Introduction
Ovarian, breast and colorectal cancers are highly
prevalent worldwide, representing a leading cause of human
mortality. Breast cancer is the most commonly diagnosed cancer in
women (1), whereas ovarian cancer
ranks third among reproductive cancers in women and accounts for
~4% of all malignancies (2).
Colorectal cancer was recently reported as the third most common
human cancer and ranked fourth in the list of cancers leading to
patient mortality (3).
Platinum-containing antineoplastic drugs are used to
treat a wide variety of cancers. Cisplatin, the most commonly
prescribed platinum-based drug, is the first line of adjuvant or
neoadjuvant treatment of ovarian cancer (4), and has shown promise in an animal
model of breast cancer (5).
Triple-negative breast cancer cell lines, and ovarian and breast
tumors lacking BRCA1 are sensitive to cisplatin (6), and a recent clinical trial is
currently assessing the effectiveness of the therapeutic use of
cisplatin in breast cancer patients (NCT03012477). The broad
efficacy of cisplatin is also extended to colon cancer models
(7).
Despite its effectiveness in cancer chemotherapy,
the development of resistance to cisplatin has been well reported
and is associated with cancer recurrence (4). This resistance is mediated, at least
in part, by sequestosome 1 (SQSTM1/p62), which upregulates the
Keap1/Nrf-2-antioxidant response element (ARE) signaling cascade
and promotes cell survival (8).
Improved knowledge of such mechanisms has facilitated further
research into combination therapies designed to prevent the
development of resistance to cisplatin.
Staurosporine is another broad-spectrum antitumor
agent that has shown effectiveness in in vitro models of
breast (9), ovarian (6) and colon cancer (10). Although the mode of action of
staurosporine is poorly understood, and may involve multiple
mechanisms (11), the generation of
reactive oxygen species (ROS) has been suggested as a mediator of
staurosporine-induced apoptosis (12). We therefore postulated that
staurosporine, by its presumed multiple modes of action, may
provide additive anticancer effects when co-administered with other
chemotherapeutic agents. Hence, we sought to investigate the
efficacy of the combination of staurosporine and cisplatin using
cell culture models of breast, ovarian, and colon cancers, and to
gain insights into the possible action mechanisms involving
SQSTM1/p62.
Materials and methods
Materials and methods
The HCT-116 colon cancer cell line and the MCF-7
breast cancer cell line were obtained from the Medical Technology
Center of the Medical Research Institute, University of Alexandria,
Egypt. Ovarian cancer cell lines (OVCAR3 and OVCAR4) were purchased
form the European Collection of Authenticated Cell Cultures (ECACC;
Porton Down, Salisbury, UK). Cisplatin was purchased from
MOLEQULE-ON (Auckland, New Zealand). Staurosporine was obtained
from Invitrogen/Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial
Institute (RPMI)-1640 media were purchased from HyClone
Laboratories, Inc./GE Healthcare Life Sciences (Logan, UT, USA).
Trypsin-EDTA, phosphate-buffered saline (PBS), and fetal bovine
serum (FBS) were purchased from Gibco BRL; Thermo Fisher
Scientific, Inc. The CCK-8 cell proliferation assessment reagent
was obtained from MOLEQULE-ON. Protein standards, associated
detection reagents, Laemmli sample buffer, precast gels, Clarity
Western ECL Substrate kit, and Trans-Blot Turbo PVDF Transfer Pack
and the ChemiDoc MP Imaging System were all purchased from Bio-Rad
Laboratories (Hercules, CA, USA). Mouse monoclonal antibody to
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; cat. no. ab9484)
and rabbit monoclonal antibody to SQSTM1/p62 (cat. no. ab109012)
were purchased from Abcam (Cambridge, UK). Horseradish peroxidase
(HRP)-conjugated goat anti-mouse IgG (H+L; cat. no. 170-6516) and
goat anti-rabbit IgG (cat. no. 170-5046) were obtained from Bio-Rad
Laboratories.
Cell culture and maintenance
HCT-116, MCF-7, OVCAR3 and OVCAR4 cells were grown
as adherent monolayers and incubated at 37°C under a humidified
atmosphere containing 5% CO2. HCT-116 and MCF-7 cells
were maintained in DMEM (10% FBS), while RPMI-1640 medium (10% FBS)
was used to culture OVCAR3 and OVCAR4 cells.
