Introduction
Gastric cancer (GC) remains one of the most common
cancers and the third leading cause of tumor-related deaths
(1). Seventy percent of the newly
detected cases and deaths from GC were in developing countries, of
which 42% occurred in China (2).
Despite advances in the treatment of GC, the prognosis of GC
remains poor. Like all other cancers, the development of GC is a
multistep process involving numerous genetic and epigenetic
alterations of growth factors and receptors, angiogenic factors,
cell cycle regulators and DNA mismatch repair genes (3). These multiple factors on genetic and
epigenetic alterations are likely to be the target in GC cancer
prevention and treatment. Therefore, identifying the molecular
mechanisms underlying the development of GC can benefit diagnosis
and treatment of this disease.
Eph receptors are the largest known subfamily of
receptor tyrosine kinases that are activated in response to binding
with Eph receptor-interacting proteins (ephrins). Eph receptors
have been divided into two groups based on their sequence homology
and their affinities for binding ephrin-A and ephrin-B ligands:
EphA and EphB receptors. In the human genome, there are nine EphAs
and five EphBs that generally bind preferentially to five ephrin-A
and three ephrin-B ligands, respectively (4,5). The
Eph receptor and their ligands are frequently overexpressed in a
variety of cancers and affect tumor growth, angiogenesis and
metastasis (6). However, Eph
signaling activities in cancer are complex and interesting in their
paradoxical effects.
EphA3, an EphA receptor subfamily member, has been
found to be aberrantly expressed in a variety of human cancers
including malignant melanoma, glioblastoma and mutated in lung and
breast cancer (7,8). Increased expression of EphA3 can
promote tumor cellular proliferation, angiogenesis, invasion and is
regarded as a promising target in cancer therapy (9). Soluble EphA3-Fc receptors and the
anti-EphA3 antibody can inhibit tumor angiogenesis and progression
(10,11). However, little is known about the
function of EphA3 in GC. Our previous study reported that the
upregulated expression of EphA3 in GC was correlated with tumor
size, distant metastasis, pTNM stage and poor prognosis. We also
observed that the expression of EphA3 was significantly and
positively associated with VEGF and microvessel density (MVD)
expression (12). In the present
study, we extended our previous study and knocked down EphA3 in
HGC-27 cells to investigate the direct role of EphA3 in GC cell
growth and angiogenesis.
Materials and methods
Cell lines and culture conditions
Human GC cell line HGC-27 and human umbilical vein
endothelial cells (HUVECs) were purchased from the Cell Bank of
Shanghai Institute of Biochemistry and Cell Biology, Chinese
Academy of Sciences (Shanghai, China). Human GC cell lines (AGS,
SGC-7901 and MGC-803) and a human gastric epithelial cell line
(GES-1) were purchased from the American Tissue Culture Collection
(ATCC; Manassas, VA, USA). GC cells or HUVECs were grown in
RPMI-1640 medium or Dulbecco's Modified Eagle's medium (DMEM;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with
10% fetal bovine serum (FBS; HyClone Laboratories; GE Healthcare
Life sciences, Chicago, IL, USA). All the media were supplemented
with 100 U/ml penicillin and 100 µg/ml streptomycin (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and maintained in
a 5% CO2 incubator at 37°C.
Reagents
Antibodies against STAT3 (1:1,000; cat. no. 9139),
p-STAT3(Tyr705) (1:1,000; cat. no. 9145), AKT (1:1,000; cat. no.
4691) and p-AKT473 (1:1,000; cat. no. 4060;) were purchased from
Cell Signaling Technology (Danvers, MA, USA). Antibodies against
EphA3 (1:200; cat. no. sc-919), CD31 (1:100; cat. no. sc-1506) and
γ-tubulin (1:500; cat. no. sc-7396;) were obtained from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). VEGF neutralizing antibody
(V4758; 2 µg/ml) was purchased from Sigma-Aldrich; Merck KGaA.
Growth factor-reduced Matrigel was obtained from BD Biosciences
(Bedford, MA, USA). Medium 200PRF and low-serum growth supplements
(LSGS) were obtained from Invitrogen; Thermo Fisher Scientific,
Inc. A VEGF ELISA Development kit was purchased from PeproTech,
Inc. (Rocky Hill, NJ, USA).
Lentiviral vector production and cell
transduction
Stable knockdown of EphA3 in HGC-27 cells was
obtained using lentivirus. In brief, pLKO.1-EphA3-shRNA or
scrambled plasmids were co-transfected with psPAX2 and pMD2.G
plasmids into 293T cells at 70% confluency. Following 48 h, viral
supernatants were collected and filtrated. Subconfluent HGC-27
cells were transduced with lentivirus in the presence of 8 mg/ml
Polybrene (Sigma-Aldrich; Merck KGaA). Between 16 to 24 h
post-transduction, the cells were replaced with fresh medium.
Following 48 h, transduced cells were selected with puromycin at 2
µg/ml for 3 days.
Preparation of tumor cell-conditioned
medium (CM)
To prepare CM, target cells shNC-HGC-27 or
shEphA3-HGC-27 were seeded and grew to 30–40% confluency. Growth
medium was replaced with serum-free RPMI-1640 medium for 24 h.
Subsequenlty, CM was harvested when cells reached 60–80%
confluency. CM was aliquoted and stored at −80°C since it was not
immediately used. In addition, shNC-HGC-27 or shEphA3-HGC-27 cells
were pretreated with DMSO or WP1066 (5 µM) in serum-free culture
medium for 24 h. The supernatants were collected as CM.
