Introduction
Oral squamous cell carcinoma (OSCC) is one of the 10
most common types of neoplasms in the USA (1). OSCC, a major factor of morbidity and
mortality among head and neck cancers, constitutes ~90% of all
cases of oral malignancies (2). At
present, the treatment methods for OSCC, primarily chemotherapy,
radiotherapy and surgery, are insufficient to overcome the issues
of drug resistance, recurrence and metastasis (3), leading to a poor prognosis and a high
mortality rate. Therefore, the investigation of the molecular
pathogenesis, including the survival mechanisms of cells under
stress, may provide prospective targets for reducing metastasis and
resistance to therapy, thereby improving the survival and prognosis
of patients with OSCC.
Autophagy, cellular ‘self-eating’, is the process of
intracellular lysosomal degradation to recycle proteins and
organelles, which is regulated by autophagy-related genes (4). Autophagy is critical to prevent the
toxic accumulation of damaged proteins and organelles, and
stabilizes the metabolism to maintain energy homeostasis and ensure
cell survival (5). Therefore,
autophagy is predominantly a pro-survival adaptive response that
enables cancer cells to withstand the unfavorable conditions to
which they are exposed, such as starvation, ischemia, hypoxia and
chemotherapy (6–8). Consequently, autophagy can promote
malignant processes after tumorigenesis (7), and facilitate chemotherapy and
radiotherapy resistance (8–11). It has been reported that resistant
cells can be re-sensitized to chemotherapy drugs by using autophagy
inhibitors or affecting the molecular regulators of autophagy
(9). The role of autophagy in
cancer metastasis is a double-edged sword, as it can promote both
pro-metastasis and anti-metastasis processes. The cellular response
to autophagy during cancer metastasis is stage-specific (12–14).
Autophagy is regarded as a potential target in cancer treatment and
may provide a promising therapeutic strategy for overcoming
resistance and enhancing the effect of chemotherapy. However, as
autophagy is a complex process involving many molecules and
pathways, the specific mechanisms and molecules involved remain
under continuous research and expansion.
Karyopherin α2 (KPNA2), which is a member of the
importin α family, plays an important role in nucleocytoplasmic
transport, as previously reported (15–18).
KPNA2 may mediate the translocation of cancer-associated functional
proteins to affect tumorigenesis (19). Additionally, KPNA2 has been
demonstrated to be involved in the translocation of various
proteins, including transcription factors or cargo proteins
associated with DNA repair and cell-cycle regulation (16). These proteins are involved in a
multitude of cellular processes, such as proliferation, apoptosis
and metastasis. Recently, the biological function of KPNA2 has been
confirmed in oncological clinical studies and cell experiments
(20–24). For example, KPNA2 can enhance the
migratory ability and viability of breast cancer cells (20,23).
In addition, the knockdown of KPNA2 can inhibit the proliferation
of cells derived from prostate and ovarian cancer (22,24).
Thus, KPNA2 is regarded as a candidate oncogene. However, the role
of KPNA2 in the progression of OSCC remains unclear and limited
information is available regarding the role of KPNA2 in the process
of autophagy. Thus, there are additional molecular mechanisms of
KPNA2 that need to be further investigated.
In the present study, we reported that KPNA2
knockdown inhibited the migration, cisplatin resistance and
autophagy of OSCC cells, and that the mechanisms involved were
associated with the blockade of p53 translocation. Collectively,
our findings may provide an original perspective for improving the
therapeutic efficacy of OSCC.
Materials and methods
Reagents
Antibodies for LC3B and KPNA2 were purchased from
Abcam (Cambridge, UK), autophagy-related gene (Atg)3, Atg5, Atg7,
SQSTM1/p62, p53 and β-actin were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Rapamycin, chloroquine
phosphate, 3-methyladenine (3-MA) and cisplatin were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Cell Counting Kit-8
(CCK-8) was acquired from Dojindo Molecular Technologies, Inc.
(Kumamoto, Japan). The catalogue numbers of the agents are listed
in Table I.
 | Table I.The catalog numbers of the agents
used in the present study. |
Table I.
The catalog numbers of the agents
used in the present study.
| Agent | Source | Catalogue
number |
|---|
| Anti-KPNA2 | Abcam | ab173295 |
| Anti-LC3B | Abcam | ab51520 |
| Anti-Atg3 | CST | 3415T |
| Anti-Atg5 | CST | 12994T |
| Anti-Atg7 | CST | 2631T |
|
Anti-SQSTM1/p62 | CST | 5114T |
| Anti-p53 | CST | 2527T |
| Anti-β-actin | CST | 4970T |
| HRP-conjugated goat
anti-rabbit IgG | ZSGB-Bio | ZB-2301 |
| TRITC-conjugated
goat anti-rabbit IgG | ZSGB-Bio | ZF-0316 |
| Rapamycin | Sigma-Aldrich | V900930-1MG |
| Chloroquine
phosphate | Sigma-Aldrich | C6628-25G |
|
3-Methyladenine | Sigma-Aldrich | M9281-100MG |
| Cisplatin | Sigma-Aldrich | P4394-25MG |
| CCK-8 | Dojindo | CK04-500T |
Cell culture
The three OSCC cell lines, CAL-27, SCC-15 and
Tca8113, were donated by the Institute of Hard Tissue Development
and Regeneration, The Second Affiliated Hospital of Harbin Medical
University (Harbin, China). Cells were cultured in RPMI-1640 medium
(HyClone Laboratories; GE Healthcare Life Science, Logan, UT, USA)
with 10% (v/v) fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 1% (v/v)
penicillin-streptomycin solution (Gibco; Thermo Fisher Scientific).
Cells were incubated at 37°C with 5% (v/v) CO2 in
humidified air.
Lentivirus transfection
KPNA2 shRNA, p53 shRNA and scramble control shRNAs
packaged in a lentiviral vector were purchased from Shanghai
GeneChem Co., Ltd. (Shanghai, China). The sequences were
synthesized and verified by Shanghai GeneChem Co., Ltd. According
to the manufacturer's protocol, cells were seeded in a 6-well plate
and incubated in infection medium with the lentiviral vectors for
12 h. The medium was then replaced with fresh complete medium. The
cells were screened with puromycin (Shanghai GeneChem Co., Ltd.) at
a dose of 2 µg/ml after 72 h transfection. The transfection
efficiency was confirmed by western blotting. The sequences of the
three shRNAs are listed in Table
II.
| Table II.The sequences of the three shRNA
insert elements. |
Table II.
The sequences of the three shRNA
insert elements.
| shRNA | Sequence |
|---|
| KPNA2-shRNA |
5′-CCGGCCGTTGATGAACCTCTTAACTCGAGTTAAGAGGTTCATCAACGGTTTTTG-3′ |
| p53-shRNA |
5′-CCGGCGGCGCACAGAGGAAGAGAATCTCGAGATTCTCTTCCTCTGTGCGCCGTTTTT-3′ |
| Scramble control
shRNA |
5′-CCGGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTG-3′ |
CCK-8 detection of cell viability
The three treated OSCC cell lines were seeded in
96-well plates (104 cells/well) and cultured in 100 µl
serum-free medium for 24 h. Then, the medium was replaced with 0,
0.01, 0.1, 1, 10 and 100 µg/ml cisplatin diluted in RPMI-1640
medium with 10% FBS (v/v) and cultured for a further 24 h.
Subsequently, the cells were incubated with 10% CCK-8 solution for
another 2 h, and the absorbance of each well was assessed at 450 nm
using a microplate reader (Bio-Rad iMark; Bio-Rad Laboratories,
Hercules, CA, USA). Each experiment was repeated three times
independently.
Wound-healing assay
The three treated OSCC cell lines were reseeded onto
a 6-well plate and cultured to 100% confluence. The cell monolayer
was scratched using a pipette tip, and the medium was replaced with
serum-free medium. The cells were incubated at 37°C in 5%
CO2 for 24 h and representative images of the wound were
captured by a digital camera installed on an inverted microscope
(Nikon Eclipse Ti-E; Nikon, Kobe, Japan). The scratch area was
quantified using ImageJ 1.46r (National Institutes of Health,
Bethesda, MA, USA). Each experiment was repeated three times
independently.
Transwell migration assay
Migration assays were performed in 6.5 mm Transwell
inserts with 8.0-µm pore polycarbonate membranes purchased from
Corning Inc. (Corning, NY, USA). A total of 1×105
treated cells/well from each of the three OSCC cell lines were
seeded in the upper chambers with 200 µl serum-free medium, while
the lower chambers were filled with 500 µl medium supplemented with
FBS. Following incubation at 37°C in 5% CO2 for 24 h,
the cells were fixed with methanol for 15 min. Cells were removed
from the upper chambers gently using cotton swabs, and the
membranes were stained with crystal violet (Beyotime Institute of
Biotechnology, Shanghai, China). The membranes were photographed
and the images were analyzed with ImageJ 1.46r (National Institutes
of Health). Each experiment was repeated three times
independently.
