Introduction
Autophagy is an evolutionarily conserved degradation
process that plays an important role in maintaining cellular
homoeostasis and metabolism. Unlike the proteasome system,
autophagy mainly disintegrates long-lived proteins and impaired
organelles, which are engulfed by autophagosomes and transported to
lysosomes for the digestion when it is induced under stress
conditions such as mitochondrial depolarization, nutrient
starvation, aggregation of toxic proteins, and pathogen infection
(1,2). It is well known that there are three
main types of autophagy: Macroautophagy (hereafter referred to as
autophagy), microautophagy and chaperone-mediated autophagy (CMA).
Microautophagy directly degrades cellular bulks via endocytic
membrane emboly, whereas CMA only delivers specifically marked
proteins to the lysosome. Macroautophagy involves the sequestration
of cytosol or cytoplasmic organelles by the formation of
autophagosomes that subsequently fuse with endosomes and eventually
with lysosomes, thereby creating autophagolysosomes or
autolysosomes, in which the cell contents are degraded (3,4).
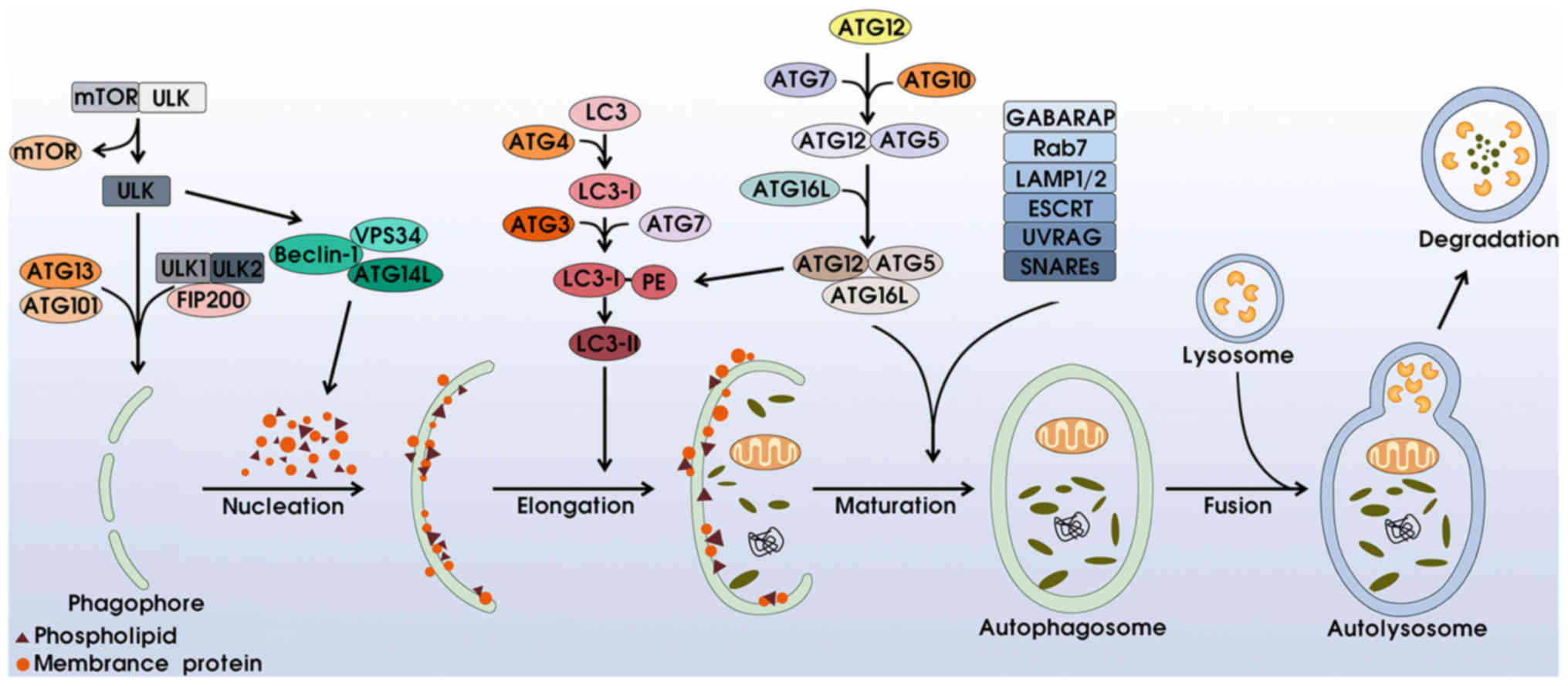
Autophagy is a multi-step process involving at least
four distinct phases: i) Initiation and nucleation: the activation
of autophagy is associated with the unc-51-like kinase (ULK)
complex and phosphatidylinositol 3-kinase (PI3K) complex. Under
stressful conditions such as hypoxia and starvation, mTOR complex 1
(mTORC1) is separated from the ULK complex, subsequently activating
ULK1/2 and phosphorylating ATG13, ATG101 and FIP200 to induce
phagophore formation. Simultaneously, the excitation of
Beclin-1-VPS34-ATG14L generates phosphatidylinositol 3-phosphate
(PI3P), motivating membrane proteins and phospholipids for
phagophore nucleation (5–7). ii) Membrane elongation and completion:
this process is mainly regulated via two ubiquitin-like conjugation
complexes. On the one hand, combining with ATG4, LC3 (microtubule
associated protein 1 light-chain 3) proteins split into LC3-I,
which are incorporated with phosphatidylethanolamine (PE) with the
help of ATG3 and ATG17, for the transformation of LC3-II, which
surrounds both the internal and external membranes of the
phagosome. In addition, the ATG12-ATG5 dipolymer conjugates with
ATG16L to form the ATG12-ATG5-ATG16L complex, which can promote LC3
transformation and accelerate transformation from phagophore to
autophagosome (8–12). iii) Maturation: the upregulation of
γ-aminobutyric acid receptor-associated proteins (GABARAP)
contributes to phagophore closure and the formation of airtight
double-membrane autophagosomes (13). Rab7, LAMP1/2 proteins, ESCRT, UVRAG
and SNAREs also participate in this procedure (1,14,15).
iv) Degradation: the outer membrane of the autophagosome fuses with
the lysosome to create an autolysosome, whereas the inner membrane
and engulfed components are degraded into small molecular nutrients
(Fig. 1).
Mounting evidence has certified that autophagy
affects cell metabolism, including cell proliferation,
differentiation, survival and apoptosis (16). Autophagy facilitates clearance of
damaged cytoplasmic components and maintains cell homeostasis when
cells are submitted to hostile environments. Nevertheless,
immoderate autophagy can induce autophagic cell death (ACD), also
known as type II programmed cell death, leading to tumor
self-degradation (17–20). In addition, excessive autophagy can
deplete mitochondria and metabolic substances, thereby trapping
cells in the state of hunger (21,22).
Autophagy can also mobilize cell survival substances to
autophagosomes for degradation, incurring the accumulation of
reactive oxygen species (ROS), caspases, and concomitant cell death
(23,24). This evidence may also explain the
antitumor mechanisms of autophagy. Notably, in blood tumors, the
role of autophagy remains elusive. In this review, we discuss the
autophagy molecular mechanism, its relative signal pathways, and
focus on its dual role in hematological malignancies, which may
provide some novel strategies for treatment of hematological
tumors.
