Introduction
Gastric cancer (GC) is one of the primary causes of
tumor-associated deaths worldwide, most cases occur in East Asia,
and the mortality rates are high (1). In China, GC represents the third most
established malignancy (2). The
incidence of GC has decreased with the improved health conditions
and the optimized medical level. However, due to the high rates of
metastasis and recurrence, the 5-year overall survival rate of
advanced GC is 20% (3). New
therapeutic strategies such as targeting molecules have been
reported in GC (4). Hence, the
study of new treatments that manage this disease is urgent.
Autophagy is an evolutionarily conserved process
that includes the degradation of intracellular organelles and
cytoplasmic components by lysosomes (5), this process is effective in
maintaining cellular homeostasis (6,7).
However, the effects of autophagy on particular cellular functions
are sometimes contradictory, and autophagy is involved in the
pathway of cell survival and cell death under metabolic stress
(8,9). It has been reported that autophagy
exerted influence on tumorigenesis and therapy (10,11).
MicroRNAs (miRNAs) are a class of endogenous, highly
conserved non-coding small RNA, which could bind to the
3′-untranslated region (3′-UTR) of the target gene and restrain
translation or accelerate their degradation, thereby regulating the
expression of the target gene at post-transcriptional or
translation level (12,13). It has been reported that miRNAs are
implicated in regulating autophagy in a variety of diseases
(14–16). Our previous study demonstrated that
miR-let-7a functions as antitumor gene and inhibits cell
proliferation, migration and invasion in GC cells (17).
Rictor (Rptor independent companion of mTOR complex
2) is a core component of mTORC2, and mTORC2 is one part of mTOR
(the mechanistic target of rapamycin) signaling pathway, mTOR
signaling pathway is related to regulation of cell autophagy
(18,19). Rictor has also been reported to be
involved in autophagy process (20). Rictor was predicted as the target
protein of miR-let-7a through exploiting bioinformatics software
including TargetScan and miRanda, and was verified by
dual-luciferase reporter assay (21–23).
In the present study, we revealed the impact of
miR-let-7a on Rictor expression and its molecular mechanism in the
regulation of autophagy in GC cell. Firstly, we found that
overexpression of miR-let-7a increased autophagic activity, while
knockdown had the reverse effect. Next, we found that Rictor was
the direct functional target of miR-let-7a. Finally, miR-let-7a
promoted autophagic activity by suppressing the activation of
Akt-mTOR pathway.
Materials and methods
Culture of human GC cells
Human GC cell lines MGC-803, SGC-7901, BGC-823, AGS
and human normal gastric mucous epithelium cell GES-1 were obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA), and were grown in RPMI-1640 medium, supplemented with 10%
fetal bovine serum (FBS) and 1% antibiotics
(penicillin-streptomycin) (all from Gibco, Carlsbad, CA, USA). All
cells were cultured in humidified incubator containing 5%
CO2 at 37°C.
Quantitative real-time PCR analysis
for mRNA and miRNA
Total RNA were isolated from cells using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. TaqMan miRNA assays were performed to
determine the expression of miR-let-7a. TaqMan miRNA reverse
transcription kit (Invitrogen) was used for cDNA synthesis and
qRT-PCR was carried out by ABI StepOne Plus (Applied Biosystems,
Foster City, CA, USA). U6 was an endogenous reference for
miR-let-7a quantification using the 2−ΔΔCt method. The
relative expression of Rictor was quantifed using SYBR-Green Master
Mix kit (Roche Diagnostics, Indianapolis, IN, USA) and β-actin was
selected as control. The specific primers were as follows:
miR-let-7a forward, 5′-GGTGAGGTAGTAGGTTGTATAGTT-3′ and reverse,
5′-CTCGCTTCGGCAGCACATATA-3′; U6 forward, 5′-CTCGCTTCGGCAGCACA-3′
and reverse, 5′-AACGCTTCACGAATTTGCGT-3′; Rictor forward,
5′-ACCGGGCTTCTGACCATTAAA-3′ and reverse, 5′TTGTATGAACCGCCGACACT-3′;
β-actin forward, 5′-AGAAAATCTGGCACCACACC-3′ and reverse,
5′-TAGCACAGCCTGGATAGCAA-3′. All data collected were in
triplicate.
Western blot analysis
RIPA buffer (Beyotime, Shanghai, China) was used to
isolate total proteins from cell lysates on ice. Equal amounts of
protein were separated by 10% SDS polyacrylamide gel
electrophoresis. Proteins on the gel were transferred to
polyvinylidene difluoride (PVDF) membranes. Then, the membranes
were blocked with Tris-buffered saline with Tween-20 (TBST)
containing 5% milk for 2 h and incubated overnight in special
primary antibodies: anti-LC3, anti-p62, anti-Rictor, anti-Akt,
anti-p-Akt, anti-mTOR, anti-p-mTOR and anti-GAPDH (dilution
1:1,000; Cell Signaling Technology, Beverly, MA, USA). After being
washed with TBST three times 15-min each, the HRR-conjugated
anti-mouse or anti-rabbit IgG antibodies (dilution 1:20,000;
Jackson ImmunoResearch, Inc., West Grove, PA, USA) were incubated
with bolted membranes at room temperature for 2 h and washed again.
The enhanced chemiluminescence detection system was used to detect
the expression of target bands.
Immunofluorescence
Cells were seeded into glass bottom cell culture
dish at 20,000 cells and cultured for 48 h. After being washed with
PBS three times, cells were fixed in 4% paraformaldehyde for 20 min
and permeabilized by 0.5% Triton X-100 for 10 min. Then, cells were
blocked with 3% bovine serum albumin for 1 h and incubated
overnight in special primary antibody: anti-LC3 (dilution 1:200).
After removing primary antibody and washing again with PBS next
day, cells were incubated with Alexa Flour 594 Rhodamine-conjugated
goat anti-rabbit (dilution 1:200; Jackson ImmunoResearch) for 1 h
and 30 min and mounted by staining with
4′,6-diamidino-2-phenylindole (DAPI) for 5 min at room temperature.
