Introduction
Breast cancer has become a worldwide health problem
for females. It is also a major cause of cancer-related deaths in
women globally (1). Current
treatment options include surgery, radiotherapy, chemotherapy,
hormonal therapy and targeted therapy, the selection of which
depends heavily on the classification of breast cancers (2–5). A
commonly used criterion is the receptor status, namely whether the
breast cancer is hormone receptor (estrogen and/or
progesterone)-positive or hormone receptor-negative. For example,
patients with estrogen receptor α (ERα)-positive tumors can be
given tamoxifen (TAM) which is designed to antagonize the function
of ER (6,7). They can also be treated with aromatase
inhibitors, such as letrozole, which inhibits the synthesis of
estrogen hormone. These patients have a better survival rate than
those with ERα-negative tumors (8).
In addition, targeted therapies, a form of precision medicine, such
as palbociclib and ribociclib, have also been developed to treat
those ER-dependent breast cancers. Although these therapies can
suppress cancer growth for a significant period of time, very often
they will be confronted with drug resistance acquired by cancer
cells, indicating that there could be unknown signaling molecules
and events in the ER cascade which warrant further exploration and
characterization.
Recently, the Piwi (P-element-induced wimpy
testis)-like proteins have gained much interest as prospective
biomarkers and targets for anticancer therapies due to their
ectopic expression in many types of cancers (9). Characterized by the presence of PAZ
and PIWI domains, Piwil proteins belong to a subclade of the
Argonaute family which also includes the AGO subclade (10). While AGO proteins are ubiquitously
found in all tissues, Piwil proteins were initially regarded to be
restricted to germline cells (11).
The first Piwi gene in Drosophila was identified by
screening for mutants affecting the asymmetric division of stem
cells (12). Subsequently,
identification of the Piwi homologs in a number of organisms has
revealed that Piwi is evolutionarily conserved (13). In humans, there are four Piwi-like
genes, namely, Piwi-like 1 (Hiwi, Piwil1), Piwi-like 2 (Hili,
Piwil2), Piwi-like 3 (Piwil3) and Piwi-like 4 (Hiwi2, Piwil4)
(14). In germ stem cells, Piwil
proteins are involved in self-renewal, retrotransposon silencing,
translational regulation and chromatin remodeling (15). Given that cancer cells share many
characteristics with germ stem cells such as rapid proliferation
and almost infinite self-renewal (11), it is therefore conceivable that
Piwil proteins could be expressed in cancer cells and that some of
the functions identified in germ stem cells could be hijacked by
cancer cells for their own survival and metastasis.
Indeed, recent studies have detected the expression
of Piwil proteins in a variety of somatic contexts, including
cancers. For example, the expression of Piwil1 was observed in
seminoma, a cancer of male germ cells (16). Furthermore, other members of the
Piwi subclade have been found in many malignant tumors, including
gastric, colon and breast cancers (9,17–21),
and usually their expression is associated with poorer prognosis. A
plethora of mechanisms have been proposed to explain the
correlation, including epigenetic and post-transcriptional
regulation (22–25). Piwil proteins can even either
physically or functionally interact with some canonical signaling
molecules and transcription factors, such as p38 and STAT3
(20,26).
Notably, a microarray profiling of breast cancer
tissues indicated that Piwil3 and Piwil4 could serve as potential
prognostic markers for breast cancer (27). Another study also revealed that
Piwil4 was abundantly expressed in many breast cancer cases,
particularly when only the triple-negative breast cancer (TNBC)
samples were considered (21). By
using a typical TNBC line, MDA-MB-231, the study revealed that
Piwil4 was involved in regulating tumor invasion and growth via the
TGFβ and FGF pathways and in facilitating immune escape of cancer
cells by suppressing the expression of MHC II. Hence, it is of
great interest to further investigate whether in ER-positive cases,
which represent a majority of the breast cancer population, Piwil4
or any other Piwi homologs play a functional role in modulating the
ER signaling events.
In the present study, we found that in ER-positive
MCF-7 breast cancer cells, 17β-estradiol increased the expression
of Piwil4, which was not observed in another ER-positive but
non-tumorigenic breast epithelial cell line, MCF-12A. Conversely,
the expression of Piwil4 was not induced by the antiestrogen TAM,
but did depend on the presence of ER, particularly ERα. We further
explored the effect of knocking down Piwil4 on ER transcriptional
activity, and found that it led to reduced expression of some ER
target genes. In functional assays, we observed that the knockdown
of Piwil4 in MCF-7 reduced 17β-estradiol-enhanced cell migration
and invasion. The knockdown also impaired the wound healing
motility of the cells, indicating that Piwil4 plays essential roles
in modulating the response of ER-positive breast cancer cells to
17β-estradiol in terms of target gene activation and migration
capacity. These findings indicated that Piwil proteins could play a
somatic role in regulating ER signaling activities, thus serving as
a potential target for treating ER-positive breast cancers.
Materials and methods
Cell culture
The ER-positive breast cancer cell line MCF-7 and
the non-tumorigenic breast cell line MCF-12A were obtained from The
American Type Culture Collection (ATCC; Manassas, VA, USA). MCF-7
cells were cultured in high-glucose Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (FBS; both
from GE Healthcare, Chicago, IL, USA). MCF-12A cells were cultured
in DMEM/F12 medium (GE Healthcare) supplemented with an FBS
cocktail of 20 ng/ml Human epidermal growth factor (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 ng/ml
cholera toxin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), 0.01
mg/ml bovine insulin (Invitrogen; Thermo Fisher Scientific, Inc.),
500 ng/ml hydrocortisone (Invitrogen; Thermo Fisher Scientific,
Inc.) and 5% horse serum (Gibco; Thermo Fisher Scientific, Inc.).
Both cell types were grown in 75-cm2 flasks (Corning
Inc., Corning, NY, USA) in a humidified incubator under standard
conditions at 37°C and 5% CO2.
To activate the ER signaling in these cells, when
the cultures reached 70% confluency, the cells were first washed
and incubated with their corresponding media containing 5%
charcoal-treated FBS (Sigma-Aldrich; Merck KGaA) for two days.
Subsequently, 10 nM 17β-estradiol (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) was added to the culture medium at indicated
time-points. Alternatively, 1 µM tamoxifen (Sigma-Aldrich; Merck
KGaA) in dimethyl sulfoxide (DMSO) was added to the culture medium
for 1 or 3 h.
siRNA transfection
A day prior to transfection, MCF-7 cells were seeded
into 6-well plates at a density of 3.5 ×105 cells/well.
Transfection of the cells with the siRNAs against Piwil4
(Sigma-Aldrich; Merck KGaA) and the siRNA Universal Negative
(Qiagen GmbH, Hilden, Germany) was performed using Lipofectamine
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. In brief, for each well, 125 µl Opti-MEM
(Invitrogen; Thermo Fisher Scientific, Inc.) containing 5 µl
Lipofectamine 3000 was mixed with 125 µl Opti-MEM containing 100
pmol siRNA in a microcentrifuge tube. Following incubation at room
temperature for 5 min, the total mixture was added to the cells in
the 6-well plate. The product information about the siRNAs used was
as follows: i) siPiwil4-1: GUUACAAAGUCCUCCGGAA; ii) siPiwil4-2:
GCUUGAAGCCCGACCAUAU; iii) esiPiwil4: EHU029551; iv) esiESR1:
EHU141651; (all from Sigma-Aldrich; Merck KGaA); and v) siGENOME
Human ESR2 SMARTpool: GGGCUGAUGUGGCGCUCAA; GUGUGAAGCAAGAUCGCUA;
CAAGAAGAUUCCCGGCUUU; CUUUAGUGGUCCAUCGCCA (Dharmacon, Lafayette, CO,
USA).
RNA extraction and quantification
Total RNAs from MCF-7 cells and MCF-12A cells were
extracted with the TRIzol reagent (Life Technologies; Thermo Fisher
Scientific, Inc.) as per the manufacturer's instructions. In brief,
in a 6-well plate, each well received 1 ml TRIzol and then, 200 µl
chloroform was added per ml of TRIzol reagent for phase separation.
Following centrifugation at 12,000 × g for 15 min at 4°C, 500 µl of
the aqueous phase was transferred to a new tube and mixed with 500
µl isopropanol for RNA precipitation. Following centrifugation, the
RNA pellet was washed with 70% ethanol and left to air dry. The RNA
pellet was dissolved in 20 µl RNAse-free water (Promega Corp.,
Madison, WI, USA) and the RNA concentration was assessed using a
NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific,
Inc.). The RNA was stored at −20°C for further use.