Western blot analysis
Cells were rinsed with phosphate-buffered saline
(PBS) and lysed in ice-cold RIPA buffer [40 mM HEPES (pH 7.4), 1%
Triton X-100, 0.1% SDS, 100 mM NaCl, 5 mM EDTA, 0.5% sodium
deoxycholate and 1 mM sodium orthovanadate] with protease and
phosphatase inhibitors. The BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.) was used to quantitate the lysates. The same
amount of protein was separated by 8–10% SDS-PAGE gel and then
transferred onto a PVDF membrane (Millipore, Bedford, MA, USA).
Following blocking with 5% non-fat dry milk in Tris-buffered saline
containing 0.1% Tween-20 (TBST), the membrane was first incubated
overnight at 4°C with anti-STAT3 antibody, anti-p-STAT3 (Tyr705)
antibody, anti-AKT antibody, anti-p-AKT473 and anti-EphA3 antibody
and the corresponding signals were detected with an enhanced
chemiluminescence kit (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) following incubation with the appropriate HRP-conjugated
secondary antibodies (1:5,000; cat. nos. ZB-2301 and ZB2305;
Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China).
Quantification of band densities on western blot films was
performed using ImageJ 1.44 software (National Institutes of
Health, Bethesda, MD, USA).
Cell proliferation assay
Cell proliferation was assessed using Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Laboratories, Kumamoto, Japan)
according to the manufacturer's instructions. In brief,
~2×103 shNC-HGC-27 and shEphA3-HGC-27 cells were seeded
into 96-well plates and incubated for the indicated time periods.
Subsequently, 10 µl of CCK-8 solution was added to each well and
incubated for 1 h. The absorbance was finally determined at a
wavelength of 450 nm with a microplate reader (Bio-Rad Laboratories
Inc., Hercules, CA, USA). Each measurement was performed in
triplicate and the experiments were repeated 3 times.
Colony formation assays
Cells were seeded into 6-well plates at a density of
1,000, 2,000 and 3,000 cells/well. Following culture for 14 days in
complete growth media, the cells were washed with PBS and stained
with crystal violet. Colonies containing 50 or more cells were
counted under a light microscope at ×40 magnification. Each
measurement was performed at least three times.
ELISA assay
Secreted VEGF in CM of shNC-HGC-27 and
shEphA3-HGC-27 was determined using a commercially available human
VEGF-A ELISA Development kit (PeproTech) according to the
manufacturer's instructions. The data were adjusted by the amount
of total protein within the cells.
In vitro tube formation assay
A 96-well plate was coated with 50 µl growth
factor-reduced Matrigel and maintained at 37°C for 2 h. Then,
growth factor-deprived HUVECs suspended in 100 µl of CM were plated
at 1×104 cells/well and incubated at 37°C and 5%
CO2 for 4 to 6 h to allow the formation of a tubular
structure. Cells were imaged and the tube lengths were counted
using the Scion Image Beta 4.02 software (Scion Corp., Fredrick,
MD, USA).
In vitro migration assay
A cell migration assay was performed using 24-well
Boyden chambers with 8.0-µm pore sizes (Corning, Inc., Corning, NY,
USA). HUVECs were plated at 5×104 cells/well in the
upper chamber of serum-free medium. The lower chamber was filled
with 500 µl CM from shNC-HGC-27 or shEphA3-HGC-27 cells. The plates
were incubated for 4 to 6 h at 37°C in 5% CO2 and
non-migrated cells were wiped away with cotton swabs. The cells
that migrated to the lower side of the filter were fixed in 4%
formaldehyde solution for 15 min and stained with 0.05% crystal
violet in PBS for 15 min. Migrated cells were counted using 10
random fields with an inverted phase contrast microscope. Filters
were counted in triplicate per experiment. For VEGF inhibition
assay, HUVEC migration ability was determined as aforementioned
after adding neutralizing antibody (0.2 µg/ml) to VEGF-A into
CM.
Xenograft models and
immunohistochemistry
Five-week-old female BALB/c nude mice were purchased
from Beijing Vital River Laboratory Animal Technology Co., Ltd.
(Beijing, China). Three mice were housed per cage under controlled
temperature and humidity (23±2°C, 50±10%), on a 12-h light/dark
cycle. Mice had access to tap water and food ad libitum. All
animal studies conformed to the relevant regulatory standards and
were approved by the Institutional Animal Care Committee of Beijing
Institute of Biotechnology. Approximately 5×106
shNC-HGC-27 or shEphA3-HGC-27 cells were suspended in 200 µl of
serum-free RPMI-1640 medium and implanted subcutaneously into the
right flank of 5-week-old female BALB/c nude mice (n=3) separately.
Tumor volumes were determined using digital calipers every 3 days
and calculated as follows: length × width × height × 0.5236. Mice
were monitored for 3 weeks, when the experiment was terminated.
Further termination points of the experiment were when the maximum
tumor volume was ~2 cm3 or when the mice showed any
signs of distress (e.g., breathing disorders, weight loss, or
immobility). At the end of the experiment the mice were
anesthetized with 1.5% isofluorane-air mixture and sacrificed by
cervical dislocation. Tumor xenografts were removed and prepared
for immunohistochemistry. Tumor sections from shNC and
shEphA3-HGC-27 mice were processed for immunohistochemical analysis
of CD31 to visualize MVD. A portion of the tumor tissue was fixed
in 10% neutral-buffered formalin, dehydrated, embedded in paraffin
and sectioned at 4- to 5-µm thickness. The sections were
deparaffinized, treated with 3% H2O2 for 15
min, and then were microwaved in 10 mM citric sodium (pH 6.0) for
15 min to unmask antigens. The sections were rinsed in PBS and then
incubated with the primary anti-CD31 goat polyclonal antibody
(1:100; cat. no. sc-1506; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) for 1 h at room temperature and washed with TBS.