Confocal microscopy (Carl Zeiss LSM710; Carl Zeiss, Jena, Germany)
was used to photograph the stained cells in a ×40 oil lens and LC3
puncta was manually quantified.
Cell transfection
The miR-let-7a mimics and miR-let-7a inhibitor
vector lentiviral constructs (GenePharma, Shanghai, China) were
modified to interfere with expression of miR-let-7a, as were
siRictor and pcDNA3.1-Rictor to interfere with expression of Rictor
from Santa Cruz Biotechnology (Dallas, TX, USA). Negative
miRNA-let-7a and scramble Rictor were also transfected as negative
controls. When MGC-803, SGC-7901 and GES-1 cells grew to 50–60%
confluency, the miR-let-7a-NC, miR-let-7a mimics and miR-let-7a
inhibitor lentiviral vectors were used to infect cells according to
different multiplicity of infection (MOI) offered by the
manufacturer. Puromycin (3 µg/ml) (Sigma, St. Louis, MO, USA) was
used to screen out stable cell lines for one week and the
expression of miR-let-7a was analyzed by qRT-PCR. siRNA against
Rictor and pcDNA3.1-Rictor plasmids were transfected into GC cells
using Lipofectamine 3000 reagent (Invitrogen).
Dual-luciferase reporter assay
The wild-type (WT) or mutated-type (MUT) 3′-UTRs of
Rictor containing miR-let-7a binding sites were amplified by PCR
and subcloned into pmirGLO Dual-Luciferase miRNA Target Expression
vectors (Promega, Madison, WI, USA). For luciferase activity
assays, HEK293T cells were seeded into a 24-well plate, then
co-transfected with Rictor 3′-UTR construct and miR-let-7a or NC
using Lipofectamine 3000 (Invitrogen). Luciferase activity was
measured by Dual-Luciferase Reporter assay kit (Promega, USA) and
normalized by Renilla luciferase overnight.
Statistical analysis
Data are presented as mean ± standard deviation (SD)
from at least three independent experiments and the Students
two-tailed t-test was used to determine whether these data are
statistically significant. Values of P<0.05 were considered
significant.
Results
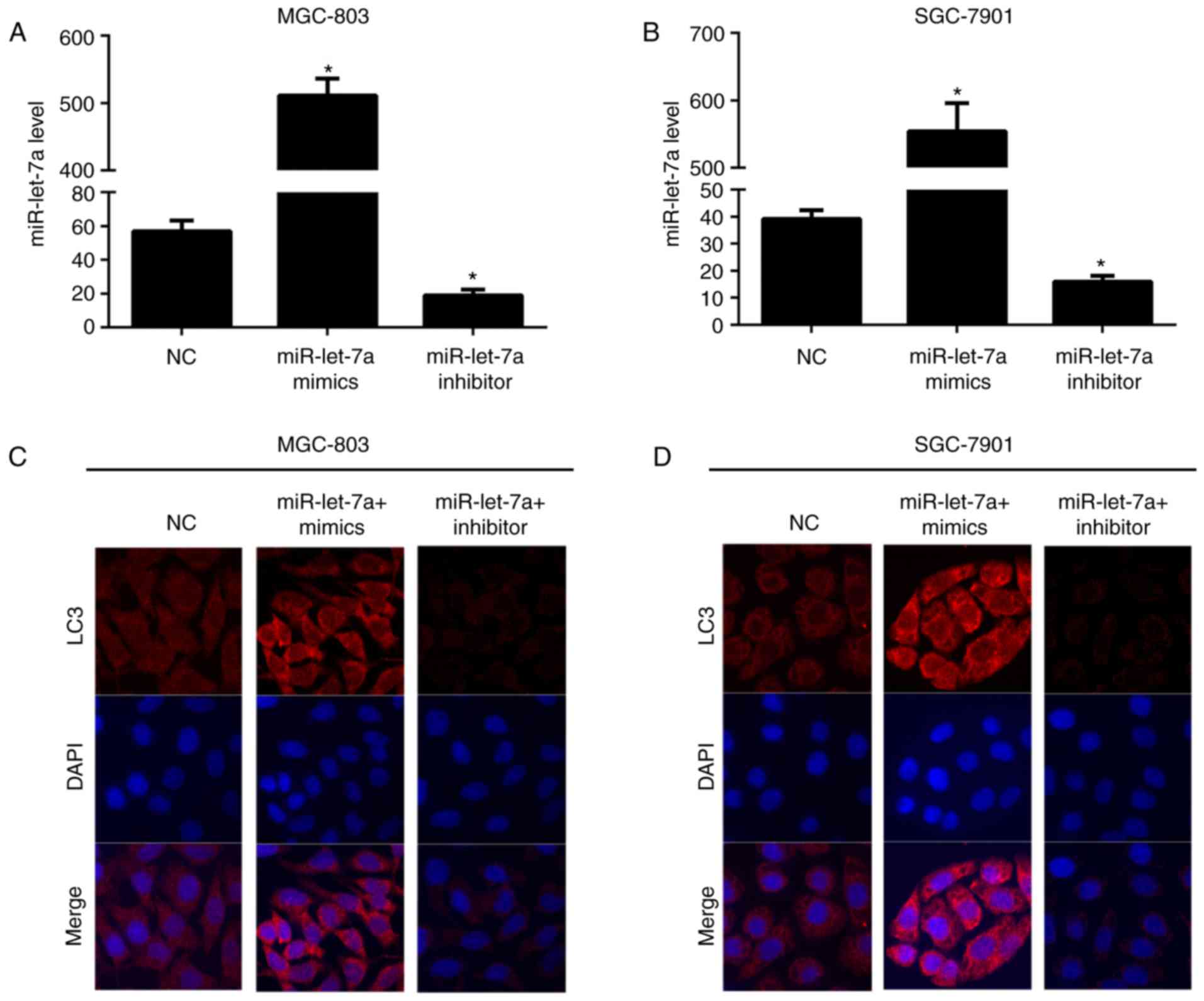
miR-let-7a affects autophagic activity
in GC cells
To elucidate the role of miR-let-7a on autophagic
activity in GC, we firstly constructed miR-let-7a mimics and
miR-let-7a inhibitors using lentiviral transfection in GC cells
(MGC-803 and SGC-7901). miRNA real-time polymerase chain reaction
(RT-PCR) was used to determine the expression of miR-let-7a
(Fig. 1A and B). As shown,
miR-let-7a was knocked downed ~65 and 60%, increased appoximately
9- and 14-fold in MGC-803 and SGC-7901 cells, respectively. Next,
we explored the effect of miR-let-7a on autophagy, confocal imaging
was employed to evaluate the number of LC3 puncta (Fig. 1C and D). It showed that the number
of LC3 puncta were markedly elevated in MGC-803 and SGC-7901 cells
which were overexpressed of miR-let-7a. However, the LC3 dot
formation was significantly reduced after knocking down miR-let-7a
both in MGC-803 and SGC-7901 cells. These results recognized
miR-let-7a as an enhancer of autophagy and it contributes to the
promotion of autophagic responses in GC cells.
