
Lee YC, Tseng CH, Chen YL and Yang CN: TCH-1030 targeting on
topoisomerase I induces S-phase arrest, DNA fragmentation, and cell
death of breast cancer cells. Breast Cancer Res Treat. 138:383–393.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeansonne DP, Koh GY, Zhang F,
Kirk-Ballard H, Wolff L, Liu D, Eilertsen K and Liu Z:
Paclitaxel-induced apoptosis is blocked by camptothecin in human
breast and pancreatic cancer cells. Oncol Rep. 25:1473–1480.
2011.PubMed/NCBI
|
|
13
|
Numbenjapon T, Wang J, Colcher D, Schluep
T, Davis ME, Duringer J, Kretzner L, Yen Y, Forman SJ and
Raubitschek A: Preclinical results of camptothecin-polymer
conjugate (IT-101) in multiple human lymphoma xenograft models.
Clin Cancer Res. 15:4365–4373. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morandi E, Severini C, Quercioli D, DArio
G, Perdichizzi S, Capri M, Farruggia G, Mascolo MG, Horn W, Vaccari
M, et al: Gene expression time-series analysis of camptothecin
effects in U87-MG and DBTRG-05 glioblastoma cell lines. Mol Cancer.
7:662008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen YL, Chueh FS, Yang JS, Hsueh SC, Lu
CC, Chiang JH, Lee CS, Lu HF and Chung JG: Antitumor effects with
apoptotic death in human promyelocytic leukemia HL-60 cells and
suppression of leukemia xenograft tumor growth by irinotecan HCl.
Environ Toxicol. 30:803–815. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yao Y, Su X, Xie Y, Wang Y, Kang T, Gou L,
Yi C and Yang J: Synthesis, characterization, and antitumor
evaluation of the albumin-SN38 conjugate. Anticancer Drugs.
24:270–277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meco D, Di Francesco AM, Cusano G, Bucci
F, Pierri F, Patriarca V, Torella AR, Pisano C and Riccardi R:
Preclinical evaluation of the novel 7-substituted camptothecin
Namitecan (ST1968) in paediatric tumour models. Cancer Chemother
Pharmacol. 70:811–822. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu LM, Zhang XR, Li XB, Yang Y, Wei HY, He
XX, Gu LQ, Huang ZS, Pommier Y and An LK: Synthesis and biological
evaluation of 6-substituted indolizinoquinolinediones as catalytic
DNA topoisomerase I inhibitors. Eur J Med Chem. 101:525–533. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fukuda Y, Kanbe M, Watanabe M, Dan K,
Matsuzaki K, Kitanaka S and Miyata S: 3EZ,20Ac-ingenol, a catalytic
inhibitor of topoisomerases, downregulates p-Akt and induces DSBs
and apoptosis of DT40 cells. Arch Pharm Res. 36:1029–1038. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou M, Liu M, He X, Yu H, Wu D, Yao Y,
Fan S, Zhang P, Shi W and Zhong B: Synthesis and biological
evaluation of novel 10-substituted-7-ethyl-10-hydroxycamptothecin
(SN-38) prodrugs. Molecules. 19:19718–19731. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu D, Shi W, Zhao J, Wei Z, Chen Z, Zhao
D, Lan S, Tai J, Zhong B and Yu H: Assessment of the
chemotherapeutic potential of a new camptothecin derivative,
ZBH-1205. Arch Biochem Biophys. 604:74–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shoemaker RH: The NCI60 human tumour cell
line anticancer drug screen. Nat Rev Cancer. 6:813–823. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rivory LP, Bowles MR, Robert J and Pond
SM: Conversion of irinotecan (CPT-11) to its active metabolite,
7-ethyl-10-hydroxycamptothecin (SN-38), by human liver
carboxylesterase. Biochem Pharmacol. 52:1103–1111. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Senter PD, Beam KS, Mixan B and Wahl AF:
Identification and activities of human carboxylesterases for the
activation of CPT-11, a clinically approved anticancer drug.
Bioconjug Chem. 12:1074–1080. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Atyabi F, Farkhondehfai A, Esmaeili F and
Dinarvand R: Preparation of pegylated nano-liposomal formulation
containing SN-38: In vitro characterization and in vivo
biodistribution in mice. Acta Pharm. 59:133–144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lansiaux A, Léonce S, Kraus-Berthier L,
Bal-Mahieu C, Mazinghien R, Didier S, David-Cordonnier MH,
Hautefaye P, Lavielle G, Bailly C, et al: Novel stable camptothecin
derivatives replacing the E-ring lactone by a ketone function are
potent inhibitors of topoisomerase I and promising antitumor drugs.