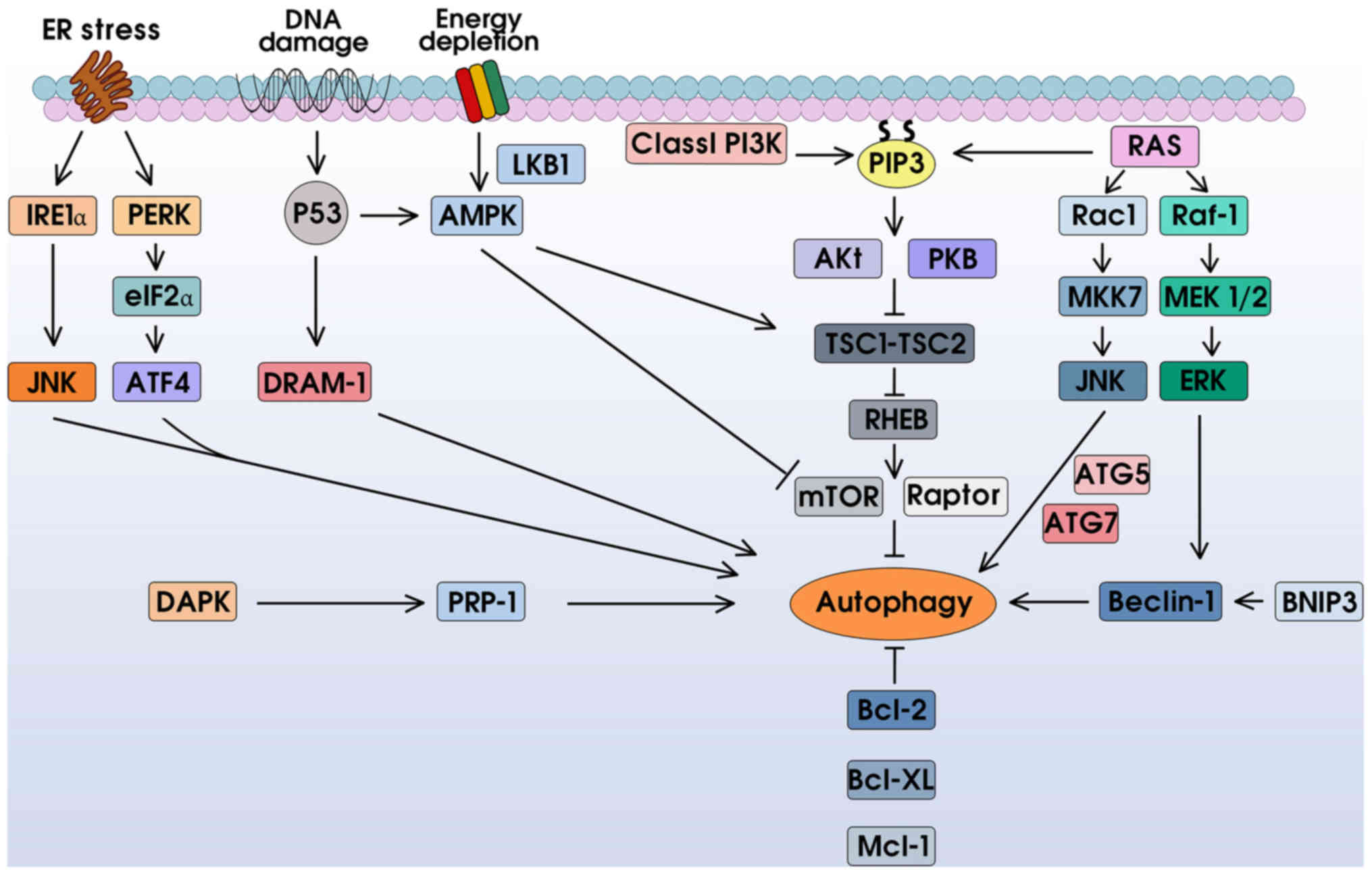
Autophagy signal pathways in cancer
The autophagy signal pathways are intricate
networks. In this section, we introduce the main molecular
regulators of autophagy. A deeper understanding of these regulatory
pathways may provide us with potential targets against
tumorigenesis (Fig. 2).
Phosphatidylinositol 3-kinase (PI3K)
signaling
The phosphatidylinositol 3-kinases are a family of
enzymes that can phosphorylate phosphoinositides of cell membranes,
and have thus been deemed as the initiators of cellular signal
transduction. PI3Ks are divided into three classes. However, only
Class I and III PI3Ks have been shown to be associated with
autophagy (25). The Class I PI3K
promotes the generation of phosphatidylinositol-3,4,5-trisphosphate
(PIP3) at the plasma membrane, thereby increasing Akt/PKB
recruitment. The activated Akt/PKB hinders its downstream TSC1-TSC2
complex, influences GTP binding, and motivates the mammalian target
of rapamycin (mTOR), which acts as a negative regulator of
autophagy by sensing variable ATPs, amino acids, and energy
metabolism (2,26). In addition, mTOR can be directly
inhibited by rapamycin treatment, leading to autophagy induction.
Conversely, Class III PI3K activates autophagy. It phosphorylates
PI to generate PI3P, which contributes to the movement of lysosomal
enzymes for component degradation (27). Previous findings have shown that the
Class III PI3K integral protein Beclin-1, which is also known as
the mammalian homolog of an essential yeast autophagy gene
(Atg6), can facilitate phagosome nucleation, maturation and
clearance of apoptotic cells (28).
Therefore, knockdown or silence of Beclin-1 inhibits autophagy and
causes cell death (29).
ER stress response
The endoplasmic reticulum (ER) is in charge of
protein folding. Under stressed circumstances, the accumulation of
immature proteins provokes ER stress response and boosts autophagy,
resulting in autophagic cell death (30). This process is mainly regulated via
the IRE1α-JNK and PERK-eIF2α-ATF4 pathways (31,32).
AMP-activated protein kinase (AMPK)
pathway
AMP-activated protein kinase is the energy sensor.
In energy-defective situations (decreased ATP/AMP ratio and
starvation), the AMPK is excited by LKB1 and p53, leading to
autophagy activation via the positive regulation of the TSC1-TSC2
complex or negative regulation of mTOR. AMPK can also directly
suppress the raptor, which is a subunit of mTORC1 that inhibits
autophagy activity (16,33).
RAS system
The relationship between RAS and autophagy is
multifaceted. In fact, RAS plays both positive and negative roles
in autophagy with different pathways. On the one hand, RAS inhibits
autophagy by directly activating Class I PI3K-Akt-mTOR1. By
contrast, RAS upregulates Raf-1/MEK/ERK- or Rac1/MKK7/JNK-dependent
autophagy activity with the help of ATG5/7 and Beclin-1 (34–36).