Western blot analysis
Cells were harvested and lysed in ice-cold buffer,
including RIPA (Beyotime Institute of Biotechnology), 1% PMSF
(Beyotime Institute of Biotechnology) and a protease inhibitor
cocktail (Roche Diagnostics GmbH, Mannheim, Germany) for 30 min to
extract the total protein. The concentration of protein was
quantified using a BCA protein assay kit (Beyotime Institute of
Biotechnology) and the protein samples were denatured. Nuclear
proteins were extracted using a nuclear protein extraction kit
(Beyotime Institute of Biotechnology). Protein samples (96 µg/lane)
were separated by 10, 12 or 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (Beyotime Institute of
Biotechnology) and blotted onto polyvinylidene difluoride membranes
(Roche Diagnostics GmbH). After blocking with 5% skimmed milk
(Beyotime Institute of Biotechnology), the membranes were incubated
with the primary antibodies overnight at 4°C. The membranes were
incubated with a horseradish peroxidase-conjugated secondary
antibody (Zhongshan Jinqiao Biological Technology, Co., Ltd.,
Beijing, China) for 1.5 h, followed by an electrochemiluminescence
reagent (Beyotime Institute of Biotechnology) for 3 min. Images of
the bands were captured with SmartChemiI (Beijing Sage Creation
Science, Co., Beijing, China) and the gray value was assessed with
ImageJ 1.46r (National Institutes of Health). Each experiment was
repeated three times independently. The antibodies used for western
blot analysis are listed in Table
III.
| Table III.The antibodies used in the present
study. |
Table III.
The antibodies used in the present
study.
| Antibody | Type | Dilution | Source | Application |
|---|
| KPNA2 | Rabbit-mAb | 1:1,000 | CST | WB |
| LC3 | Rabbit-mAb | 1:1,000 | Abcam | WB |
| Atg3 | Rabbit-mAb | 1:1,000 | CST | WB |
| Atg5 | Rabbit-mAb | 1:1,000 | CST | WB |
| Atg7 | Rabbit-mAb | 1:1,000 | CST | WB |
| SQSTM1/P62 | Rabbit-mAb | 1:1,000 | CST | WB |
| β-actin | Rabbit-mAb | 1:1,000 | CST | WB |
| P53 | Rabbit-mAb | 1:160 | Abcam | ICC/IF |
| HRP-conjugated goat
anti-rabbit IgG | Goat-mAb | 1:20,000 | ZSGB-Bio | WB |
| TRITC-conjugated
goat anti-rabbit IgG | Goat-mAb | 1:50 | ZSGB-Bio | ICC/IF |
Transmission electron microscopy
(TEM)
Cells were collected, fixed with 2.5% glutaraldehyde
overnight at 4°C and post-fixed with 1% osmium acid for 3 h. The
cells were dehydrated in a graded ethanol series (30, 50, 70, 80,
90, 95 and 100%) followed by a treatment with absolute acetone for
20 min after each incubation. The samples were incubated in a 3:1
mixture of absolute acetone and Spurr resin (Shanghai GenMed, Co.,
Ltd., Shanghai, China) (1 h), a 1:1 mixture of absolute acetone and
Spurr resin (1 h), a 1:3 mixture of absolute acetone and Spurr
resin (1 h) once again, and a final Spurr resin incubation (4 h).
The samples were embedded in Spurr resin at room temperature
overnight, then heated to 70°C for 9 h in capsules. Finally, the
samples were cut into 70-nm-thick sections using an ultramicrotome
(Leica Ultracut R; Leica Microsystems GmbH, Wetzlar, Germany), and
the specimens were double-stained with uranyl acetate and lead
citrate for 10 min each. The samples were observed and images were
captured using a transmission electron microscope (JEM-1230; JEOL,
Ltd., Tokyo, Japan).
Immunofluorescence assay
Treated cells were seeded onto glass slides and
cultured for 24 h. Then, cells were fixed with 4% paraformaldehyde
for 10 min and permeabilized with 0.2% Triton X-100 (Beyotime
Institute of Biotechnology) for 5 min on ice. After blocking with
5% normal goat serum (NGS; Wuhan Boster Biological Technology, Co.,
Ltd., Wuhan, China) for 1 h, slides were incubated with primary
antibodies overnight at 4°C, followed by incubation with
TRITC-conjugated secondary antibodies (Zhongshan Jinqiao Biological
Technology, Co., Ltd., Beijing, China) for 1.5 h in the dark.
Subsequent to mounting with DAPI (Beyotime Institute of
Biotechnology) for nuclear staining, the slides were covered on
microscope slides with anti-quench mount reagents (Beyotime
Institute of Biotechnology). The slides were examined and
photographed using a fluorescence microscope (Nikon Eclipse Ci-S;
Nikon). The antibodies used in these experiments are listed in
Table III.
Statistical analysis
The data are presented as the means ± standard
deviation (SD). Differences between the groups were examined using
a one-way ANOVA in GraphPad Prism 5 software (GraphPad Software,
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
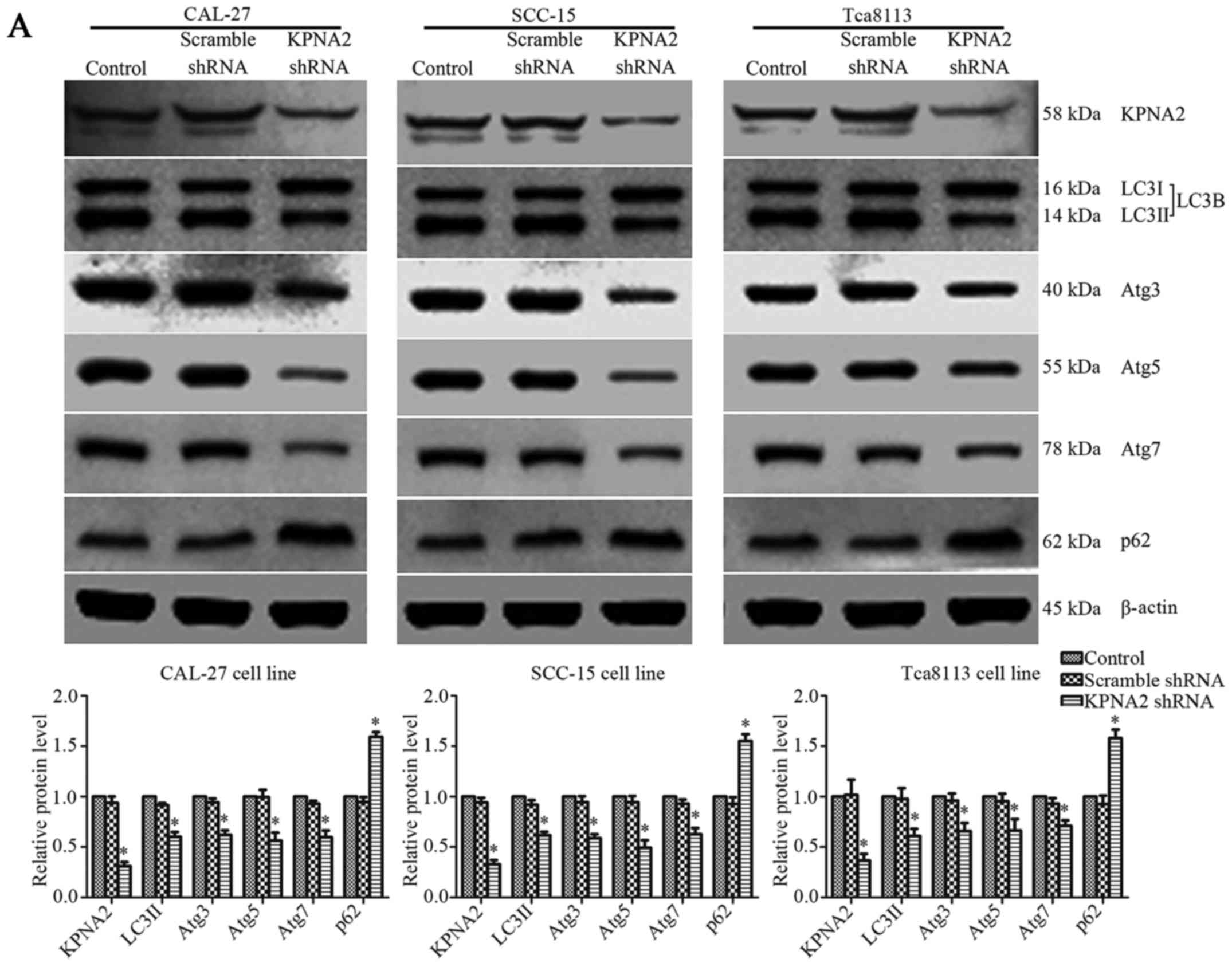
Lentiviral transfection of shRNA can
effectively knock down the expression of KPNA2 and p53
The efficiency of shRNA lentivirus transfection was
evaluated at the protein level by western blotting. KPNA2-targeted
shRNA significantly downregulated the level of KPNA2 protein
expression in CAL-27, SCC-15 and Tca8113 cells by 68.9±4.1,
67.1±4.1 and 63.4±6.7%, respectively (P<0.05, n=3).