Biotinylated anti-goat IgG (1:100; cat. no. B-205; Zhongshan Golden
Bridge Biotechnology Co., Ltd.) was applied for 30 min at room
temperature. Color was developed following 10 min of incubation
with the 3,3′-diaminobenzidine. For MVD quantification, the number
of CD31-positive vessels/x100 field were counted in 5 randomly
selected fields. The data are represented as the number of
CD31-positive microvessels/x200 microscopic field.
Statistical analysis
Statistical analysis was performed using SPSS 13.0
(SPSS, Inc., Chicago, IL, USA) and the results were analyzed
statistically using Student's t-test for comparisons between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Comparison of EphA3 expression
levels
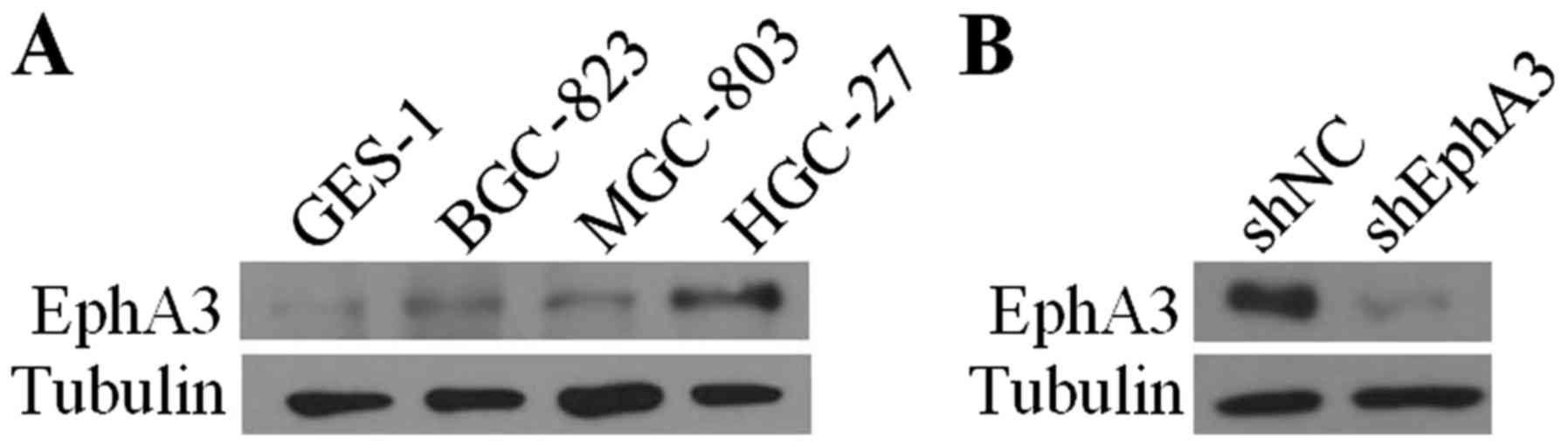
To investigate the molecular effects of EphA3 in GC,
we initially examined the expression level of EphA3 in GC cells
compared with the human gastric epithelial mucosa cells GES-1 using
western blot analysis. The results revealed that EphA3 expression
was upregulated in three distinct differentiated GC cell lines
MGC-803, BGC-823 and HGC-27 (Fig.
1A). With the highest levels of EphA3 expression, the HGC-27
cell line was selected to knock down endogenous EphA3. To establish
stable cell lines, EphA3 expression was effectively inhibited in
HGC-27 cells infected with lentiviral particles containing EphA3
shRNA compared to those infected with control shRNA vector
(Fig. 1B).
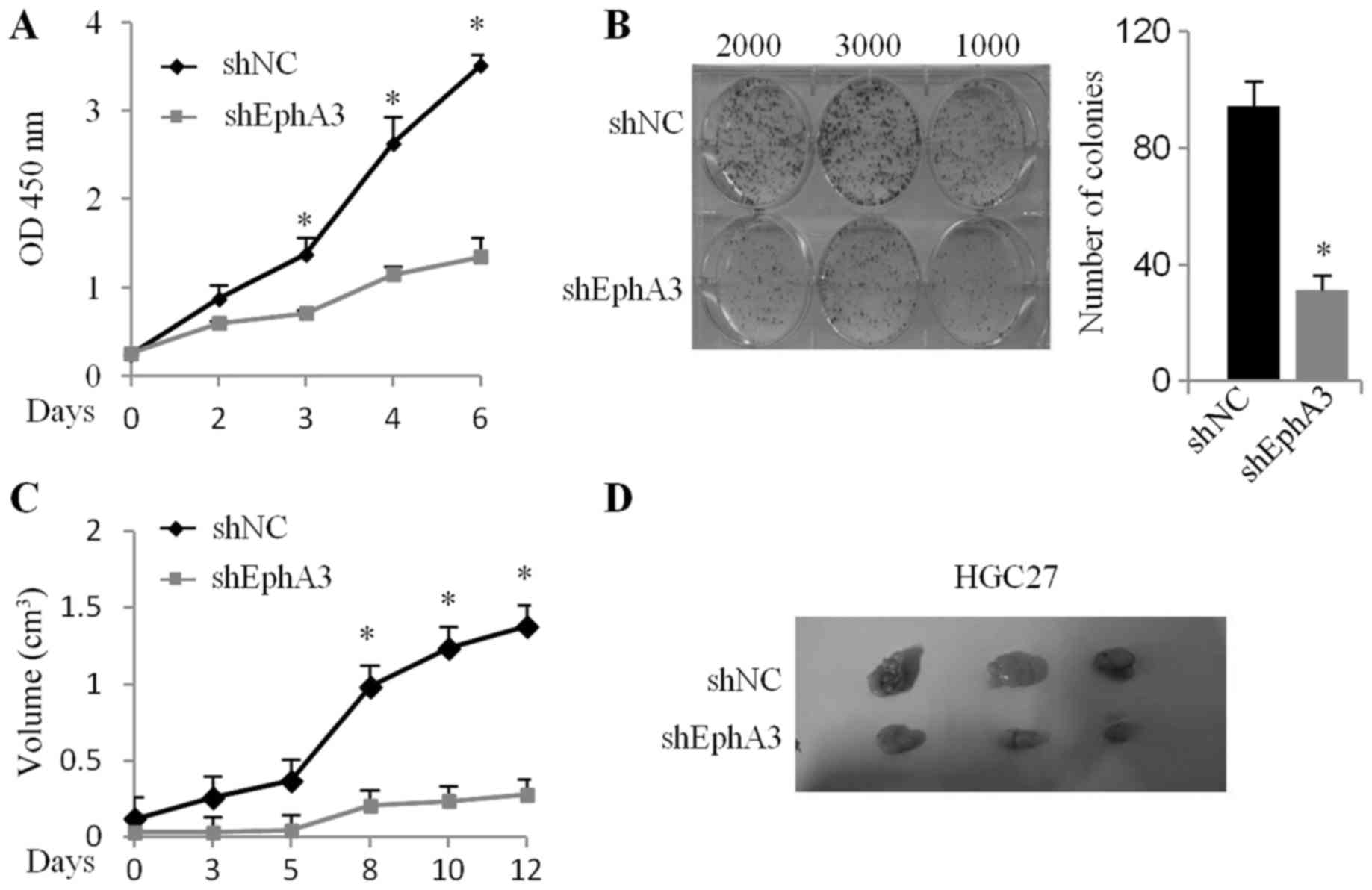
EphA3 knockdown inhibits the growth of
HGC-27 cells in vitro and in vivo