The Bcl-2 family
The Bcl-2 family is divided into three types, based
on their BH domains and functions. Bcl-2, Bcl-xL and myeloid cell
leukemia sequence-1 (MCL-1) act as anti-apoptosis pathways, while
BAX-like proteins (BAX, BAK) and BH3-only proteins (BNIP3) promote
apoptosis (2). Similarly, these
proteins play a bidirectional role in autophagy. On the one hand,
the BNIP3 can integrate with Bcl-2, which correspondingly decreases
Bcl-2 binding to Beclin-1, thereby liberating Beclin-1 for
autophagy induction (37,38). By contrast, Bcl-2, Bcl-xL and MCL-1
inhibit autophagy.
Other autophagy networks
Death-associated protein kinase (DAPK) is associated
with autophagy in cancer. Hyperactive DAPK influences its related
protein kinase DRP-1, which increases plasma membrane flexing and
blebbing, enabling phagocytosis (39). In addition to the aforementioned
p53-AMPK network, DNA damage can trigger autophagy through
p53-dependent damage-regulated autophagy modulator-1 (DRAM-1)
activation (40).
In summary, autophagy networks are sophisticated.
Dysfunctions of these signal pathways are involved in
tumorigenesis, and targeting signal transduction proteins may
provide potential therapeutic schemes for cancer.
Autophagy in hematological malignancies
Several studies have demonstrated the important role
of autophagy in blood tumors. Autophagy participates in the
regulation of hematopoietic stem cell (HSC) differentiation, and
the downregulation of autophagy would facilitate leukemic
transformation from normal HSC (41). Recently, the potential anti-leukemic
effect of tetrandrine was proven through the promotion of the
autophagy-dependent megakaryocytic differentiation of leukemia
cells (42). In addition, Schläfli
et al revealed that autophagy scaffold protein ALFY was, not
only involved in APL cell differentiation induced by ATRA, but also
contributed to degradation of oncoprotein PML-RARα (43). However, autophagy also played a
cytoprotective role and led to chemoresistance in leukemia. In
lymphoma, on the one hand, autophagy was capable of suppressing
lymphomagenesis through the modulation of autophagy proteins and
signal pathways. On the other hand, activated autophagy acted as
the mechanism of lymphoma cell survival, and additional autophagy
inhibitors enhanced antitumor activity (44). The survival of myeloma cells was
also dependent on autophagy for protein degradation and energy
recycling, which potentially results in drug resistance. For
example, Zhang et al reported that bortezomib induced
pro-survival autophagy, which reduced the chemosensitivity of
bortezomib in myeloma cells (45),
whereas excessive autophagy may lead to autophagic cell death,
providing an available approach for myeloma treatment (46). Therefore, we discussed the dual role
of autophagy mediated by conventional chemotherapeutics and
summarized promising strategies associated with autophagy
modulation in Table I.
 | Table I.Therapeutic compounds-induced
autophagy and its outcomes in hematological malignancies. |
Table I.
Therapeutic compounds-induced
autophagy and its outcomes in hematological malignancies.
| Compounds | Signal
pathways | Types of tumor | Outcome | Additional
treatment | Refs. |
|---|
| Fludarabine | Mcl-1-BECN1, Bcl-2,
AMPK | Leukemia | Cell
death/survival | Obatoclax | 48 |
| Metformin | AMPK-mTOR | B/T-lymphoma,
AML | Cell death | Doxorubicin,
temsirolimus, sorafenib | 52–53 |
| Sertraline | Not mentioned | AML | Cell death | Not mentioned | 54 |
| ATRA | Bcl-2, mTOR | APL | Cell death | Everolimus,
rapamycin | 55–58 |
|
As2O3 | MEK-ERK,
Beclin-1 | Leukemia | Cell death | Not mentioned | 59–60 |
| Idarubicin | AMPK-mTOR,
Akt-mTOR | Leukemia | Cell death | Not mentioned | 61 |
| Dexamethasone | PML-Akt | Leukemia, multiple
myeloma | Cell death | Rapalogues | 62–63 |
|
2-Deoxy-5-azacytidine | Intrinsic
pathway | CML | Cell death | Not mentioned | 83 |
| AG490 (TKI) | STAT3-Mcl-1 | PEL | Cell survival | Bafilomycin,
2-phenylethynesulfonamide | 64,66 |
| Imatinib | PI3K-AKT-mTOR,
Beclin-1 | CML | Cell
survival/death | 3-MA, CQ,
NVP-BEZ235 | 67–72 |
| Interferon | JAK1-STAT1, NF-κB,
BECN1 | CML | Cell survival | CQ | 73 |
| Cytarabine | Akt-mTOR, AMPK,
ERK | Leukemia | Cell survival | Bafilomycin,
CQ | 74 |
| Doxorubicin,
Melphalan | Bcl-2-Beclin-1,
Beclin-1-PI3KC3 | MM | Cell survival | HCQ, 3-MA | 75 |
| Bortezomib | JNK-Bcl2,
Bcl-2-Beclin-1, Beclin-1-PI3KC3 | B-ALL,
B-lymphoma | Cell survival | CQ, 3-MA,
SP600125 | 76–77 |
| Daunorubicin | MEK-ERK | Myeloid
leukemia | Cell survival | U0126 | 78 |
| Dasatinib | AMPK | CLL, Ph+
leukemia | Cell
survival/death | Not mentioned | 79–80 |
| Sorafenib | mTOR | MM | Cell survival | 3-MA, CQ,
ABT737 | 81 |
| L-asparaginase | ROS-p53 | ALL | Cell survival | CQ | 82 |
| 5-Azacitidine | Beclin-1 | MDS | Cell survival | CQ, leupeptin | 84 |
Autophagy as a pro-death
mechanism
The pro-death role of autophagy is vital for
suppressing tumor progression and enhancing antitumor response.
Fludarabine (Fd), a nucleoside analog that inhibits DNA/RNA
synthesis and DNA repair, is widely used for chronic lymphocytic
leukemia (CLL). Mahoney et al observed fludarabine-triggered
autophagy activity in CLL cells. However, the inhibition of
autophagy did not enhance or diminish cell death induced by
fludarabine (47). Recently, a
study by Sharma et al reported that fludarabine induced
BECN-dependent autophagy by decreasing Mcl-1 expression, resulting
in autophagic cell death in Fd-sensitive leukemic B cells (48). However, in Fd-resistant leukemic
cells, Mcl-1 expression was increased, which inhibited
BECN-dependent autophagic cell death. The Bcl-2 family also
contributed to promoting cell survival via sequestering BECN.
Moreover, activated AMPK-dependent autophagy assisted Fd-resistant
cells escape cell death. Of note, adding the BCL-2 inhibitor
obatoclax can induce cell death in both Fd-sensitive and
Fd-resistant leukemic cells. The findings suggest that combining
autophagy repressors such as Mcl-1, Bcl-2 and AMPK inhibitors, with
fludarabine may improve the curative effect for CLL patients,
especially those who acquire Fd-resistance after several cycles of
fludarabine chemotherapy.
Metformin, an anti-diabetic agent, has already been
proven to play a positive role in cancer control, including acute
leukemia (47–50). Wang et al reported that
metformin enhanced the anti-leukemic effect of sorafenib by
triggering mTOR-dependent autophagy in FLT3-ITD-positive AML, which
suggested the combination strategy of the two drugs in AML
treatment (51). Metformin also
induced a dose-dependent inhibition of lymphoma cell proliferation
in vitro and in vivo via the negative control of the
mTOR signal pathway (52).