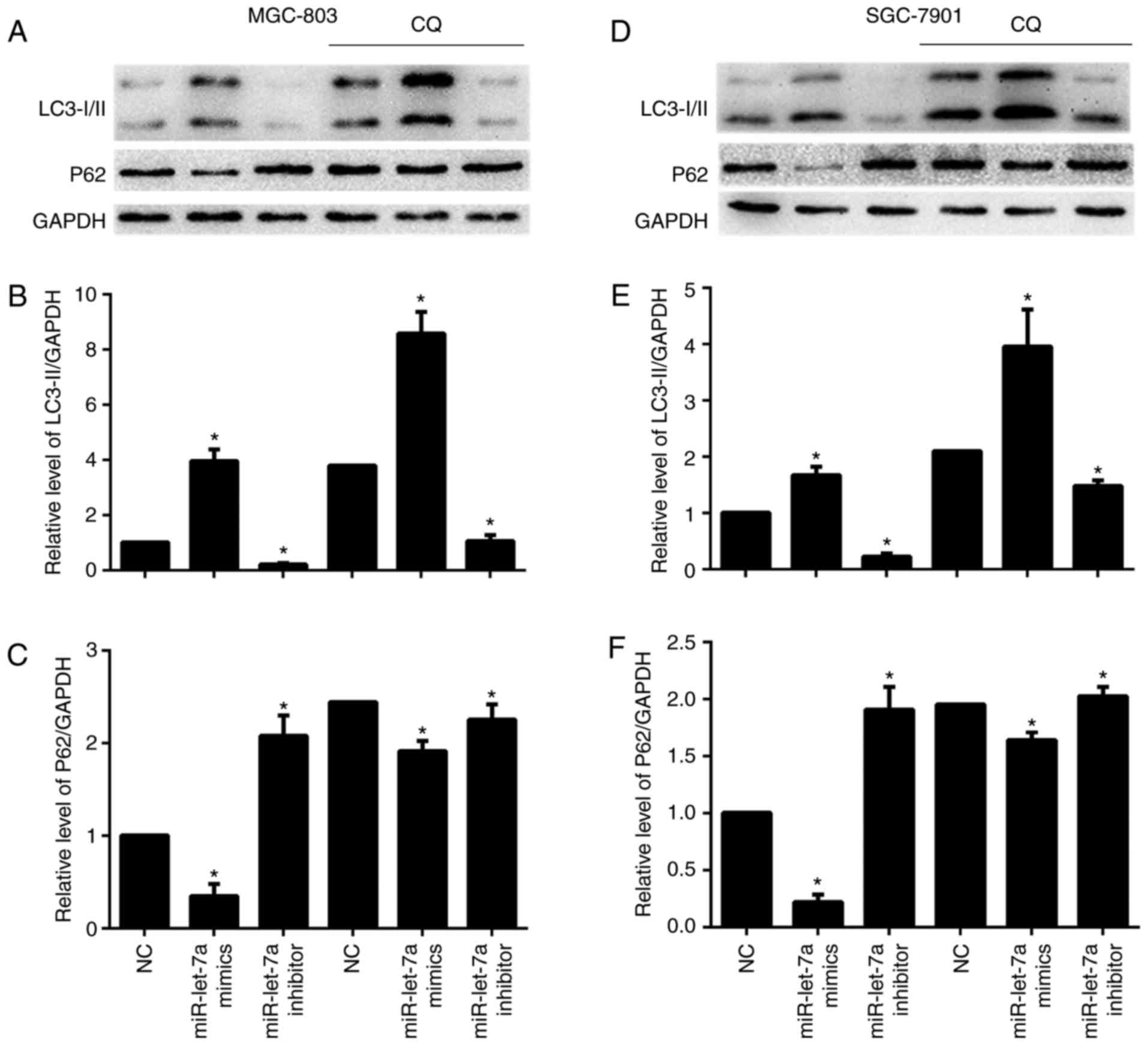
miR-let-7a affects autophagic flux in
GC cells
The conversion of LC3-I to LC3-II and p62 protein
expression were detected by western blotting. LC3-I scattered in
the cytoplasm, and upon autophagy induction, LC3-I was conjugated
with phosphatidylethanolamine (PE) to form LC3-II and localized to
autophagosomes and adventitia. LC3-II is always retained on the
autophagic membrane until it is fused to lysosomes. We found that
LC3-II/GAPDH ratios were increased after overexpressing miR-let-7a
and were decreased after downregulating miR-let-7a, these results
were consistent with previous LC3 puncta formation assay (Fig. 2A and B, and D and E). P62 is a
selective autophagic-lysosomal degradation substrate, total
cellular p62 protein levels reflect autophagic activity. When
autophagy occurs, p62 levels are decreased, while autophagic
activity is inhibited, p62 protein is accumulated. As expected,
overexpression of miR-let-7a led to reduced p62 protein levels, but
inhibition of miR-let-7a resulted in an increase of p62 expression
(Fig. 2A, C, D and F). Then, we
performed an autophagic flux assay to determine whether
autophagosome accumulation resulted from increased autophagic
activity or due to a block in downstream degradation. Chloroquine
(CQ), a lysosomotropic reagent, was used to block autophagosome
degradation in MGC-803 and SGC-7901 cells. As shown, lipidated
LC3-II was significantly increased both in NC and miR-let-7a
transfected cells by CQ (Fig. 2A and B
and D and E). Similarly, p62 protein levels were also increased
in miR-let-7a transfected cells treated with CQ (Fig. 2A, C, D and F). These results
demonstrate that miR-let-7a affected autophagic activity in GC
cells.