To determine whether EphA3 knockdown affected the
proliferation of HGC-27 cells, cell growth was determined using
CCK-8 and colony formation assays. EphA3 knockdown in HGC-27 cells
significantly impaired cellular proliferation compared with the
control cells (Fig. 2A).
Furthermore, colony formation capacity was also reduced by EphA3
knockdown (Fig. 2B). We further
examined the effects of EphA3 on tumor growth using a subcutaneous
xenograft model of HGC-27 cells in mice. EphA3 knockdown did not
induce any toxicity in mice, however, we observed a significant
decrease (39.1%) in mean tumor volume in nude mice implanted with
shEphA3-HGC-27 cells compared with that implanted with shNC-HGC-27
×enografts (Fig. 2C and D). These
results indicated that loss of EphA3 expression inhibited the
proliferation of HGC-27 in vitro and in vivo.
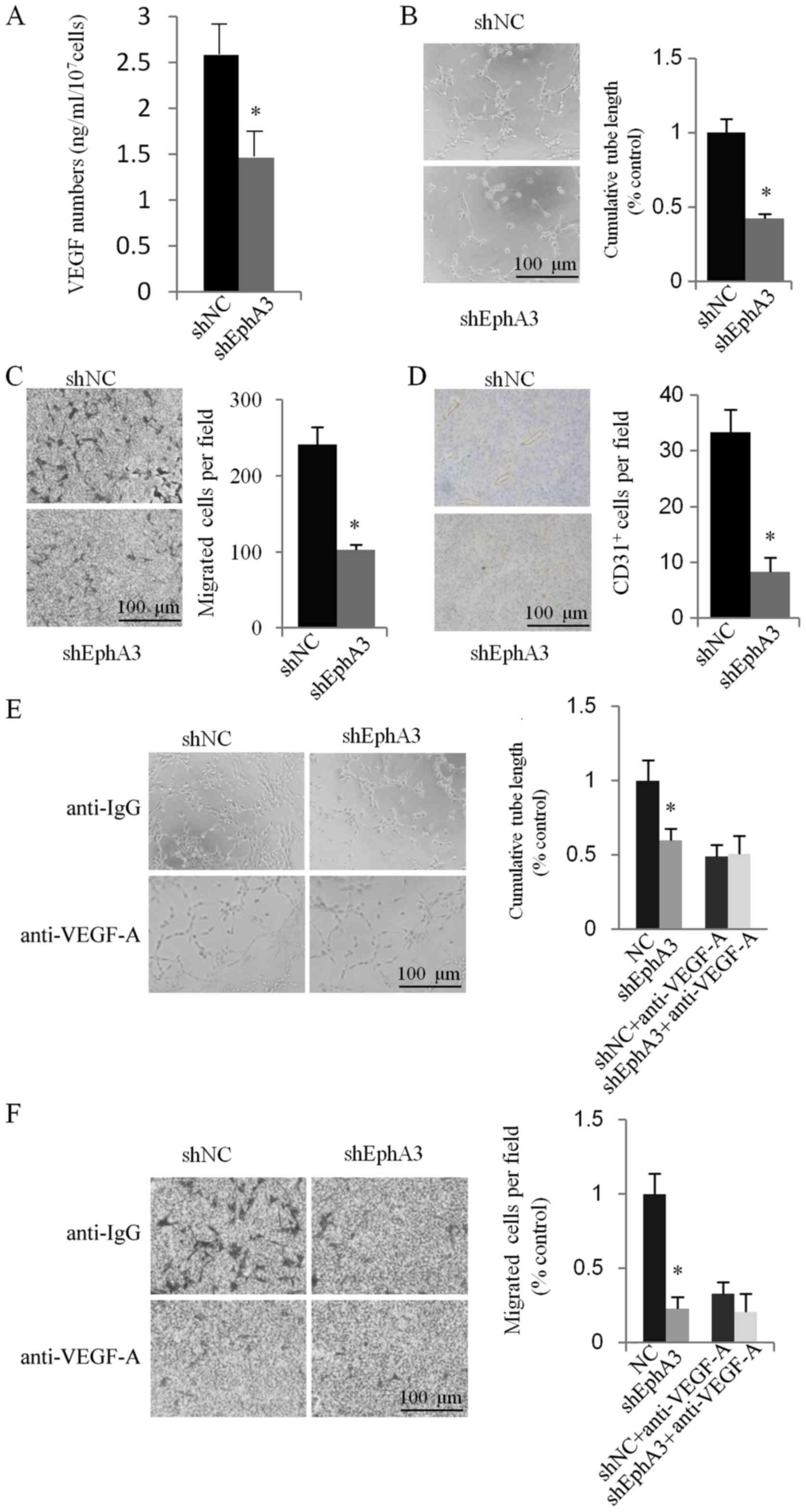
EphA3 knockdown inhibits tube
formation and angiogenesis in vitro and in vivo through the
regulation of the expression of VEGF
In our previous study, we provided clinical evidence
that EphA3 expression was significantly and positively associated
with the expression of VEGF and MVD status. These findings
indicated that EphA3 may be involved in the angiogenic process of
GC. To obtain direct evidence on whether EphA3 regulated the
expression of VEGF and was involved in angiogenesis, an ELISA assay
was performed to determine secreted VEGF-A in the CM of HGC-27
cells. The results revealed that EphA3 knockdown reduced VEGF
secretion in HGC-27 cells (Fig.