Metformin-induced autophagy enhanced the anti-lymphoma effect of
doxorubicin and temsirolimus (mTOR inhibitor), offering a novel
therapeutic strategy.
Sertraline, a widely used antidepressant drug, was
found to induce leukemia cell death mediated by autophagy and
apoptosis. It also enhanced the sensitivity of chemotherapy drugs,
thereby providing potential alternative strategies for leukemia
treatment (53).
Trocoli et al observed that all-trans
retinoic acid (ATRA) can promote autophagy by reducing the activity
of Bcl-2 and mTOR in acute promyelocytic leukemia (APL). In
addition, the upregulation of Beclin-1 contributed to the stability
of mature APL cells in a non-autophagic manner, although
autophagy-related cell differentiation was not explored (54). Notably, by combining ATRA with
RAD001 (mTOR1C inhibitor), Nishioka et al observed step-down
cell growth and enhanced cell differentiation of AML cells
(55). Similarly, Isakson et
al reported that ATRA-dependent autophagy induced the
degradation of PML-RARα, which is known to create abnormal
granulocytic differentiation, via inhibition of the mTOR pathway as
well (56). Thus, the simultaneous
use of ATRA and mTOR suppressant may be a valid protocol for APL
patients.
Arsenic trioxide (As2O3),
another metalloid poison that has conventionally been used in APL
patients, also induced cell death. We previously reported that
As2O3 led to cell growth arrest by means of
both apoptosis and Beclin-1-dependent autophagy (58). Goussetis et al also
documented a MEK-ERK-mediated autophagic cell death in leukemia
cells (59). The studies
demonstrated As2O3-induced autophagy as the
mechanism of cell death.
Autophagy in response to idarubicin served as a
pro-death mechanism in leukemic cells as well. Idarubicin
restrained mTOR either by upregulation of its upstream inhibitor
AMPK, or downregulation of its activator Akt, thus leading to
autophagic cell death. Pharmacological impairment (bafilomycin A1
or chloroquine) of autophagy partially reduced the cytotoxicity of
idarubicin towards REH cells, which highlighted its cell-killing
function (60).
Dexamethasone (Dex) also contributed to cell death
in lymphoid leukemia and multiple myeloma. Several experiments
observed Dex-induced autophagic cell death through the marked
upregulation of promyelocytic leukemia protein (PML) and
PML-dependent Akt dephosphorylation (61,62).
In depth studies on PML and Akt-dependent intracellular pathways
may provide optimized use of Dex in the treatment of lymphoid
malignancies.
Autophagy as a pro-survival
mechanism
Besides its potential to induce cell death,
autophagy is appropriated to serve as a cell survival mechanism as
well.
For example, the tyrosine kinase inhibitor AG490 was
found to induce both apoptosis and autophagy by inhibiting STAT3,
reducing the expression of HSF1/HSP70, and downregulating Mcl-1 in
primary effusion lymphoma (PEL) cells. A combination of AG490 with
autophagy inhibitor bafilomycin is believed to enhance the
cytotoxic effect (63,64). The HSP70 and its main transcription
factor HSF1 have been reported as essential for PEL survival by
affecting protein folding and cell stability, and HSP70 inhibition
via 2-phenylethynesulfonamide could block the autophagic process
and induce immunogenic cell death (65). Therefore, a combination of AG490 and
HSP inhibitors may provide a potential treatment strategy. Another
TKI, imatinib (IM), was also reported to activate a cytoprotective
autophagy in chronic myeloid leukemia (CML) (66). Rothe et al identified that
CML stem/progenitor cells obtained imatinib resistance due to its
intensive pro-survival activities associated with elevated
autophagy gene transcripts and proteins, especially ATG4B (67). Mancini et al describe a
survival role of autophagy in imatinib-treated Bcr-Abl-positive
cells, possibly connected with ER stress and following responses
(68). The group of Howard Hughes
Medical Institute suggested that imatinib-induced autophagy was
associated with the Bcr-Abl/PI3K/Akt/FOXO4/ATF5/mTOR pathways
(69). Recently, the combination of
NVP-BEZ235 (dual PI3K/mTOR inhibitor) and imatinib was reported to
significantly inhibit CML cell growth and proliferation, as well as
enhance sensitivity to imatinib in imatinib-resistant CML cells
(70). Inversely, imatinib-induced
autophagy also isolated the mosaic gene Bcr-Abl in
autophagosomes through the regulation of Beclin-1, which prevented
disease progression (71).
Therefore, autophagy inhibition of the aforementioned specific
autophagic pathways would enhance the treatment effect of imatinib
in CML.
Zhu et al also reported a pro-survival role
of autophagy in CML treated with interferon. Interferon-1 activated
both JAK1-STAT1 and NF-κB pathways, leading to increasing
BECN1-ATG5-ATG7, one of the major regulators of the classical
autophagy pathway. Inhibiting autophagy by using chloroquine
enhanced the cytotoxic effect of interferon-1 (72).
Cytarabine was reported to increase the
phosphorylation of AMPK/ERK, inhibit Akt and attenuate mTOR
activity, thereby inducing cytoprotective autophagy in leukemic
cells. Pharmacological (bafilomycin A1 and chloroquine treatment)
or genetic (siRNA downregulation of either LC3b or autophagic cargo
receptor p62) impairment of autophagy markedly increased cell death
by accumulating DNA fragmentation and mitochondrial damage as well
as activating oxidative stress, thus providing the combination
strategy of cytarabine and autophagy inhibitor in leukemia
treatment (73).
Notably, doxorubicin and melphalan were shown to
motivate autophagy in multiple myeloma (MM) cells, which provided
an adaptive condition for evading DNA-damaging stimulus. The
increasing Beclin-1-PI3KC3 complex and Bcl-2 disintegrated from
Beclin-1, functioned as a cell survival mechanism in this process.
Thus, the inhibition of autophagy with DNA-damaging agents likely
augmented anti-myeloma activities without additional side effects
(74). In cases of acute
lymphocytic leukemia (ALL) treated with bortezomib, cytoprotective
autophagy was also associated with the balance between the
Bcl-2-Beclin-1 complex and the Beclin-1-PI3KC3 complex, as its
downregulation by chloroquine potentiated anti-ALL activity
(75). The aforementioned results
indicated a pivotal role of Bcl-2-Beclin-1-PI3KC3 complexes in
autophagy and provided a novel target to improve clinical efficacy.
Another experiment provided evidence of autophagic cell
proliferation in aggressive B-cell lymphoma managed with
bortezomib. In that study, ER stress-associated IRE1-JNK was
stimulated, followed by phosphorylated Bcl-2 and elevated LC3-II,
which was widely used for autophagic flex surveillance. Certainly,
the addition of JNK inhibitor SP600125 enhanced the anti-lymphoma
effects of bortezomib (76).
Anthracycline daunorubicin (DNR) revealed potent
leukemia killing activity by DNA damage. However, DNR-induced
autophagy was inversely cytoprotective, and authors of that study
identified the activation of MAPK (MEK-ERK) in DNR-treated myeloid
leukemia cells (77). Obviously,
adding MEK1/2 inhibitor U0126 to daunorubicin would enhance
chemotherapeutic response and reduce drug toxicities.