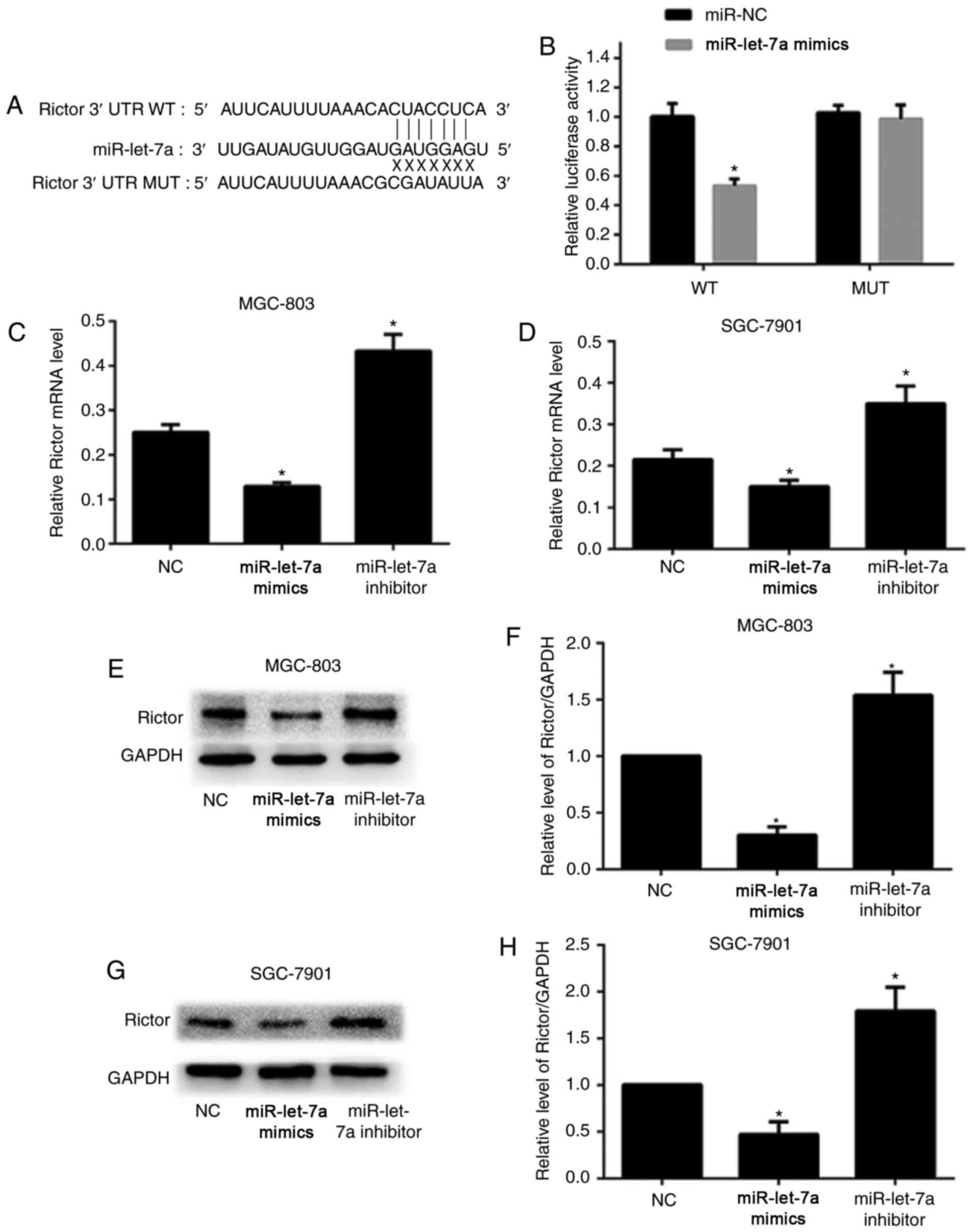
miR-let-7a directly binds to the 3′
UTR of Rictor
In order to clarify the specific mechanism of
miR-let-7a-induced autophagy, bioinformatics software including
TargetScan and miRanda were employed to predict the potential
binding sequences in the 3′-untranslated region (3′-UTR) of target
genes. As shown, the 3′-UTR of human Rictor mRNA resides underlying
miR-let-7a binding sequences (Fig.
3A). Furthermore, the wild-type (WT) or mutated-type (MUT)
sequences in the 3′-UTR of Rictor that miR-let-7a targets were
designed to construct luciferase reporter vector and co-transfected
with either miR-let-7a or NC control. As shown, overexpression of
miR-let-7a markedly reduced luciferase activity when HEK293T cells
were co-transfected with WT 3′-UTR of Rictor compared to NC. In
contrast, this inhibitory effect of miR-let-7a on luciferase
activity was abrogated when HEK293T cells were co-transfected with
MUT (Fig. 3B).
We performed RT-PCR assay and western blot analysis
to further confirm whether miR-let-7a suppressed the expression of
Rictor in GC cells. As shown, overexpression of miR-let-7a led to a
significant decrease both in mRNA and protein levels in MGC-803 and
SGC-7901 cells compared with NC. However, inhibition of miR-let-7a
notably increased the levels of Rictor mRNA and protein (Fig. 3C-H). Therefore, the data above
provided strong evidence that Rictor is the target of miR-let-7a,
and miR-let-7a inhibits the expression of Rictor by directly
binding to its 3′-UTR.
