Introduction
Lung cancer is the most life threatening cancer in
the world as well as in the United States due to high occurrences
of metastasis and drug resistance. In 2017, 2,22,500 Americans were
estimated to be diagnosed with lung cancer, and 1,55,870 were
estimated to die of lung cancer, accounting for 27 and 25% in total
estimated deaths in males and females, respectively (1). Non-small cell lung cancers (NSCLCs)
account for 84% of all lung cancer cases, and the 5-year survival
rate (15% for male and 21% for female) is extremely poor (2). Therefore, identification of promising
molecular target is required for the effective prevention and
therapy of NSCLCs.
Epithelium-specific ETS-1 (ESE-1) protein, also
identified as ELF3 (3), ERT
(4), ESX (5) and PDEF (6), belongs to an ETS transcription factor
superfamily and is mainly expressed in epithelial-rich tissues
including the lung and gut (7).
ESE-1 regulates the expression of target genes that determine
cancer phenotype including cell division, differentiation and
apoptosis (8). However, the effects
of ESE-1 on cancer are extremely complex. ESE-1 expression is
associated with poor prognosis in colorectal cancer patients
(9) while ESE-1 downregulation was
found to be related to reduced survival in ovarian cancer (10) and ESE-1 inhibits the invasion of
oral squamous cell carcinoma (11).
In an in vitro condition, ESE-1 knockdown inhibited cell
proliferation in colon cancer cells (9) while ESE-1 was found to facilitate cell
growth arrest and apoptosis in the colon cancer cells (12). ESE-1 expression also led to two
conflicting results in prostate cancer cells. For example, ESE-1
expression mediated transforming phenotypes (13) while ESE-1 repressed
androgen-receptor action and growth/migration of prostate tumors
(14). These contradictory results
strongly support a dichotomous effect of ESE-1 depending on the
cellular context.
In lung tissue, ESE-1 plays a key role in the
proliferation and differentiation during airway inflammation and
injured airway epithelium repair (15). A high level of ESE-1 expression was
detected in the developing fetal and adult lung tissues as well
(3). The aim of this study was to
investigate whether ESE-1 modulates lung cancer development using
human NSCLC cells and a xenograft mouse model. Here, we report that
ESE-1 possesses tumor suppressive activity in lung cancer.
Materials and methods
Materials
Human NSCLC cell lines (A549, H358, H1299 and H1703)
were purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). TGF-β (cat. no. 240-B) was purchased from
R&D System, Inc. (Minneapolis, MN, USA). Antibodies for Smad2
(cat. no. 5339), Smad3 (cat. no. 9513), p-smad2 (cat. no. 8828),
p-smad3 (cat. no. 9520), E-cadherin (cat. no. 3195), N-cadherin
(cat. no. 4061), Snail (cat. no. 3879), ZO-1 (cat. no. 5406), CDK4
(cat. no. 12790), CDK6 (cat. no. 3136), p27 (cat. no. 3688), MMP-9
(cat. no. 13667), PCNA (cat. no. 2586), cleaved caspase-3 (cat. no.
9661) and β-actin (cat. no. 5125s) were purchased from Cell
Signaling Technology, Inc. (Beverly, MA, USA). Antibodies for ESE-1
(cat. no. SC-376055), VEGF (cat. no. SC-507), Smad4 (cat. no.
SC-7966), and siRNA for Smad4 (cat. no. SC-29484) were purchased
from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-V5 (cat. no.
46–0708) was purchased from Invitrogen; Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). Bcl2 antibody (cat. no. 610538) was
purchased from BD Biosciences (San Jose, CA, USA). All chemicals
were purchased from Thermo Fisher Scientific or VWR International
LLC (Radnor, PA, USA) unless otherwise specified.
Cell culture and establishment of
stable cell lines
Human NSCLC cells were grown in DMEM/F12 media
supplemented with 10% fetal bovine serum (FBS). H1299 and H1703
stable cell lines overexpressing LacZ and ESE-1 were generated by
transient transfection with expression vectors and selected with
G418 (800 µg/ml for selection and 400 µg/ml for maintenance).
Cloning of ESE-1 promoters
Human ESE-1 promoter (Reference: NC_018912.2; +1 ~
+713 bp; +1 ~ +1,500 bp) was amplified with primers as indicated in
Table I. PCR products were then
inserted into the pGL3-basic vector by using In-Fusion cloning
method (Clontech Laboratories, Inc., Mountain View, CA, USA). The
pGL3-basic vector was linearized with FastDigest HindIII
enzyme (Thermo Fisher Scientific, Inc.) and cleaned up with
QIAquick PCR purification kit (Qiagen, Inc., Germantown, MD, USA).
Internal deletion and point mutation constructs (both single and
double) were cloned using the QuickChange Mutagenesis kit (Thermo
Fisher Scientific, Inc.) as previously described (16).
 | Table I.Sequences of primers used for cloning
of the ESE-1 promoters. |
Table I.
Sequences of primers used for cloning
of the ESE-1 promoters.