Additionally, p53-targeted shRNA reduced the level of p53 protein
expression in CAL-27, SCC-15 and Tca8113 cells by 70.5±2.6,
70.8±2.0 and 71.8±1.9%, respectively (P<0.05, n=3).
Representative bands, and histograms of the relative protein
expression level are displayed in Figs.
3A and 6C.
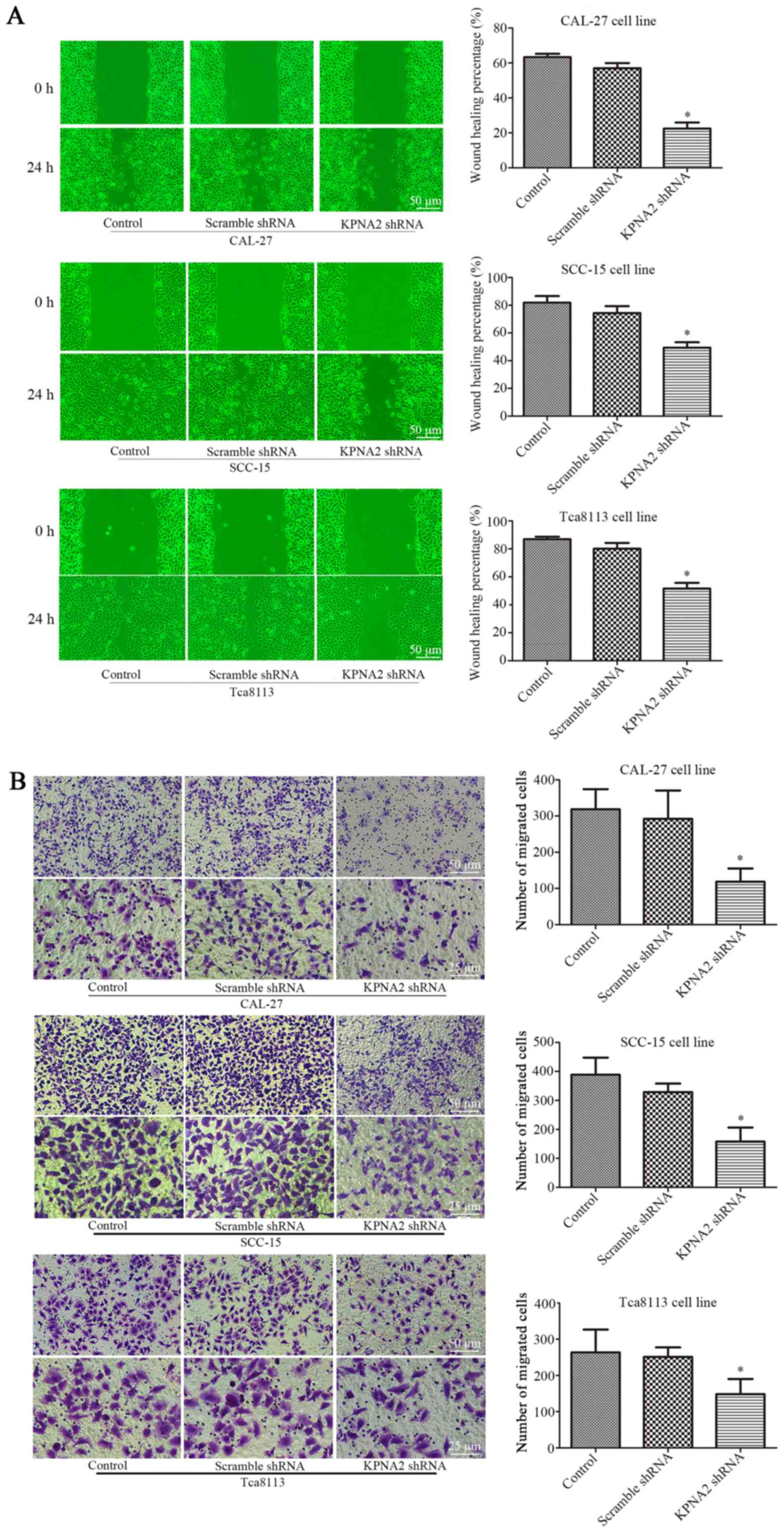
Knockdown of KPNA2 inhibits the
migration of OSCC cells
To determine the role of KPNA2 in the migration of
autophagic cells, KPNA2 was downregulated in the three OSCC cell
lines. Wound healing and Transwell assays were performed for each
group of cells treated with 100 nM rapamycin for 24 h (25,26).
As displayed in Fig. 1A the wound
healing percentage was reduced from 63.3±1.9 to 22.4±3.5%, from
81.9±4.8 to 49.4±4.0% and from 87.0±1.7 to 51.7±4.1% in the
KPNA2-knockdown cells at 24 h, in CAL-27, SCC-15 and Tca8113 cells,
respectively, compared with the corresponding control groups
(P<0.05, n=3). Furthermore, as demonstrated in Fig. 1B, the knockdown of KPNA2 reduced the
number of migrating cells by 63.8, 59.4 and 43.8% in CAL-27, SCC-15
and Tca8113 cells, respectively, consistent with the results of the
wound healing assay (P<0.05, n=3). These data demonstrated that
the knockdown of KPNA2 significantly inhibited cell migration in
the OSCC cell lines.
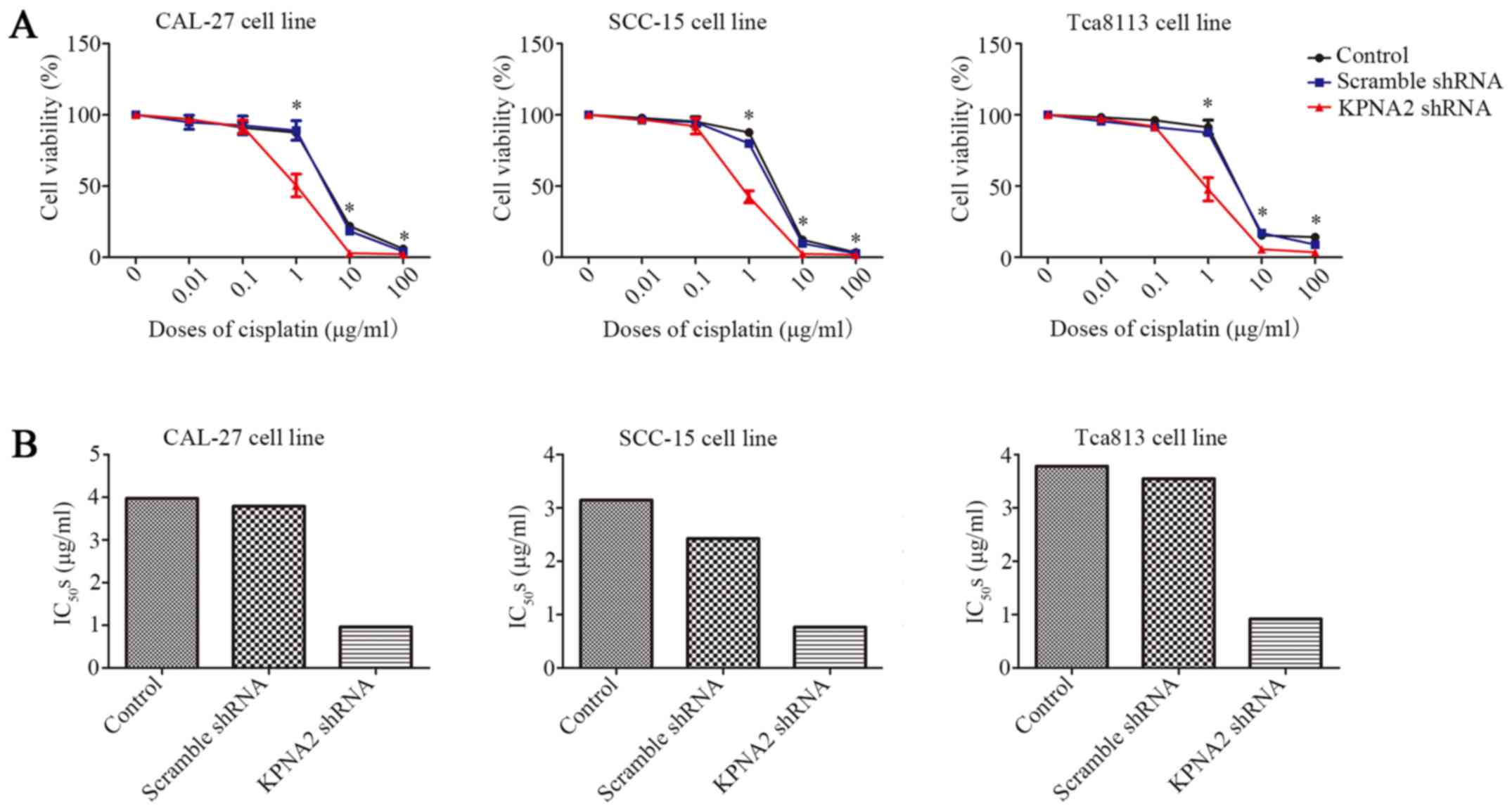
Knockdown of KPNA2 reduces the
cisplatin resistance of OSCC cells
In addition to cell migration, we also examined the
effect of KPNA2 on the resistance to cisplatin using a CCK-8 assay.