The example of the autophagic pro-survival role in
CLL originated from the observation that dasatinib was capable of
sustaining stable cellular metabolism from AMPK activation and
consequent autophagy motivation, which was also the mechanism of
drug resistance (78). However,
Morita et al reported that dasatinib induced
autophagy-dependent cell death in Bcr-Abl-positive leukemia cells
with central nervous system (CNS) infiltration in vitro and
in vivo, confirming the important mechanism of autophagy for
CNS leukemia treatment (79).
Of note, sorafenib regulated the mTOR or Mcl-1
pathway in multiple myeloma with different outcomes (80): on the one hand, mTOR inhibition by
sorafenib induced cytoprotective autophagy; on the other hand,
sorafenib-triggered downregulation of Mcl-1 was essential to cell
death, and the addition of ABT737 (Bcl-2 antagonist) improved the
efficacy of sorafenib. As a result, a combination of sorafenib with
autophagy inhibitor and ABT737 is a potentially new treatment
strategy in multiple myeloma.
Recently, Takahashi et al demonstrated that
L-asparaginase (L-asp)-triggered autophagy contributed to ALL cell
survival by eliminating accumulated DNA damage and injured
mitochondria with the help of a ROS-p53 loop, indicating that
combination treatment with an autophagy inhibitor enhanced
anti-leukemic effects of L-asp and overcame drug resistance for ALL
patients (81).
Thus, the aforementioned studies fully demonstrated
the dual role of autophagy in hematological malignancies, of which
induction or inhibition may improve the therapeutic efficacy
depending on the types of antitumor agents and pathways of stress
responses.
Conclusions and perspectives
Autophagy, an evolutionarily conserved degradation
process, has recently been proven to play a dual role in cancer, by
triggering autophagic cell death or by protecting tumor cells from
harsh conditions and DNA damage. From the research discussed above
in this review, autophagy can be triggered via various conventional
chemotherapeutic agents in blood tumors, which results in different
effects. Indeed, the outcome of autophagy can be diametrically
opposite. Some agent-induced autophagy promotes tumor cell death
and prevents disease progression (47–62),
whereas in some circumstances, activated autophagy acts as a
protective mechanism, and adding autophagy inhibitors enhances
treatment efficacy (72–77,80,81).
Furthermore, in a few cases, agent-induced autophagy fills both
pro-death and pro-survival roles (66–71,78,79,82,83).
Thus, therapeutic development in blood tumors targeting autophagy
remains to be elucidated. It is well known that, the autophagy
signal transduction pathway is intricate, and upregulation or
downregulation of autophagy is hampered through diverse molecular
regulators at multiple levels. Therefore, the role of autophagy
modulated by these drugs depends on cell types, differentiation
status, and signal transformation pathways (84,85).
We present different changes of autophagy signaling
pathways induced by conventional chemotherapeutic agents and
potential combination treatment strategies in Table I, which may improve the therapeutic
effect and provide novel targets for the treatment of hematologic
malignancies. However, several basic questions remain to be
elucidated.
First, induction or inhibition of autophagy is a
complicated process because of its dual role in different types of
blood tumors with different chemotherapy regimens. Another
significant challenge is to determine concrete molecular mechanisms
in the process, and interpret the crosstalk between autophagy
pathways and intracellular responses. Fully understanding these
issues may provide more precise treatment involving the addition of
autophagy regulators such as mTOR repressor, AMPK agonist, and
hydroxychloroquine to increase efficacy and overcome drug
resistance. Additionally, some studies did report that autophagy
contributed to cell protection and its pharmacological inhibition
improved drug sensitivity. Further research is required to
determine whether autophagy inhibitors directly affect cell fate or
affect it in a more indirect manner.
In the near future, with awareness of the molecular
mechanism and accurate targets underlying this complex process,
clinicians will identify patients who are likely to benefit from
treatment involving autophagy regulation by agonists or
blockers.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (nos. 81500110, 81370645 and
81670178), the National Key Research and Development Program of
China (no. 2016YFC090150X) and the Research Project for Practice
Development of National TCM Clinical Research Bases (no.
JDZX2015113).
Available data and materials
All data generated or analyzed during this study are
included in this published article.
Author's contributions
JSH and YLS conceived and designed the study. JSH
and WJY researched related literatures and drafted the manuscript.
QWB and LH reviewed and revised the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of
interest.
Glossary
Abbreviations
Abbreviations:
|
ATG
|
autophagy-related genes
|
|
AMPK
|
AMP-activated protein kinase
|
|
ATRA
|
all-trans retinoic acid
|
|
As2O3
|
arsenic trioxide
|
|
ACD
|
autophagic cell death
|
|
ALL
|
acute lymphocytic leukemia
|
|
APL
|
acute promyelocytic leukemia
|
|
CQ
|
chloroquine
|
|
CML
|
chronic myeloid leukemia
|
|
CLL
|
chronic lymphocytic leukemia
|
|
CMA
|
chaperone-mediated autophagy
|
|
DAPK
|
death-associated protein kinase
|
|
DRAM-1
|
damage-regulated autophagy modulator
1
|
|
Dex
|
dexamethasone
|
|
DNR
|
daunorubicin
|
|
ER
|
endoplasmic reticulum
|
|
Fd
|
fludarabine
|
|
GABARAP
|
γ-aminobutyric acid
receptor-associated proteins
|
|
HCQ
|
hydroxychloroquine
|
|
HSC
|
hematopoietic stem cell
|
|
HSP
|
heat shock protein
|
|
IM
|
imatinib
|
|
LC3
|
microtubule-associated protein 1
light-chain 3
|
|
3-MA
|
3-methyladenine
|
|
MM
|
multiple myeloma
|
|
MDS
|
myelodysplastic syndrome
|
|
mTOR
|
mammalian target of rapamycin
|
|
PI3P
|
phosphatidylinositol 3-phosphate
|
|
PIP3
|
phosphatidylinositol
(3,4,5)-trisphosphate
|
|
PE
|
phosphatidylethanolamine
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
PML
|
primary effusion lymphoma
|
|
ROS
|
reactive oxygen species
|
|
TKI
|
tyrosine kinase inhibitor
|
|
ULK
|
unc-51-like kinase
|
References
|
1
|
Dong Z, Liang S, Hu J, Jin W, Zhan Q and
Zhao K: Autophagy as a target for hematological malignancy therapy.
Blood Rev. 30:369–380. 2016. View Article : Google Scholar
|
|
2
|
Helgason GV, Holyoake TL and Ryan KM: Role
of autophagy in cancer prevention, development and therapy. Essays
Biochem. 55:133–151. 2013. View Article : Google Scholar
|
|
3
|
Duffy A, Le J, Sausville E and Emadi A:
Autophagy modulation: A target for cancer treatment development.
Cancer Chemother Pharmacol. 75:439–447. 2015. View Article : Google Scholar
|
|
4
|
Klionsky DJ: The molecular machinery of
autophagy: Unanswered questions. J Cell Sci. 118:7–18. 2005.