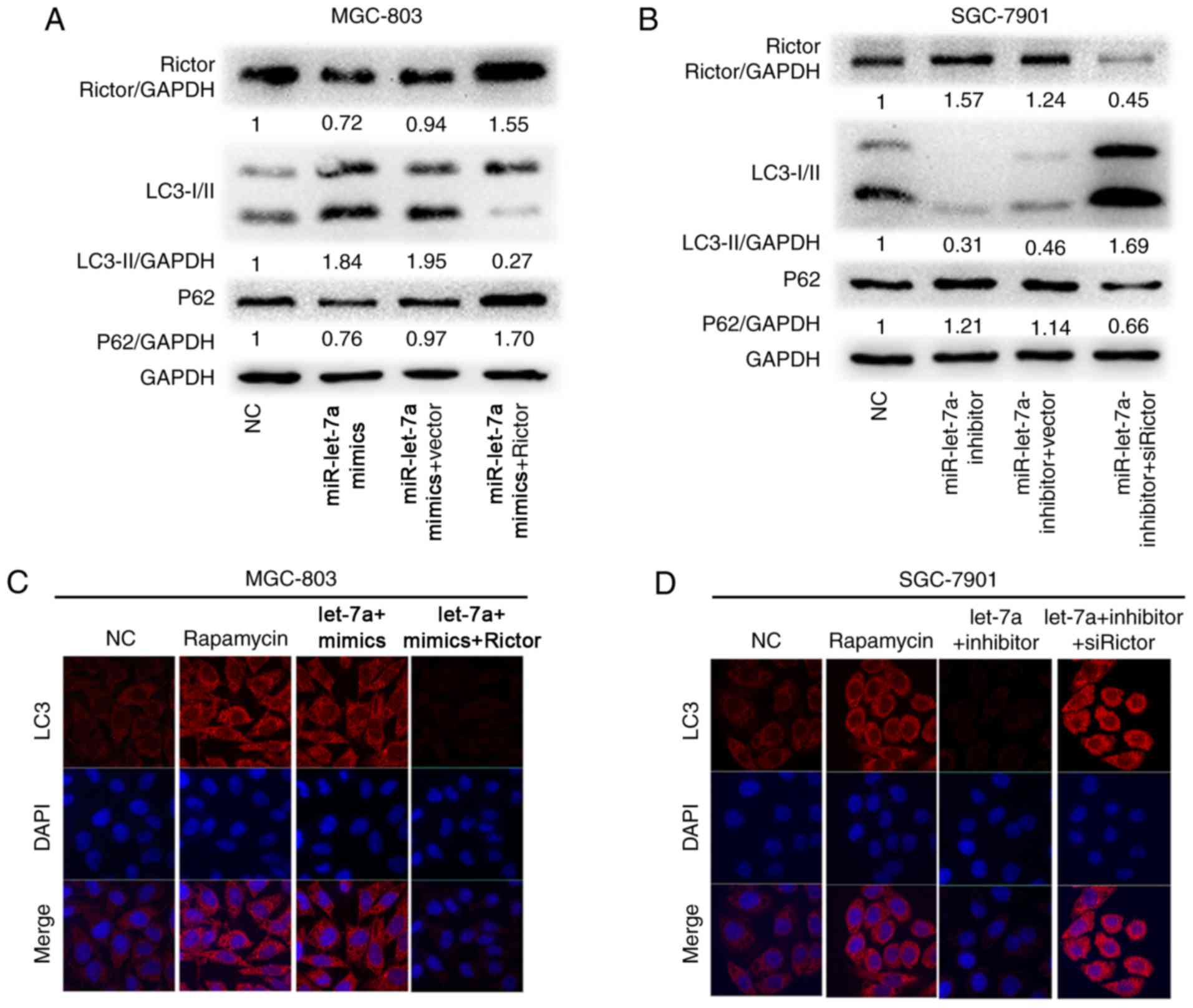
Rictor reverses autophagic activity
regulated by miR-let-7a
To explore whether the effect of miR-let-7a on GC
cell autophagic activity was mediated by Rictor, we performed
‘rescue experiments’. GC cell lines MGC-803 and SGC-7901 were
co-transfected with miR-let-7a overexpression lentivirus and either
pcDNA3.1-Rictor plasmids or empty vector, worthwhile, other MGC-803
and SGC-7901 cells were co-transfected with miR-let-7a supression
lentivirus and either siRictor or empty vector, respectively. As
shown, upregulated Rictor expression reversed the promotion of
autophagic activity caused by the overexpression of miR-let-7a, the
results suggested that LC3-II/GAPDH ratios were decreased and p62
protein levels were increased in MGC-803 and SGC-7901 cells
(Fig. 4A and B). Simultaneously, we
found that the similar rescue effect was observed in MGC-803 and
SGC-7901 cells where downregulated Rictor expression reversed the
supression of autophagic activity caused by the knockdown of
miR-let-7a (Fig. 4A and B).
Rapamycin, as a potent immunosuppressive and
antitumor agent (24), is commonly
used in autophagy study. As a control, we found that miR-let-7a
enhanced the activity of autophagy as well as rapamycin by confocal
imaging in MGC-803 cells, and LC3 dots formation was reversed upon
co-expression with Rictor (Fig.
4C). Inhibition of endogenous miR-let-7a led to the opposite
effect (Fig. 4D). These results
show that Rictor is the vital functional element in
miR-let-7a-mediated autophagic response.