Mol Pharmacol. 72:311–319. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu H, Lu H, Liao L, Zhang X, Gong T and
Zhang Z: Lipid nanoparticles loaded with
7-ethyl-10-hydroxycamptothecin-phospholipid complex: In vitro and
in vivo studies. Drug Deliv. 22:701–709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou S, Li N, Wang X, Li C, Tian F, Ren S,
Zhang Y, He Y, Qiu Z, Zhao D, et al: In vitro cytotoxicity,
pharmacokinetics and tissue distribution in rats of MXN-004, a
novel conjugate of polyethylene glycol and SN38. Xenobiotica.
44:562–569. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Demarquay D, Huchet M, Coulomb H,
Lesueur-Ginot L, Lavergne O, Camara J, Kasprzyk PG, Prévost G and
Bigg DC: BN80927: A novel homocamptothecin that inhibits
proliferation of human tumor cells in vitro and in vivo. Cancer
Res. 64:4942–4949. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hyatt JL, Tsurkan L, Morton CL, Yoon KJ,
Harel M, Brumshtein B, Silman I, Sussman JL, Wadkins RM and Potter
PM: Inhibition of acetylcholinesterase by the anticancer prodrug
CPT-11. Chem Biol Interact 157–158. 1–252. 2005.
|
|
31
|
Dodds HM, Hanrahan J and Rivory LR: The
inhibition of acetylcholinesterase by irinotecan and related
camptothecins: Key structural properties and experimental
variables. Anticancer Drug Des. 16:239–246. 2001.PubMed/NCBI
|
|
32
|
Patnaik A, Papadopoulos KP, Tolcher AW,
Beeram M, Urien S, Schaaf LJ, Tahiri S, Bekaii-Saab T, Lokiec FM,
Rezaï K, et al: Phase I dose-escalation study of EZN-2208
(PEG-SN38), a novel conjugate of poly(ethylene) glycol and SN38,
administered weekly in patients with advanced cancer. Cancer
Chemother Pharmacol. 71:1499–1506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wadkins RM, Bearss D, Manikumar G, Wani
MC, Wall ME and Von Hoff DD: Topoisomerase I-DNA complex stability
induced by camptothecins and its role in drug activity. Curr Med
Chem Anticancer Agents. 4:327–334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Motwani M, Sirotnak FM, She Y, Commes T
and Schwartz GK: Drg1, a novel target for modulating sensitivity to
CPT-11 in colon cancer cells. Cancer Res. 62:3950–3955.
2002.PubMed/NCBI
|
|
35
|
Liu YQ, Li WQ, Morris-Natschke SL, Qian K,
Yang L, Zhu GX, Wu XB, Chen AL, Zhang SY, Nan X, et al:
Perspectives on biologically active camptothecin derivatives. Med
Res Rev. 35:753–789. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li J, Ouyang Y, Zhang X, Zhou W, Wang F,
Huang Z, Wang X, Chen Y, Zhang H and Fu L: Effect of HM910, a novel
camptothecin derivative, on the inhibition of multiple myeloma cell
growth in vitro and in vivo. Am J Cancer Res. 5:1000–1016.
2015.PubMed/NCBI
|
|
37
|
Ohmori T, Podack ER, Nishio K, Takahashi
M, Miyahara Y, Takeda Y, Kubota N, Funayama Y, Ogasawara H, Ohira
T, et al: Apoptosis of lung cancer cells caused by some anti-cancer
agents (MMC, CPT-11, ADM) is inhibited by bcl-2. Biochem Biophys
Res Commun. 192:30–36. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zuco V, De Cesare M, Zaffaroni N, Lanzi C
and Cassinelli G: PLK1 is a critical determinant of tumor cell
sensitivity to CPT11 and its inhibition enhances the drug antitumor
efficacy in squamous cell carcinoma models sensitive and resistant
to camptothecins. Oncotarget. 6:8736–8749. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cao Y, Jin ZX, Tong XP, Yue S, Sakai T,
Kawanami T, Sawaki T, Miki M, Iwao H, Nakajima A, et al:
Synergistic effects of topoisomerase I inhibitor, SN38, on
Fas-mediated apoptosis. Anticancer Res. 30:3911–3917.
2010.PubMed/NCBI
|
|
40
|
Di Francesco AM, Riccardi A, Barone G,
Rutella S, Meco D, Frapolli R, Zucchetti M, DIncalci M, Pisano C,
Carminati P, et al: The novel lipophilic camptothecin analogue
gimatecan is very active in vitro in human neuroblastoma: A
comparative study with SN38 and topotecan. Biochem Pharmacol.
70:1125–1136. 2005. View Article : Google Scholar : PubMed/NCBI
|