As displayed in Fig. 2A KPNA2
knockdown significantly decreased cell viability in a series of
cisplatin concentrations, particularly 1 µg/ml, whereas
transfection with the scramble control shRNA had little effect on
cell vitality compared with untransfected cells. Subsequently,
IC50 for each group was calculated, as presented in
Fig. 2B and Table IV. KPNA2 knockdown reduced the
IC50 from 3.97 to 0.96 µg/ml in CAL-27 cells, from 3.14
to 0.76 µg/ml in SCC-15 cells and from 3.78 to 0.92 µg/ml in
Tca8113 cells. These data revealed that KPNA2 knockdown
significantly reduced cisplatin resistance in the OSCC cell
lines.
| Table IV.IC50s of KPNA2-knockdown
cells. |
Table IV.
IC50s of KPNA2-knockdown
cells.
|
| Control | Scramble shRNA | KPNA2 shRNA |
|---|
| CAL-27 | 3.971 | 3.791 | 0.9596 |
| SCC-15 | 3.144 | 2.426 | 0.7639 |
| Tca8113 | 3.781 | 3.549 | 0.9181 |
KPNA2 knockdown inhibits autophagy in
OSCC cells
The effect of KPNA2 on autophagy remains unclear.
Following KPNA2 knockdown in three OSCC cell lines, western
blotting and TEM were used to detect the extent of autophagy
induced by treatment with rapamycin in each group. Representative
western blotting bands and gray values for autophagy-related
proteins are displayed in Fig. 3A.
KPNA2 knockdown induced a reduction in the relative level of LC3II
by 39.7±4.7, 38.3±3.4, and 39.0±7.1%; in Atg3 by 37.9±4.4, 41.1±3.8
and 34.0±8.0%; in Atg5 by 43.5±7.8, 50.5±7.4 and 33.6±11.4%; and in
Atg7 by 40.3±6.7, 37.1±6.0 and 29.0±5.5%, and reduced the extent of
SQSTM1/p62 degradation in CAL-27, SCC-15 and Tca8113 cells (n=3).
The relative level of SQSTM1/p62 protein in the control group cells
was 159.3±4.8, 155.1±6.8 and 158.1±8.5% of that in the
KPNA2-knockdown cells in the three OSCC cell lines (P<0.05,
n=3). The results of western blot analysis revealed that KPNA2
knockdown induced the suppression of autophagy in the three OSCC
cell lines at the protein level.
In order to further investigate autophagy at the
cellular microstructure level, TEM was performed for each group of
cells. The results from TEM are displayed in Fig. 3B. As identified with TEM, the cells
with KPNA2 knockdown displayed a decreased number of autolysosomes
in the cytoplasm, the characteristic vacuole-like structures
indicated by red arrows in Fig. 3B.
The results of TEM demonstrated that KPNA2 knockdown suppressed
autophagy in the three OSCC cell lines at a microstructure level,
consistent with the results of western blot analysis. These data
demonstrated that KPNA2 knockdown inhibited the process of
autophagy in OSCC cells.
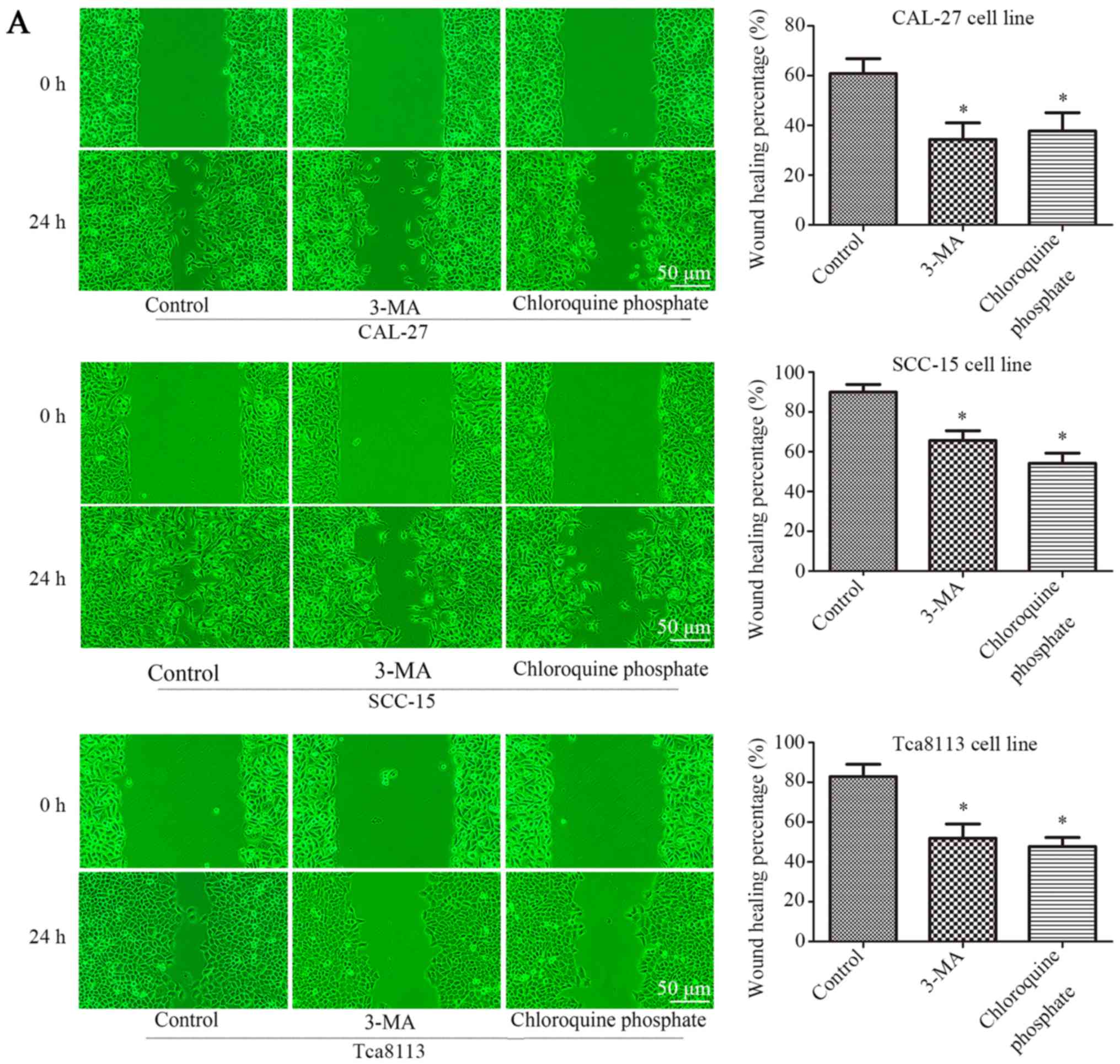
Inhibition of autophagy decreases the
migration and cisplatin resistance of OSCC cells
The three cell lines were treated with 100 nM
rapamycin for 24 h. When 3-MA (2 mM) (27) or chloroquine phosphate (10 µM)
(28) were used in combination with
rapamycin, the rate of cell migration was significantly reduced. As
displayed in Fig. 4A, 3-MA or
chloroquine phosphate treatment reduced the rate of wound closure.
Wound-healing was reduced from 60.8±6.0 to 34.36±6.7% (3-MA) or
37.8±7.4 (chloroquine phosphate) in CAL-27 cells, 89.9±3.9 to
65.6±5.0% (3-MA) or 54.2±5.0% (chloroquine phosphate) in SCC-15
cells, and 82.8±6.2 to 51.9±7.2% (3-MA) or 47.7±4.6% (chloroquine
phosphate) in Tca8113 cells (P<0.05, n=3). Fig. 4B presents the results of a Transwell
assay, in which 3-MA or chloroquine phosphate treatment reduced the
number of migrating cells by ~43.1% (3-MA) and 40.5% (chloroquine
phosphate), 37.8% (3-MA) and 39.2% (chloroquine phosphate), 35.5%
(3-MA) and 44.6% (chloroquine phosphate) in the three OSCC cell
lines (P<0.05, n=3). These data demonstrated that the inhibition
of autophagy can decrease the rate of cell migration in OSCC cells.
Subsequently, whether the inhibition of autophagy affected the
cisplatin resistance of OSCC cells was further evaluated with a
CCK-8 assay. As displayed in Fig.
4C 3-MA or chloroquine phosphate treatment decreased the cell
viability of the three cell lines in combination with a range of
cisplatin concentrations, particularly 1 µg/ml, according to the
results of the CCK-8 assay. Additionally, the IC50 was
reduced from 2.72 to 1.37 µg/ml (3-MA) and 1.08 µg/ml (chloroquine
phosphate) in CAL-27 cells, 3.64 to 1.44 µg/ml (3-MA) and 1.12
µg/ml (chloroquine phosphate) in SCC-15 cells, and 4.00 to 1.14
µg/ml (3-MA) and 0.85 µg/ml (chloroquine phosphate) in Tca8113
cells, as displayed in Fig. 4D and
Table V. These data demonstrated
that the inhibition of autophagy decreased cisplatin resistance of
OSCC cells.
| Table V.IC50s of cells treated
with 3-MA or chloroquine phosphate. |
Table V.