Cell proliferation
Cells were trypsinized, counted and seeded in
96-well tissue culture plates at a density of 1×104
cells/well. The seeded cells were incubated under standard culture
conditions overnight. The next day, media were removed from the
wells and fresh media were added onto the double negative wells and
the control cells (designated 100% cell proliferation). Serial
dilutions of cisplatin and staurosporine were prepared from stock
solutions prepared in dimethyl sulfoxide (DMSO) in DMEM or
RPMI-1640 as appropriate. Cells were then treated with cisplatin or
staurosporine (1–100 µg/ml) or cisplatin + staurosporine (100
µg/ml). Plates were incubated for 72 h at 37°C under 5%
CO2 before assessment of proliferation.
Cell proliferation was assessed using the CCK-8 kit
by incubating control and treated cells with 10 µl of CCK-8 reagent
for 1 h and reading the absorbance at 490 nm using a multi-plate
reader. Cell proliferation was then calculated as a percentage of
the control cell proliferation by subtracting the mean absorbance
of the blank wells from all values, dividing the absorbance values
of the treated cells by the mean value of the control cells, and
multiplying by 100. Results are presented as concentration-response
relationships.
Cell morphology analysis
Cells were trypsinized, counted, and seeded at a
density of 2×105 cells/well in 6-well tissue culture
plates. Plated cells were then incubated overnight at 37°C under 5%
CO2. The next day, cells were incubated with fresh
medium containing staurosporine (100 µg/ml), cisplatin (100 µg/ml),
or cisplatin + staurosporine for 24 h. Photomicrographs using
IRMECO IM-5000 trinocular inverted biological microscope (IRMECO
GmBH & Co. KG, Geesthacht, Germany) linked to ToupView software
(ToupTek Photonics, Hangzhou, China) from different microscopic
fields were captured to assess the effect of cell treatment on cell
morphology. Plates were then re-incubated under the same conditions
for another 24 h, and photomicrographs were captured again from
different microscopic fields to assess the impact of prolonged
incubation with the drugs on morphology in comparison to the
corresponding control cells.
Western blotting
HCT-116, MCF-7, OVCAR3 and OVCAR4 cells were
trypsinized and counted, and 1×106 cells were plated in
9-cm, round tissue culture dishes and allowed to adhere overnight
at 37°C under 5% CO2. The next day, the cells were
incubated for 24 h with medium (control), cisplatin (100 µg/ml),
staurosporine (100 µg/ml), or cisplatin + staurosporine (100 µg/ml
each). Control and treated cells were washed with ice-cold PBS,
scraped, and incubated in 100 µl of ice-cold lysis buffer
containing protease inhibitor cocktail. Lysates were spun for 10
min at the maximum speed (13,400 rpm) in a cooling centrifuge and
the protein concentration was estimated in the supernatant using
the ABC protein estimation kit. Then, 10 µg of protein was loaded
into each well of precast gels, electrophoresed and transferred to
polyvinylidene difluoride (PVDF) membranes. Membranes were blocked
with 5% skim milk in PBS-Tween (PBST) buffer and probed with an
anti-p62 antibody (Abcam). Anti-GAPDH was used for normalization
and to ensure equal protein loading. Membranes were washed three
times in PBST, incubated with the appropriate secondary antibody,
washed again, and the signal was detected using the Bio-Rad ECL
detection kit. Antibody dilutions were performed according to the
instructions in the datasheet.
Statistical analysis
Data analysis was performed using GraphPad Prism
version 7 (GraphPad Software, Inc., La Jolla, CA, USA).
Multivariate statistical comparisons were performed using one-way
analysis of variance (ANOVA). Multiple comparisons were conducted
between the control group and all other groups, and Dunnett's post
hoc test was employed whenever the P-value was statistically
significant (Dunnett's post hoc test is recommended when comparing
multiple groups to a control group). P≤0.05 was considered
statistically significant.
