Introduction
Liver cancer is one of the most common causes of
cancer-related deaths worldwide. Due to the high incidence in
sub-Saharan Africa and eastern Asia, liver cancer has become an
important medical problem in these areas (1). Despite advances in liver cancer
prevention, detection, diagnosis and treatment, the prognosis of
liver cancer remains unsatisfactory (2). There are still a considerable number
of liver cancer cases that are diagnosed at a stage with distant
metastasis, at which point it is not possible to perform radical
surgery (2). The targeted
therapeutic drug sorafenib can significantly improve
progression-free survival and overall survival of patients with
advanced liver cancer (3). However,
the expensive cost and occurrence of drug resistance limit the wide
application of sorafenib. Thus, identification of novel and
effective drugs to manage this disease is urgently required.
Hepatocyte growth factor (HGF) and its tyrosine
kinase receptor (RTK) c-Met have been revealed to exert diverse
physiological effects during embryogenesis and morphogenesis
(4). Normally, activation of c-Met
by HGF induces a variety of cellular responses, which are critical
for liver development and regeneration (5). However, dysregulation of HGF/c-Met
signaling is implicated in liver cancer carcinogenesis and
progression (6). Previous studies
reported that c-Met was overexpressed in liver cancer tissue and
was closely associated with portal vein invasion and early
recurrence of liver cancer (7,8).
Binding of the HGF to c-Met induced phosphorylation of c-Met and
activation of multiple downstream signaling pathways, including
mitogen-activated protein kinase and phosphoinositide-3 kinase
pathways, resulting in enhancement of motility and invasiveness of
a variety of types of tumor cell (9). Therefore, identification of novel
agents that target the HGF/c-Met axis may present a promising
therapeutic strategy for liver cancer treatment (10).
Ginkgo biloba L., also known as ginkgo, is an
ancient gymnosperm species that is widely distributed in China.
Ginkgolic acid (GA) is the main botanical component extracted from
the seed coat of Ginkgo biloba L. As a mixture of phenolic
acids, GA possesses a wide range of bioactive properties and can
exert diverse pharmacological activities (11,12).
Several monomer structures of GA have been identified (13). C15:1 is one of the most abundant GAs
in Ginkgo biloba L. extract (14). Recently, a number of studies have
indicated the anticancer activity of GA in a variety of cancer
types and the less toxic reaction on non-cancerous cells (15–18).
However, to the best of our knowledge, whether GA can inhibit the
invasion of liver cancer cells and the underlying mechanisms
remains unknown. The aim of the present study was to investigate
the effects of GA on the migration and invasion abilities of liver
cancer cells and identify the underlying molecular mechanism.
Materials and methods
Cell culture and reagents
The human liver cancer cell line HepG2 was purchased
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
and cultured in Dulbecco's modified Eagle's medium containing 10%
FBS (HyClone Laboratories; GE Healthcare Life Sciences, Logan, UT,
USA) and 1% penicillin/streptomycin. Experiments were conducted
with cells at <25 passages. GA (C15:1;
C22H34O3) was obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany) and initially
dissolved in pure methanol as a stock solution of 1 mM. Working
dilutions of GA were performed with culture medium immediately
before use. Recombinant human HGF was purchased from R&D
Systems (Minneapolis, MN, USA). Primary antibodies against
E-cadherin (cat. no. 3195, rabbit), N-cadherin (cat. no. 13116,
rabbit), ZO-1 (cat. no. 13663, rabbit), vimentin (cat. no. 5741,
rabbit), HGF (cat. no. 52445, rabbit), c-Met (cat. no. 8198,
rabbit) and phosphorylated c-Met (p-c-Met) (cat. no. 3077, rabbit)
were purchased from Cell Signaling Technology (Danvers, MA, USA);
mouse anti-MMP-2 (cat. no. ab86607), mouse anti-MMP-9 (cat. no.
ab58803) and mouse anti-β-actin (cat. no. ab8226) were obtained
from Abcam (Cambridge, MA, USA); and secondary antibodies (goat
anti-rabbit IgG-HRP; cat. no. sc-2004; goat anti-mouse IgG-HRP;
cat. no. 2005) were obtained from Santa Cruz Biotechnology (Santa
Cruz, CA, USA).