IC50s of cells treated
with 3-MA or chloroquine phosphate.
|
| Control | 3-MA | Chloroquine
phosphate |
|---|
| CAL-27 | 2.718 | 1.371 | 1.081 |
| SCC-15 | 3.642 | 1.443 | 1.121 |
| Tca8113 | 4.001 | 1.14 | 0.8534 |
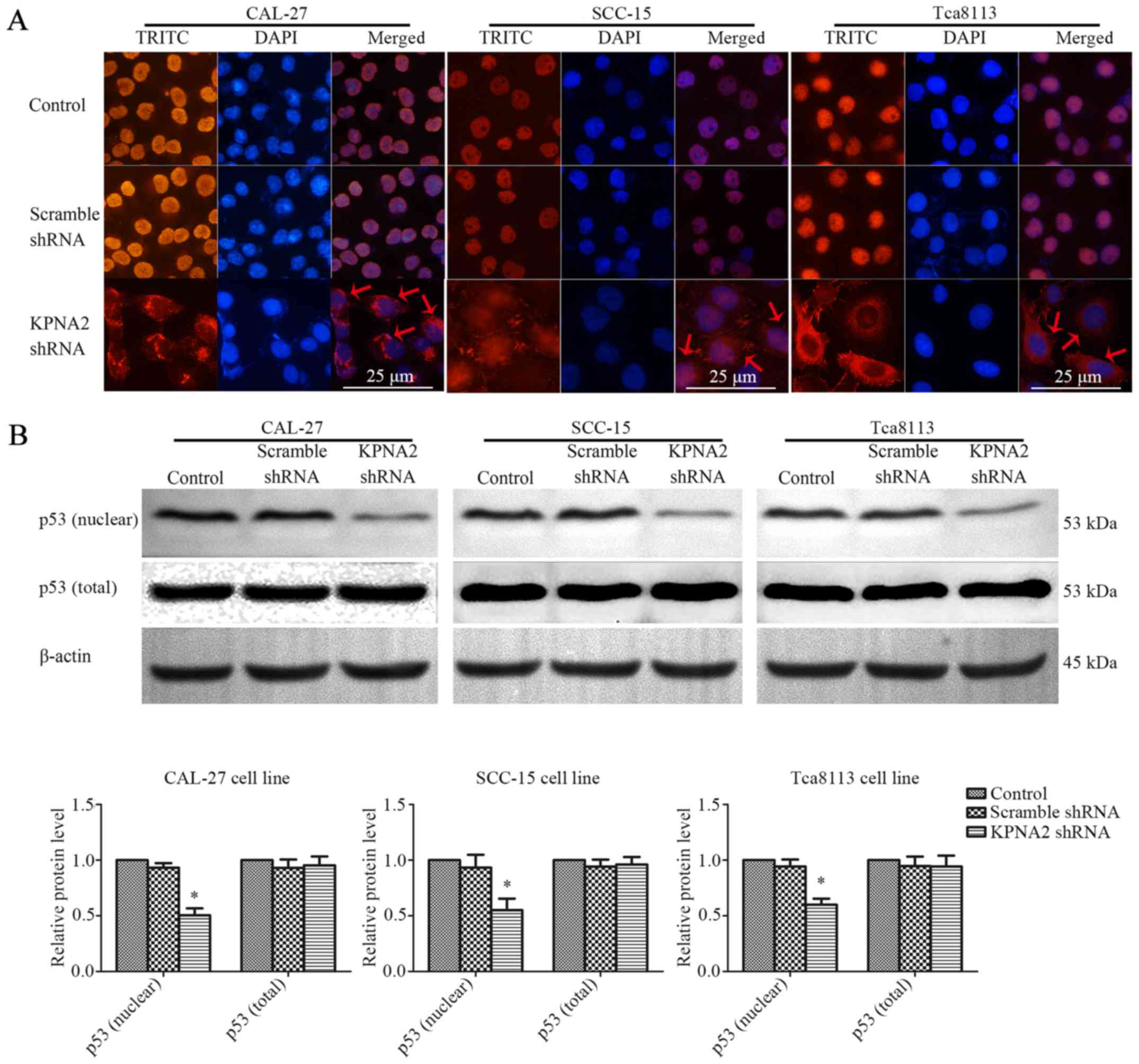
KPNA2 knockdown inhibits p53 nuclear
translocation
KPNA2 plays an important role in the nuclear import
of transcription factors, including p53 (15). The interaction between KPNA2 and p53
was assessed by an immunofluorescence assay and western blot
analysis of nuclear or total p53 protein in the three OSCC cell
lines. The results of immunofluorescence are displayed in Fig. 5A. p53 protein localized to the
nucleus of the control cells, whereas KPNA2 knockdown caused the
accumulation of p53 protein in the cytoplasm. As displayed in
Fig. 5B, KPNA2 knockdown only
marginally affected the total p53 protein level. However, KPNA2
knockdown reduced the level of nuclear p53 by 49.6±6.3, 44.9±10.3
and 40.1±5.5% in the three OSCC cell lines (P<0.05, n=3). These
data demonstrated that KPNA2 knockdown interrupted p53 nuclear
import in OSCC cells.
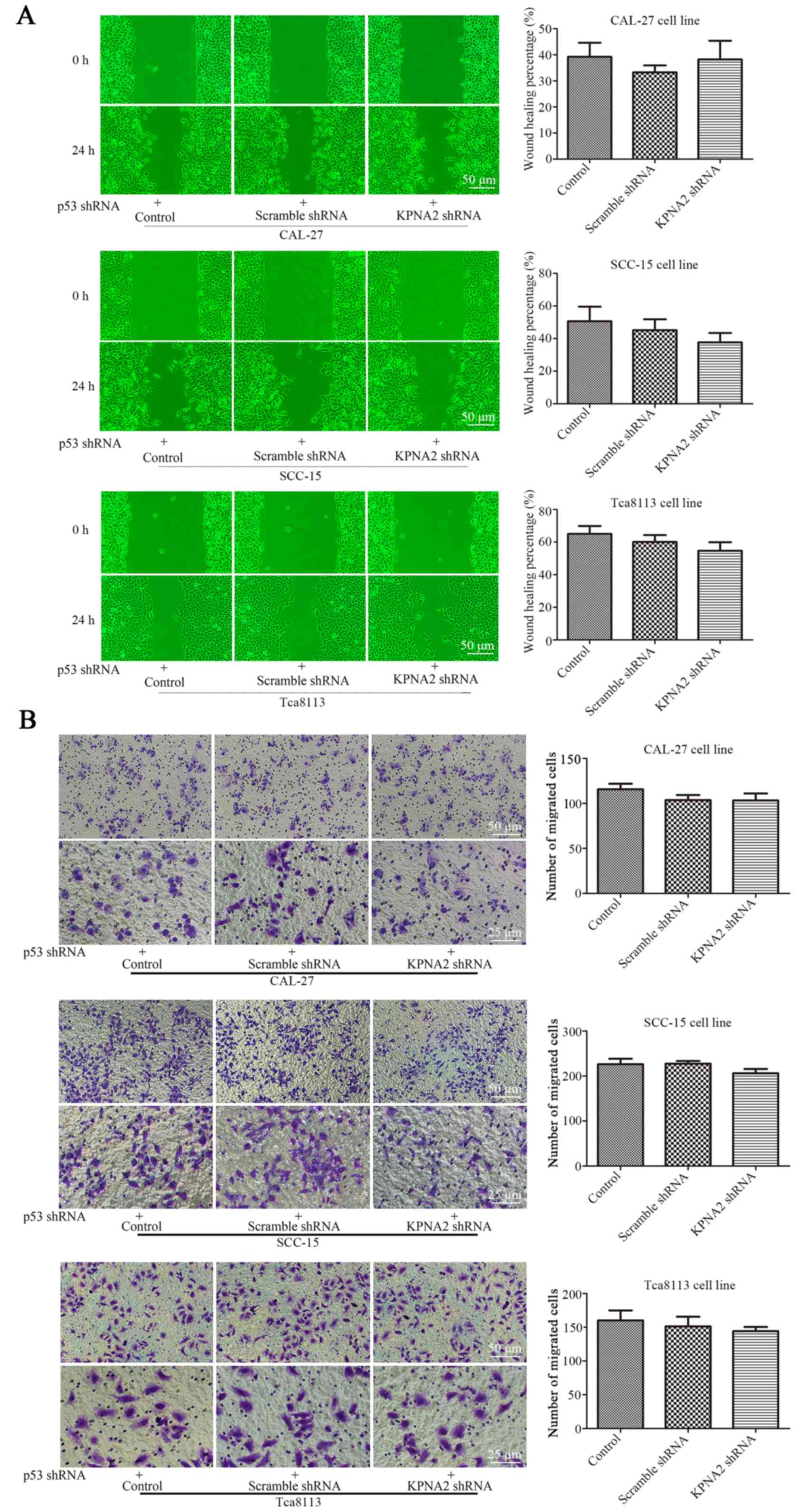
KPNA2 knockdown does not inhibit
autophagy in p53-knockdown cells
To further validate the roles of KPNA2 and p53 in
the process of autophagy, p53 was knocked down in the three OSCC
cell lines. Cell migration, cisplatin resistance and the level of
autophagy were detected in the p53− OSCC cells with or
without KPNA2 knockdown. As displayed in Fig. 6A, despite the downregulation of
KPNA2, there was no significant difference in the wound closure
among the groups of cells in the three p53− OSCC cell
lines (P>0.05, n=3). The results were confirmed by a Transwell
assay as displayed in Fig. 6B,
indicating that the number of migrating cells in the
KPNA2-knockdown groups did not differ from that in the control
groups for the three p53− OSCC cell lines. These data
demonstrated that KPNA2 knockdown failed to inhibit cell migration
in the p53-knockdown cells of the three OSCC cell lines (P>0.05,
n=3). Furthermore, in Fig. 6D, a
western blot analysis for autophagy-related proteins including
LC3I/II, Atg3, Atg5, Atg7 and SQSTM1/p62 identified no difference
among the groups for the three p53− OSCC cell lines
(P>0.05, n=3). The results were confirmed by TEM, as displayed
in Fig. 6E, indicating that the
number of autolysosomes was similar among the groups in the three
p53− OSCC cell lines (P>0.05, n=3). These data
confirmed that KPNA2 knockdown failed to inhibit autophagy in
p53-knockdown cells of the three OSCC cell lines. Collectively, it
was demonstrated that the KPNA2 knockdown-induced inhibition of
autophagy and suppression of cell migration are both
p53-dependent.