Results
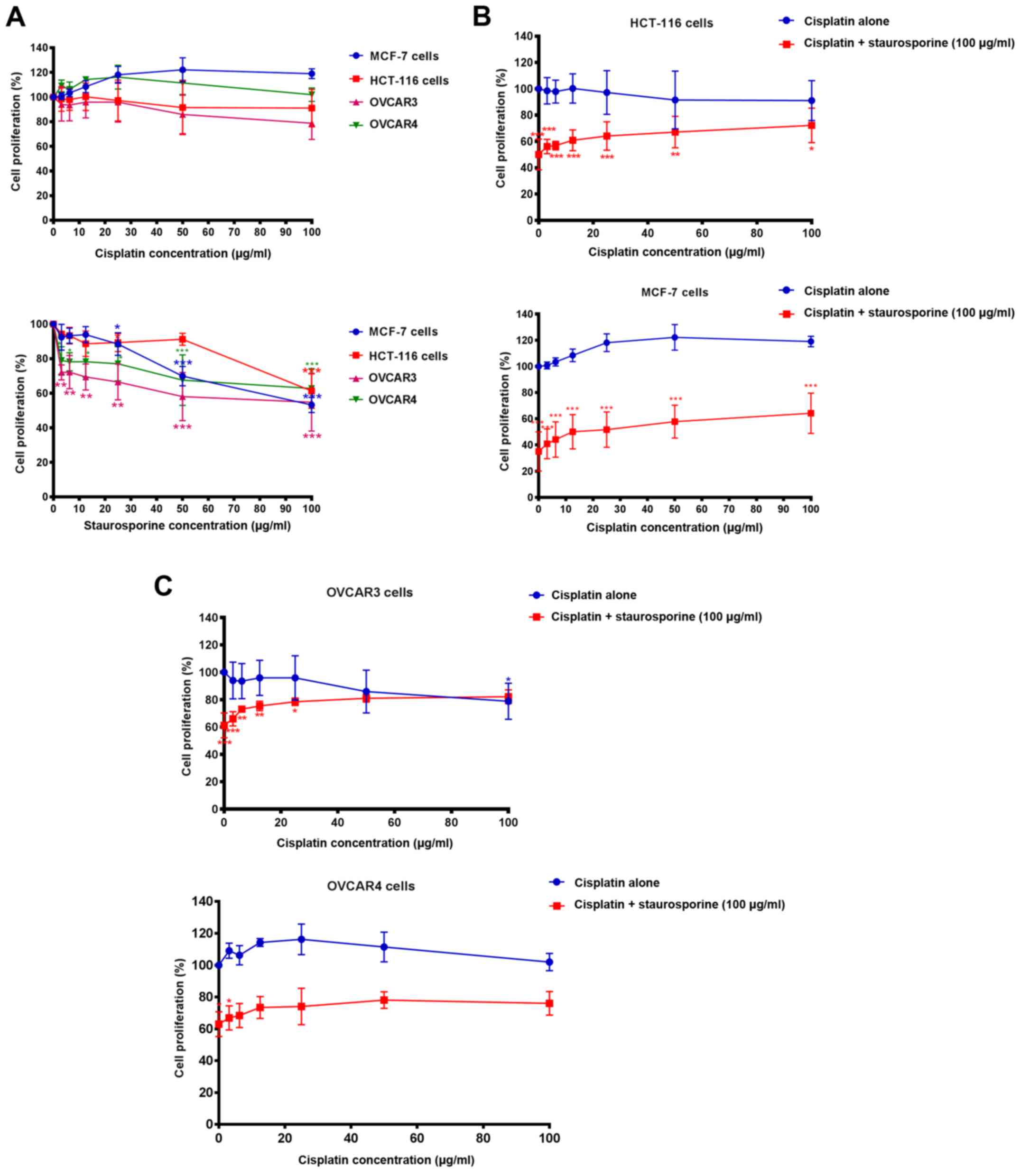
Chemoresistance of breast, colon, and
ovarian cancer cell lines was reversed in the presence of
staurosporine
Cisplatin alone failed to significantly inhibit cell
proliferation in comparison to the corresponding negative control
following 72 h of incubation at concentrations up to 100 µg/ml.
OVCAR3 was the only cell line that showed a partial response to the
antiproliferative effect of cisplatin (~20% inhibition, though this
was not statistically significant) at the highest tested
concentration of the drug (100 µg/ml) (Fig. 1A, top graph). Staurosporine
treatment, in contrast, resulted in a significant inhibition of
MCF-7 (P<0.001), OVCAR3 and OVCAR4 cell proliferation when
compared to the corresponding negative control; this inhibition was
more significant at higher drug concentrations (P<0.001)
(Fig. 1A, bottom graph). Although
HCT-116 cells appeared to be more resistant to staurosporine at low
concentrations, they were more susceptible at the highest
concentration of the drug (100 µg/ml; P<0.001) (Fig. 1A, bottom graph). Fig. 1B and C shows the effect of the
co-treatment of staurosporine (100 µg/ml) with cisplatin.