Reverse transcription-polymerase chain
reaction (RT-PCR) and quantitative polymerase chain reaction
(qPCR)
For the reverse transcription reaction using the
GoScript RT system (Promega Corp.), 3 µg of total RNA was
pretreated with 1 µl Turbo DNase I (Ambion; Thermo Fisher
Scientific, Inc.) at 37°C for 30 min to eliminate residual genomic
DNAs. Following inactivation of DNase I with 5 mM EDTA at 70°C for
10 min, total RNA was mixed with 1 µl oligo(dT) and incubated at
70°C for 5 min. Following cooling on ice for 5 min, the sample was
mixed with 9 µl of a Master Mix which contained 4 µl 5 X Reverse
Transcription (RT) buffer, 2.5 µl 25 mM MgCl2, 1 µl
dNTP, 0.5 µl RNase inhibitor and 1 µl RT enzyme. The reaction was
performed using a Thermal Cycler (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) at 25°C for 5 min followed by 42°C for 60 min
and inactivated at 70°C for 15 min. The synthesized cDNAs were
stored at −20°C for further use.
For the amplification of target genes, PCR was
employed with the use of GoTaq DNA polymerase (Promega Corp.). A
mixture of reaction components was prepared on ice from the
following: 2.5 µl 10 X Standard Taq Reaction Buffer, 0.5 µl 10 mM
dNTPs, 0.5 µl 10 µM forward primer, 0.5 µl 10 µM reverse primer, 1
µl GoTaq DNA polymerase, 1 µl of template cDNA and diluted with
nuclease-free water to 25 µl. The reaction was performed on the
thermal cycler (Bio-Rad Laboratories, Inc.) using the following
cycling conditions: 30 cycles at 95°C for 20 sec, 60°C for 20 sec,
and 72°C for 30 sec. The PCR products were then separated and
analyzed in a 2% TAE agarose gel.
qPCR was performed in 96-well reaction plates (Life
Technologies; Thermo Fisher Scientific, Inc.) on a real-time
thermal cycler (7900HT Fast Real-Time PCR System; Applied
Biosystems; Thermo Fisher Scientific, Inc.). Each well received 0.5
µl of cDNA template, 5 µl of 2 X GoTaq qPCR Master Mix, 0.25 µl 10
µM forward primer and 0.25 µl 10 µM reverse primer. The following
reaction conditions were used. An initial polymerase activation at
95°C for 2 min followed by 40 cycles of denaturation at 95°C for 3
sec, annealing and extension at 60°C for 30 sec and one cycle of
dissociation. β-actin mRNA expression was assessed and served as a
reference gene which other mRNAs were normalized to. The relative
abundance of mRNA was determined by the ∆∆Cq method (28). A two-way Student's t-test for each
gene was performed, and significant difference analyses were
calculated using 95% confidence levels. The primer sequences for
genes tested are listed below in the format of ‘forward + reverse’:
β-actin, ggcatgggtcagaaggatt + aggtgtggtgccagattttc; GAPDH,
tgccatgggtggaatcatattgg + gaaggtgaaggtcggagtcaacg; Piwil1,
cactgatggaagccagaagactc + gatcgttatcctcacatcctctcc; Piwil2,
gtatcagcagagaagtggacaagc + agtcccaaagactgaggtgttcc; Piwil3,
atatctcgtcctcagtgggttg + tccactctccgctcttttagtg; Piwil4,
aatgctcgctttgaactagagac + attttggggtagtccacattaaat; Greb1,
gagtggacaatgaggaagaggaag + ctcgttggaaatggagacaagg; Pgr,
cttaatcaactaggcgagag + aagctcatccaagaatactg; Cxcl12,
attcttcgaaagccatgttgc + tttctccaggtactcctgaatcc; Tff1,
ccatggacaacaaggtgatctgc + acagcagcccttatttgcacac; Nanog,
cacttctgcagagaagagtgtcg + gtagctgaggttcaggatgttgg; c-Myc,
tcagagtctggatcaccttctg + ggttgttgctgatctgtctcag; Oct4,
gtgaagctggagaaggagaagc + gtcgtttggctgaataccttcc; Sox2,
ccctgcagtacaactccatgac + tggagtgggaggaagaggtaac; Calcr,
ccagtacatgatggcctgcaac + agtacacggccctggtaatagc; Ccnd1,
gcatgttcgtggcctctaag + cgtgtttgcggatgatctgt; vimentin,
ggctcgtcaccttcgtgaat + gagaaatcctgctctcctcgc; N-cadherin,
acatcttcagcgacgatagttcc + ctaattccatggcagtctggttcc; Esr1,
ggatacgaaaagaccgaagagg + gccaggctgttcttcttagagc; Esr2,
tccctggtgtgaagcaagatc + atccttcacacgaccagactcc.
Protein preparation and
immunoblotting
The MCF-7 cells were lysed in RIPA Buffer (Thermo
Fischer Scientific, Inc.) supplemented with cOmplete™ protease
inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) and
phosphatase inhibitors (Thermo Fischer Scientific, Inc.). The
protein concentration was determined using the Bradford protein
assay (Bio-Rad Laboratories, Inc.). Protein denaturation was
performed by the addition of β-mercaptoethanol and 4 X
NuPAGE® LDS Sample Buffer followed by 95°C for 10 min.
Subsequently, 30 µg of lysates were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (4–20% SDS-PAGE) and
transferred onto PVDF membranes (Bio-Rad Laboratories, Inc.). The
membranes were incubated with primary antibodies overnight at 4°C,
rinsed 3 times with TBS-Tween followed by incubation with
horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (H+L)
secondary antibody (1:200; cat. no. A16072; Thermo Fisher
Scientific, Inc.) or goat anti-rabbit IgG (H+L) secondary antibody
(1:200; cat. no. 31460; Thermo Fisher Scientific, Inc.). The
immunoreactive bands were chemiluminescently detected using
CL-Xposure™ Film (Thermo Fisher Scientific, Inc.). The primary
antibodies used in the present study were anti-Piwil4 (1:100; cat.
no. ab21869; Abcam, Cambridge, UK) and anti-β actin (1:200; cat.
no. sc47778; Santa Cruz Biotechnology, Inc.).
Immunofluorescence
MCF-7 cells were seeded onto glass coverslips in 5%
charcoal stripped FBS DMEM. Following 24 h, ethanol and
17β-estradiol were administered to the control and treatment groups
respectively for another 24 h. The cells were rinsed twice with
cold PBS, fixed in situ using 4% paraformaldehyde and
permeabilized with 0.1% Triton X-100 in PBS. Cells were then rinsed
thrice with PBS and blocked with 1% BSA (GE Healthcare) for 30 min.
This was followed by incubation with primary antibody Piwil4
(1:100; cat. no. ab21869; Abcam) in 0.2% BSA for 2 h, 3 PBS washes
and incubation with Alexa488-conjugated anti-rabbit IgG (1:200;
cat. no. A11008; Invitrogen; Thermo Fisher Scientific, Inc.) in
0.2% BSA for 1 h in the dark. Counterstaining was performed with
Hoechst 33342 (1:20,000; Sigma-Aldrich; Merck KGaA) in PBS for 5
min and rinsed with PBS thrice and desalted. Coverslips were left
to dry and mounted onto glass slides using Prolong Gold Antifade
medium (Molecular Probes; Thermo Fisher Scientific, Inc.). Images
were captured using the Nikon Eclipse TE2000-S inverted microscope
at ×400 (Nikon Corp., Tokyo, Japan).
Transwell™ migration assay
The migration capability of transfected MCF-7 cells
was studied using 8-mm Transwell migration chambers with 8-µm pores
(Corning Inc.). At 24 h post-transfection, the cell medium was
removed, then the cells were rinsed once with serum-free DMEM and
maintained in DMEM supplemented with 5% charcoal stripped FBS
(Sigma-Aldrich; Merck KGaA) for another 24 h before trypsinizing.
Prior to the seeding of cells, the bottom surface of each migration
chamber was coated with 20 µg/ml fibronectin (Corning Inc.) for 1
h. Cells (5 ×105) were seeded into the upper chambers in
200 µl of DMEM supplemented with 5% of charcoal stripped FBS. DMEM
(600 µl) with 50% FBS was added to the lower chambers. Ethanol and
17β-estradiol (Santa Cruz Biotechnology) was added to the medium of
the control and treatment groups, respectively. The cells were
allowed to migrate for 24 h. Chambers were then rinsed in cold PBS.