In conclusion, these data demonstrated that KPNA2
plays an important role in regulating cell migration and cisplatin
resistance by altering the level of autophagy in OSCC cells by
mediating p53 nucleocytoplasmic translocation.
Discussion
In the present study, the role of KPNA2 knockdown in
the migration, cisplatin resistance and autophagy of OSCC cells was
examined in vitro. It was identified that the knockdown of
KPNA2 inhibited cell migration and autophagy, and decreased
cisplatin resistance through the inhibition of p53
nucleocytoplasmic translocation. Therefore, this study may present
a novel insight into the clinical role of KPNA2 in the treatment of
OSCC.
According to recent research, KPNA2 has been
regarded as a potential diagnostic biomarker in multiple cancers,
and the aberrant overexpression of KPNA2 in tumor tissue is
frequently associated with a poor prognosis or adverse outcome, as
reviewed by Christiansen and Dyrskjøt (16). This has been confirmed in clinical
trials with various types of malignant tumors, including lung,
breast, hepatocellular, gastric, colorectal, ovarian and prostate
carcinoma (19,29–32).
However, the effect of KPNA2 on the oncogenesis and progression of
OSCC remains unclear. Previous studies focused more on the
pro-survival and pro-proliferation functions of KPNA2, whereas the
present study investigated the impact of KPNA2 knockdown on the
metastasis and cisplatin resistance of OSCC cells. The knockdown of
KPNA2, a nuclear transport protein regulating the translocation of
target proteins, was expected to alter the nuclear levels of
several proteins implicated in the processes of DNA repair, cell
cycle and transcriptional regulation of a range of target genes in
cancer cells (16). Additionally,
it has been confirmed in several types of cancer cell lines that
KPNA2 knockdown decreased proliferation and increased apoptosis and
cell mobility, and that increasing the level of KPNA2 may enhance
proliferation (18,22,23,33,34).
However, the role of KPNA2 in the process of autophagy is unclear
and remains to be further studied.
In the present study, western blot analysis of
autophagy-related proteins and TEM of cellular microstructure
revealed that KPNA2 knockdown significantly reduced the level of
autophagy in OSCC cells. It is established that autophagy is a
protective cellular process that occurs frequently in oncogenesis
and chemotherapy. Generally, autophagy supports the survival of
cancer cells during the stress of anticancer therapies, such as
chemotherapy, radiotherapy or targeted agents, thus promoting
resistance (35–37). A range of evidence indicates that
chemotherapy resistance develops following autophagy. Increased
levels of autophagy after chemotherapy have been detected in
patients with a poor prognosis, indicating that autophagy may
enhance chemoresistance (38).
Previous research has indicated that autophagy induction by
chemotherapy may promote the resistance of cancer cells to a range
of anti-neoplastic drugs, including paclitaxel, tamoxifen and
epirubicin (39–41). As autophagy degrades various types
of substrates into small molecules for reuse by the cell, autophagy
has the potential to provide fuel for almost all aspects of central
carbon metabolism (42–44). In brief, the mechanism for autophagy
participation in chemotherapy resistance may be that autophagy
supplies a multitude of metabolic and biosynthetic pathways,
providing tremendous metabolic plasticity to tumor cells and thus
ensuring their survival under chemotherapeutic stress. In addition,
there is a wealth of preclinical evidence supporting the notion
that the inhibition of autophagy can improve clinical outcomes in
cancer treatment, especially in solid tumors (45,46).
Thus, there is sufficient evidence to suggest that the inhibition
of autophagy can reduce the resistance to chemotherapy. The results
of the present study confirmed that inhibiting autophagy with
anti-autophagy agents, such as 3-MA and chloroquine phosphate,
significantly decreased the cisplatin resistance of OSCC cells.
Furthermore, the results of the present study demonstrated that
KPNA2 knockdown reduced the cisplatin resistance of OSCC cells. In
the cells transfected with KPNA2 shRNA, the viability of the
cisplatin-treated group was reduced at all concentrations, and the
cisplatin IC50 was significantly decreased. Given that
KPNA2 knockdown inhibits autophagy, as determined in the present
study, it is reasonable to conclude that the reduction of cisplatin
resistance by KPNA2 knockdown may be associated with the inhibition
of autophagy.
As aforementioned, the role of autophagy in
metastasis is controversial. In previous studies, evidence has
suggested that the role of autophagy in metastasis can be both
facilitative and inhibitive, and that the role of autophagy in
metastasis depends on the context and stage (12–14).
The anti-metastatic function of autophagy may be related to the
inhibition of necrosis and the mediation of autophagic cell death.
In addition, autophagy promotes metastasis by enhancing the
adaptability of cancer cells in response to stress (47). According to previous research,
autophagy protects cancer cells from anoikis or apoptosis, promotes
the dormancy of cancer cell to avoid detection or destruction by
the immune system and promotes the survival of cancer stem cells
(47–51). As previously described, autophagy
can also promote cancer metastasis depending on the context,
therefore an inhibitor of autophagy can simultaneously inhibit
metastasis and increase the cytotoxicity of the anti-metastatic
agents, and efficiently block tumor invasion and metastasis
(52–54). Additionally, according to the
present study, cell migration was reduced in OSCC cells treated
with anti-autophagy agents, including 3-MA or chloroquine
phosphate, which was consistent with previous research that was
referred above. Furthermorre, the present study revealed
anti-metastatic effects of KPNA2 knockdown on OSCC cells. We
identified that OSCC cells transfected with KPNA2 shRNA exhibited
reduced cell migration, using wound healing and Transwell assays.
Since KPNA2 knockdown has been demonstrated to suppress autophagy
in the present study, it can be initially suggested that the
anti-metastatic function of KPNA2 knockdown may be mediated by
autophagy inhibition. Furthermore, substantial evidence has
indicated that the level of KPNA2 itself affects the rate of cell
migration, which has been confirmed in several cancer cell lines
(19,31,34,55,56).
This is dependent on the nuclear transport function of KPNA2, as
several KPNA2-regulating proteins are associated with the movements
of cellular components and cell migration. However, the
relationship between KPNA2 knockdown-induced decrease in cell
migration and the inhibition of autophagy is still unverified and
remains to be further investigated. Although the results of the
present study indicated that KPNA2 plays a role in cancer
metastasis, cisplatin resistance and autophagy, the underlying
mechanisms for the exact function of KPNA2 still require further
research.
In the present study, in order to understand the
molecular mechanisms underlying the role of KPNA2 knockdown in
autophagy and metastasis, further experiments considered the effect
on p53 which plays a critical role in autophagy. Not only the
regulatory functions of p53 itself, but also the subcellular
localization of p53 are particularly important in the process of
autophagy. In response to the stressors that cells are faced with,
such as metabolic or oxidative stress, and DNA damage, p53 is
activated to regulate the transcription of genes, or act through a
non-transcriptional mechanism, in order to either assist in stress
adaptation or to remove irreparable cells through mediating
apoptosis or senescence. One of the components of the
transcriptional responses mediated by the p53 gene is the
activation of autophagy (57). It
has been reported that p53 regulates autophagy depending on the
context. The transcriptional mechanism of p53 in regulating
autophagy is widely recognized. According to previous research, the
expression of several target genes induced by p53 were involved in
autophagy, including DRAM, ISG20L1, Ei24, Bax and PUMA. Negatively
regulating the mTOR signaling pathway is another mechanism through
which p53 promotes autophagy (58).