View Article : Google Scholar
|
|
5
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar
|
|
6
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar
|
|
7
|
Devereaux K, Dall'Armi C, Alcazar-Roman A,
Ogasawara Y, Zhou X, Wang F, Yamamoto A, De Camilli P and Di Paolo
G: Regulation of mammalian autophagy by class II and III PI
3-kinases through PI3P synthesis. PLoS One. 8:e764052013.
View Article : Google Scholar
|
|
8
|
Petibone DM, Majeed W and Casciano DA:
Autophagy function and its relationship to pathology, clinical
applications, drug metabolism and toxicity. J Appl Toxicol.
37:23–37. 2017. View
Article : Google Scholar
|
|
9
|
Hamasaki M, Shibutani ST and Yoshimori T:
Up-to-date membrane biogenesis in the autophagosome formation. Curr
Opin Cell Biol. 25:455–460. 2013. View Article : Google Scholar
|
|
10
|
Fujita N, Itoh T, Omori H, Fukuda M, Noda
T and Yoshimori T: The Atg16L complex specifies the site of LC3
lipidation for membrane biogenesis in autophagy. Mol Biol Cell.
19:2092–2100. 2008. View Article : Google Scholar
|
|
11
|
Hanada T, Noda NN, Satomi Y, Ichimura Y,
Fujioka Y, Takao T, Inagaki F and Ohsumi Y: The Atg12-Atg5
conjugate has a novel E3-like activity for protein lipidation in
autophagy. J Biol Chem. 282:37298–37302. 2007. View Article : Google Scholar
|
|
12
|
Geng J and Klionsky DJ: The Atg8 and Atg12
ubiquitin-like conjugation systems in macroautophagy. ‘Protein
modifications: Beyond the usual suspects’ review series. EMBO Rep.
9:859–864. 2008. View Article : Google Scholar
|
|
13
|
Weidberg H, Shvets E, Shpilka T, Shimron
F, Shinder V and Elazar Z: LC3 and GATE-16/GABARAP subfamilies are
both essential yet act differently in autophagosome biogenesis.
EMBO J. 29:1792–1802. 2010. View Article : Google Scholar
|
|
14
|
Chua CE, Gan BQ and Tang BL: Involvement
of members of the Rab family and related small GTPases in
autophagosome formation and maturation. Cell Mol Life Sci.
68:3349–3358. 2011. View Article : Google Scholar
|
|
15
|
Liang C, Lee JS, Inn KS, Gack MU, Li Q,
Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al:
Beclin1-binding UVRAG targets the class C Vps complex to coordinate
autophagosome maturation and endocytic trafficking. Nat Cell Biol.
10:776–787. 2008. View Article : Google Scholar
|
|
16
|
Pan H, Chen L, Xu Y, Han W, Lou F, Fei W,
Liu S, Jing Z and Sui X: Autophagy-associated immune responses and
cancer immunotherapy. Oncotarget. 7:21235–21246. 2016.
|
|
17
|
Stellrecht CM, Vangapandu HV, Le XF, Mao W
and Shentu S: ATP directed agent, 8-chloro-adenosine, induces AMP
activated protein kinase activity, leading to autophagic cell death
in breast cancer cells. J Hematol Oncol. 7:232014. View Article : Google Scholar
|
|
18
|
Baehrecke EH: Autophagy: Dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar
|
|
19
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar
|
|
20
|
Bursch W: The autophagosomal-lysosomal
compartment in programmed cell death. Cell Death Differ. 8:569–581.
2001. View Article : Google Scholar
|
|
21
|
Lin L and Baehrecke EH: Autophagy, cell
death, and cancer. Mol Cell Oncol. 2:e9859132015. View Article : Google Scholar
|
|
22
|
Scott RC, Juhász G and Neufeld TP: Direct
induction of autophagy by Atg1 inhibits cell growth and induces
apoptotic cell death. Curr Biol. 17:1–11. 2007. View Article : Google Scholar
|
|
23
|
Yu L, Wan F, Dutta S, Welsh S, Liu Z,
Freundt E, Baehrecke EH and Lenardo M: Autophagic programmed cell
death by selective catalase degradation. Proc Natl Acad Sci USA.
103:4952–4957. 2006. View Article : Google Scholar
|
|
24
|
Nezis IP, Shravage BV, Sagona AP, Lamark
T, Bjørkøy G, Johansen T, Rusten TE, Brech A, Baehrecke EH and
Stenmark H: Autophagic degradation of dBruce controls DNA
fragmentation in nurse cells during late Drosophila melanogaster
oogenesis. J Cell Biol. 190:523–531. 2010. View Article : Google Scholar
|
|
25
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar
|
|
26
|
Hanada M, Feng J and Hemmings BA:
Structure, regulation and function of PKB/AKT - a major therapeutic
target. Biochim Biophys Acta. 1697:3–16. 2004. View Article : Google Scholar
|
|
27
|
Yu X, Long YC and Shen HM: Differential
regulatory functions of three classes of phosphatidylinositol and
phosphoinositide 3-kinases in autophagy. Autophagy. 11:1711–1728.
2015. View Article : Google Scholar
|
|
28
|
McKnight NC and Zhenyu Y: Beclin 1, an
essential component and master regulator of PI3K-III in health and
disease. Curr Pathobiol Rep. 1:231–238. 2013. View Article : Google Scholar
|
|
29
|
Boya P, González-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar
|
|
30
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar
|
|
31
|
Moretti L, Yang ES, Kim KW and Lu B:
Autophagy signaling in cancer and its potential as novel target to
improve anticancer therapy. Drug Resist Updat. 10:135–143. 2007.
View Article : Google Scholar
|
|
32
|
Ron D: Translational control in the
endoplasmic reticulum stress response. J Clin Invest.
110:1383–1388. 2002. View Article : Google Scholar
|
|
33
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar
|
|
34
|
Schmukler E, Kloog Y and Pinkas-Kramarski
R: Ras and autophagy in cancer development and therapy. Oncotarget.
5:577–586. 2014. View Article : Google Scholar
|
|
35
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar
|
|
36
|
Byun JY, Yoon CH, An S, Park IC, Kang CM,
Kim MJ and Lee SJ: The Rac1/MKK7/JNK pathway signals upregulation
of Atg5 and subsequent autophagic cell death in response to
oncogenic Ras. Carcinogenesis. 30:1880–1888. 2009. View Article : Google Scholar
|
|
37
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar
|
|
38
|
Levine B, Sinha SC and Kroemer G: Bcl-2
family members: Dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar
|
|
39
|
Inbal B, Bialik S, Sabanay I, Shani G and
Kimchi A: DAP kinase and DRP-1 mediate membrane blebbing and the
formation of autophagic vesicles during programmed cell death. J
Cell Biol. 157:455–468. 2002. View Article : Google Scholar
|
|
40
|
Zeng X, Yan T, Schupp JE, Seo Y and
Kinsella TJ: DNA mismatch repair initiates 6-thioguanine-induced
autophagy through p53 activation in human tumor cells. Clin Cancer
Res. 13:1315–1321. 2007. View Article : Google Scholar
|
|
41
|
Auberger P and Puissant A: Autophagy, a
key mechanism of oncogenesis and resistance in leukemia. Blood.