Cell proliferation assays
The effect of GA on HepG2 cell proliferation was
assessed by an MTT assay. HepG2 cells were seeded in 96-well plates
(3,000 cells/well) and incubated for 12 h. After treatment with GA
at different concentrations (0, 5, 10, 25, 50 and 100 µM) at the
indicated time-points (12, 24, 36 and 48 h), 20 µl of 5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich, Merck KGaA) was added to each well and cells were
incubated for an additional 4 h at 37°C (19). Then 150 µl DMSO (Sigma-Aldrich;
Merck KGaA) per well was added to dissolve the crystals. Cell
proliferation was assessed by measuring the absorbance at 490 nm
using a 96-well plate spectrophotometer (Thermo Fisher Scientific,
Inc., Waltham, MA, USA).
Wound healing migration assays
Wound healing migration assays were conducted in
order to assess the migration ability of HepG2 cells. HepG2 cells
were seeded in 6-well plates and grown to 80% confluence in
complete medium. After serum-starvation for 12 h, the cells were
then treated with GA (0, 25 and 50 µM) for 24 h. Subsequently, a
sterile 200-ml pipette tip was used to make a wound across the cell
culture monolayer. Discarding the medium and washing three times
with PBS removed floating cells. Multiple images of the
matched-pair wound regions were obtained immediately after wounding
(0 h) and after 36 h using a light microscope (Nikon Corp., Tokyo,
Japan) at a magnification of ×100. The migration distance was
determined by calculating the area of the cell gap at the indicated
time-points (0 and 36 h).
Cell invasion assays
Cell invasion assays were performed using
Matrigel-coated Transwell inserts (8-µm pore size; BD Biosciences,
Franklin Lakes, NJ, USA) according to the manufacturer's
recommendation. Cells (5×104) were added to the upper
chamber and incubated in the presence of GA (0, 25 and 50 µM) or GA
plus HGF (5 ng/ml) in serum-free medium. Complete medium (500 µl)
was added to the lower chamber. After 48 h in culture, cells in the
upper chamber were carefully removed with a cotton-tipped swab. The
invaded cells were fixed and stained with 0.1% crystal violet
solution. Quantification of invasion was calculated by counting
stained invaded cells in at least 10 randomly selected fields using
a light microscope (Nikon Corp.) at a magnification of ×200. Each
experiment was performed in triplicate to confirm the
reproducibility of the data.
Western blot assays
Tissues or cells were lysed in RIPA Lysis Buffer
(Beyotime Institute of Biotechnology, Guangzhou, China). Following
protein quantification with BCA (Pierce; Thermo Fisher Scientific,
Inc.), samples were subjected to 10–12.5% SDS-PAGE and transferred
to PVDF membranes. After blocking with 5% BSA, the membranes were
incubated with the primary antibodies (dilutions for antibodies:
mmp-2, 1:800; mmp-9, 1:750; E-cadherin, 1:1,500; ZO-1, 1:1,000;
vimentin, 1:750; N-cadherin, 1:1,000; β-actin, 1:1,000; c-Met,
1:800; p-c-Met, 1:1,000) and then with species-specific secondary
antibodies (1:5,000). β-actin was used as an internal loading
control. Protein bands were detected on a ChemiDoc XRS imaging
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using
enhanced chemiluminescence (EMD Millipore, Billerica, MA, USA).