miR-let-7a regulates the Akt-mTOR
signaling pathway
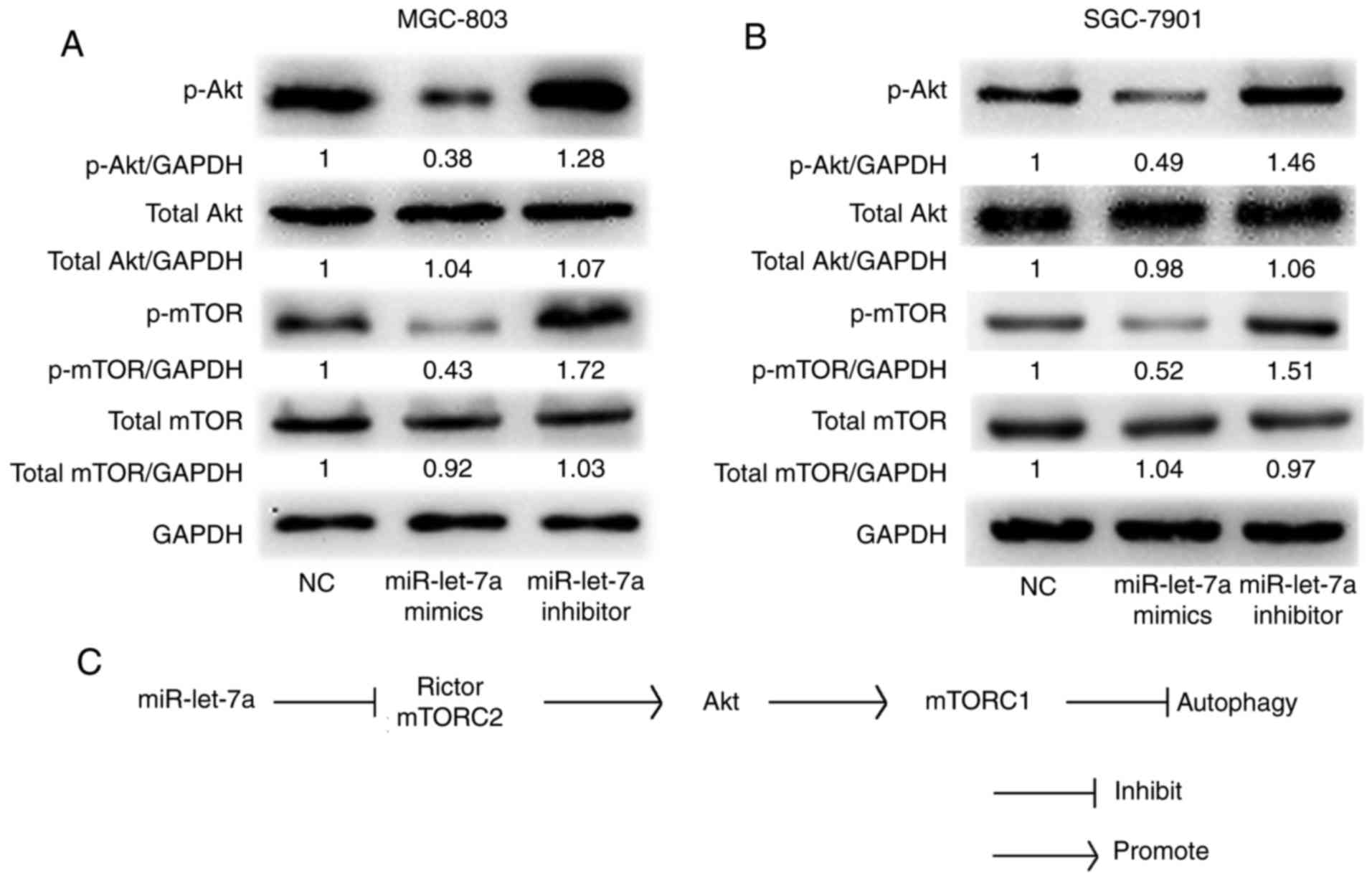
It has been reported that Akt-mTOR signaling pathway
is involved in autophagic activity and Rictor plays an important
role in the Akt-mTOR signaling pathway (21). We confirmed that Akt and mTOR
phosphorylations were involved in miR-let-7a-mediated autophagy in
GC cells by western blot analysis. As shown, both the protein
levels of phosphorylated Akt and phosphorylated mTOR were
significantly decreased by upregulating miR-let-7a and increased by
downregulating miR-let-7a in MGC-803 and SGC-7901 cells. However,
the total protein levels were almost unchanged (Fig. 5A and B). These data indicated that
there is a direct link between miR-let-7a and Akt-mTOR signaling
pathway. Therefore, we confirm that miR-let-7a regulates autophagic
activity throughout the Akt-mTOR signaling pathway in GC cells.
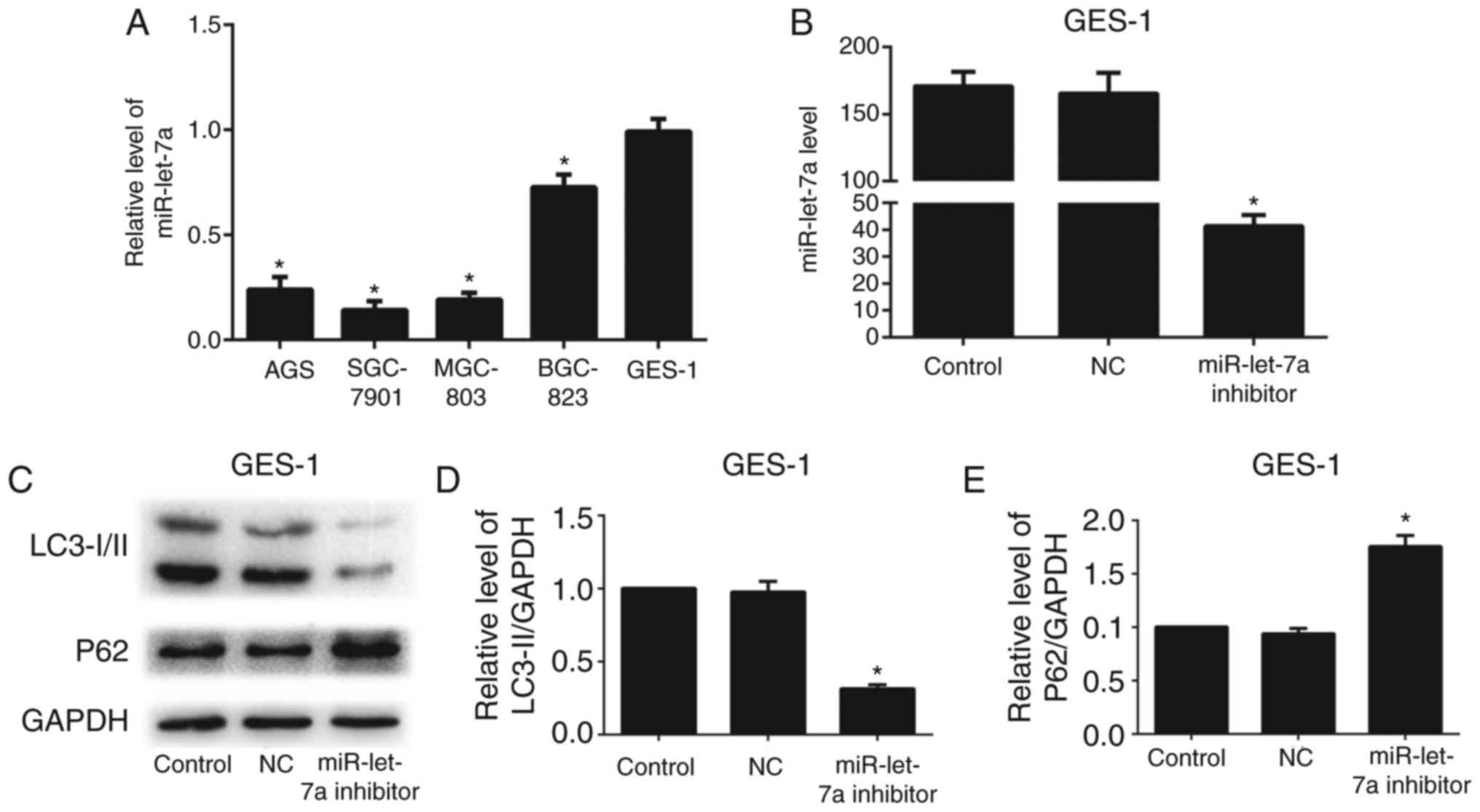
Knockdown of miR-let-7a suppresses human normal
gastric mucous epithelium cell autophagic activity. To investigate
whether miR-let-7a affects autophagy in human normal gastric mucous
epithelium cells (GES-1), we detected the expression level of
miR-let-7a in GC cells and GES-1. We found that the expression of
miR-let-7a in GES-1 cells was relatively higher than that in GC
cells (Fig. 6A). Thus, we
transfected GES-1 cells with miR-let-7a inhibitor or NC. Successful
knockdown of miR-let-7a was confirmed by RT-PCR (Fig. 6B). The expression of LC3-II was
reduced, and P62 was relatively higher compared with control or NC
after knockdown of miR-let-7a by western blotting (Fig. 6C-E). These results suggest that
miR-let-7a affects autophagy in human normal gastric mucous
epithelium cells.
Discussion
Accumulating evidence has demonstrated that miRNAs
are implicated in modulating autophagic activity through targeting
their mRNAs or regulating other signalling pathways (25–27).
In our previous study, we reported that miR-let-7a inhibits cell
proliferation, migration and invasion in gastric cancer (GC) cells
(17). However, it is not clear how
miR-let-7a suppresses GC cells by influencing the autophagy
pathways. In the present study, we provide insightful evidence to
identify miR-let-7a as a powerful enhancer of autophagy in GC
cells. Here, we discovered that miR-let-7a promotes autophagy by
directly targeting the autophagy-related gene Rictor and that
miR-let-7a could directly pair to the 3′-UTR sequence of Rictor,
leading to translational repression in GC cells. By suppressing the
motivation of Akt-mTOR signalling pathway, miR-let-7a induces
autophagic activity and regulates the growth of GC development.
Therefore, we introduced miR-let-7a as a potent autophagy inducer
which inhibited Rictor-mediated Akt-Mtor activity (Fig. 5C). Combining with previous finding
(17), miR-let-7a attributed to
autophagy a pro-death role in GC cell lines MGC-803 and SGC-7901.
This further optimized the mechanisms of miR-Let7a to serve as a