Co-treatment with staurosporine helped to overcome cisplatin
chemoresistance in HCT-116 (Fig.
1B, top graph), MCF-7 (Fig. 1B,
bottom graph), OVCAR3 (Fig. 1C, top
graph) and OVCAR4 cells (Fig. 1C,
bottom graph). In most cases, the differences between the
proliferation rates of cells co-treated with cisplatin and
staurosporine and the corresponding control cells were
statistically significant; MCF-7 and HCT-1116 cells in particular.
A marked restoration of chemoresistance was observed only in the
case of OVCAR3 cells at the highest concentration of cisplatin +
staurosporine, where there was no significant difference between
the proliferation rate of cells treated with both drugs and the
negative control (100% cell proliferation), and the pattern of
cisplatin effect alone was restored (Fig. 1C, top graph).
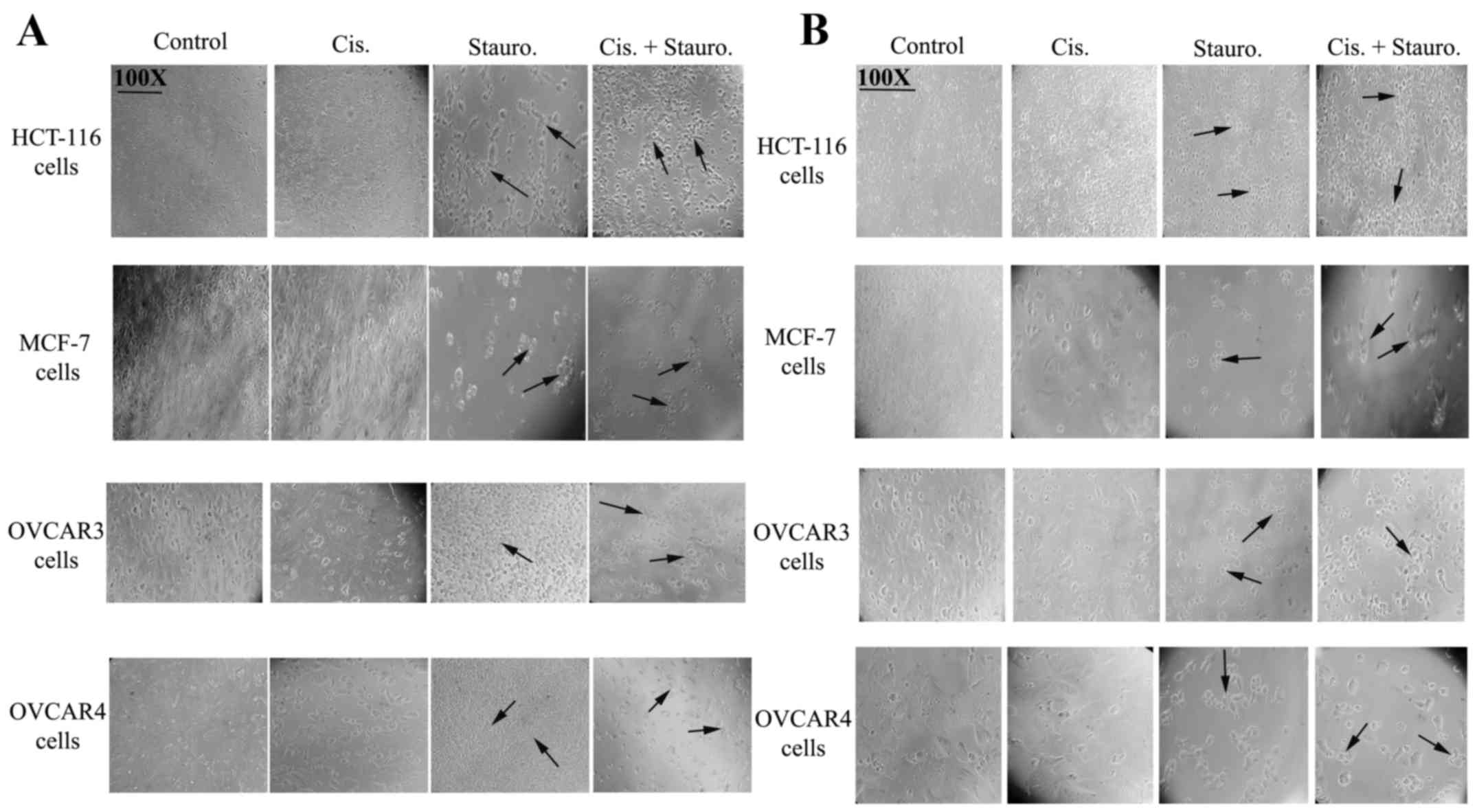
Microscopic analysis confirms
cisplatin chemoresistance and staurosporine-induced sensitization
to cisplatin
The photomicrographs presented in Fig. 2 show the effects of cisplatin,
staurosporine, or a combination of the two on cell morphology and
viability. Control treatments (incubation with DMEM or RPMI-1640)
appeared to increase cell density after 48 h of incubation, with
cells exhibiting normal morphology. Cells incubated with cisplatin
for 24 h (Fig. 2A) or 48 h
(Fig. 2B) showed normal morphology
with no evidence of apoptosis. Incubation of all cell lines with
staurosporine for 24 or 48 h resulted in altered morphology (black
arrows) indicative of a marked loss of normal cell structure and
reduced cell density, with increasing morphological evidence of
cell death. Incubation of cells with both drugs (100 µg/ml of each