3A), indicating that EphA3 may be involved in regulating the
formation of new blood vessels in endothelial cells. To determine
whether the CM from shNC-HGC-27 and shEphA3-HGC-27 cells could
induce the tube formation of HUVECs, we performed a tube formation
assay in growth factor-reduced Matrigel in vitro. We
observed that the CM from shNC-HGC-27 and shEphA3-HGC-27 cells was
able to significantly induce HUVEC tube formation in 6-h incubation
time-periods. However, tube formation of HUVECs was significantly
reduced in CM from the shEphA3-HGC-27 cells (Fig. 3B). Since the migration and invasion
of endothelial cells through basement membranes is a crucial step
in the development of new blood vessels, we assessed the effect of
CM from HGC-27 on endothelial cell HUVEC migration. As displayed in
Fig. 3C, HUVECs cultured with the
CM from the NC-HGC-27 cells migrated faster compared to the
shEphA3-HGC-27 cells.
To further examine the effect of EphA3 on
angiogenesis in vivo, we identified the MVD status in HGC-27
cell xenograft nude mice by immunohistochemical staining with
anti-CD31 antibody. EphA3 knockdown significantly reduced
microvessel formation in the shEphA3-HGC-27 tumors relative to that
in the shNC-HGC-27 control (Fig.
3D). These results indicated that blockade of EphA3 activity
suppressed VEGF expression and impaired the angiogenic phenotype of
GC HGC-27 cells in vitro and in vivo.
The VEGF signaling pathway plays a crucial role in
tumor angiogenesis, thereby promoting endothelial cell
proliferation, migration and capillary tube formation. To further
elucidate the role of VEGF in EphA3-mediated angiogenesis
modulation, neutralization of the VEGF antibody was used to
antagonize the functions of VEGF. We found that when VEGF
neutralizing antibody was used to neutralize VEGF in the culture
supernatants of HUVEC cells from the CM of shNC-HGC-27 and
shEphA3-HGC-27 cells, the tube formation and migration of the HUVEC
cells induced by EphA3 were markedly attenuated in vitro
(Fig. 3E and F). These results
indicated that EphA3-dependent VEGF expression played an important
role in the process of EphA3-regulated angiogenesis.
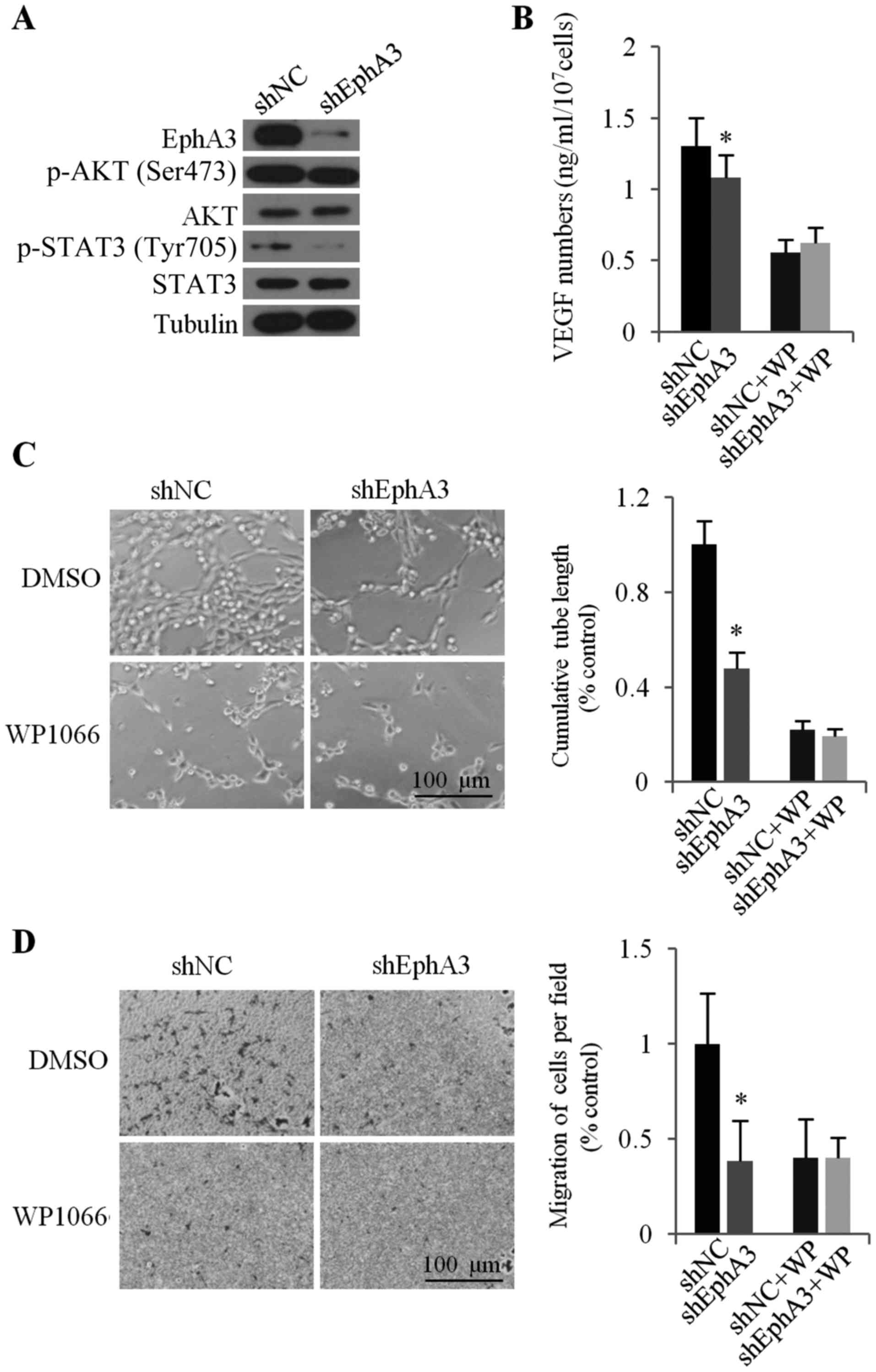
The JAK2/STAT3 signaling pathway is
involved in EphA3-mediated VEGF expression and angiogenesis
Our present study indicated that knockdown of EphA3
expression reduced the tube formation and migration of HUVECs and
tumor angiogenesis by inhibiting the secretion of VEGF. To further
understand the regulation of VEGF expression by EphA3, we
investigated the main signaling pathways related to the expression
of VEGF. Accumulating evidence is defining STAT3 signaling as an
important pathway for upregulation of the expression of VEGF and
tumor angiogenesis. In order to investigate whether changes in
EphA3 levels would affect the phosphorylation of STAT3, we assessed
the changes in STAT3 phosphorylation by western blotting following
knockdown of EphA3 in HGC-27 cells. Our data indicated that after
EphA3 knockdown, p-STAT3 (Tyr705) levels were significantly