tumor-suppressor in GC, as well as its clinical value.
LC3, microtubule-associated protein 1 light chain 3,
is autophagic membrane-labeled protein. There are two forms of LC3
protein in the cell, the LC3-I and LC3-II. The formation of LC3-II
is considered to be a good marker for monitoring the occurrence of
autophagic activity (28). P62,
ubiquitin binding protein, is a marker protein that reflects
autophagic activity (29). When
autophagy occurs, p62 levels are decreased, while autophagic
activity is inhibited, p62 protein is accumulated (30). mTOR, the mechanistic target of
rapamycin, functions as a sensor that respond to many metabolic
events to regulate cell growth and homeostasis including the
control of autophagy. mTOR exists in the form of two complexes,
which are rapamycin sensitive mTORC1 and rapamycin resistant mTORC2
(31). Numerous studies indicate
that miRNAs play significant roles in cancer development by acting
on mTOR itself, mTOR pathway or the key factors within the mTOR
pathway.
Rictor, as the crucial component of mTORC2, plays an
essential role in regulating and activating Akt phosphorylation
(32). It has been reported that
activated Akt further regulates cell growth, apoptosis and
autophagy by triggering the phosphorylation activation of mTOR,
including GC (21,33,34).
The Akt-mTOR signaling pathway is a dominant negative signaling
pathway that is resistant to autophagy (35). In addition, a preliminary study
disclosed that Akt-mTOR activation was positively correlated with
Rictor in CNE and HeLa cells (21).
In line with previous studies, we provided evidence that Rictor
rescued miR-let-7a-induced autophagic activity, which suggested a
direct link between miR-let-7a and Rictor. Moreover, the
phosphorylation of Akt and mTOR could be damaged by upregulation of
miR-let-7a and be strengthened by downregulation of miR-let-7a.
These results show that miR-let-7a regulates autophagy by directly
targeting Rictor via Akt-mTOR signaling pathway in GC cells.
There is no denying that the present study has
certain limitations. Our research was incomplete and limited, what
we discovered cannot represent GC. We only verified the hypothesis
in gastric cell lines MGC-803 and SGC-7901, other gastric cell
lines, such as BGC-823, AGS and so on, were not included in the
research. Our data showed that miR-let-7a promote autophagy through
Rictor/Akt-mTOR pathway, however, the interaction between cytokines
in cancer cells is very complex, we cannot rule out other signal
pathways affected by miR-let-7a which may influence Akt-mTOR
expression.
Taken together, the present study demonstrated that
miR-let-7a works as a promoter at aspect of autophagy in GC cells,
and Rictor is its direct target gene. In addition, we revealed that
miR-let-7a may play its role in promoting autophagy through
Rictor/Akt-mTOR pathway. To the best of our knowledge, that
miR-let-7a targets Rictor has never been reported in GC cells.