drug) resulted in the loss of cell morphology and a concomitant
reduction in viable cells, suggesting additive effects or that
staurosporine co-treatment resulted in the sensitization of cells
to cisplatin.
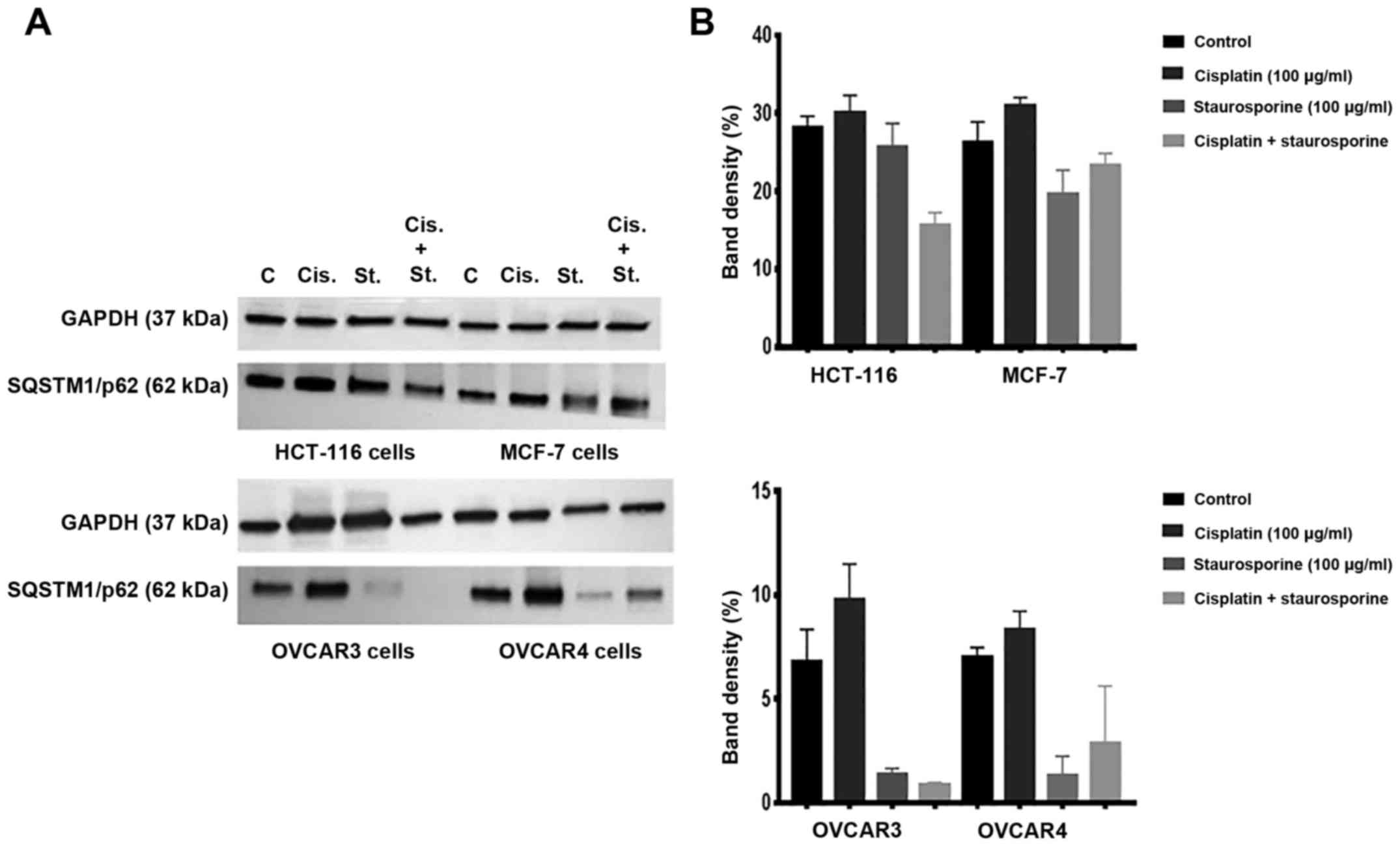
Western blotting reveals the
contrasting effects of cisplatin and staurosporine on p62
levels
Fig. 3A (western
blots) and B (band densitometry charts) show the level of p62
protein in HCT-116, MCF-7, OVCAR3 and OVCAR4 cells cultured under
control conditions or incubated with cisplatin, staurosporine, or a
combination of the two for 24 h. Whereas cisplatin exposure
resulted in an increase in the level of p62 in all cell types
compared to the corresponding control cells, staurosporine
treatment caused an apparent decrease in p62 levels.
Co-administration of staurosporine with cisplatin abrogated the
cisplatin-mediated increase in p62 levels in all cell lines when
compared to either the corresponding control cells or those treated
with cisplatin alone, although these data were not statistically
significant as the results are representative of 2 replicates of
independent experiments. Therefore, statistical comparisons were
not possible for this part. GAPDH levels remained consistent,
indicating equal protein loading.
Discussion
The anticancer drugs staurosporine and cisplatin
have been extensively studied to investigate their antitumor
potential. The mechanism of action of both drugs is believed to
involve production of reactive oxygen species (ROS) (13–16).
Although cancers do respond to staurosporine in most cases,
resistance develops in models where cisplatin is used. In the
present study, we investigated the potential of single or combined