| Primer name | Sequence |
|---|
|
ESE1-promoter-713-F |
5′-CGATCTAAGTAAGCTTGGGAAGAACTTTCAAGCAGAG-3′ |
|
ESE1-promoter-713-R |
5′-CCGGAATGCCAAGCTTGGCTTTATAGTGTGTCCCCTG-3′ |
|
ESE1-promoter-1500-F |
5′-CGATCTAAGTAAGCTTCTGGGCGACAGAGCGAGAC-3′ |
|
ESE1-promoter-1500-R |
5′-CCGGAATGCCAAGCTTGGCTTTATAGTGTGTCCCCTG-3′ |
|
ESE1-promoter-1500-ΔD-F |
5′-GGAAGAGATCTTAGGGATTATTACCTCATTTTTTATAG-3′ |
|
ESE1-promoter-1500-ΔD-R |
5′-GTTTCCCCATCTATAAAAAATGAGGTAATAATCCCTAAG-3′ |
|
ESE1-promoter-1500-ΔP-F |
5′-CGGGCTGAGTCCATAGAGACATGGGGGACTC-3′ |
|
ESE1-promoter-1500-ΔP-R |
5′-CCCTGTGGGCAGAGTCCCCCATGTCTCTATGG-3′ |
|
ESE1-promoter-1500-ΔD&P-F |
5′-GGAAGAGATCTTAGGGATTATTACCTCATTTTTTATAG-3′ |
|
ESE1-promoter-1500-ΔD&P-R |
5′-GTTTCCCCATCTATAAAAAATGAGGTAATAATCCCTAAG-3′ |
|
ESE1-promoter-1500-mD-F |
5′-GGGATTATTAAATTTTTTCAACCTCATTTTTTATAG-3′ |
|
ESE1-promoter-1500-mD-R |
5′-CTATAAAAAATGAGGTTGAAAAAATTTAATAATCCC-3′ |
|
ESE1-promoter-1500-mP-F |
5′-GAGACAACCATTTTCATGGGGGAC-3′ |
|
ESE1-promoter-1500-mP-R |
5′-GTCCCCCATGAAAATGGTTGTCTC-3′ |
|
ESE1-promoter-1500-mD&P-F |
5′-GGGATTATTAAATTTTTTCAACCTCATTTTTTATAG-3′ |
|
ESE1-promoter-1500-mD&P-R |
5′-CTATAAAAAATGAGGTTGAAAAAATTTAATAATCCC-3′ |
Transfection and reporter gene
assay
ESE-1 promoters were transiently transfected using
PolyJet™ DNA transfection reagent (SignaGen Laboratories,
Rockville, MD, USA) according to the manufacturer's instructions.
The cells were harvested in 1X luciferase lysis buffer. The
luciferase activity was measured and normalized to the pRL-null
luciferase activity using a Dual-Luciferase assay kit (Promega,
Madison, WI, USA) as previously described (16).
SDS-PAGE and western blot
analysis
SDS-PAGE and western blot analysis were performed as
we previously described (16). The
cell lysates were extracted using RIPA buffer and 25 µg of protein
was loaded onto SDS-polyacrylamide gel (8–12% depending on
molecular size of each protein). After transferring onto a nylon
membrane, the extracted proteins were incubated with the primary
antibodies (1:1,000). The antibody information was described in
Materials and methods. Chemiluminescence was detected with Pierce
ECL Western blotting substrate (Thermo Fisher Scientific, Inc.) and
visualized using the ChemiDoc MP Imaging system (Bio-Rad
Laboratories, Hercules, CA, USA).
Semi-quantitative RT PCR
Semi-quantitative RT-PCR was performed as we
previously described (16). The
primer sequences for PCR are as follows: ESE-1, forward 5′-ttagcaac
tacttcagtgcgatgt-3′ and reverse 5′-gttcttctccacttggtagctgat-3′;
Snail, forward 5′-CCTCCCTGTCAGATGAGGAC-3′ and reverse
5′-CCAGGCTGAGGTATTCCTTG-3′; GAPDH, forward
5′-GGGCTGCTTTTAACTCTGGT-3′ and reverse
5′-TGGCAGGTTTTTCTAGACGG-3′.
Soft agar assay
The bottom layer containing 1% agar and culture
medium was prepared in the 6-well culture plate. A mixture of agar
and cells (5,000 cells/well) were placed, solidified and incubated
in a 37°C humidified CO2 incubator. Then, a layer of
growth medium (200 µl) was added over the upper layer of agar to
prevent desiccation every 2 days. Cells were grown for 2 weeks and
stained overnight with nitro-blue tetrazolium chloride solution at
37°C. Images were captured by ChemiDoc MP Imaging system (Bio-Rad
Laboratories).
Invasion assay
Cell invasion was analyzed using an invasion assay
kit (Corning Inc., Corning, NY, USA) according to the
manufacturer's instructions. Briefly, cells were suspended and
plated in an upper invasion chamber (24-wells) of a Transwell with
Matrigel. The lower chamber was added with 10% FBS medium. After 12
h of incubation, the cells in the chamber were removed with a
cotton swab. After staining, images were captured under an inverted
microscope (Nikon Eclipse Ti; Nikon Corp., Tokyo, Japan).
Scratch wound assays
The cells (5×104 cells/well) were seeded
onto 6-well tissue culture plates and grown overnight. A linear
wound was generated using a sterile 20-µl pipette tip. After 22 h,
images were captured by an inverted microscope (Nikon Eclipse Ti;
Nikon).
Cell cycle distribution and analysis
of apoptosis
Cell cycle analysis was performed as we previously
described (17). The cells were
collected through typsin-EDTA treatment and stained with propidium
iodide (PI). After washing, the cell cycle distribution was
analyzed using FACS flow cytometry at the University of Maryland
MPRI Flow Cytometry and Cell Sorting Facility. Apoptosis was
measured using Cell Death Detection ELISA (Roche Diagnostics,
Indianapolis, IN, USA) according to the manufacturer's
instructions.
Xenograft study
The animal study was approved by the Animal Care and
Use Committee (Ajou University, Suwon, Korea). Fourteen male BALB/c
nude mice aged 6 weeks with average weight of 25 g were purchased
from Harlan Laboratories, Ltd., (Bicester, UK) and maintained ad
libitum at 23°C temperature and 12-h light/dark
cycle. The mice were injected s.c. in the left flank with H1299
stable cell lines overexpressing LacZ and ESE-1 (5×106
cells mixed with Matrigel, 1:1). After cell injection, the tumor
formation was monitored twice a week and tumor size was measured
with a caliper (Mitutoyo, Utsunomiya, Japan) and the volumes were
estimated using the formula: a × b2 × 0.52, where ‘a’ is
the longest and ‘b’ is the shortest diameter. Thirty five days
after injection, the mice were sacrificed by carbon dioxide
asphyxiation and the tumors were harvested and subjected to
immunohistochemistry and western blot analysis.