Cells were fixed with 100% methanol and stained with 0.2% crystal
violet. Cotton buds were used to remove the cells in the upper
chamber. Chambers were left to dry overnight before imaging the
migratory cells using the Nikon SMZ1500 inverted microscope (Nikon
Corp.).
Cell invasion assay
The BD BioCoat Matrigel Invasion Chambers (BD
Biosciences, Franklin Lakes, NJ, USA) used for the assay were first
thawed to room temperature. To hydrate the chambers, 20 µl
serum-free DMEM was added to the upper chamber while 600 µl PBS
containing 20 µg/ml fibronectin was added to the lower chamber.
Fibronectin was used as an additional chemoattractant for the less
invasive cell lines. The chambers were then maintained at 37°C in
5% CO2 for 2 h. Each upper chamber then received 5
×104 transfected MCF-7 cells suspended in 200 µl
serum-free medium while 600 µl DMEM with 50% FBS was added to the
lower chamber. Ethanol or 10 nM 17β-estradiol was added to the
upper and lower chambers of mock-treated or treated samples,
respectively. The cells were then incubated at 37°C in 5%
CO2 for 24 h before being fixed and stained as described
in the aforementioned migration assay.
Wound healing assay
Twenty-four hours post transfection, 4
×104 MCF-7 cells in a total volume of 70 µl DMEM
containing 5% charcoal stripped FBS were seeded into each side of a
2-well cell culture insert (SPL Life Sciences, Pocheon, Korea)
attached onto the surface of a 12-well plate. The cell culture
inserts were removed after 24 h leaving an area without cells
analogous to a wound. The well was then rinsed with serum-free
medium and 1 ml of 5% charcoal stripped medium with ethanol or
17β-estradiol was added. Wound healing images were captured using
the Nikon Eclipse TE2000-S inverted microscope (Nikon Corp.) at
time-points of 0, 12 and 24 h. The Nikon TI O2 inverted microscope
(Nikon Corp.) was used to create the wound healing video by
compiling images captured every 15 min for a duration of 24 h.
Statistical analysis
For statistical analysis of multiple groups in a
dataset, one-way ANOVA was first performed in Excel. When the null
hypothesis was rejected, we then performed a post hoc Student's
t-test with Bonferroni correction to further analyze the
statistical significance between two groups in the dataset. For the
data set comprised of 3 groups (3 comparisons to be made), the
Bonferroni-adjusted P-value thresholds were 0.0167 and 0.0033 to
denote the significance levels of 5 and 1%, respectively. For the
dataset comprised of 4 groups (6 comparisons to be made), the
adjusted thresholds were 0.00833 and 0.00167 to denote the
significance levels of 5 and 1%, respectively.
Results
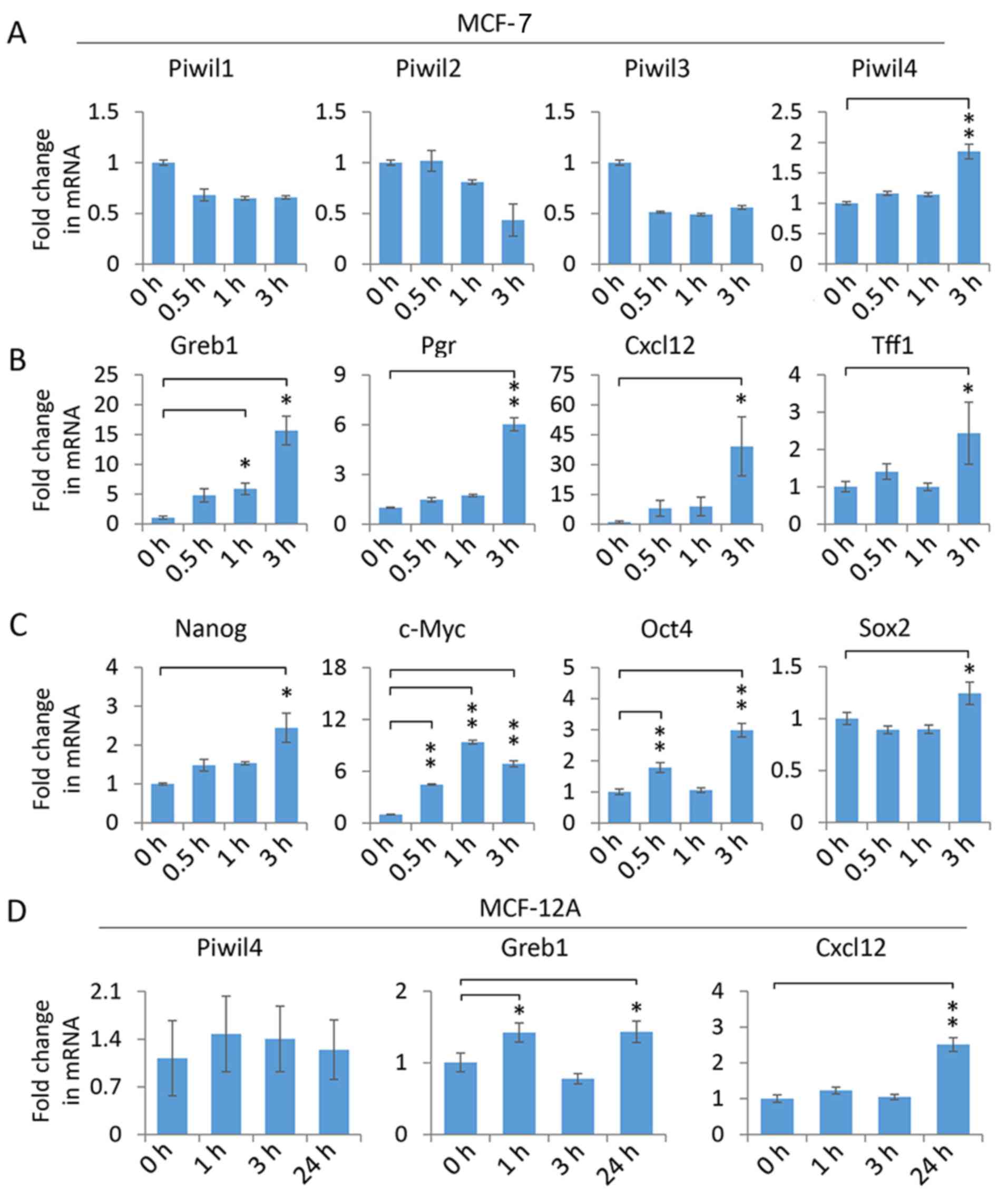
Piwi-like 4 expression is upregulated
by 17β-estradiol in MCF-7 cells
To investigate whether Piwi homologs are involved in
the ER signaling pathway, we first characterized the expression of
Piwil1-4 in MCF-7 cells, which represent an ER-positive and
epithelial-like breast cancer cell line. The quantitative PCR
(qPCR) measurement of Piwil1-4 transcripts revealed that Piwil4
expression was significantly elevated following a 3-h treatment
with 10 nM 17β-estradiol, while other Piwi homologs were not
induced by the treatment (Fig. 1A),
indicating Piwil4 could be specifically upregulated by the
activated ER. To verify that the dose and duration of 17β-estradiol
treatment applied above could indeed activate ER signaling in MCF-7
cells, we assessed the transcript levels of some canonical ER
target genes, including Greb1, Pgr, Cxcl12 and Tff1 (29–33).
As anticipated, 17β-estradiol could significantly upregulate these
well-established targets (Fig. 1B).
In addition, it has been demonstrated that 17β-estradiol could
increase the expression of stemness-related genes in breast cancer
cells (34–36). Consistently, our experiments
confirmed that Nanog, c-Myc, Oct4 and Sox2 transcript levels were
elevated in 17β-estradiol-treated MCF-7 cells (Fig. 1C), indicating that ER signaling was
potently activated.
To investigate whether the 17β-estradiol-induced
upregulation of Piwil4 is related to the tumorigenic cellular
context of MCF-7 cells, we examined another ER-positive yet
non-tumorigenic breast epithelial cell line, MCF-12A (37). Notably, Piwil4 was not induced by
17β-estradiol after a 3-h treatment. Indeed, even after a 24-h
treatment, no upregulation of Piwil4 was observed, while Greb1 and
Cxcl12 were significantly induced (Fig.
1D). This observation indicated that the
17β-estradiol-upregulated Piwil4 may be restricted to tumorigenic
breast cells, hence raising an intriguing question as to whether
Piwil4 is involved in tumorigenic ER signaling events.