Therefore, targeting miR-let-7a/Rictor/autophagy pathway in
clinical treatment of GC requires further study and
exploration.
Acknowledgements
The present study was funded by the Natural Science
Foundation of Jiangsu Province [grant no. BK20131447 (DA13)], the
‘Medical ZhongDianRenCai Project’ of Jiangsu Province (grant no.
RC2011059), and the ‘333 Project’ of Jiangsu Province [grant no.
BRA2013280 (RS13)].
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang L, Parkin DM, Ferlay J, Li L and Chen
Y: Estimates of cancer incidence in China for 2000 and projections
for 2005. Cancer Epidemiol Biomarkers Prev. 14:243–250.
2005.PubMed/NCBI
|
|
3
|
de Martel C, Forman D and Plummer M:
Gastric cancer: Epidemiology and risk factors. Gastroenterol Clin
North Am. 42:219–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song IS, Oh NS, Kim HT, Ha GH, Jeong SY,
Kim JM, Kim DI, Yoo HS, Kim CH and Kim NS: Human ZNF312b promotes
the progression of gastric cancer by transcriptional activation of
the K-ras gene. Cancer Res. 69:3131–3139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pietrocola F, Izzo V, Niso-Santano M,
Vacchelli E, Galluzzi L, Maiuri MC and Kroemer G: Regulation of
autophagy by stress-responsive transcription factors. Semin Cancer
Biol. 23:310–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang C and Jung JU: Autophagy genes as
tumor suppressors. Curr Opin Cell Biol. 22:226–233. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moreau K, Luo S and Rubinsztein DC:
Cytoprotective roles for autophagy. Curr Opin Cell Biol.
22:206–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang MC, Wu AG, Huang YZ, Shao GL, Ji SF,
Wang RW, Yuan HJ, Fan XL, Zheng LH and Jiao QL: Autophagic
regulation of cell growth by altered expression of Beclin 1 in
triple-negative breast cancer. Int J Clin Exp Med. 8:7049–7058.
2015.PubMed/NCBI
|
|
11
|
Sui H, Shi C, Yan Z and Li H: Combination
of erlotinib and a PARP inhibitor inhibits growth of A2780 tumor
xenografts due to increased autophagy. Drug Des Devel Ther.
9:3183–3190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tanzer A and Stadler PF: Molecular
evolution of a microRNA cluster. J Mol Biol. 339:327–335. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Selbach M, Schwanhäusser B, Thierfelder N,
Fang Z, Khanin R and Rajewsky N: Widespread changes in protein
synthesis induced by microRNAs. Nature. 455:58–63. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Frankel LB, Wen J, Lees M, Høyer-Hansen M,
Farkas T, Krogh A, Jäättelä M and Lund AH: microRNA-101 is a potent
inhibitor of autophagy. EMBO J. 30:4628–4641. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brest P, Lapaquette P, Souidi M, Lebrigand
K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF,
Hébuterne X, et al: A synonymous variant in IRGM alters a binding
site for miR-196 and causes deregulation of IRGM-dependent
xenophagy in Crohn's disease. Nat Genet. 43:242–245. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kovaleva V, Mora R, Park YJ, Plass C,
Chiramel AI, Bartenschlager R, Döhner H, Stilgenbauer S, Pscherer
A, Lichter P, et al: miRNA-130a targets ATG2B and DICER1 to inhibit
autophagy and trigger killing of chronic lymphocytic leukemia
cells. Cancer Res. 72:1763–1772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang R, Yang C, Ma X, Wang Y, Luo D, Huang
C, Xu Z, Liu P and Yang L: MiR-let-7a inhibits cell proliferation,
migration, and invasion by down-regulating PKM2 in gastric cancer.
Oncotarget. 7:5972–5984. 2016.PubMed/NCBI
|
|
18
|
Sarbassov DD, Ali SM, Kim DH, Guertin DA,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: Rictor, a
novel binding partner of mTOR, defines a rapamycin-insensitive and
raptor-independent pathway that regulates the cytoskeleton. Curr
Biol. 14:1296–1302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang N, Wu J, Qiu W, Lyu Q, He J, Xie W,
Xu N and Zhang Y: MiR-15a and miR-16 induce autophagy and enhance
chemosensitivity of Camptothecin. Cancer Biol Ther. 16:941–948.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wan G, Xie W, Liu Z, Xu W, Lao Y, Huang N,
Cui K, Liao M, He J, Jiang Y, et al: Hypoxia-induced MIR155 is a
potent autophagy inducer by targeting multiple players in the MTOR
pathway. Autophagy. 10:70–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu K, Huang J, Xie M, Yu Y, Zhu S, Kang
R, Cao L, Tang D and Duan X: MIR34A regulates autophagy and
apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy.
10:442–452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Korkmaz G, Le Sage C, Tekirdag KA, Agami R
and Gozuacik D: miR-376b controls starvation and mTOR
inhibition-related autophagy by targeting ATG4C and BECN1.
Autophagy. 8:165–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Faller WJ, Jackson TJ, Knight JR, Ridgway
RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB,
et al: mTORC1-mediated translational elongation limits intestinal
tumour initiation and growth. Nature. 517:497–500. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu J, Wang Y, Tan X and Jing H: MicroRNAs
in autophagy and their emerging roles in crosstalk with apoptosis.
Autophagy. 8:873–882. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mazumder A, Bose M, Chakraborty A,
Chakrabarti S and Bhattacharyya SN: A transient reversal of
miRNA-mediated repression controls macrophage activation. EMBO Rep.
14:1008–1016. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget.
6:8474–8490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kimura S, Fujita N, Noda T and Yoshimori
T: Monitoring autophagy in mammalian cultured cells through the
dynamics of LC3. Methods Enzymol. 452:1–12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Sun Y and Zhao A: MicroRNA-134
suppresses cell proliferation in gastric cancer cells via targeting
of GOLPH3. Oncol Rep. 37:2441–2448. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ying J, Xu Q, Liu B, Zhang G, Chen L and
Pan H: The expression of the PI3K/AKT/mTOR pathway in gastric
cancer and its role in gastric cancer prognosis. Onco Targets Ther.
8:2427–2433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|