decreased (Fig. 4A). To examine
whether STAT3 activity was involved in EphA3-mediated VEGF
expression and angiogenesis, JAK2/STAT3 inhibitor WP1066 was used
to inhibit the JAK2/STAT3 signaling pathway of HGC-27 cells before
collection of CM. The results revealed that pretreatment of cells
with WP1066 reduced the expression of VEGF and the difference of
the VEGF level between shNC-HGC-27 and shEphA3-HGC-27 was also
significantly decreased (Fig. 4B).
In addition, EphA3-mediated HUVEC migration and tube formation were
also attenuated by pretreatment with WP1066 (Fig. 4C and D). Collectively, EphA3
appeared to act through the JAK2/STAT3 signaling pathway to enhance
the expression of VEGF and angiogenesis in human GC HGC-27
cells.
Discussion
In GC, growing evidence has revealed that some Eph
receptors were aberrantly expressed and were associated with cancer
progression, metastasis and poor prognosis. For example, the EphA1
protein has been significantly associated with the depth of
invasion of GC (13). The
expression of EphA2 was revealed to be upregulated in GC compared
to that in normal mucosa and was consistent with tumor TNM stage.
EphA2 knockdown could inhibit GC cell proliferation and invasion
in vitro and in vivo (14). EphA7 overexpression has been
observed in gastric carcinoma specimens and is related to the
pathogenesis and development of GC (15).
EphA3 was first identified in a pre-B acute
lymphoblastic leukemia cell line and has also been reported to be
expressed in sarcomas, lung cancer, melanoma and glioblastoma.
EphA3 is currently one of the most promising therapeutic targets
(9). Numerous studies support
multiple tumor-promoting roles for EphA3 in a range of solid and
hematological cancers, including glioblastoma-initiating cells and
leukemic stem cells. Recently, we observed that in GC the
upregulated expression of EphA3 was positively associated with the
expression of VEGF, MVD as well as poor prognosis (12). Thus, EphA3 may play important roles
in angiogenesis and prognosis of GC. However, the precise role of
EphA3 in GC progression remains unknown.
To elucidate the biological functions of EphA3 in GC
tumorigenesis, we knocked down EphA3 expression in HGC-27 cells.
Knockdown of EphA3 markedly reduced cell viability and
proliferation in vitro and inhibited tumor growth in in
vivo assays. Furthermore, the present study confirmed that
EphA3 contributed to tumor angiogenesis in GC and was mediated by
STAT3-dependent expression of VEGF.
Angiogenesis, a process of neovascular formation, is
essential for the progression and metastasis of tumors (16). In recent years, ephrin ligands with
Eph receptors have been identified as contributors to tumor
angiogenesis. Studies revealed that EphA2-null ECs and
EphA2-deficient mice failed to undergo vascular assembly in
vitro and angiogenesis in vivo (17). Additionally, studies have shown that
injections of soluble EphA2 and EphA3-Fc receptors or EphB4
extracellular domains into tumor-bearing mice were able to inhibit
tumor angiogenesis and growth in vivo (11,18,19,20).