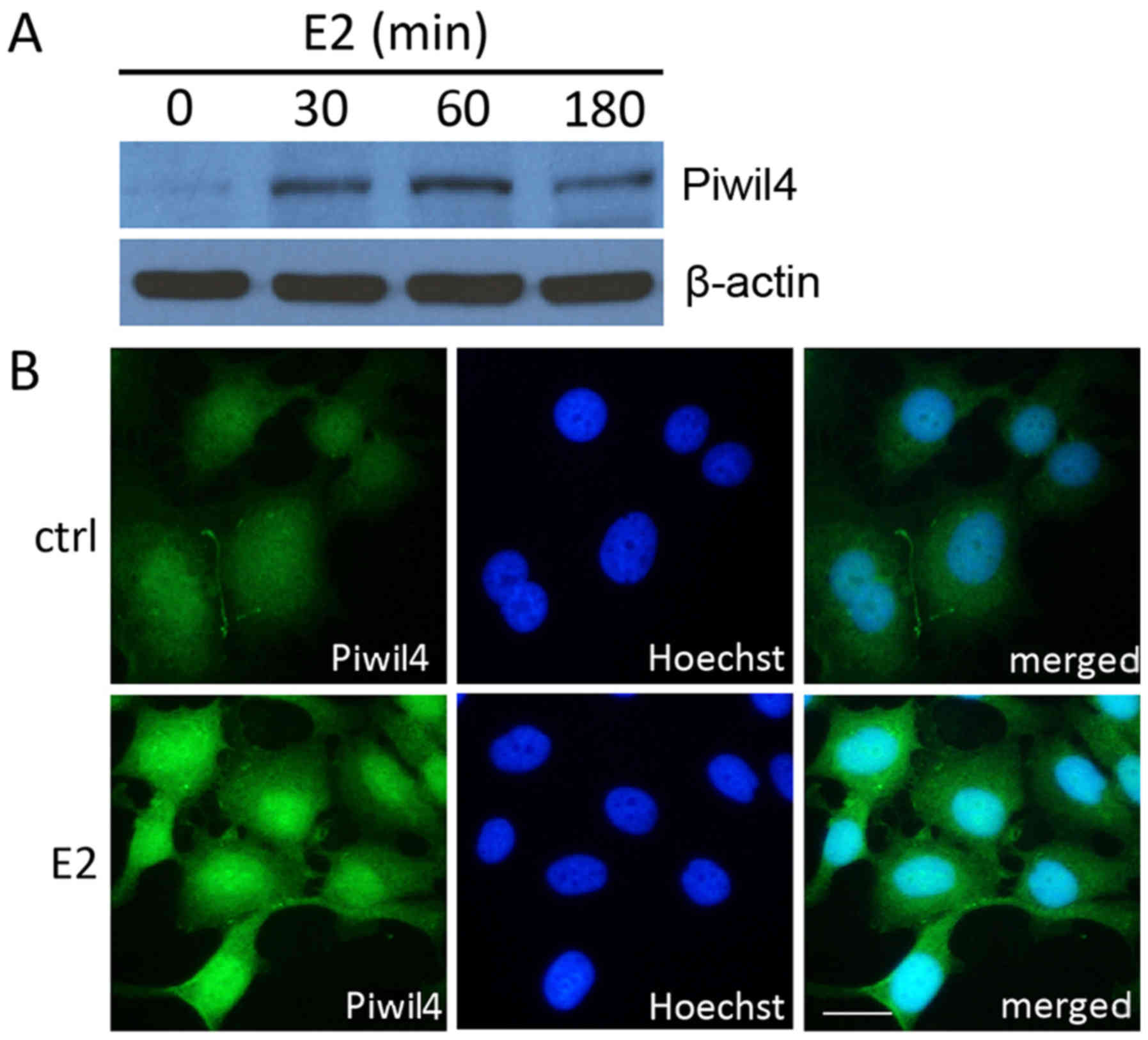
Piwil4 is elevated by 17β-estradiol
and enriched in the nucleus of MCF-7 cells
Given that the Piwil4 transcript level was
upregulated by 17β-estradiol in MCF-7 cells, we further assessed
whether the protein level of Piwil4 was similarly increased. As
anticipated, the western blot analysis revealed that Piwil4 could
be detected by a specific antibody in 17β-estradiol-treated cells
(Fig. 2A). In addition, the
immunofluorescence staining showed that in the absence of
17β-estradiol, Piwil4 was only faintly labeled and homogeneously
distributed in the nucleus and cytoplasm of MCF-7 cells, and that
17β-estradiol could elevate the level of Piwil4. Notably, Piwil4
was mostly enriched in the nuclei of 17β-estradiol-treated cells
(Fig. 2B), indicating a nuclear
role, possibly related to transcriptional regulation, of Piwil4 in
the ER signaling pathway.
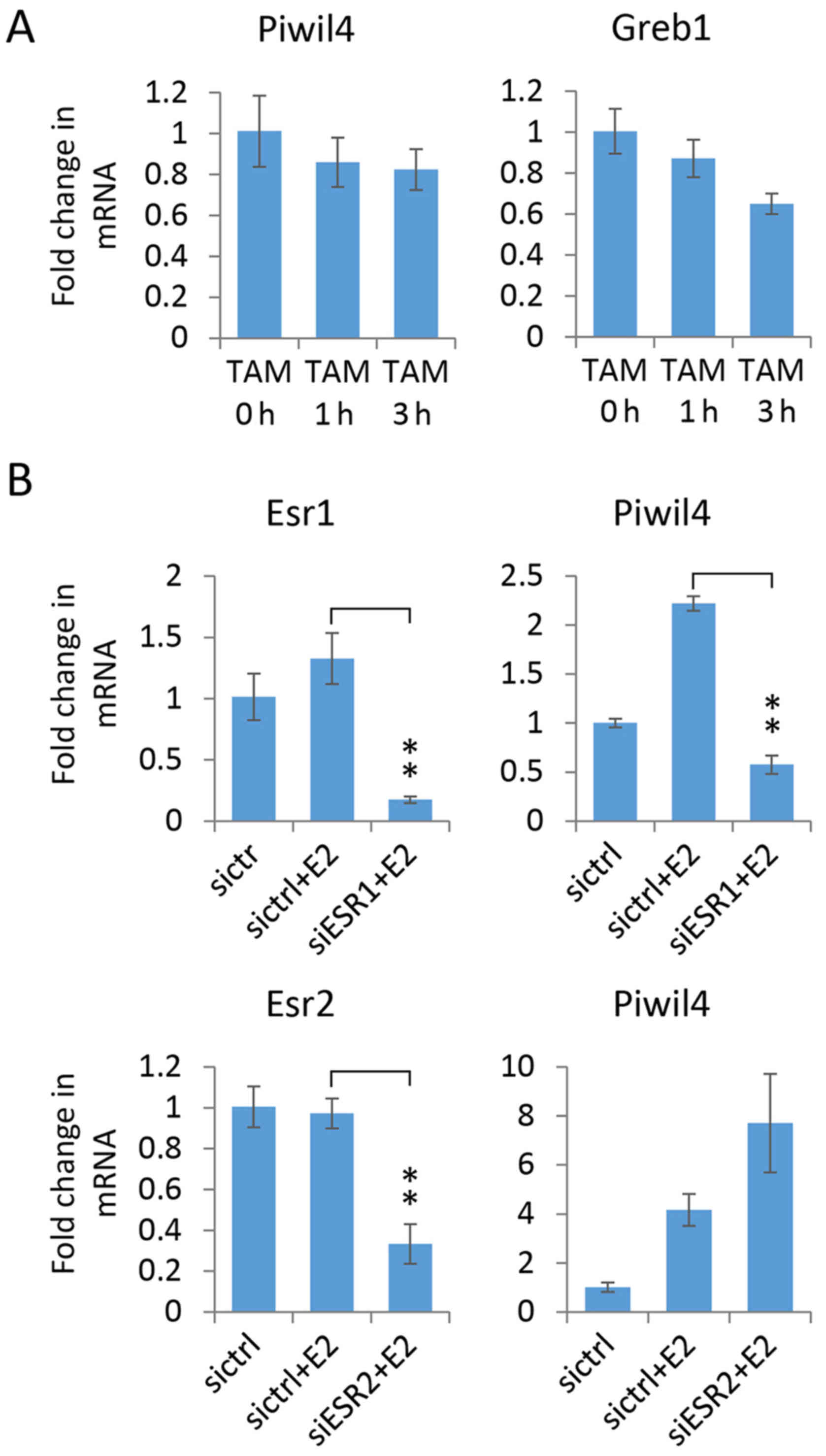
Piwil4 induction is dependent on
estrogen/ERα signaling
To investigate whether estrogen binding to ER is
required for Piwil4 expression induction, MCF-7 cells were treated
with TAM, a selective estrogen receptor modulator (SERM) (6,7).