administration of both drugs to inhibit cellular proliferation in
different human cancer cell line models (ovarian, colon, and
breast) in an attempt to understand the paradoxical responses
observed despite the possibility that a similar mode of action
underlies the therapeutic effect of both drugs.
Obvious resistance was observed in all cell lines
tested when treated with increasing doses of cisplatin. Resistance
to cisplatin was reported in colon cancer in a recent study
(17), which reported upregulation
of the PI3K pathway and increased levels of X-linked inhibitor of
apoptosis (XIAP) as a mediator of this resistance. Similarly,
cisplatin resistance was also recently reported in breast cancer
(18), and multiple studies have
reported the occurrence of cisplatin resistance in ovarian cancer
(19–21). Increasing concentrations of
staurosporine, in contrast, reduced the proliferation of all cell
types tested. These results were confirmed by microscopy, which
revealed a loss of normal cell morphology and confirmed the ability
of both drugs in combination to inhibit cell proliferation. These
observations are in concordance with the reported efficacy of
staurosporine in inhibiting the growth of breast cancer cells
(9). The antitumor potential of
staurosporine in HT-29 colon cancer cells has also been reported,
with high, but not low, concentrations of the drug associated with
higher expression of carcinoembryonic antigen (CEA). In contrast,
low concentrations were associated with lower expression of CEA in
C22.20 cells (a sub-line of HT-29), suggesting that staurosporine
could be a potent antitumor agent as well as a sensitizer for
biomarkers that may aid in diagnosis (22,23).