Whether p53 is localized in the nucleus or cytoplasm plays a
particular role in the regulation of autophagy. There is evidence
that p53 in the nucleus can facilitate autophagy in a
transcription-dependent or -independent manner, whereas p53 in the
cytoplasm inhibits autophagy induction; this has been confirmed in
glioblastoma and colon cancer cell lines (59–61).
As the effect of p53 on autophagy depends on its localization, the
nuclear import and export of p53 are tightly regulated. A component
of the nuclear transporter mediating the import of p53 is the
importin-α family (62). KPNA2, a
member of the importin-α family, is involved in the
nucleocytoplasmic transport of a number of critical transcription
factors, such as p53, E2F1 and PLAG1 (16). In the present study, an
immunofluorescence analysis of OSCC cells with KPNA2 knockdown was
conducted. The results confirmed the p53-importing function of
KPNA2. Furthermore, there is evidence that the truncated form of
importin-α can impair the nuclear import of p53 to result in the
cytoplasmic accumulation of p53 (20,63–66).
Another previous study demonstrated that the level of KPNA2
affected not only p53, but also the downstream targets of p53
(19). The present study has
revealed that KPNA2 knockdown may suppress autophagy via the
disruption of p53 nuclear import in OSCC cells, and these results
are consistent with those of previous studies. Therefore, we
hypothesized that the reduction of cell migration induced by KPNA2
knockdown may be partly associated with the autophagy inhibition
caused by the interruption of p53 nuclear translocation.
Furthermore, it has been confirmed in recent research that blocking
the intra-nuclear transport of p53 suppresses autophagy, resulting
in a reduction in epithelial-mesenchymal transition-related
migration and invasion (59).
Furthermore, the present study also illustrated that KNPA2
knockdown cannot affect the level of autophagy and the cell
migration ability following p53 knockdown. The results of the
present study confirmed that the autophagy inhibition and cell
migration reduction induced by KPNA2 knockdown was p53-dependent,
and that occured through the mechanism of p53 translocation
blockade.
In conclusion, the present study demonstrated that
the migration and cisplatin resistance of OSCC cells can be
influenced by the level of KPNA2. By hindering the nuclear import
of p53, the downregulation of KPNA2, a component of the key import
proteins, can interfere with the process of autophagy, resulting in
a reduction in cisplatin resistance and cell migration, inducing a
significant decrease in the malignant characteristics of OSCC
cells. The results of the present study indicated that KPNA2, a
widely recognized potential marker of prognosis and therapeutic
sensitivity, can be regarded as a therapeutic target aimed at
suppressing metastasis and chemoresistance in OSCC that is worth
studying for therapeutic interventions. However, the potential
participation of unknown factors or pathways as underlying
mechanisms of anti-autophagy, anti-metastasis and chemotherapy
resensitization effects of KPNA2 knockdown in OSCC cells requires
further investigation.
Acknowledgements
Not applicable.
Funding
The present study was partly funded by the National
Natural Science Foundation of China (grant nos. 81570951 and
81500816), the Postgraduate Research Innovation Fund of Harbin
Medical University (grant no. YJSCX2016-49HYD), the Special
Foundation for Sino-Russian Translational Medicine Research Center
of Harbin Medical University (grant nos. CR201412 and CR201504),
the Natural Science Foundation of Heilongjiang Province of China
(grant no. H2015103) and the Science Foundation of the Second
Affiliated Hospital of Harbin Medical University (grant no.
CX2016-20).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. Still, more details about the
datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
FL and LG conceived and designed the study. FL, ZS
and YZ performed the experiments. XC, YL and BZ were also involved
in the conception of the study. FL wrote the paper. FL, XC, YL and
BZ reviewed and edited the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
OSCC
|
oral squamous cell carcinoma
|
|
KPNA2
|
karyopherin alpha 2
|
|
3-MA
|
3-methyladenine
|
|
CCK-8
|
Cell Counting Kit-8
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
PVDF
|
polyvinylidene difluoride
|
|
ECL
|
electrochemiluminescence
|
|
TEM
|
transmission electron microscopy
|
|
IC50
|
the half maximal inhibitory
concentration
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sarode GS, Sarode SC, Maniyar N, Anand R
and Patil S: Oral cancer databases: A comprehensive review. J Oral
Pathol Med. 2017. View Article : Google Scholar :
|
|
3
|
Gharat SA, Momin M and Bhavsar C: Oral
squamous cell carcinoma: Current treatment strategies and
nanotechnology-based approaches for prevention and therapy. Crit
Rev Ther Drug Carrier Syst. 33:363–400. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
White E, Mehnert JM and Chan CS:
Autophagy, metabolism, and cancer. Clin Cancer Res. 21:5037–5046.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lorin S, Hamai A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang X, Overholtzer M and Thompson CB:
Autophagy in cellular metabolism and cancer. J Clin Invest.
125:47–54. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Villar VH, Merhi F, Djavaheri-Mergny M and
Duran RV: Glutaminolysis and autophagy in cancer. Autophagy.
11:1198–1208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kumar A, Singh UK and Chaudhary A:
Targeting autophagy to overcome drug resistance in cancer therapy.
Future Med Chem. 7:1535–1542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang Z, Zhou L, Chen Z, Nice EC and Huang
C: Stress management by autophagy: Implications for
chemoresistance. Int J Cancer. 139:23–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gomes LR, Vessoni AT and Menck CFM:
Microenvironment and autophagy cross-talk: Implications in cancer
therapy. Pharmacol Res. 107:300–307. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14:482015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mowers EE, Sharifi MN and Macleod KF:
Autophagy in cancer metastasis. Oncogene. 36:1619–1630. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marcucci F, Ghezzi P and Rumio C: The role
of autophagy in the cross-talk between epithelial-mesenchymal
transitioned tumor cells and cancer stem-like cells. Mol Cancer.
16:32017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yasuhara N and Kumar PK: Aptamers that
bind specifically to human KPNA2 (importin-α1) and efficiently
interfere with nuclear transport. J Biochem. 160:259–268. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christiansen A and Dyrskjøt L: The
functional role of the novel biomarker karyopherin α 2 (KPNA2) in
cancer. Cancer Lett. 331:18–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Teng SC, Wu KJ, Tseng SF, Wong CW and Kao
L: Importin KPNA2, NBS1, DNA repair and tumorigenesis. J Mol
Histol. 37:293–299. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao Li, Yu L, Li CM, Li Y, Jia BL and
Zhang B: Karyopherin α2 induces apoptosis in tongue squamous cell
carcinoma CAL-27 cells through the p53 pathway. Oncol Rep.
35:3357–3362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang CI, Chien KY, Wang CL, Liu HP, Cheng
CC, Chang YS, Yu JS and Yu CJ: Quantitative proteomics reveals
regulation of karyopherin subunit alpha-2 (KPNA2) and its potential
novel cargo proteins in nonsmall cell lung cancer. Mol Cell
Proteomics. 11:1105–1122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gluz O, Wild P, Meiler R, Frick M, Ting E,
Mohrmann S, Schuett G, Dahl E, Fuchs T, Herr A, et al: Nuclear
karyopherin alpha2 expression predicts poor survival in patients
with advanced breast cancer irrespective of treatment intensity.
Int J Cancer. 123:1433–1438. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sakai M, Sohda M, Miyazaki T, Suzuki S,
Sano A, Tanaka N, Inose T, Nakajima M, Kato H and Kuwano H:
Significance of karyopherin-{alpha} 2 (KPNA2) expression in
esophageal squamous cell carcinoma. Anticancer Res. 30:851–856.
2010.PubMed/NCBI
|
|
22
|
Mortezavi A, Hermanns T, Seifert HH,
Baumgartner MK, Provenzano M, Sulser T, Burger M, Montani M,
Ikenberg K, Hofstädter F, et al: KPNA2 expression is an independent
adverse predictor of biochemical recurrence after radical
prostatectomy. Clin Cancer Res. 17:1111–1121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Noetzel E, Rose M, Bornemann J, Gajewski
M, Knüchel R and Dahl E: Nuclear transport receptor
karyopherin-alpha2 promotes malignant breast cancer phenotypes in
vitro. Oncogene. 31:2101–2114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang L, Wang HY, Li JD, Wang JH, Zhou Y,
Luo RZ, Yun JP, Zhang Y, Jia WH and Zheng M: KPNA2 promotes cell
proliferation and tumorigenicity in epithelial ovarian carcinoma
through upregulation of c-Myc and downregulation of FOXO3a. Cell
Death Dis. 4:e7452013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rezabakhsh A, Ahmadi M, Khaksar M,
Montaseri A, Malekinejad H, Rahbarghazi R and Garjani A: Rapamycin
inhibits oxidative/nitrosative stress and enhances angiogenesis in
high glucose-treated human umbilical vein endothelial cells: Role
of autophagy. Biomed Pharmacother. 93:885–894. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ko JH, Yoon SO, Lee HJ and Oh JY:
Rapamycin regulates macrophage activation by inhibiting NLRP3
inflammasome-p38 MAPK-NFkappaB pathways in autophagy- and
p62-dependent manners. Oncotarget. 8:40817–40831. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu Z, Huang CM, Shao Z, Zhao XP, Wang M,
Yan TL, Zhou XC, Jiang EH, Ke Liu and Shang ZJ: Autophagy induced
by areca nut extract contributes to decreasing cisplatin toxicity
in oral squamous cell carcinoma cells: Roles of reactive oxygen
species/AMPK signaling. Int J Mol Sci. 18:E5242017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jia L, Wang J, Wu T, Wu J, Ling J and
Cheng B: In vitro and in vivo antitumor effects of
chloroquine on oral squamous cell carcinoma. Mol Med Rep.