Quantitative PCR
After the designated treatment, total cell RNA was
isolated using TRIzol reagent (Invitrogen, Thermo Fisher
Scientific, Inc.) and cDNA was synthesized using a PrimeScript RT
reagent kit (Takara Biotechnology Co., Ltd., Dalian, China)
according to the manufacturer's instructions. Quantitative PCR was
then performed with an iQ5 Multicolor Real-Time PCR Detection
System (Bio-Rad Laboratories, Inc.) using a SYBR Green reagent
(Takara Biotechnology Co., Ltd.). The cycling conditions were as
follows: Denaturing at 95°C for 10 min followed by 35 cycles of
denaturing at 95°C for 10 min, 38 cycles of denaturing at 95°C for
20 sec, annealing and extension at 60°C for 1 min. The primer
sequences used in this study are listed in Table I. Relative expression of the sample
genes was calculated using the ΔΔCq method (20) with β-actin as the endogenous
control.
 | Table I.Primers for real-time PCR. |
Table I.
Primers for real-time PCR.
| Genes | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| MMP-2 |
GATGATGCCTTTGCTCGTGC |
CAAAGGGGTATCCATCGCCA |
| MMP-9 |
GAGACCGGTGAGCTGGATAG |
TACACGCGAGTGAAGGTGAG |
| HGF |
TTTGCCTTCGAGCTATCGGG |
TGATCCCAGCGCTGACAAAT |
| E-cadherin |
ATTCTGATTCTGCTGCTCTTG |
AGTCCTGGTCCTCTTCTCC |
| N-cadherin |
ACAACAGACCTGAGTTCTTACAC |
TTGGAGCCTGAGACACGATT |
| Vimentin |
AATGACCGCTTCGCCAAC |
CCGCATCTCCTCCTCGTAG |
| ZO-1 |
GAGATGAACGGGCTACGC |
GAGACTGCCATTGCTTGG |
| β-actin |
CATCACTATCGGCAATGAGC |
GACAGCACTGTGTTGGCATA |
Immunofluorescence analysis
After the designated treatment, cells were fixed
with 4% ice-cold methanol and permeabilized with 0.5% Triton X-100.
Cells were then blocked with 1% bovine serum albumin (BSA) followed
by incubation with a primary antibody against HGF (dilution 1:200)
at 4°C overnight. After washing with PBS, cells were incubated with
a goat anti-rabbit FITC (green) IgG antibody (cat. no. ZF-0311;
ZSGB-BIO Inc., Beijing, China) at 1:200 dilutions for 1 h at room
temperature and then washed with PBS again. Subsequently, the cells
were stained with DAPI in order to visualize the nuclei. Images
were captured with a fluorescence microscope (Nikon Eclipse Ti-s;
Nikon Corp.) using the appropriate excitation wavelength at a
magnification of ×200.
In vivo study
Animal experimental protocols were approved by the
Ethics Committee of The Second Affiliated Hospital of Medical
College, Xi'an Jiaotong University (Xi'an, Shaanxi, China). Twenty
6-week-old male BALB/c nude mice were supplied by and housed in the
Animal Center at the Medical College, Xi'an Jiaotong University.
The mice were housed in a ventilated, temperature-controlled
(22–24°C) and standardized sterile animal room with a 12-h
light/dark cycle, and free access to food and water. A single-cell
suspension (30 µl) of HepG2 cells (suspended in HBSS, containing
1×106 cells) was inoculated subcutaneously into the back
of the BALb/c nude mice. One week after inoculation, mice were
randomly divided into two cohorts with 10 mice in each group. One
cohort received the vehicle (100 µl saline) by oral gavage and the
other was orally administrated GA (suspended in saline, 50 mg/kg)
daily for 5 weeks. The tumor volume was monitored throughout the
experiment and calculated using the following formula: V (tumor
volume)=S (shorter diameter)2 × L (longer diameter)
×0.5. At the end of the experiment, the mice were euthanized by
CO2 asphyxiation (the CO2 flow rate used for
euthanasia was 30% of the chamber's volume/min) and the tumor
samples were harvested. Tissue protein was prepared and subjected
to western blot assays as previously described.
Statistical analysis
Data are presented as the mean ± standard deviation.