Immunohistochemistry
Immunohistochemistry was performed to analyze the
expression of ESE-1, PCNA and cleaved caspase-3. Paraffin tissue
sections (4 µm thick) were briefly deparaffinized with xylene and
rehydrated through alcohol, and washed in distilled water. After
H2O2-induced inactivation for endogenous
peroxidase activity and antigen retrieval in pepsin (Dako,
Carpinteria, CA, USA) at 37°C for 30 min, the tissue sections were
incubated with primary antibodies for ESE-1 (1:100), PCNA (1:4,000)
and cleaved caspase-3 (1:2,000) overnight at 4°C. After washing,
signals were detected with 3,3-diaminobenzidine tetrahydrochloride
(DAB) and the sections were counterstained with hematoxylin.
Statistical analysis
Statistical analysis was performed by Student's
t-test. Data represent mean ± SD from three replicates.
Results
Basal expression of ESE-1 in human
NSCLC cells
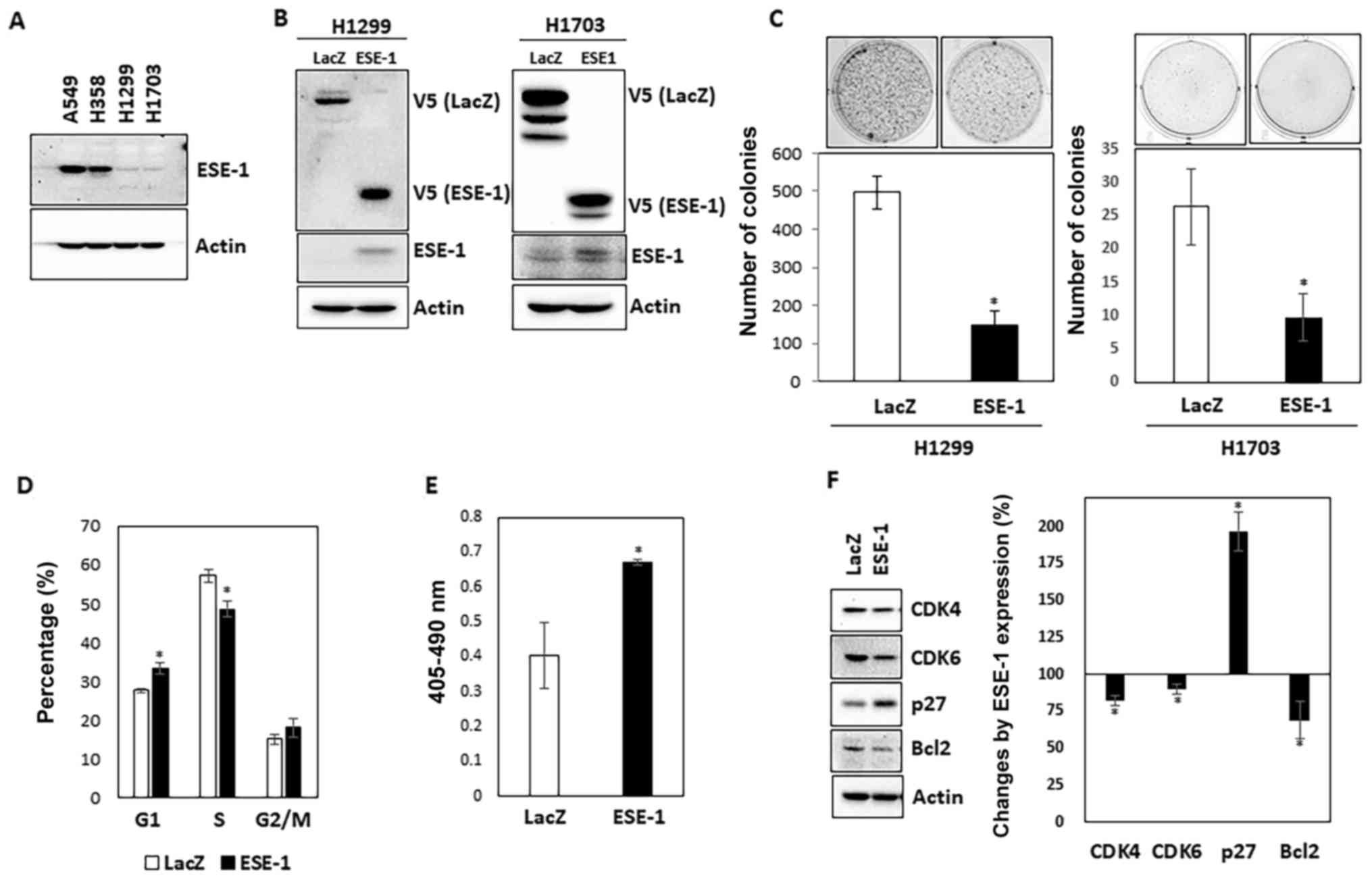
First, we determined the basal expression of ESE-1
in four different human NSCLC cell lines (A549, H358, H1299 and
H1703). As shown in Fig. 1A, A549
and H358 were ESE-1 abundant whereas H1299 and H1703 expressed low
levels of ESE-1. We generated H1299 and H1703 stable cell lines
overexpressing LacZ (control) and ESE-1 by transfecting V5-tag-LacZ
(control) and the V5-tag-ESE-1 expression vector into these cell
lines and subsequent selection with G418. Overexpression of LacZ
and ESE-1 were validated through V5 expression as well as
endogenous ESE-1 expression (Fig.
1B.)
ESE-1 suppresses anchorage-independent
growth of human NSCLC cells
Next, we performed soft agar assay to ascertain
whether ESE-1 overexpression affects the anchorage-independent
growth of H1299 and H1703 stable cell lines. As shown in Fig. 1C, the number of colonies was 3-fold
higher in the LacZ-expressing H1299 cells compared to the number of
colonies noted in the ESE-1-expressing H1299 cells. Similar result
was obtained from ESE-1-overexpressing H1703 cells. The result
indicates that ESE-1 possesses anti-growth activity in human NSCLC
cells.
‘To investigate whether retarded growth by ESE-1
overexpression is associated with cell growth arrest and apoptosis,
cell cycle distribution and the fraction of apoptotic cells were
compared between LacZ- and ESE-1-expressing H1299 stable cells.