However, a marked regain of resistance was observed in OVCAR3 cells
that were co-treated with cisplatin and staurosporine. This pattern
of response should draw attention to the importance of further
investigation of the impact of cellular characteristics on
application of the current hypothesis of using staurosporine (in
our case), or other drugs to sensitize cancer cells to
platinum-based chemotherapy when chemoresistance is a clinical
concern. For example, deciding on drug dose could be an important
factor that may have a major influence on sensitization of cells
without possible regain of resistance.
The reported antitumor effects of staurosporine are
thought to involve the modulation of cell cycle progression and the
induction of anti-survival pathways, as reviewed previously
(23). Sequestosome 1 (p62), a
protein that participates in a survival pathway involving Keap1 and
Nrf2, is thought to contribute to cisplatin resistance by allowing
translocation of Nrf2 to the nucleus, thereby upregulating
antioxidant gene expression in addition to other reported
mechanisms (8,24). In this context, because ROS
generation has been proposed as a central mediator of the
anticancer effects of cisplatin and the Keap1/Nrf2 system is
considered to be one possible inducer of cisplatin resistance in
cancer cells (25), staurosporine
could contribute to a reduction in cisplatin resistance in cancer
cells.
In the present study, cisplatin treatment resulted
in elevation of p62 levels compared to control cells, whereas
staurosporine treatment resulted in an obvious reduction of p62
levels in all three cell types, thereby potentiating the effects of
cisplatin. These results suggest that staurosporine could sensitize
cancer cells to cisplatin via a mechanism involving the
downregulation of p62. It is possible that co-administration of
both drugs may reduce cell proliferation and overcome cisplatin
resistance in other cancer models.
It is of note to mention that despite reporting p62
as a potential target by which staurosporine could sensitize
different types of human cancer cells to cisplatin, this hypothesis
should be further investigated and verified in a more comprehensive
research model. For example, the role of p62 as a pivotal mediator
of cisplatin chemoresistance should be investigated along with the
signaling pathways where it functions; the Keap/Nrf/ROS axis, for
instance. The influence of upregulation and downregulation of p62
in these types of human cancer cells could be thoroughly
investigated by employing gene expression analysis to spot other
biomarkers that could be crucial mediators of chemoresistance
either independently or by working side-by-side with p62. In
addition, experimental models could be used to investigate the
potential correlation between p62 upregulation and downregulation
and major cell survival pathways for better understanding of the
involvement of p62 in the regulation of cell survival and
proliferation where staurosporine and cisplatin are used, or,
perhaps, other treatment regimens. In this respect, this avenue of
research warrants further investigation.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
KA analyzed the data, carried out western blotting
experiments and contributed to manuscript writing. OSEM designed
the study, carried out cell proliferation experiments and
photomicrography and contributed to the manuscript writing. Both
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Ghoncheh M, Pournamdar Z and Salehiniya H:
Incidence and mortality and epidemiology of breast cancer in the
world. Asian Pac J Cancer Prev. 17:43–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ai Z, Lu Y, Qiu S and Fan Z: Overcoming
cisplatin resistance of ovarian cancer cells by targeting
HIF-1-regulated cancer metabolism. Cancer Lett. 373:36–44. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Y, Han F, Cao LH, Li C, Wang JW, Li
Q, Zheng W, Guo ZX, Li AH and Zhou JH: Dose-response relationship
in cisplatin-treated breast cancer xenografts monitored with
dynamic contrast-enhanced ultrasound. BMC Cancer. 15:1362015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wahba HA and El-Hadaad HA: Current
approaches in treatment of triple-negative breast cancer. Cancer
Biol Med. 12:106–116. 2015.PubMed/NCBI
|
|
7
|
Son DJ, Hong JE, Ban JO, Park JH, Lee HL,
Gu SM, Hwang JY, Jung MH, Lee DW, Han SB and Hong JT: Synergistic
inhibitory effects of cetuximab and cisplatin on human colon cancer
cell growth via inhibition of the ERK-dependent EGF receptor
signaling pathway. Biomed Res Int. 2015:3975632015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xia M, Yu H, Gu S, Xu Y, Su J, Li H, Kang
J and Cui M: p62/SQSTM1 is involved in cisplatin resistance in
human ovarian cancer cells via the Keap1-Nrf2-ARE system. Int J
Oncol. 45:2341–2348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xue LY, Chiu SM and Oleinick NL:
Staurosporine-induced death of MCF-7 human breast cancer cells: A
distinction between caspase-3-dependent steps of apoptosis and the
critical lethal lesions. Exp Cell Res. 283:135–145. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
del Solar V, Lizardo DY, Li N, Hurst JJ,
Brais CJ and Atilla-Gokcumen GE: Differential regulation of
specific sphingolipids in colon cancer cells during
staurosporine-induced apoptosis. Chem Biol. 22:1662–1670. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Manns J, Daubrawa M, Driessen S, Paasch F,
Hoffmann N, Löffler A, Lauber K, Dieterle A, Alers S, Iftner T, et
al: Triggering of a novel intrinsic apoptosis pathway by the kinase
inhibitor staurosporine: Activation of caspase-9 in the absence of
Apaf-1. FASEB J. 25:3250–3261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Olguin-Albuerne M, Dominguez G and Morén
J: Effect of staurosporine in the morphology and viability of
cerebellar astrocytes: Role of reactive oxygen species and NADPH
oxidase. Oxid Med Cell Longev. 2014:6783712014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marullo R, Werner E, Degtyareva N, Moore
B, Altavilla G, Ramalingam SS and Doetsch PW: Cisplatin induces a
mitochondrial-ROS response that contributes to cytotoxicity
depending on mitochondrial redox status and bioenergetic functions.