Each experiment was performed at least three times. All
quantitative data were analyzed using SPSS (version 15.0; SPSS,
Inc., Chicago, IL, USA). Student's t-test was performed to assess
the difference between two groups. Two-way analysis of variance was
used to analyze data between groups and post-hoc Tukey's test was
used for multiple comparisons, and P<0.05 was considered to
indicate a statistically significant difference.
Results
GA suppresses the proliferation,
migration and invasion abilities of HepG2 cells
Firstly, the effect of GA on the migration ability
of HepG2 cells was investigated. The concentration of GA used was
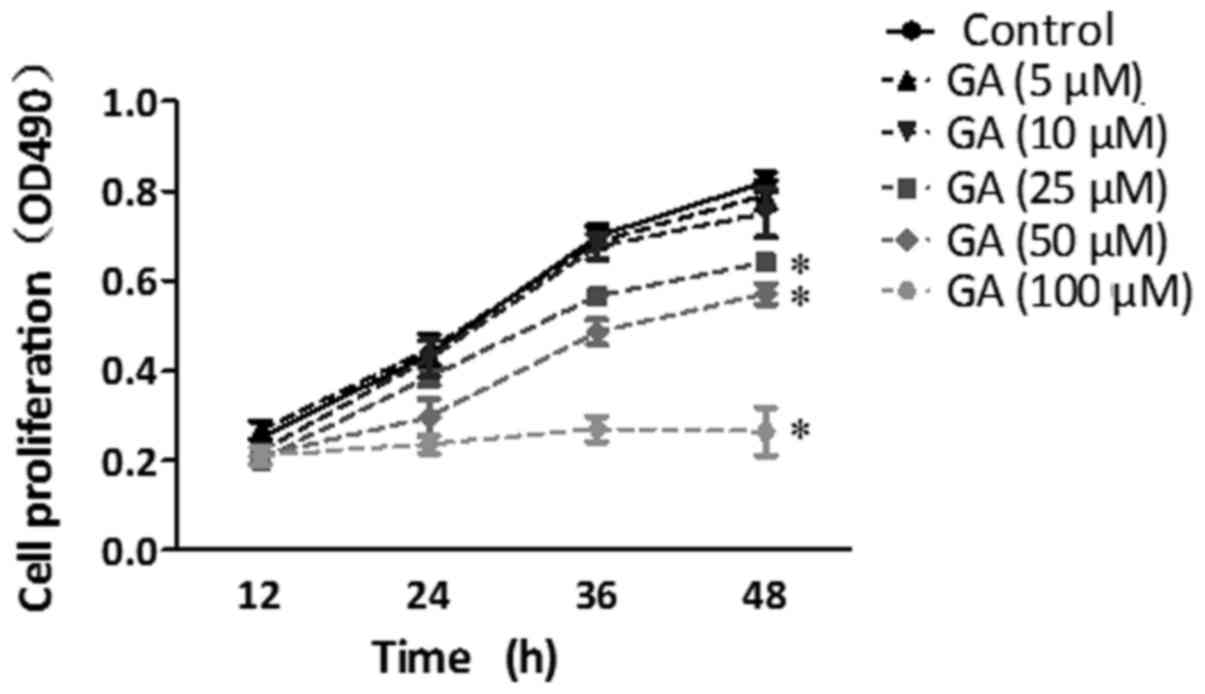
determined according to a previous study (15) and by cell proliferation experiments.