However, qPCR demonstrated that the expression of Piwil4, as well
as Greb1, was not elevated by the treatment (Fig. 3A), indicating that binding of ER
alone is not sufficient to increase the Piwil4 transcript level;
the event is ligand-specific.
Of the two ERs, ERα (Esr1) and ERβ
(Esr2), MCF-7 cells mainly express the former (38). Hence, to elucidate whether ER,
particularly ERα, is essential for estrogen-induced Piwil4
expression, we used specific siRNA pools to knock down ERα and ERβ
in MCF-7 cells which were then treated with 17β-estradiol.
Markedly, we found that 17β-estradiol-induced Piwil4 upregulation
was abolished in ERα-depleted cells, but not in ERβ-depleted cells
(Fig. 3B). The expression of Piwil4
was even higher in ERβ-depleted cells, supporting the notion that
ERβ may antagonize ERα as previously suggested (38,39).
In summary, these findings indicated that estrogen/ERα signaling is
required for Piwil4 upregulation in MCF-7 cells.
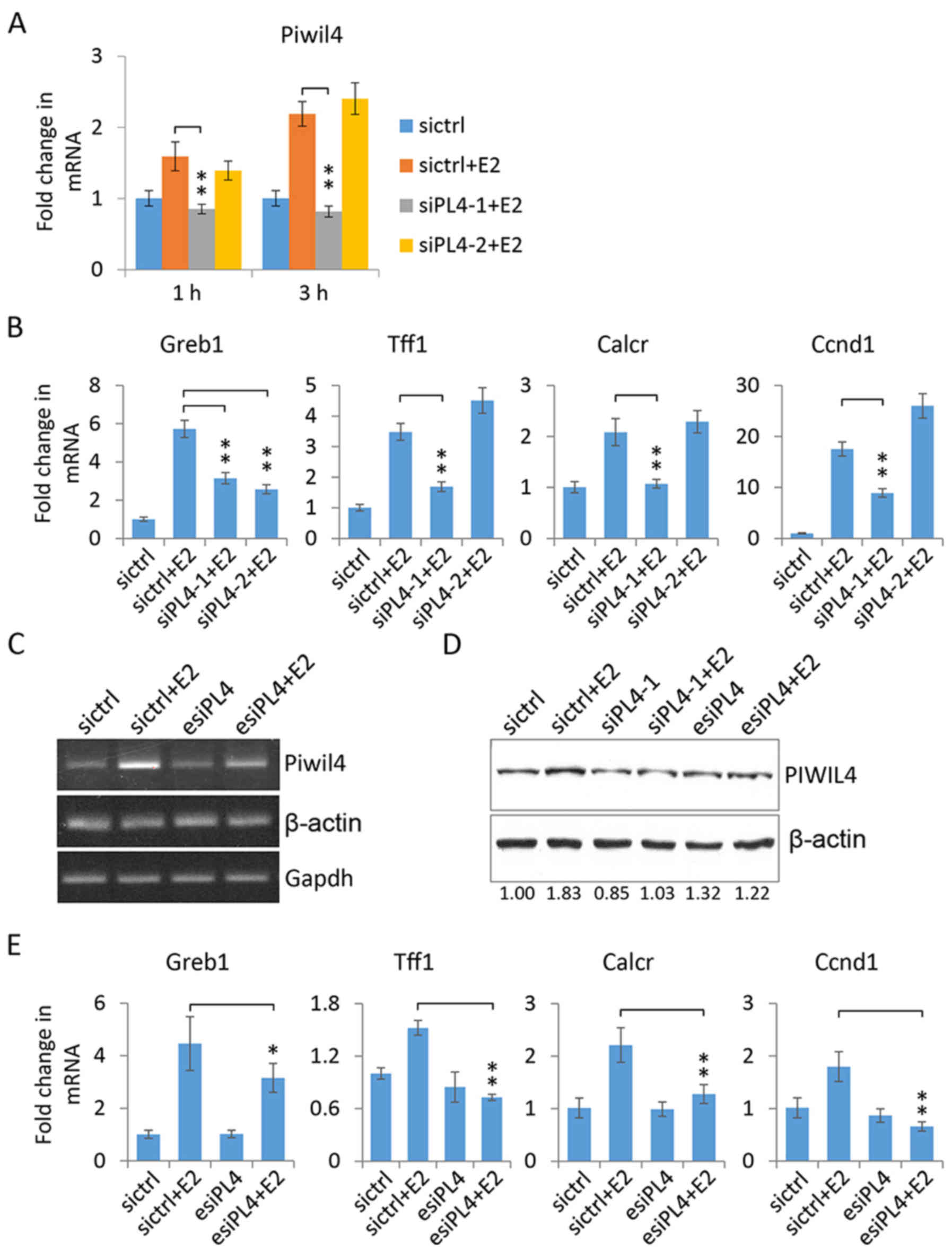
Piwil4 knockdown affects the
transcription of ER target genes in MCF-7 cells
To investigate the role of Piwil4 in regulating ER
signaling in MCF-7 cells, we performed the loss-of-function assays
by using two gene-specific siRNAs, siPL4-1 and siPL4-2. The qPCR
verification revealed that siPL4-1 could efficiently suppress
Piwil4 expression at both the 1- and 3-h time-points following
17β-estradiol treatment, while siPL4-2 demonstrated a very modest
knockdown effect only at the 1-h time-point (Fig. 4A). Subsequently, when we assessed
the transcript levels of some ER target genes following 3-h
17β-estradiol treatment, it was noted that Greb1 (33), Tff1 (40), Calcr (31,41)
and Ccnd1 (42,43) were significantly induced in MCF-7
cells. Notably, their upregulation was largely abolished by the
suppression of Piwil4 using siPL4-1, but not siPL4-2 except for
Greb1 (Fig. 4B), indicating that
the transcriptional activation of some ER target genes was
dependent on the expression of Piwil4.
To rule out the possibility of an off-target effect
of siPL4-1, we used a siRNA pool (esiPL4) consisting of four
different siRNAs against Piwil4. The reverse transcription PCR
(RT-PCR) revealed that 17β-estradiol could significantly increase
the transcript level of Piwil4 and the induction was largely
abolished by esiPL4 (Fig. 4C).
Immunoblotting confirmed that esiPL4, as well as siPL4-1, could
suppress the induced expression of Piwil4 at the protein level
(Fig. 4D). Consistently, esiPL4
transfection significantly reduced the 17β-estradiol-induced
expression of Greb1, Tff1, Calcr and Ccnd1 in MCF-7 cells (Fig. 4E), confirming the potential
involvement of Piwil4 in regulating ER target gene expression.