Cell cycle analysis data showed that ESE-1 overexpression resulted
in a significant increased percentage of cells in the G1 phase
(LacZ, 27.7±0.5 vs. ESE-1, 33.3±1.3%) and a decrease in the S phase
(LacZ, 57.2±1.7 vs. ESE-1, 48.6±2.0%), and no significant change in
the G2 phase (LacZ, 15.0±1.2 vs. ESE-1, 18.1±2.4%) (Fig. 1D). In addition, the number of
apoptotic cells was increased by 1.7-fold in the ESE-1-expressing
H1299 cells compared to that noted in the control cells (Fig. 1E). We also measured the expression
of several cell cycle- and apoptosis-regulating marker proteins
using western blot analysis. As shown in Fig. 1F, ESE-1 expression led to a decrease
in CDK4 and CDK6 and an increase in p27. We also found that ESE-1
overexpression decreased Bcl2 expression.
ESE-1 suppresses invasion and
migration of human NSCLC cells
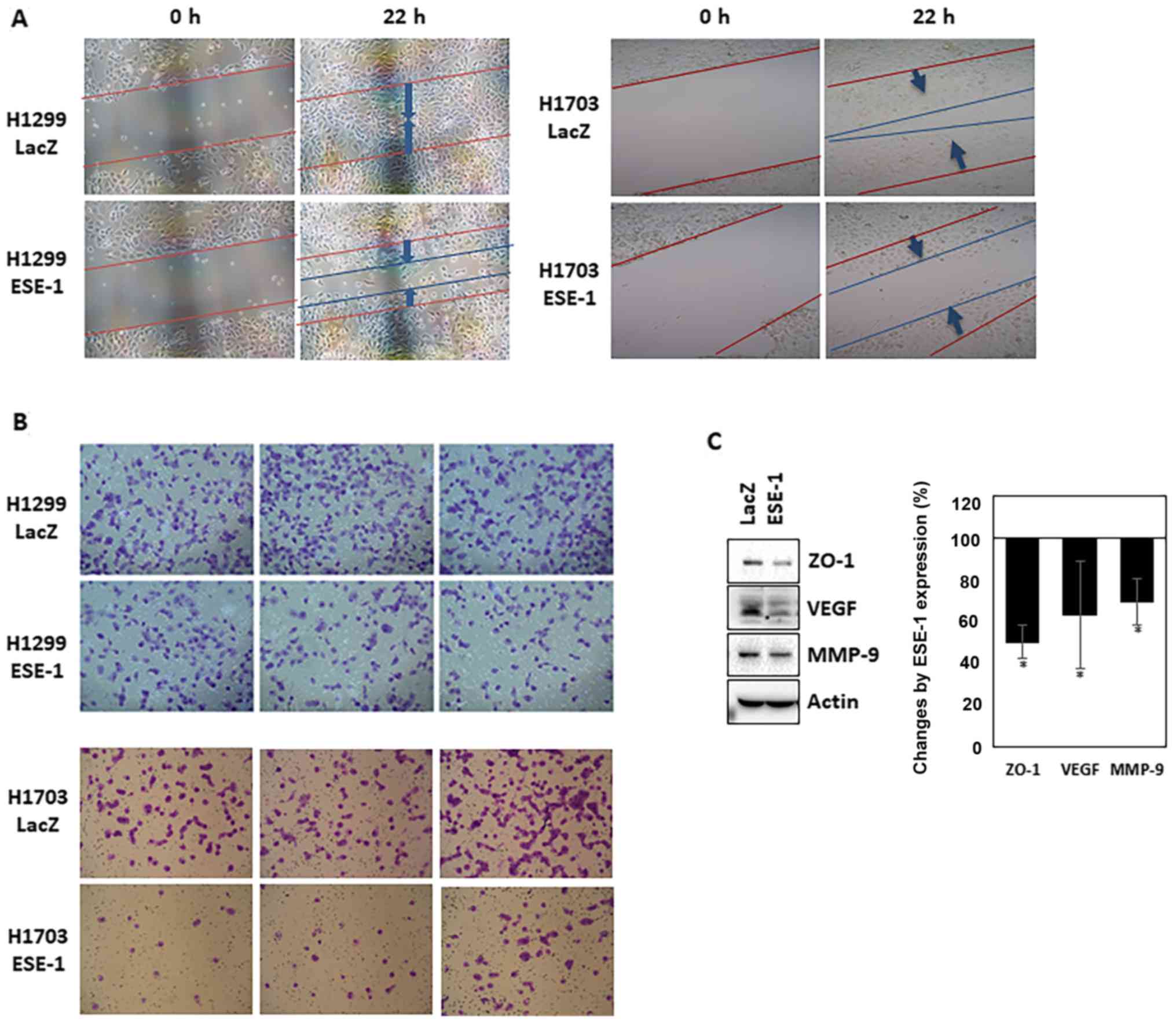
Based on our finding that ESE-1 possesses
tumor-suppressive activity, we further explored whether ESE-1
possesses anti-metastatic activity by performing in vitro
cell invasion and migration assays. As indicated in Fig. 2A, the wound healing assay indicated
that ESE-1-expressing cells had reduced migration capacity than
that of the control cells. In addition, the ESE-1-expressing stable
cell line showed lower invasive ability when compared to the
control cells (Fig. 2B). These
results imply that ESE-1 overexpression inhibits the metastatic
activity of human NSCLC cells in vitro. Western blot
analysis showed that expression of MMP-9 was decreased in the
ESE-1-expressing cells compared to that noted in the control cells
(Fig. 2C). An angiogenic protein
(VEGF) and a tight junction protein (ZO-1) were also downregulated
by ESE-1 overexpression (Fig.
2C).
ESE-1 is a target of TGF-β in human
NSCLC cells
To further study the possible anti-metastatic
mechanism mediated by ESE-1, we aimed to ascertain whether
epithelial-mesenchymal transition (EMT) influences the expression
of ESE-1. Since TGF-β is a major trigger of EMT, we treated ESE-1
abundant A549 and H358 cells with TGF-β in a low serum (0.5%)
condition. Treatment of TGF-β to A549 cells led to morphological
changes characteristic of EMT (Fig.
3A) which was accompanied by increased phosphorylation of Smad2
and Smad3 (Fig. 3B, top panel).