Two recent studies revealed ephrin-B2 as a key regulator in VEGFR-2
and VEGFR-3 endocytosis, with consequences for the development and
tumor angiogenesis (21,22). Recently, a study in multiple myeloma
(MM) demonstrated that EphA3 was overexpressed in bone marrow ECs,
and that EphA3 knockdown inhibited the adhesion, migration and
angiogenesis in vitro of ECs of MM patients (23,24).
Our present study provided evidence that EphA3 knockdown in HGC-27
cells inhibited tube formation and migration of EC in vitro.
We also revealed that vessel density was lower in HGC-27 tumors
following knockdown of EphA3 compared with the control group.
Collectively, our results demonstrated a role for EphA3 in
promoting angiogenesis in GC.
The balance of various proangiogenic stimulators and
angiogenesis inhibitors regulates the angiogenic process. Tumor
angiogenesis begins with the activation of ECs by a few specific
angiogenic factors, among which VEGF is a potent angiogenic
molecule responsible for tumor progression and metastasis through
its enhancement of angiogenesis (25,26).
In hepatocellular carcinoma cells, EphA3 knockdown was found to
decrease the invasiveness of cells by regulating VEGF protein
expression and proteolytic activity (27). In the present study, the effect of
EphA3 on VEGF secretion and expression in GC cells was assessed by
ELISA. Our data revealed that EphA3 knockdown inhibited VEGF
production in HGC-27 cells. Furthermore, VEGF was involved in the
effects of EphA3 on tube formation and migration in HUVECs cultured
in vitro since a neutralizing anti-VEGF antibody reduced
angiogenesis. These results demonstrated that EphA3 played a role
in tumor angiogenesis by regulating VEGF expression in HGC-27
cells.
It is well recognized that VEGF is regulated by many
signaling pathways, such as the ERK1/2 and JAK/STAT3 pathways
(28,29). STAT3 is a critical transcription
activator and has been demonstrated to be very important for cancer
progression. Accumulating evidence supports a pivotal role of
constitutive Stat3 activity in upregulating VEGF expression and
tumor angiogenesis (30,31). Many studies have concentrated on
STAT3 as a potential target for cancer therapy and found that STAT3
inhibition effectively blocked production of VEGF and tumor
angiogenesis (32,33). Previously, Eph family members,
including EphA1, EphA5, EphB2, EphB3 and EphB4, have been reported
to constitutively activate STAT3 (34). A study has shown that ephrinB1
interacted with STAT3 in a tyrosine phosphorylation-dependent
manner, resulting in the phosphorylation and enhanced
transcriptional activation of STAT3 (35). This study revealed that EphA3
knockdown negatively regulated STAT3 activity. Blocking the
JAK/STAT3 signaling pathway with WP1066 effectively impaired the
promotion of VEGF expression, HUVEC tube formation and migration
caused by EphA3 expression. These results indicated that the
effects of EphA3 on angiogenesis of HGC-27 cells were dependent on
STAT3-mediated VEGF production.
Despite the challenges presented by the complex
biology of the Eph receptors and ephrins, this system still
represents promising new therapeutic targets for the inhibition of
angiogenesis and tumorigenesis. In the present study, the results
demonstrated that EphA3 promoted HGC-27 tumor cell growth and
angiogenesis through the STAT3/VEGF pathway, indicating that EphA3
may be an effective indicator for prognosis and a potential target
for GC therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (nos. 81372740, 81372770,
81672604 and 81772750) and the National Key Research and
Development Program of China (no. 2016YFC1202402).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SHL and BW designed the study; JW wrote the
manuscript; JGZ revised the manuscript; XYL and JW performed the
experiments including in vitro experiments, xenograft
models, immunohistochemical staining and statistical analysis; FH
and PW participated in cell culture, cell transfection and western
blot assay. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All animal studies conformed to the relevant
regulatory standards and were approved by the Institutional Animal
Care Committee of Beijing Institute of Biotechnology.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in
china.2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nagini S: Carcinoma of the stomach: A
review of epidemiology, pathogenesis, molecular genetics and
chemoprevention. World J Gastrointest Oncol. 4:156–169. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pasquale EB: Eph-ephrin promiscuity is now
crystal clear. Nat Neurosci. 7:417–418. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pasquale EB: Eph receptor signalling casts
a wide net on cell behavior. Nat Rev Mol Cell Biol. 6:462–475.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pasquale EB: Eph receptors and ephrins in
cancer: Bidirectional signalling and beyond. Nat Rev Cancer.
10:165–180. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valsesia A, Rimoldi D, Martinet D,
Ibberson M, Benaglio P, Quadroni M, Waridel P, Gaillard M, Pidoux
M, Rapin B, et al: Network-guided analysis of genes with altered
somatic copy number and gene expression reveals pathways commonly
perturbed in metastatic melanoma. PLoS One. 6:e183692011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wood LD, Calhoun ES, Silliman N, Ptak J,
Szabo S, Powell SM, Riggins GJ, Wang TL, Yan H, Gazdar A, et al:
Somatic mutations of GUCY2F, EPHA3, and NTRK3 in human cancers. Hum
Mutat. 27:1060–1061. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Janes PW, Slape CI, Farnsworth RH,
Atapattu L, Scott AM and Vail ME: EphA3 biology and cancer. Growth
Factors. 32:176–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Day BW, Stringer BW, Al-Ejeh F, Ting MJ,
Wilson J, Ensbey KS, Jamieson PR, Bruce ZC, Lim YC, Offenhäuser C,
et al: EphA3 maintains tumorigenicity and is a therapeutic target
in glioblastoma multiforme. Cancer Cell. 23:238–248. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brantley DM, Cheng N, Thompson EJ, Lin Q,
Brekken RA, Thorpe PE, Muraoka RS, Cerretti DP, Pozzi A, Jackson D,
et al: Soluble Eph A receptors inhibit tumor angiogenesis and
progression in vivo. Oncogene. 21:7011–7026. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xi HQ, Wu XS, Wei B and Chen L: Aberrant
expression of EphA3 in gastric carcinoma: Correlation with tumor
angiogenesis and survival. J Gastroenterol. 47:785–794. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, Dong Y, Wang X, Ma H, Sheng Z, Li
G, Lu G, Sugimura H and Zhou X: Expression of EphA1 in gastric
carcinomas is associated with metastasis and survival. Oncol Rep.