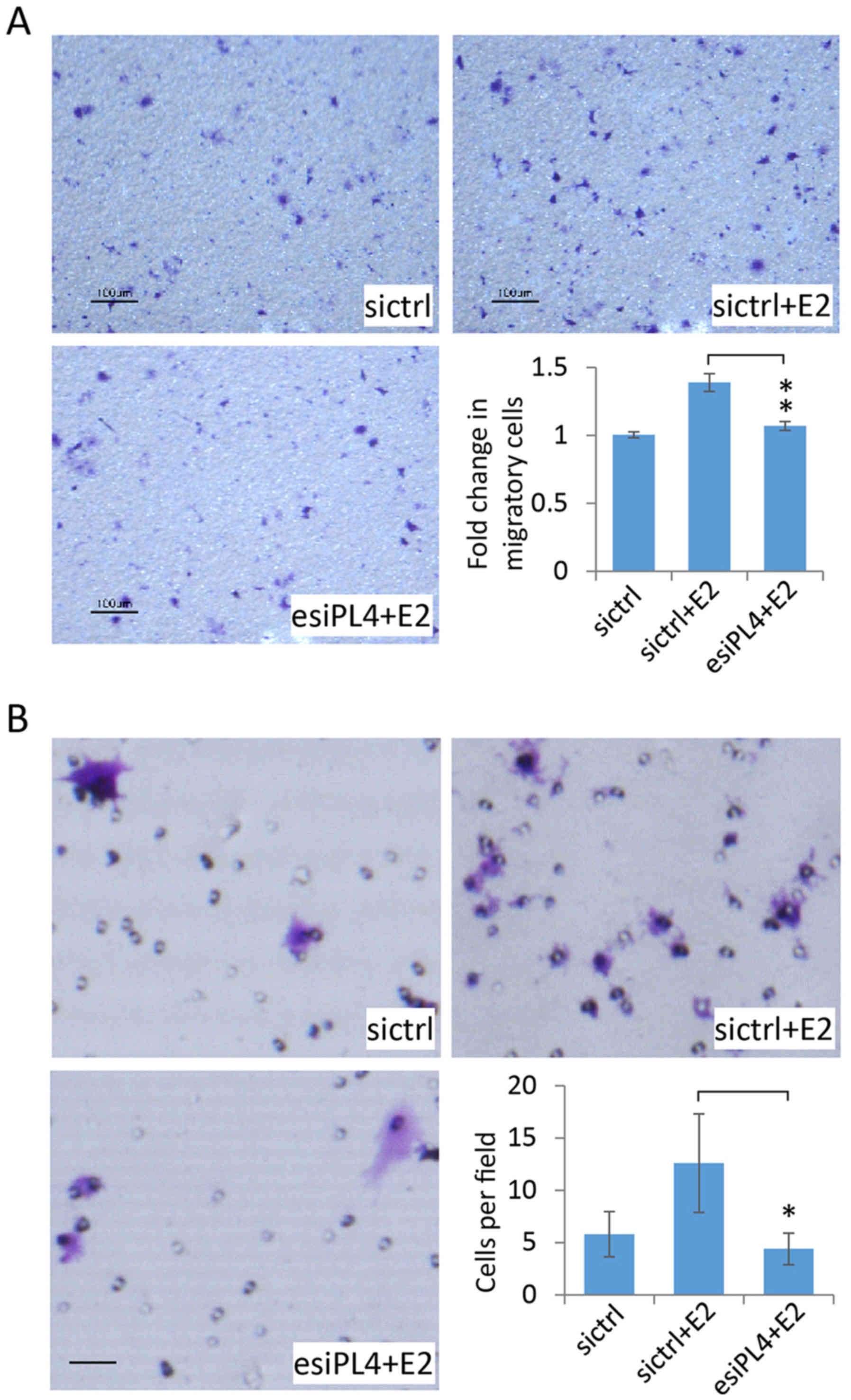
Piwil4 knockdown decreases the
migration and invasion of MCF-7 cells
We further employed esiPL4 to assess the role of
Piwil4 in the regulation of the migration capability of MCF-7
cells. The transfected cells were seeded into the 8-µm Transwell
inserts. Similar to previous studies on the effect of estrogen on
breast cancer cell migration (44–46),
17β-estradiol enhanced the migration of control siRNA
(sictrl)-transfected MCF-7 cells across the substrate (Fig. 5A, upper panels). Notably, when
Piwil4 was knocked down in MCF-7 cells by the transfection of
esiPL4, 17β-estradiol could no longer promote cell migration
(Fig. 5A, lower left panel). The
counting of migratory cells on the lower surface of Transwell
inserts confirmed this observation (Fig. 5A, lower right panel), indicating
that Piwil4 could be involved in 17β-estradiol-mediated MCF-7
migration.
We then investigated whether Piwil4 could regulate
the invasion behavior of MCF-7 cells by using the Matrigel-coated
chamber. As MCF-7 is not a highly invasive breast cancer cell line,
the lower surface of the chamber was additionally coated with
fibronectin which has been shown to promote the invasion of MCF-7
cells (47). We then observed that
estradiol treatment could enhance the cell invasion as previously
reported (36) (Fig. 5B, upper panels). In agreement with
the migration assay (Fig. 5A),
Piwil4 knockdown significantly reduced the invasion of MCF-7 cells
(Fig. 5B, lower panels). This was
reminiscent of a previous study that reported an essential role of
Piwil4 in regulating the invasion capability of a TNBC cell line,
MDA-MB-231 (21).
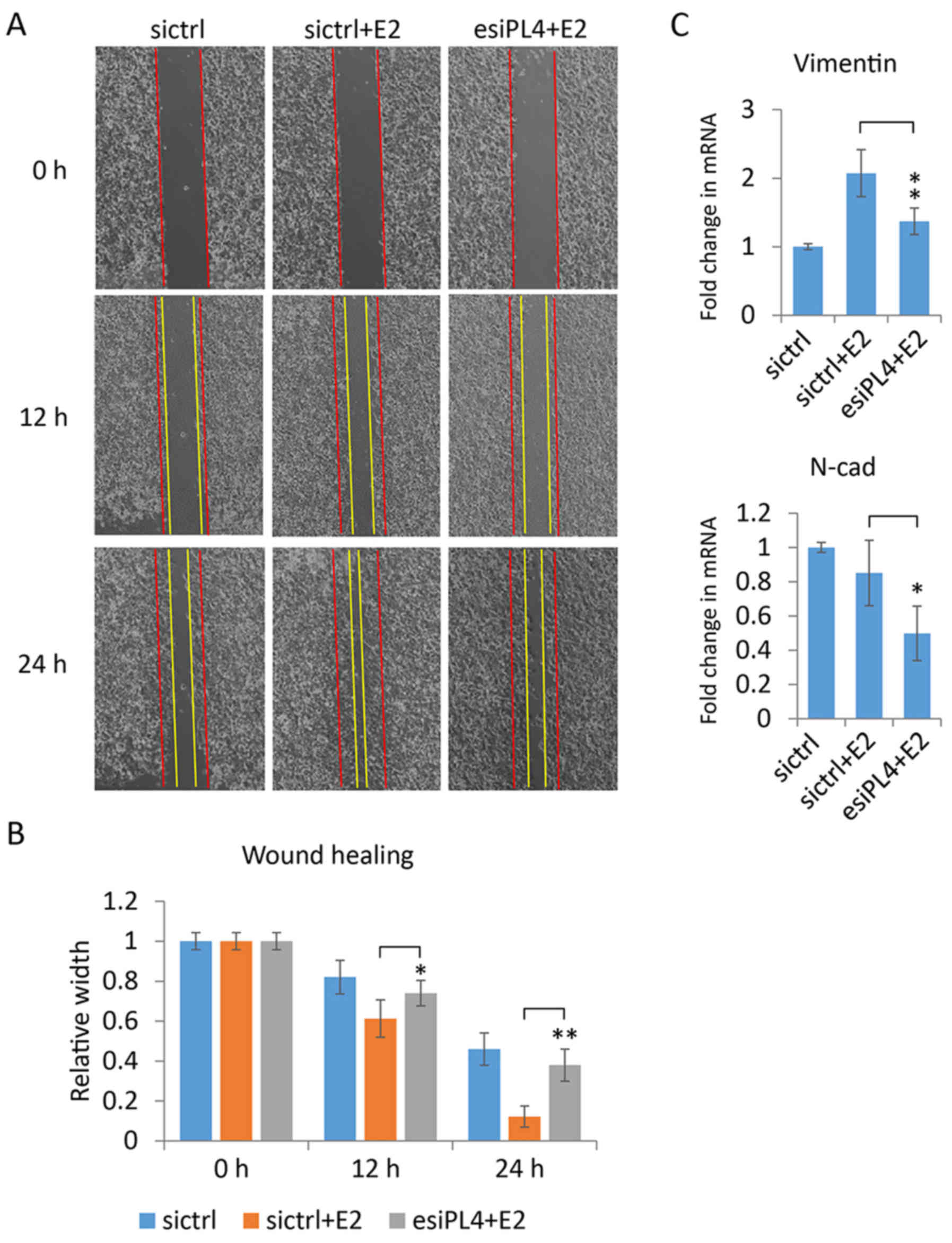
Piwil4 knockdown decreases the
motility of MCF-7 cells in wound healing assays
To further assess the role of Piwil4 in the motility
of MCF-7 cells, we performed wound healing assays by using esiPL4.
Consistent to previous studies (44,45,48),
it was noted that estrogen could enhance the motility and wound
healing process of breast cancer cells, as evidenced by the almost
complete filling of the gap by the 17β-estradiol-treated MCF-7
cells in a time-dependent manner (Fig.
6A, first two columns). Markedly, when Piwil4 was suppressed in
MCF-7 cells, 17β-estradiol enhanced the motility of the cells less
effectively (Fig. 6A, last column).
The measurement demonstrated that the wound gap was significantly
wider in Piwil4-knockdown cells than in control-transfected cells
12- and 24-h following the scratch (Fig. 6B), implying an essential role of
Piwil4 in regulating the motility of MCF-7 cells.