16:5779–5786. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alshareeda AT, Negm OH, Green AR, Nolan
CC, Tighe P, Albarakati N, Sultana R, Madhusudan S, Ellis IO and
Rakha EA: KPNA2 is a nuclear export protein that contributes to
aberrant localisation of key proteins and poor prognosis of breast
cancer. Br J Cancer. 112:1929–1937. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Altan B, Yokobori T, Mochiki E, Ohno T,
Ogata K, Ogawa A, Yanai M, Kobayashi T, Luvsandagva B, Asao T and
Kuwano H: Nuclear karyopherin-alpha2 expression in primary lesions
and metastatic lymph nodes was associated with poor prognosis and
progression in gastric cancer. Carcinogenesis. 34:2314–2321. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang L, Zhou Y, Cao XP, Lin JX, Zhang L,
Huang ST and Zheng M: KPNA2 is a potential diagnostic serum
biomarker for epithelial ovarian cancer and correlates with poor
prognosis. Tumour Biol. 39:10104283177062892017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou LN, Tan Y, Li P, Zeng P, Chen MB, Ye
Tian and Zhu YQ: Prognostic value of increased KPNA2 expression in
some solid tumors: A systematic review and meta-analysis.
Oncotarget. 8:303–314. 2017.PubMed/NCBI
|
|
33
|
Wang CI, Wang CL, Wang CW, Chen CD, Wu CC,
Liang Y, Tsai YH, Chang YS, Yu JS and Yu CJ: Importin subunit
alpha-2 is identified as a potential biomarker for non-small cell
lung cancer by integration of the cancer cell secretome and tissue
transcriptome. Int J Cancer. 128:2364–2372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsai MM, Huang HW, Wang CS, Lee KF, Tsai
CY, Lu PH, Chi HC, Lin YH, Kuo LM and Lin KH: MicroRNA-26b inhibits
tumor metastasis by targeting the KPNA2/c-jun pathway in human
gastric cancer. Oncotarget. 7:39511–39526. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Buchser WJ, Laskow TC, Pavlik PJ, Lin HM
and Lotze MT: Cell-mediated autophagy promotes cancer cell
survival. Cancer Res. 72:2970–2979. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rebecca VW and Amaravadi RK: Emerging
strategies to effectively target autophagy in cancer. Oncogene.
35:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang M, Zeng P, Kang R, Yu Y, Yang L, Tang
D and Cao L: S100A8 contributes to drug resistance by promoting
autophagy in leukemia cells. PLoS One. 9:e972422014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ajabnoor GM, Crook T and Coley HM:
Paclitaxel resistance is associated with switch from apoptotic to
autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis.
3:e2602012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qadir MA, Kwok B, Dragowska WH, To KH, Le
D, Bally MB and Gorski SM: Macroautophagy inhibition sensitizes
tamoxifen-resistant breast cancer cells and enhances mitochondrial
depolarization. Breast Cancer Res Treat. 112:389–403. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guo JY, Teng X, Laddha SV, Ma S, Van
Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD and White E:
Autophagy provides metabolic substrates to maintain energy charge
and nucleotide pools in Ras-driven lung cancer cells. Genes Dev.
30:1704–1717. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Guo JY and White E: Autophagy, metabolism,
and cancer. Cold Spring Harb Symp Quant Biol. 81:73–78. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kimmelman AC and White E: Autophagy and
tumor metabolism. Cell Metab. 25:1037–1043. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rangwala R, Chang YC, Hu J, Algazy KM,
Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT and
Troxel AB: Combined MTOR and autophagy inhibition: Phase I trial of
hydroxychloroquine and temsirolimus in patients with advanced solid
tumors and melanoma. Autophagy. 10:1391–1402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Levy JMM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yao D, Wang P, Zhang J, Fu L, Ouyang L and
Wang J: Deconvoluting the relationships between autophagy and
metastasis for potential cancer therapy. Apoptosis. 21:683–698.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Debnath J: Detachment-induced autophagy
during anoikis and lumen formation in epithelial acini. Autophagy.
4:351–353. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Herrero-Martin G, Hoyer-Hansen M,
Garcia-Garcia C, Fumarola C, Farkas T, López-Rivas A and Jäättelä
M: TAK1 activates AMPK-dependent cytoprotective autophagy in
TRAIL-treated epithelial cells. EMBO J. 28:677–685. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sosa MS, Bragado P and Aguirre-Ghiso JA:
Mechanisms of disseminated cancer cell dormancy: An awakening
field. Nat Rev Cancer. 14:611–622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ojha R, Bhattacharyya S and Singh SK:
Autophagy in cancer stem cells: A potential link between
chemoresistance, recurrence, and metastasis. Biores Open Access.
4:97–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare
S, Kondo S, Kondo Y, Yu Y, Mills GB, et al: The tumor suppressor
gene ARHI regulates autophagy and tumor dormancy in human
ovarian cancer cells. J Clin Invest. 118:3917–3929. 2008.PubMed/NCBI
|
|
53
|
Maes H, Kuchnio A, Peric A, Moens S, Nys
K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et
al: Tumor vessel normalization by chloroquine independent of
autophagy. Cancer Cell. 26:190–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhao X, Fang Y, Yang Y, Qin Y, Wu P, Wang
T, Lai H, Meng L, Wang D, Zheng Z, et al: Elaiophylin, a novel
autophagy inhibitor, exerts antitumor activity as a single agent in
ovarian cancer cells. Autophagy. 11:1849–1863. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhou J, Dong D, Cheng R, Wang Y, Jiang S,
Zhu Y, Fan L, Mao X, Gui Y, Li Z, et al: Aberrant expression of
KPNA2 is associated with a poor prognosis and contributes to OCT4
nuclear transportation in bladder cancer. Oncotarget.
7:72767–72776. 2016.PubMed/NCBI
|
|
56
|
Takada T, Tsutsumi S, Takahashi R, Ohsone
K, Tatsuki H, Suto T, Kato T, Fujii T, Yokobori T and Kuwano H:
KPNA2 over-expression is a potential marker of prognosis and
therapeutic sensitivity in colorectal cancer patients. J Surg
Oncol. 113:213–217. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
White E: Autophagy and p53. Cold Spring
Harb Perspect Med. 6:a0261202016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu J, Zhang C, Hu W and Feng Z: Tumor
suppressor p53 and its mutants in cancer metabolism. Cancer Lett.
356:197–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lu Y, Xiao L, Liu Y, Wang H, Li H, Zhou Q,
Pan J, Lei B, Huang A and Qi S: MIR517C inhibits autophagy
and the epithelial-to-mesenchymal (-like) transition phenotype in
human glioblastoma through KPNA2-dependent disruption of TP53
nuclear translocation. Autophagy. 11:2213–2232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Maiuri MC, Galluzzi L, Morselli E, Kepp O,
Malik SA and Kroemer G: Autophagy regulation by p53. Curr Opin Cell
Biol. 22:181–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Galluzzi L, Morselli E, Kepp O, Maiuri MC
and Kroemer G: Defective autophagy control by the p53 rheostat in
cancer. Cell Cycle. 9:250–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
O'Brate A and Giannakakou P: The
importance of p53 location: Nuclear or cytoplasmic zip code? Drug
Resist Updat. 6:313–322. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim IS, Kim DH, Han SM, Chin MU, Nam HJ,
Cho HP, Choi SY, Song BJ, Kim ER, Bae YS and Moon YH: Truncated
form of importin alpha identified in breast cancer cell inhibits
nuclear import of p53. J Biol Chem. 275:23139–23145. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kau TR and Silver PA: Nuclear transport as
a target for cell growth. Drug Discov Today. 8:78–85. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jamali T, Jamali Y, Mehrbod M and Mofrad
MR: Nuclear pore complex: Biochemistry and biophysics of
nucleocytoplasmic transport in health and disease. Int Rev Cell Mol
Biol. 287:233–286. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dickmanns A, Kehlenbach RH and Fahrenkrog
B: Nuclear pore complexes and nucleocytoplasmic transport: From
structure to function to disease. Int Rev Cell Mol Biol.
320:171–233. 2015. View Article : Google Scholar : PubMed/NCBI
|