TGF-β also decreased the expression of E-cadherin and increased the
expression of N-cadherin and Snail in the A549 and H358 cells
(Fig. 3B, bottom panels). Notably,
TGF-β treatment decreased expression of ESE-1 in these two cell
lines (Fig. 3B, bottom panels).
Treatment of TGF-β to A549 cells also showed time-dependent
downregulation of ESE-1 protein (Fig.
3C) and ESE-1 mRNA (Fig. 3D).
The time-point that ESE-1 began to decrease was earlier (4 h) than
the time point that EMT marker proteins (E-cadherin and N-cadherin)
were changed. These data propose a potential link between ESE-1
downregulation and EMT. Finally, we produced two ESE-1 promoter
clones spanning to −1,500/+1 and −713/+1 and measured the
transcriptional activity (Fig. 3E).
The results indicate that treatment of TGF-β suppressed luciferase
activity by 43.3 and 45.8% in the −1,500/+1 and −173/+1 clones,
respectively. These data indicate that ESE-1 is an inverse
downstream target protein of TGF-β during EMT.
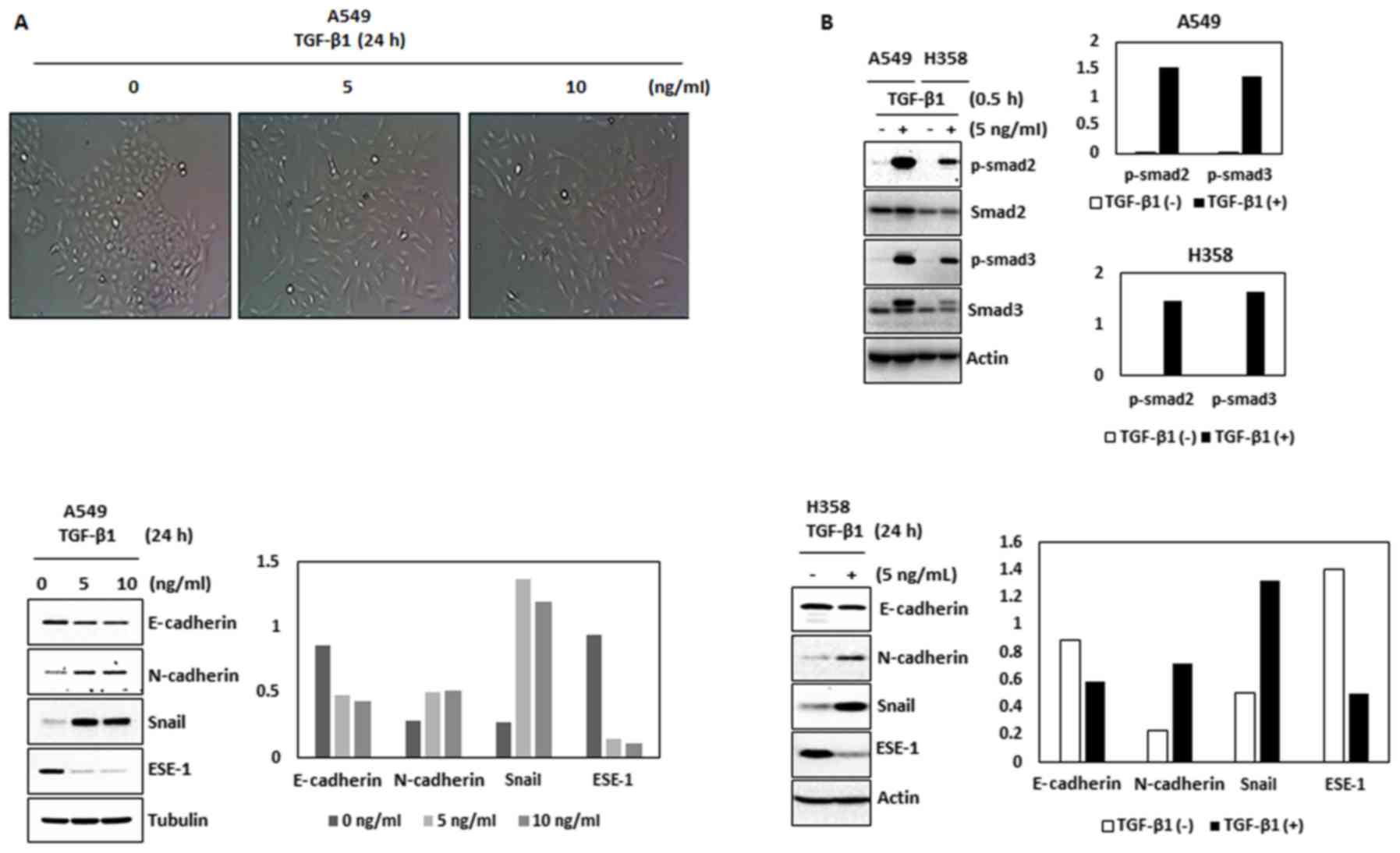 | Figure 3.ESE-1 is a target of TGF-β during EMT
in human NSCLC cells. (A) A549 cells were treated with TGF-β1 (0,
5, and 10 ng/ml) for 24 h. The images were captured by a
microscope. (B) A549 and H358 cells were treated with TGF-β1 for
the indicated doses for 30 min and 24 h. The cell lysates were
subjected to western blot analysis for the indicated antibodies.