As vimentin has been reported to promote motility in
several breast cancer cell lines, including MCF-7 and MDA-MB-231
(49,50), we further evaluated whether the
observed effect of Piwil4 depletion on MCF-7 motility (Fig. 6A) was associated to vimentin. The
qPCR data revealed that 17β-estradiol significantly increased the
mRNA level of vimentin, and that the knockdown of Piwil4 abolished
the upregulation of vimentin (Fig.
6C). We also assessed the transcript level of another
mesenchymal marker, N-cadherin (21). Notably, the knockdown of Piwil4
reduced its transcript level in 17β-estradiol-treated cells
(Fig. 6C). These observations could
explain the suppressed motility of Piwil4-depleted MCF-7 cells
(Fig. 6A and B).
Discussion
In the present study, we examined the expression
profile of Piwi homologs in an ER-positive breast cancer cell line,
MCF-7, and we recorded an estrogen hormone-induced upregulation of
Piwil4, but not of other Piwi homologs.
Typically, once activated by the estrogen hormone,
ER will translocate into the nucleus, associate with a cohort of
coactivators, bind to the estrogen response element (ERE) and
promote the transcription of target genes related to cell
proliferation and growth (51).
Indeed, the prediction with PROMO revealed that the proximal
promoter region of Piwil4 contains the potential binding
sites of some classic transcription factors associated with ER,
including AP-1 (52), C/EBP-β
(53) and ETS1 (54). In addition, it is also conceivable
that Piwil4 may be upregulated by an alternative, non-genomic ER
signaling cascade. In that case, the hormone receptor could
directly interact and activate mitogen-activated protein kinases
(MAPK), phosphatidylinositol 3-OH kinase (PI3K) and tyrosine
kinases (55–57), which could subsequently initiate
downstream events to regulate cellular functions.
Notably, Piwil4 was not similarly upregulated by
17β-estradiol in another ER-positive but non-tumorigenic breast
cell line, MCF-12A cells, raising an interesting question whether
the induced expression of Piwil4 is restricted to tumorigenic
cells. In fact, a recent study revealed a globally higher level of
Piwil4 in breast cancer samples than in normal breast tissues
(21). Hence, it is conceivable
that Piwil4 could play some essential role in tumorigenic signaling
activities.
Indeed, in Piwil4-depleted MCF-7 cells, some ER
target genes were significantly less induced, including Tff1,
Greb1, Ccnd1 and Calcr (Fig. 4),
indicating that Piwil4 was involved in modulating ER signaling.
Similarly, a recent study identified a global alteration in gene
expression in Piwil4-depleted MDA-MB-231 cells (21). A subsequent pathway analysis
revealed that the most affected genes were enriched in the TGFβ and
FGF pathways. It would be interesting to further explore the
distinct populations of Piwil4-regulated targets in different types
of breast cancers so that the novel function of Piwil4 can be
precisely targeted.
Furthermore, by knocking down Piwil4 in MCF-7 cells,
our functional assays also revealed that Piwil4 was involved in
regulating the migration and invasion of breast cancer cells, and
that the regulation could be achieved via two mesenchymal markers,
vimentin and N-cadherin (Figs. 5
and 6). This observation indicated
that Piwil4 could be targeted to limit the migratory ability of
cancer cells, and to even prevent the epithelial-mesenchymal
transition. Indeed, in the TNBC cell line, MDA-MB-231, the
depletion of Piwil4 was also reported to reduce the expression of
N-cadherin and metastasis (21).
In summary, the present study revealed an
estrogen-induced upregulation of Piwil4 in breast cancer cells. The
loss-of-function assays further indicated that Piwil4 may be
involved in regulating the expression of ER target genes and the
migratory ability of breast cancer cells. Future studies are
warranted to characterize the potential interaction between Piwil4
and canonical ER signaling components so that Piwil4 can be further
exploited to treat breast cancer.
Acknowledgements
We would like to thank Dr George Wai-Cheong Yip at
the National University of Singapore for donating the MCF-12A
cells.
Funding
The present study was supported by the Singapore MOE
Tier 1 R-181-000-163-112 and the Singapore NMRC CBRG-NIG
R-181-000-171-551 to QH. CSS and CW were supported by the NUS
Postgraduate Scholarships.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
ZSLH and JYL performed the experiments. CSS, CW and
LZT initiated the project and provided technical support for
immunoblotting and immunofluorescence. QH designed the experiments,
analyzed the data and wrote the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
DeSantis C, Ma J, Bryan L and Jemal A:
Breast cancer statistics, 2013. CA Cancer J Clin. 64:52–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wiggans RG, Woolley PV, Smythe T, Hoth D,
Macdonald JS, Green L and Schein PS: Phase-II tlial of tamoxifen in
advanced breast cancer. Cancer Chemother Pharmacol. 3:45–48. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carey LA, Dees EC, Sawyer L, Gatti L,
Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML and Perou
CM: The triple negative paradox: Primary tumor chemosensitivity of
breast cancer subtypes. Clin Cancer Res. 13:2329–2334. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Higgins MJ and Baselga J: Targeted
therapies for breast cancer. J Clin Invest. 121:3797–3803. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fisher B, Costantino JP, Wickerham DL,
Cecchini RS, Cronin WM, Robidoux A, Bevers TB, Kavanah MT, Atkins
JN, Margolese RG, et al: Tamoxifen for prevention of breast cancer:
Report of the national surgical adjuvant breast and bowel project
P-1 study. J Natl Cancer Inst. 97:1652–1662. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seth P, Krop I, Porter D and Polyak K:
Novel estrogen and tamoxifen induced genes identified by SAGE
(Serial Analysis of Gene Expression). Oncogene. 21:836–843. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaiyesimi IA, Buzdar AU, Decker DA and
Hortobagyi GN: Use of tamoxifen for breast cancer: Twenty-eight
years later. J Clin Oncol. 13:513–529. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ross RJ, Weiner MM and Lin H: PIWI
proteins and PIWI-interacting RNAs in the soma. Nature.
505:353–359. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meister G: Argonaute proteins: Functional
insights and emerging roles. Nat Rev Genet. 14:447–459. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suzuki R, Honda S and Kirino Y: PIWI
expression and function in cancer. Front Genet. 3:2042012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cox DN, Chao A, Baker J, Chang L, Qiao D
and Lin H: A novel class of evolutionarily conserved genes defined
by piwi are essential for stem cell self-renewal. Genes Dev.
12:3715–3727. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deng W and Lin H: miwi, a murine
homolog of piwi, encodes a cytoplasmic protein essential for
spermatogenesis. Dev Cell. 2:819–830. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sasaki T, Shiohama A, Minoshima S and
Shimizu N: Identification of eight members of the argonaute family
in the human genome. Genomics. 82:323–330. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thomson T and Lin H: The biogenesis and
function PIWI proteins and piRNAs: Progress and prospect. Annu Rev
Cell Dev Biol. 25:355–376. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qiao D, Zeeman AM, Deng W, Looijenga LH
and Lin H: Molecular characterization of hiwi, a human
member of the piwi gene family whose overexpression is
correlated to seminomas. Oncogene. 21:3988–3999. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He G, Chen L, Ye Y, Xiao Y, Hua K,
Jarjoura D, Nakano T, Barsky SH, Shen R and Gao JX: Piwil2
expressed in various stages of cervical neoplasia is a potential
complementary marker for p16INK4a. Am J Transl Res.
2:156–169. 2010.PubMed/NCBI
|
|
18
|
Lee JH, Jung C, Javadian-Elyaderani P,
Schweyer S, Schütte D, Shoukier M, Karimi-Busheri F, Weinfeld M,
Rasouli-Nia A, Hengstler JG, et al: Pathways of proliferation and
antiapoptosis driven in breast cancer stem cells by stem cell
protein piwil2. Cancer Res. 70:4569–4579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu W, Jiang X and Zhang Z: Expression of
PSCA, PIWIL1, and TBX2 in endometrial adenocarcinoma. Onkologie.