129:547–552. 2017. View Article : Google Scholar
|
|
42
|
Liu T, Zhang Z, Yu C, Zeng C, Xu X, Wu G,
Huang Z and Li W: Tetrandrine antagonizes acute megakaryoblastic
leukemia growth by forcing autophagy-mediated differentiation. Br J
Pharmacol. 174:4308–4328. 2017. View Article : Google Scholar
|
|
43
|
Schläfli AM, Isakson P, Garattini E,
Simonsen A and Tschan MP: The autophagy scaffold protein ALFY is
critical for the granulocytic differentiation of AML cells. Sci
Rep. 7:129802017. View Article : Google Scholar
|
|
44
|
Pierdominici M, Barbati C, Vomero M,
Locatelli SL, Carlo-Stella C, Ortona E and Malorni W: Autophagy as
a pathogenic mechanism and drug target in lymphoproliferative
disorders. FASEB J. 28:524–535. 2014. View Article : Google Scholar
|
|
45
|
Zhang H, Pang Y, Ma C, Li J, Wang H and
Shao Z: ClC5 decreases the sensitivity of multiple myeloma cells to
bortezomib via promoting pro-survival autophagy. Oncol Res. Sep
11–2017.(Epub ahead of print). doi:
10.3727/096504017X15049221237147. View Article : Google Scholar
|
|
46
|
Yun Z, Zhichao J, Hao Y, Ou J, Ran Y, Wen
D and Qun S: Targeting autophagy in multiple myeloma. Leuk Res.
59:97–104. 2017. View Article : Google Scholar
|
|
47
|
Mahoney E, Lucas DM, Gupta SV, Wagner AJ,
Herman SE, Smith LL, Yeh YY, Andritsos L, Jones JA, Flynn JM, et
al: ER stress and autophagy: New discoveries in the mechanism of
action and drug resistance of the cyclin-dependent kinase inhibitor
flavopiridol. Blood. 120:1262–1273. 2012. View Article : Google Scholar
|
|
48
|
Sharma A, Singh K, Mazumder S, Hill BT,
Kalaycio M and Almasan A: BECN1 and BIM interactions with MCL-1
determine fludarabine resistance in leukemic B cells. Cell Death
Dis. 4:e6282013. View Article : Google Scholar
|
|
49
|
Zakikhani M, Dowling RJ, Sonenberg N and
Pollak MN: The effects of adiponectin and metformin on prostate and
colon neoplasia involve activation of AMP-activated protein kinase.
Cancer Prev Res. 1:369–375. 2008. View Article : Google Scholar
|
|
50
|
Wang LW, Li ZS, Zou DW, Jin ZD, Gao J and
Xu GM: Metformin induces apoptosis of pancreatic cancer cells.
World J Gastroenterol. 14:7192–7198. 2008. View Article : Google Scholar
|
|
51
|
Wang F, Liu Z, Zeng J, Zhu H, Li J, Cheng
X, Jiang T, Zhang L, Zhang C, Chen T, et al: Metformin
synergistically sensitizes FLT3-ITD-positive acute myeloid leukemia
to sorafenib by promoting mTOR-mediated apoptosis and autophagy.
Leuk Res. 39:1421–1427. 2015. View Article : Google Scholar
|
|
52
|
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX,
Shen ZX, Chen SJ, Chen Y and Zhao WL: Therapeutic metformin/AMPK
activation blocked lymphoma cell growth via inhibition of mTOR
pathway and induction of autophagy. Cell Death Dis. 3:e2752012.
View Article : Google Scholar
|
|
53
|
Xia D, Zhang YT, Xu GP, Yan WW, Pan XR and
Tong JH: Sertraline exerts its antitumor functions through both
apoptosis and autophagy pathways in acute myeloid leukemia cells.
Leuk Lymphoma. 58:1–10. 2017. View Article : Google Scholar
|
|
54
|
Trocoli A, Mathieu J, Priault M, Reiffers
J, Souquère S, Pierron G, Besançon F and Djavaheri-Mergny M:
ATRA-induced upregulation of Beclin 1 prolongs the life span of
differentiated acute promyelocytic leukemia cells. Autophagy.
7:1108–1114. 2011. View Article : Google Scholar
|
|
55
|
Nishioka C, Ikezoe T, Yang J, Gery S,
Koeffler HP and Yokoyama A: Inhibition of mammalian target of
rapamycin signaling potentiates the effects of all-trans retinoic
acid to induce growth arrest and differentiation of human acute
myelogenous leukemia cells. Int J Cancer. 125:1710–1720. 2009.
View Article : Google Scholar
|
|
56
|
Isakson P, Bjørås M, Bøe SO and Simonsen
A: Autophagy contributes to therapy-induced degradation of the
PML/RARA oncoprotein. Blood. 116:2324–2331. 2010. View Article : Google Scholar
|
|
57
|
Eriksen AB, Torgersen ML, Holm KL,
Abrahamsen G, Spurkland A, Moskaug JØ, Simonsen A and Blomhoff HK:
Retinoic acid-induced IgG production in TLR-activated human primary
B cells involves ULK1-mediated autophagy. Autophagy. 11:460–471.
2015. View Article : Google Scholar
|
|
58
|
Qian W, Liu J, Jin J, Ni W and Xu W:
Arsenic trioxide induces not only apoptosis but also autophagic
cell death in leukemia cell lines via up-regulation of Beclin-1.
Leuk Res. 31:329–339. 2007. View Article : Google Scholar
|
|
59
|
Goussetis DJ, Altman JK, Glaser H, McNeer
JL, Tallman MS and Platanias LC: Autophagy is a critical mechanism
for the induction of the antileukemic effects of arsenic trioxide.
J Biol Chem. 285:29989–29997. 2010. View Article : Google Scholar
|
|
60
|
Ristic B, Bosnjak M, Arsikin K, Mircic A,
Suzin-Zivkovic V, Bogdanovic A, Perovic V, Martinovic T,
Kravic-Stevovic T, Bumbasirevic V, et al: Idarubicin induces
mTOR-dependent cytotoxic autophagy in leukemic cells. Exp Cell Res.
326:90–102. 2014. View Article : Google Scholar
|
|
61
|
Grandér D, Kharaziha P, Laane E,
Pokrovskaja K and Panaretakis T: Autophagy as the main means of
cytotoxicity by glucocorticoids in hematological malignancies.
Autophagy. 5:1198–1200. 2009. View Article : Google Scholar
|
|
62
|
Laane E, Tamm KP, Buentke E, Ito K,
Kharaziha P, Oscarsson J, Corcoran M, Björklund AC, Hultenby K,
Lundin J, et al: Cell death induced by dexamethasone in lymphoid
leukemia is mediated through initiation of autophagy. Cell Death
Differ. 16:1018–1029. 2009. View Article : Google Scholar
|
|
63
|
Granato M, Chiozzi B, Filardi MR, Lotti
LV, Di Renzo L, Faggioni A and Cirone M: Tyrosine kinase inhibitor
tyrphostin AG490 triggers both apoptosis and autophagy by reducing
HSF1 and Mcl-1 in PEL cells. Cancer Lett. 366:191–197. 2015.