(C) A549 cells were treated with TGF-β1 (5 ng/ml) for the indicated
times. The cell lysates were subjected to western blot analysis for
E-cadherin, N-cadherin, ESE-1, Snail and actin. (D) A549 cells were
treated with TGF-β1 (5 ng/ml) for the indicated times. RNA was
isolated and semi-quantitative reverse transcriptase (RT)-PCR was
performed for ESE-1, Snail and GAPDH. (E) Two different sizes of
the ESE-1 promoter (−1,500/+1 and −173/+1) were cloned as described
in Materials and methods and transfected into A549 cells and
luciferase activity was measured. Data represent mean ± SD from
three replicates. *P<0.05 vs. the control. NSCLC, non-small cell
lung cancer; ESE-1, epithelial-specific ETS-1; TGF-β, transforming
growth factor β; EMT, epithelial-mesenchymal transition. |
ESE-1 suppresses formation and
development of tumors in vivo
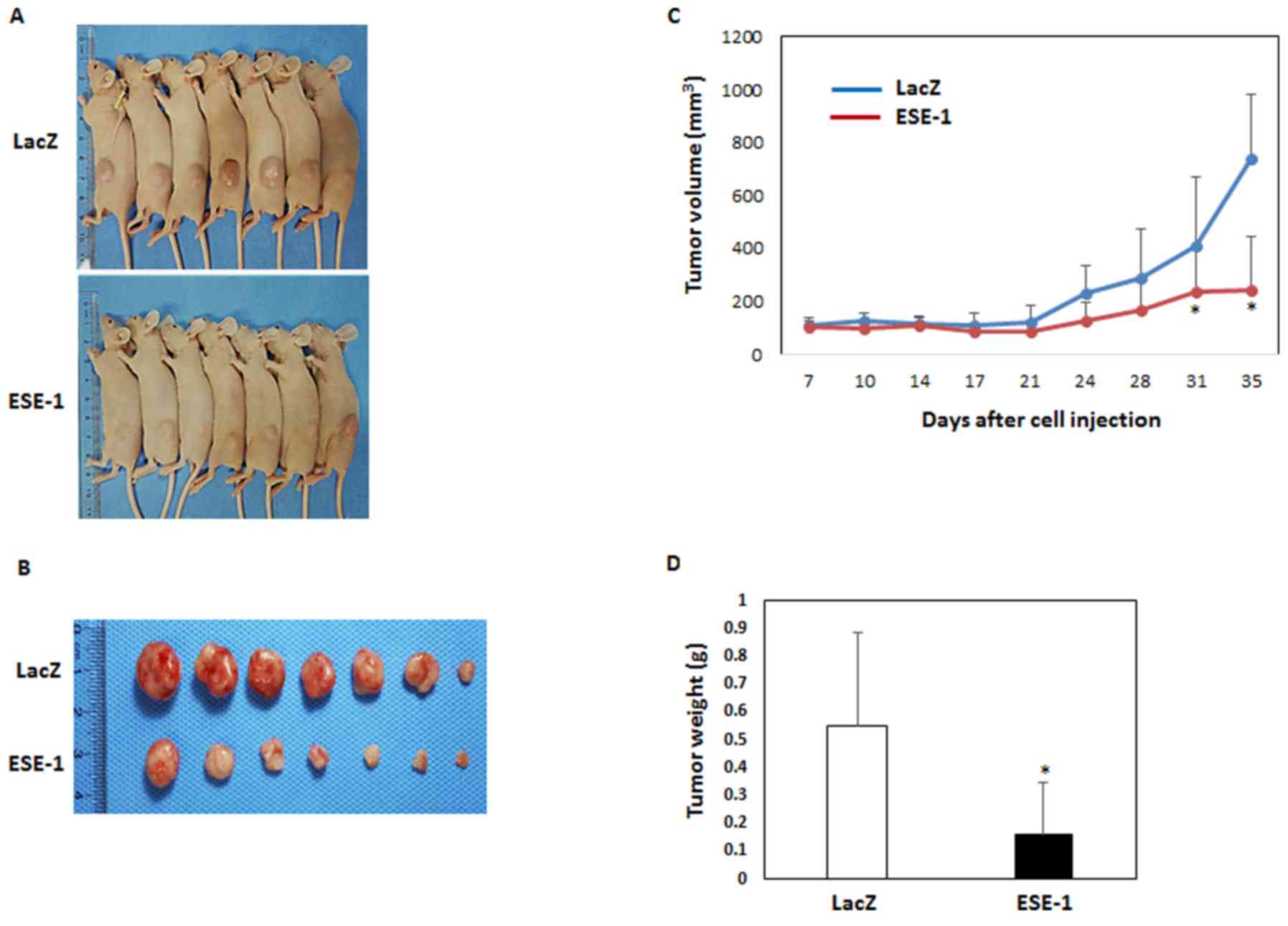
Finally, we verified the anti-cancer activity of
ESE-1 using a xenograft model. The LacZ- or ESE-1-expressing H1299
stable cell lines were subcutaneously injected into the flank of
nude mice. Fig. 4A shows
representative images of the mice of each group. The mice implanted
with ESE-1-expressing cells had smaller tumors than those noted in
the control mice (Fig. 4A and B).
Consistent with these images, the volume and weight of the tumors
were significantly decreased in the ESE-1-expressing group compared
with the control group (Fig. 4C and
D). On day 35, we isolated the tumor tissues and performed
immunohistochemistry with antibodies against ESE-1, PCNA and
cleaved caspase 3. Expression of ESE-1 in the tumor tissues was
associated with a decrease in PCNA (proliferating marker) and an
increase in cleaved caspase 3 (Fig.
4E). Western blot analysis indicated that the expression of
cleaved caspase-3 was increased in ESE-1-expressing tumor tissues
(Fig. 4F). Taken together, ESE-1
exhibits anticancer activity in vivo, similar to what was
observed in vitro.
Discussion
Due to the fact that ESE-1 is constitutively
expressed in terminally differentiated epithelial cells (7,18) and
that 80% of cancer is epithelial origin carcinoma, the significance
of this protein was broadly proposed during progression in
malignant neoplasms of epithelial origin. However, the effects of
ESE-1 on tumorigenesis are controversial depending on the type of
cancer (9–11). Therefore, further extensive studies
concerning the association of ESE-1 with cancer progression are
required using diverse cancer models.
Lung cancer is the most life-threatening cancer due
to the difficulty of early detection and poor prognosis. In
particular, NSCLC is associated with a poor survival rate due to
the acquired resistance of patients to therapeutic drugs. Targeted
therapy is a new therapeutic paradigm to cure cancer by targeting
specific and promising molecular targets. Recently, Wang et
al reported that Elf3 (ESE-1) facilitates the growth and
metastasis of lung cancer cells (19). We also explored the possibility of
using ESE-1 as a molecular target for lung cancer in the present
study.