33:241–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu Y, Zhang K, Li C, Yao Y, Tao D, Liu Y,
Zhang S and Ma Y: Piwil2 suppresses p53 by inducing phosphorylation
of signal transducer and activator of transcription 3 in tumor
cells. PLoS One. 7:e309992012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Z, Liu N, Shi S, Liu S and Lin H: The
role of PIWIL4, an argonaute family protein, in breast cancer. J
Biol Chem. 291:10646–10658. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sugimoto K, Kage H, Aki N, Sano A,
Kitagawa H, Nagase T, Yatomi Y, Ohishi N and Takai D: The induction
of H3K9 methylation by PIWIL4 at the p16Ink4a locus. Biochem
Biophys Res Commun. 359:497–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He X, Chen X, Zhang X, Duan X, Pan T, Hu
Q, Zhang Y, Zhong F, Liu J, Zhang H, et al: An Lnc RNA
(GAS5)/SnoRNA-derived piRNA induces activation of TRAIL gene by
site-specifically recruiting MLL/COMPASS-like complexes. Nucleic
Acids Res. 43:3712–3725. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu D, Fu H, Zhou H, Su J, Zhang F and Shen
J: Effects of Novel ncRNA molecules, p15-piRNAs, on the methylation
of DNA and histone H3 of the CDKN2B promoter region in U937 cells.
J Cell Biochem. 116:2744–2754. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhong F, Zhou N, Wu K, Guo Y, Tan W, Zhang
H, Zhang X, Geng G, Pan T, Luo H, et al: A SnoRNA-derived piRNA
interacts with human interleukin-4 pre-mRNA and induces its decay
in nuclear exosomes. Nucleic Acids Res. 43:10474–10491.
2015.PubMed/NCBI
|
|
26
|
Jiang S, Zhao L, Lu Y, Wang M, Chen Y, Tao
D, Liu Y, Sun H, Zhang S and Ma Y: Piwil2 inhibits keratin 8
degradation through promoting p38-induced phosphorylation to resist
Fas-mediated apoptosis. Mol Cell Biol. 34:3928–3938. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Krishnan P, Ghosh S, Graham K, Mackey JR,
Kovalchuk O and Damaraju S: Piwi-interacting RNAs and PIWI genes as
novel prognostic markers for breast cancer. Oncotarget.
7:37944–37956. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Horwitz KB, Koseki Y and McGuire WL:
Estrogen control of progesterone receptor in human breast cancer:
Role of estradiol and antiestrogen. Endocrinology. 103:1742–1751.
1978. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Henry JA, Nicholson S, Hennessy C, Lennard
TW, May FE and Westley BR: Expression of the oestrogen regulated
pNR-2 mRNA in human breast cancer: Relation to oestrogen receptor
mRNA levels and response to tamoxifen therapy. Br J Cancer.
61:32–38. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Frasor J, Danes JM, Komm B, Chang KC,
Lyttle CR and Katzenellenbogen BS: Profiling of estrogen up- and
down-regulated gene expression in human breast cancer cells:
Insights into gene networks and pathways underlying estrogenic
control of proliferation and cell phenotype. Endocrinology.
144:4562–4574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Journé F, Body JJ, Leclercq G, Nonclercq D
and Laurent G: Estrogen responsiveness of IBEP-2, a new human cell
line derived from breast carcinoma. Breast Cancer Res Treat.
86:39–53. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rae JM, Johnson MD, Scheys JO, Cordero KE,
Larios JM and Lippman ME: GREB 1 is a critical regulator of hormone
dependent breast cancer growth. Breast Cancer Res Treat.
92:141–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jung JW, Park SB, Lee SJ, Seo MS, Trosko
JE and Kang KS: Metformin represses self-renewal of the human
breast carcinoma stem cells via inhibition of estrogen
receptor-mediated OCT4 expression. PLoS One. 6:e280682011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang C, Mayer JA, Mazumdar A, Fertuck K,
Kim H, Brown M and Brown PH: Estrogen induces c-myc gene
expression via an upstream enhancer activated by the estrogen
receptor and the AP-1 transcription factor. Mol Endocrinol.
25:1527–1538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun Y, Wang Y, Fan C, Gao P, Wang X, Wei G
and Wei J: Estrogen promotes stemness and invasiveness of
ER-positive breast cancer cells through Gli1 activation. Mol
Cancer. 13:1372014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Van Zijl C, Lottering ML, Steffens F and
Joubert A: In vitro effects of 2-methoxyestradiol on MCF-12A and
MCF-7 cell growth, morphology and mitotic spindle formation. Cell
Biochem Funct. 26:632–642. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Treeck O, Lattrich C, Springwald A and
Ortmann O: Estrogen receptor beta exerts growth-inhibitory effects
on human mammary epithelial cells. Breast Cancer Res Treat.
120:557–565. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Paruthiyil S, Parmar H, Kerekatte V, Cunha
GR, Firestone GL and Leitman DC: Estrogen receptor beta inhibits
human breast cancer cell proliferation and tumor formation by
causing a G2 cell cycle arrest. Cancer Res. 64:423–428. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Prest SJ, May FE and Westley BR: The
estrogen-regulated protein, TFF1, stimulates migration of human
breast cancer cells. FASEB J. 16:592–594. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Nakamura M, Mori I, Takeda K,
Nakamura Y, Utsunomiya H, Yoshimura G, Sakurai T and Kakudo K:
Calcitonin receptor gene and breast cancer: Quantitative analysis
with laser capture microdissection. Breast Cancer Res Treat.
83:109–117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arnold A and Papanikolaou A: Cyclin D1 in
breast cancer pathogenesis. J Clin Oncol. 23:4215–4224. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Roy PG and Thompson AM: Cyclin D1 and
breast cancer. Breast. 15:718–727. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mathew AC, Rajah TT, Hurt GM, Abidi Abbas
SM, Dmytryk JJ and Pento JT: Influence of antiestrogens on the
migration of breast cancer cells using an in vitro wound model.
Clin Exp Metastasis. 15:393–399. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Malek D, Gust R and Kleuser B:
17-Beta-estradiol inhibits transforming-growth-factor-beta-induced
MCF-7 cell migration by Smad3-repression. Eur J Pharmacol.
534:39–47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zheng S, Huang J, Zhou K, Zhang C, Xiang
Q, Tan Z, Wang T and Fu X: 17β-Estradiol enhances breast cancer
cell motility and invasion via extra-nuclear activation of
actin-binding protein ezrin. PLoS One. 6:e224392011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li CL, Yang D, Cao X, Wang F, Hong DY,
Wang J, Shen XC and Chen Y: Fibronectin induces
epithelial-mesenchymal transition in human breast cancer MCF-7
cells via activation of calpain. Oncol Lett. 13:3889–3895. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Park SJ, Kim JG, Kim ND, Yang K, Shim JW
and Heo K: Estradiol, TGF-β1 and hypoxia promote breast cancer
stemness and EMT-mediated breast cancer migration. Oncol Lett.
11:1895–1902. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mendez MG, Kojima S and Goldman RD:
Vimentin induces changes in cell shape, motility, and adhesion
during the epithelial to mesenchymal transition. FASEB J.
24:1838–1851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu CY, Lin HH, Tang MJ and Wang YK:
Vimentin contributes to epithelial-mesenchymal transition cancer
cell mechanics by mediating cytoskeletal organization and focal
adhesion maturation. Oncotarget. 6:15966–15983. 2015.PubMed/NCBI
|
|
51
|
Gross JM and Yee D: How does the estrogen
receptor work? Breast Cancer Res. 4:62–64. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dahlman-Wright K, Qiao Y, Jonsson P,
Gustafsson JÅ, Williams C and Zhao C: Interplay between AP-1 and
estrogen receptor α in regulating gene expression and proliferation
networks in breast cancer cells. Carcinogenesis. 33:1684–1691.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dong J, Tsai-Morris CH and Dufau ML: A
novel estradiol/estrogen receptor alpha-dependent transcriptional
mechanism controls expression of the human prolactin receptor. J
Biol Chem. 281:18825–18836. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kalet BT, Anglin SR, Handschy A,
O'Donoghue LE, Halsey C, Chubb L, Korch C and Duval DL:
Transcription factor Ets1 cooperates with estrogen receptor α to
stimulate estradiol-dependent growth in breast cancer cells and
tumors. PLoS One. 8:e688152013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Simoncini T, Mannella P, Fornari L, Caruso
A, Varone G and Genazzani AR: Genomic and non-genomic effects of
estrogens on endothelial cells. Steroids. 69:537–542. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Huan J, Wang L, Xing L, Qin X, Feng L, Pan
X and Zhu L: Insights into significant pathways and gene
interaction networks underlying breast cancer cell line MCF-7
treated with 17β-Estradiol (E2). Gene. 533:346–355. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Raffo D, Pontiggia O, de Kier Joffé Bal E
and Simian M: Non-genomic actions of estradiol and 4-OH-tamoxifen
on murine breast cancer cells. Oncol Rep. 33:439–447. 2015.
View Article : Google Scholar : PubMed/NCBI
|