View Article : Google Scholar
|
|
64
|
Germain M, Nguyen AP, Le Grand JN, Arbour
N, Vanderluit JL, Park DS, Opferman JT and Slack RS: MCL-1 is a
stress sensor that regulates autophagy in a developmentally
regulated manner. EMBO J. 30:395–407. 2011. View Article : Google Scholar
|
|
65
|
Granato M, Lacconi V, Peddis M, Lotti LV,
Di Renzo L, Gonnella R, Santarelli R, Trivedi P, Frati L, D'Orazi
G, et al: HSP70 inhibition by 2-phenylethynesulfonamide induces
lysosomal cathepsin D release and immunogenic cell death in primary
effusion lymphoma. Cell Death Dis. 4:e7302013. View Article : Google Scholar
|
|
66
|
Crowley LC, Elzinga BM, O'Sullivan GC and
McKenna SL: Autophagy induction by Bcr-Abl-expressing cells
facilitates their recovery from a targeted or nontargeted
treatment. Am J Hematol. 86:38–47. 2011. View Article : Google Scholar
|
|
67
|
Rothe K, Lin H, Lin KB, Leung A, Wang HM,
Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM and Jiang X:
The core autophagy protein ATG4B is a potential biomarker and
therapeutic target in CML stem/progenitor cells. Blood.
123:3622–3634. 2014. View Article : Google Scholar
|
|
68
|
Mancini M, Leo E, Campi V, Castagnetti F,
Zazzeroni L, Gugliotta G, Santucci MA and Martinelli G: A
calpain-cleaved fragment of β-catenin promotes BCRABL1+
cell survival evoked by autophagy induction in response to
imatinib. Cell Signal. 26:1690–1697. 2014. View Article : Google Scholar
|
|
69
|
Sheng Z, Ma L, Sun JE, Zhu LJ and Green
MR: BCR-ABL suppresses autophagy through ATF5-mediated regulation
of mTOR transcription. Blood. 118:2840–2848. 2011.
View Article : Google Scholar
|
|
70
|
Xin P, Li C, Zheng Y, Peng Q, Xiao H,
Huang Y and Zhu X: Efficacy of the dual PI3K and mTOR inhibitor
NVP-BEZ235 in combination with imatinib mesylate against chronic
myelogenous leukemia cell lines. Drug Des Devel Ther. 11:1115–1126.
2017. View Article : Google Scholar
|
|
71
|
Elzinga BM, Nyhan MJ, Crowley LC,
O'Donovan TR, Cahill MR and McKenna SL: Induction of autophagy by
Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates
Bcr-Abl protein. Am J Hematol. 88:455–462. 2013. View Article : Google Scholar
|
|
72
|
Zhu S, Cao L, Yu Y, Yang L, Yang M, Liu K,
Huang J, Kang R, Livesey KM and Tang D: Inhibiting autophagy
potentiates the anticancer activity of IFN1@/IFNα in chronic
myeloid leukemia cells. Autophagy. 9:317–327. 2013. View Article : Google Scholar
|
|
73
|
Bosnjak M, Ristic B, Arsikin K, Mircic A,
Suzin-Zivkovic V, Perovic V, Bogdanovic A, Paunovic V, Markovic I,
Bumbasirevic V, et al: Inhibition of mTOR-dependent autophagy
sensitizes leukemic cells to cytarabine-induced apoptotic death.
PLoS One. 9:e943742014. View Article : Google Scholar
|
|
74
|
Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang
Y, Dong B and Chen X: Targeting autophagy augments in vitro and in
vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer
Res. 17:3248–3258. 2011. View Article : Google Scholar
|
|
75
|
Wang Z, Zhu S, Zhang G and Liu S:
Inhibition of autophagy enhances the anticancer activity of
bortezomib in B-cell acute lymphoblastic leukemia cells. Am J
Cancer Res. 5:639–650. 2015.
|
|
76
|
Granato M, Santarelli R, Lotti LV, Di
Renzo L, Gonnella R, Garufi A, Trivedi P, Frati L, D'Orazi G,
Faggioni A, et al: JNK and macroautophagy activation by bortezomib
has a pro-survival effect in primary effusion lymphoma cells. PLoS
One. 8:e759652013. View Article : Google Scholar
|
|
77
|
Han W, Sun J, Feng L, Wang K, Li D, Pan Q,
Chen Y, Jin W, Wang X, Pan H, et al: Autophagy inhibition enhances
daunorubicin-induced apoptosis in K562 cells. PLoS One.
6:e284912011. View Article : Google Scholar
|
|
78
|
Marignac Martinez VL, Smith S, Toban N,
Bazile M and Aloyz R: Resistance to Dasatinib in primary chronic
lymphocytic leukemia lymphocytes involves AMPK-mediated energetic
re-programming. Oncotarget. 4:2550–2566. 2013.
|
|
79
|
Morita M, Nishinaka Y, Kato I, Saida S,
Hiramatsu H, Kamikubo Y, Heike T, Nakahata T and Adachi S:
Dasatinib induces autophagy in mice with Bcr-Abl-positive leukemia.
Int J Hematol. 105:335–340. 2017. View Article : Google Scholar
|
|
80
|
Kharaziha P, De Raeve H, Fristedt C, Li Q,
Gruber A, Johnsson P, Kokaraki G, Panzar M, Laane E, Osterborg A,
et al: Sorafenib has potent antitumor activity against multiple
myeloma in vitro, ex vivo, and in vivo in the 5T33MM mouse model.
Cancer Res. 72:5348–5362. 2012. View Article : Google Scholar
|
|
81
|
Takahashi H, Inoue J, Sakaguchi K, Takagi
M, Mizutani S and Inazawa J: Autophagy is required for cell
survival under L-asparaginase-induced metabolic stress in acute
lymphoblastic leukemia cells. Oncogene. 36:4267–4276. 2017.
View Article : Google Scholar
|
|
82
|
Schnekenburger M, Grandjenette C, Ghelfi
J, Karius T, Foliguet B, Dicato M and Diederich M: Sustained
exposure to the DNA demethylating agent, 2-deoxy-5-azacytidine,
leads to apoptotic cell death in chronic myeloid leukemia by
promoting differentiation, senescence, and autophagy. Biochem
Pharmacol. 81:364–378. 2011. View Article : Google Scholar
|
|
83
|
Romano A, Giallongo C, La Cava P,
Parrinello NL, Chiechi A, Vetro C, Tibullo D, Di Raimondo F, Liotta
LA, Espina V, et al: Proteomic analysis reveals autophagy as
pro-survival pathway elicited by long-term exposure with
5-azacitidine in high-risk myelodysplasia. Front Pharmacol.
8:2042017. View Article : Google Scholar
|
|
84
|
Evangelisti C, Evangelisti C, Chiarini F,
Lonetti A, Buontempo F, Neri LM, McCubrey JA and Martelli AM:
Autophagy in acute leukemias: A double-edged sword with important
therapeutic implications. Biochim Biophys Acta. 1853:14–26. 2015.
View Article : Google Scholar
|
|
85
|
Ekiz HA, Can G and Baran Y: Role of
autophagy in the progression and suppression of leukemias. Crit Rev
Oncol Hematol. 81:275–285. 2012. View Article : Google Scholar
|