In the initial step of our studies, we observed
endogenous expression of ESE-1 in four different human NSCLC cell
lines. As a result, A549 and H358 were identified as ESE-1 abundant
cells, whereas H1299 and H1703 were ESE-1 null cells. We did not
ascertain why H1299 and H1703 NSCLC cells show very low expression
of ESE-1 at the basal level. We speculate that the different basal
expression might be a result of genetic/epigenetic difference in
the process of ESE-1 gene transcription. This is supported
by differential expression of ESE-1 in different human colorectal
cancer cells as we previously reported (20). However, we do not exclude the
possibility that there could be differential autocrine regulation
of ESE-1 by TGF-β among these cell lines as lung tumor cells
secrete TGF-β which may repress ESE-1 expression.
We selected the H1299 cell line for further studies
due to its potent tumorigenic and metastatic property (21). Using soft agar assay, we identified
ESE-1 as a tumor suppressor in human NSCLC cells. As a part of a
mechanistic link, we observed increased G1 arrest and apoptosis in
ESE-1-expressing cells which were consistent with the upregulation
or downregulation of cell cycle- and apoptosis-regulating proteins
(Fig. 1F).
Regarding a role of ESE-1 in metastasis, Yeung et
al recently reported that ESE-1 is a negative regulator of EMT
and metastasis in ovarian cancer (10). Upregulation of ESE-1 inhibited
ovarian tumor progression with increased expression of epithelial
markers and decreased expression of mesenchymal markers (10). This is consistent with our
observation, supporting the anti-metastatic activity of ESE-1 with
suppression of cell invasion and migration and downregulation of
MMP-9, VEGF and ZO-1 (Fig. 2).
Epithelial-mesenchymal transition (EMT) is a dynamic
process that allows epithelial cells to lose polarity and transit
into mesenchymal characteristics. Activation of EMT transcription
factors leads to downregulation of E-cadherin (epithelial marker),
upregulation of N-cadherin and vimentin (mesenchymal markers), loss
of cell adhesion and promotion of cell mobility and invasion
(22). TGF-β facilitates EMT
through the Smad-dependent pathway known as the canonical pathway
(23). Besides typical Smad
activation, TGF-β also induces ERK, JNK/p38, PI3K/AKT, and Rho-like
GTPases activation, known as the non-canonical or Smad-independent
pathway (24). Several studies
support a potential role of ESE-1 in EMT. For example, ESE-1
reciprocally regulates expression of ZEB1/ZEB2, dependent on ERK
activity in breast cancer cells (25) and ESE-1 increases expression of
TGF-β II receptor in breast cancer cells (26).
To elucidate the anti-metastatic mechanism, we
focused on the involvement of ESE-1 in EMT of lung cancer cells.
Our significant finding was that ESE-1 is a negative downstream
target of TGF-β signaling. Time-dependent downregulation of ESE-1
was detected in the presence of TGF-β (Fig. 3C and D). To investigate if this
downregulation is associated with transcriptional repression, we
cloned the ESE-1 promoter spanning −1,500 to +1 and −713 to +1. The
results indicated that TGF-β suppresses transcription of the
ESE-1 gene. Since Smads are major mediators of TGF-β
signaling and we identified two Smad responsive elements (SRE) in
the promoter (Fig. 3E), we
hypothesized that Smad may mediate TGF-β-induced downregulation of
the ESE-1 gene. However, TGF-β-induced repression of
luciferase activities was identical in the two promoters where one
contained SRE and the other did not (Fig. 3E). In addition, luciferase assays
using internal deletion and point mutated clones lacking distal and
proximal SRE (Table I) showed no
difference in the TGF-β effect (data not shown). Knockdown of
Smad2, Smad3 and Smad4 using siRNA and western blot data also
indicated Smad-independent downregulation of ESE-1 by TGF-β (data
not shown). Therefore, we tested several non-canonical pathways
(ERK, p38 MAPK, JNK, RAS, GSK3, PI3K and NF-ĸB) using selective
inhibitors. None of the pathways were responsible for ESE-1
downregulation by TGF-β (data not shown).
Concerning the potential mechanism by which ESE-1
suppresses lung cancer progression, we propose several
cancer-related downstream target genes of ESE-1. One of the targets
is MMP-9 which was downregulated by ESE-1 through ETS-binding site
on the MMP-9 promoter in squamous cell carcinoma (11). ESE-1 downregulates OCT4, a
transcription factor, involved in stem cell pluripotency of NCCIT
human embryonic carcinoma cells (27). Another interesting target gene we
propose is wnt/β-catenin as we previously observed that ESE-1
physically interacts with β-catenin (20) and suppresses transcriptional
activity of β-catenin/LEF (data not shown). Since β-catenin and APC
mutations are uncommon in NSCLC, this mechanism could be promising
to elucidate the mechanism of ESE-1 in regard to the suppression of
lung cancer progression. Therefore, further research is warranted
to elucidate further detailed mechanisms of ESE-1 downregulation by
TGF-β and molecular target mediating anticancer activity of ESE-1
in human NSCLC cells.
In conclusion, we identified ESE-1 as a tumor
suppressor in human NSCLC. ESE-1 represses anchorage-independent
growth and invasion and migration of human NSCLC cells. ESE-1 is a
target of TGF-β during EMT. In addition, ESE-1 suppressed the
formation and development of tumors in vivo.
Acknowledgements
The authors thank Jihye Lee for her technical
supports.
Funding
The present study was supported by a grant from the
National Research Foundation of Korea (SRC, no. 2011–0030043: no.
2018R1A2B3009008).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SHL and CHK conceived and designed the study. ZL,
BSL, TH, YX and HJK performed the experiments. ZL, BSL and TH wrote
the paper. SHL reviewed and edited the manuscript. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Animal Care and Use Committee (Ajou University, Korea).
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schiller JH, Gandara DR, Goss GD and Vokes
EE: Non-small-cell lung cancer: Then and now. J Clin Oncol.
31:981–983. 2013.doi: 10.1200/JCO.2012.47.5772. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tymms MJ, Ng AY, Thomas RS, Schutte BC,
Zhou J, Eyre HJ, Sutherland GR, Seth A, Rosenberg M, Papas T, et
al: A novel epithelial-expressed ETS gene, ELF3: Human and murine
cDNA sequences, murine genomic organization, human mapping to
1q32.2 and expression in tissues and cancer. Oncogene.