PLoS One. 8:e811622013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhaleh H, Azadbakht M and Pour Bidmeshki
A: Possible involvement of calcium channels and plasma membrane
receptors on Staurosporine-induced neurite outgrowth. Bosn J Basic
Med Sci. 12:20–25. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang XD, Gillespie SK and Hersey P:
Staurosporine induces apoptosis of melanoma by both
caspase-dependent and -independent apoptotic pathways. Mol Cancer
Ther. 3:187–197. 2004.PubMed/NCBI
|
|
16
|
Gil J, Almeida S, Oliveira CR and Rego AC:
Cytosolic and mitochondrial ROS in staurosporine-induced retinal
cell apoptosis. Free Radic Biol Med. 35:1500–1514. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xiong Z, Fu Z, Shi J, Jiang X and Wan H:
HtrA1 down-regulation induces cisplatin resistance in colon cancer
by increasing XIAP and activating PI3K/Akt pathway. Ann Clin Lab
Sci. 47:264–270. 2017.PubMed/NCBI
|
|
18
|
Cataldo A, Cheung DG, Balsari A, Tagliabue
E, Coppola V, Iorio MV, Palmieri D and Croce CM: miR-302b enhances
breast cancer cell sensitivity to cisplatin by regulating E2F1 and
the cellular DNA damage response. Oncotarget. 7:786–797. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen J, Solomides C, Parekh H, Simpkins F
and Simpkins H: Cisplatin resistance in human cervical, ovarian and
lung cancer cells. Cancer Chemother Pharmacol. 75:1217–1227. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eckstein N: Platinum resistance in breast
and ovarian cancer cell lines. J Exp Clin Cancer Res. 30:912011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang J and Wu GS: Role of autophagy in
cisplatin resistance in ovarian cancer cells. J Biol Chem.
289:17163–17173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aquino A, Prete SP, Baier S, Cappelletti
D, Greiner JW, De Vecchis L, Graziani G and Bonmassar E:
Staurosporine increases carcinoembryonic antigen expression in a
human colon cancer cell line. J Chemother. 12:167–172. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aquino A, Prete SP, Balduzzi A, Formica V,
Fossile E, Bonmassar L, Concolino F, Bonmassar E and Graziani G:
Treatment of peripheral blood with staurosporine increases
detection of circulating carcinoembryonic antigen positive tumor
cells. Int J Cancer. 100:119–121. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan XY, Zhang Y, Zhang JJ, Zhang LC, Liu
YN, Wu Y, Xue YN, Lu SY, Su J and Sun LK: p62/SQSTM1 as an
oncotarget mediates cisplatin resistance through activating
RIP1-NF-κB pathway in human ovarian cancer cells. Cancer Sci.
108:1405–1413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brozovic A, Ambriovic-Ristov A and Osmak
M: The relationship between cisplatin-induced reactive oxygen
species, glutathione, and BCL-2 and resistance to cisplatin. Crit
Rev Toxicol. 40:347–359. 2010. View Article : Google Scholar : PubMed/NCBI
|