24:1577–1584. 2010.PubMed/NCBI
|
|
14
|
Nakamura R, Kataoka H, Sato N, Kanamori M,
Ihara M, Igarashi H, Ravshanov S, Wang YJ, Li ZY, Shimamura T, et
al: EPHA2/EFNA1 expression in human gastric cancer. Cancer Sci.
96:42–47. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Li G, Ma H, Bao Y, Wang X, Zhou H,
Sheng Z, Sugimura H, Jin J and Zhou X: Differential expression of
EphA7 receptor tyrosine kinase in gastric carcinoma. Hum Pathol.
38:1649–1656. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brantley-Sieders DM, Fang WB, Hicks DJ,
Zhuang G, Shyr Y and Chen J: Impaired tumor microenvironment in
EphA2-deficient mice inhibits tumor angiogenesis and metastatic
progression. FASEB J. 19:1884–1886. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dobrzanski P, Hunter K, Jones-Bolin S,
Chang H, Robinson C, Pritchard S, Zhao H and Ruggeri B:
Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist.
Cancer Res. 64:910–919. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kertesz N, Krasnoperov V, Reddy R,
Leshanski L, Kumar SR, Zozulya S and Gill PS: The soluble
extracellular domain of EphB4 (sEphB4) antagonizes EphB4-EphrinB2
interaction, modulates angiogenesis, and inhibits tumor growth.
Blood. 107:2330–2338. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martiny-Baron G, Korff T, Schaffner F,
Esser N, Eggstein S, Marmé D and Augustin HG: Inhibition of tumor
growth and angiogenesis by soluble EphB4. Neoplasia. 6:248–257.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sawamiphak S, Seidel S, Essmann CL,
Wilkinson GA, Pitulescu ME, Acker T and Acker-Palmer A: Ephrin-B2
regulates VEGFR2 function in developmental and tumour angiogenesis.
Nature. 465:487–491. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Nakayama M, Pitulescu ME, Schmidt
TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U,
et al: Ephrin-B2 controls VEGF-induced angiogenesis and
lymphangiogenesis. Nature. 465:483–486. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Caivano A, La Rocca F, Laurenzana I,
Annese T, Tamma R, Famigliari U, Simeon V, Trino S, De Luca L,
Villani O, et al: Epha3 acts as proangiogenic factor in multiple
myeloma. Oncotarget. 8:34298–34309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
La Rocca F, Airoldi I, Di Carlo E, Marotta
P, Falco G, Simeon V, Laurenzana I, Trino S, De Luca L, Todoerti K,
et al: EphA3 targeting reduces in vitro adhesion and invasion and
in vivo growth and angiogenesis of multiple myeloma cells. Cell
Oncol (Dordr). 40:483–496. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iruela-Arispe ML and Dvorak HF:
Angiogenesis: A dynamic balance of stimulators and inhibitors.
Thromb Haemost. 78:672–677. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nicosia RF: What is the role of vascular
endothelial growth factor-related molecules in tumor angiogenesis?
Am J Pathol. 153:11–16. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu CY, Yang ZX, Zhou L, Huang ZZ, Zhang
HT, Li J, Tao KS and Xie BZ: High levels of EphA3 expression are
associated with high invasive capacity and poor overall survival in
hepatocellular carcinoma. Oncol Rep. 30:2179–2186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Milanini J, Viñals F, Pouysségur J and
Pagès G: p42/p44 MAP kinase module plays a key role in the
transcriptional regulation of the vascular endothelial growth
factor gene in fibroblasts. J Biol Chem. 273:18165–19172. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Niu G, Wright KL, Huang M, Song L, Haura
E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, et al:
Constitutive Stat3 activity up-regulates VEGF expression and tumor
angiogenesis. Oncogene. 21:2000–2008. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang S: Regulation of metastases by
signal transducer and activator of transcription 3 signaling
pathway: Clinical implications. Clin Cancer Res. 13:1362–1366.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei D, Le X, Zheng L, Wang L, Frey JA, Gao
AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL and Xie K: Stat3
activation regulates the expression of vascular endothelial growth
factor and human pancreatic cancer angiogenesis and metastasis.
Oncogene. 22:319–329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheong JH, Hong SY, Zheng Y and Noh SH:
Eupatilin inhibits gastric cancer cell growth by blocking
STAT3-mediated VEGF expression. J Gastric Cancer. 11:16–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Q, Briggs J, Park S, Niu G, Kortylewski
M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, et al:
Targeting Stat3 blocks both HIF-1 and VEGF expression induced by
multiple oncogenic growth signaling pathways. Oncogene.
24:5552–5560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Uan ZL, Guan YJ, Wang L, Wei W, Kane AB
and Chin YE: Central role of the threonine residue within the p+1
loop of receptor tyrosine kinase in STAT3 constitutive
phosphorylation in metastatic cancer cells. Mol Cell Biol.
24:9390–9400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bong YS, Lee HS, Carim-Todd L, Mood K,
Nishanian TG, Tessarollo L and Daar IO: ephrinB1 signals from the
cell surface to the nucleus by recruitment of STAT3. Proc Natl Acad
Sci USA. 104:17305–17310. View Article : Google Scholar : PubMed/NCBI
|