15:2449–2462. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi SG, Yi Y, Kim YS, Kato M, Chang J,
Chung HW, Hahm KB, Yang HK, Rhee HH, Bang YJ and Kim SJ: A novel
ets-related transcription factor, ERT/ESX/ESE-1, regulates
expression of the transforming growth factor-β type II receptor. J
Biol Chem. 273:110–117. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang CH, Scott GK, Kuo WL, Xiong X,
Suzdaltseva Y, Park JW, Sayre P, Erny K, Collins C, Gray JW, et al:
: ESX: A structurally unique Ets overexpressed early during human
breast tumorigenesis. Oncogene. 14:1617–1622. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oettgen P, Finger E, Sun Z, Akbarali Y,
Thamrongsak U, Boltax J, Grall F, Dube A, Weiss A, Brown L, et al:
PDEF, a novel prostate epithelium-specific ets transcription
factor, interacts with the androgen receptor and activates
prostate-specific antigen gene expression. J Biol Chem.
275:1216–1225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oettgen P, Alani RM, Barcinski MA, Brown
L, Akbarali Y, Boltax J, Kunsch C, Munger K and Libermann TA:
Isolation and characterization of a novel epithelium-specific
transcription factor, ESE-1, a member of the ets family. Mol Cell
Biol. 17:4419–4433. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brembeck FH, Opitz OG, Libermann TA and
Rustgi AK: Dual function of the epithelial specific ets
transcription factor, ELF3, in modulating differentiation.
Oncogene. 19:1941–1949. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang JL, Chen ZF, Chen HM, Wang MY, Kong
X, Wang YC, Sun TT, Hong J, Zou W, Xu J, et al: Elf3 drives
β-catenin transactivation and associates with poor prognosis in
colorectal cancer. Cell Death Dis. 5:e12632014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yeung TL, Leung CS, Wong KK,
Gutierrez-Hartmann A, Kwong J, Gershenson DM and Mok SC: ELF3 is a
negative regulator of epithelial-mesenchymal transition in ovarian
cancer cells. Oncotarget. 8:16951–16963. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Iwai S, Amekawa S, Yomogida K, Sumi T,
Nakazawa M, Yura Y, Nishimune Y and Nozaki M: ESE-1 inhibits the
invasion of oral squamous cell carcinoma in conjunction with MMP-9
suppression. Oral Dis. 14:144–149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee SH, Bahn JH, Choi CK, Whitlock NC,
English AE, Safe S and Baek SJ: ESE-1/EGR-1 pathway plays a role in
tolfenamic acid-induced apoptosis in colorectal cancer cells. Mol
Cancer Ther. 7:3739–3750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Longoni N, Sarti M, Albino D, Civenni G,
Malek A, Ortelli E, Pinton S, Mello-Grand M, Ostano P, D'Ambrosio
G, et al: ETS transcription factor ESE1/ELF3 orchestrates a
positive feedback loop that constitutively activates NF-kappaB and
drives prostate cancer progression. Cancer Res. 73:4533–4547. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shatnawi A, Norris JD, Chaveroux C, Jasper
JS, Sherk AB, McDonnell DP and Giguère V: ELF3 is a repressor of
androgen receptor action in prostate cancer cells. Oncogene.
33:862–871. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oliver JR, Kushwah R, Wu J, Pan J, Cutz E,
Yeger H, Waddell TK and Hu J: Elf3 plays a role in regulating
bronchiolar epithelial repair kinetics following Clara
cell-specific injury. Lab Invest. 91:1514–1529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SH, Bahn JH, Whitlock NC and Baek SJ:
Activating transcription factor 2 (ATF2) controls tolfenamic
acid-induced ATF3 expression via MAP kinase pathways. Oncogene.
29:5182–5192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SH, Cekanova M and Baek SJ: Multiple
mechanisms are involved in 6-gingerol-induced cell growth arrest
and apoptosis in human colorectal cancer cells. Mol Carcinog.
47:197–208. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ng AY, Waring P, Ristevski S, Wang C,
Wilson T, Pritchard M, Hertzog P and Kola I: Inactivation of the
transcription factor Elf3 in mice results in dysmorphogenesis and
altered differentiation of intestinal epithelium. Gastroenterology.
122:1455–1466. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang H, Yu Z, Huo S, Chen Z, Ou Z, Mai J,
Ding S and Zhang J: Overexpression of ELF3 facilitates cell growth
and metastasis through PI3K/Akt and ERK signaling pathways in
non-small cell lung cancer. Int J Biochem Cell Biol. 94:98–106.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang X and Lee SH: Identification of ESE1
as a β-Catenin Binding Protein. Anticancer Res. 36:2697–2703.
2016.PubMed/NCBI
|
|
21
|
Singla AK, Downey CM, Bebb GD and Jirik
FR: Characterization of a murine model of metastatic human
non-small cell lung cancer and effect of CXCR4 inhibition on the
growth of metastases. Oncoscience. 2:263–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sinh ND, Endo K, Miyazawa K and Saitoh M:
Ets1 and ESE1 reciprocally regulate expression of ZEB1/ZEB2,
dependent on ERK1/2 activity, in breast cancer cells. Cancer Sci.
108:952–960. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang J, Lee C, Hahm KB, Yi Y, Choi SG and
Kim SJ: Over-expression of ERT(ESX/ESE-1/ELF3), an ets-related
transcription factor, induces endogenous TGF-beta type II receptor
expression and restores the TGF-beta signaling pathway in Hs578t
human breast cancer cells. Oncogene. 19:151–154. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park SW, Do HJ, Ha WT, Han MH, Yang HM,
Lee SH, Song H, Kim NH and Kim JH: Transcriptional regulation of
OCT4 by the ETS transcription factor ESE-1 in NCCIT human embryonic
carcinoma cells. Biochem Biophys Res Commun. 450:984–990. 2014.
View Article : Google Scholar : PubMed/NCBI
|