The proliferation of HepG2 cells was suppressed by GA treatment at
a concentration of more than 25 µM (Fig. 1). HepG2 cells were pre-treated with
GA (25 and 50 µM) for 24 h and then wound healing migration assays
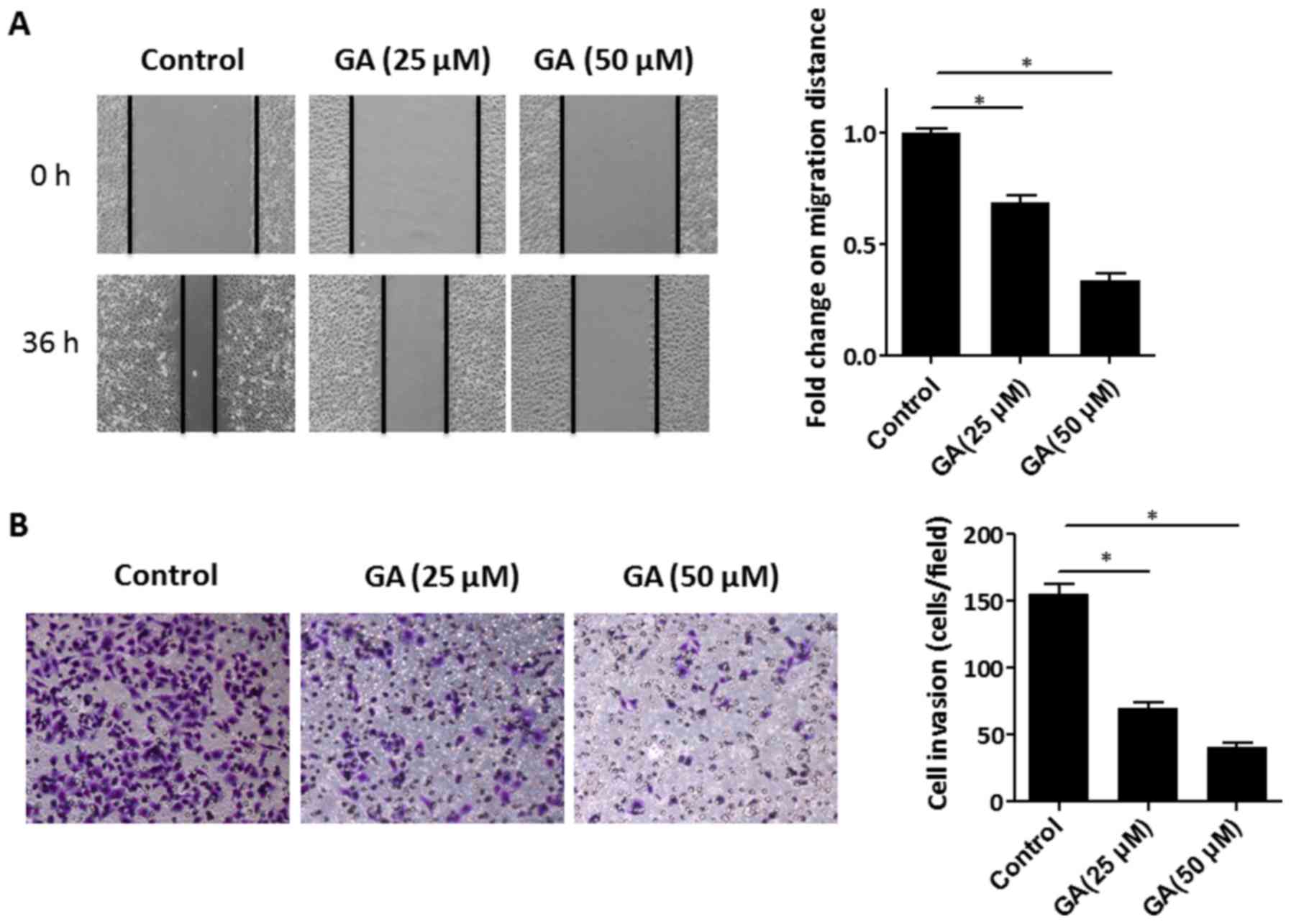
were performed. As shown in Fig.
2A, the migration capacity of HepG2 cells was impaired by 25
and 50 µM GA intervention compared to that of the control cells (0
µM GA), as determined by the cell migration distance. To further
assess the effect of GA on the invasion capacity of HepG2 cells, a
Matrigel invasion assay was conducted. The results revealed that
there was a significant reduction in the number of invaded cells
after the HepG2 cells were treated with 25 or 50 µM GA (Fig. 2B). Altogether, the data demonstrated
that GA inhibited the migration and invasion of HepG2 cells in
vitro.
GA inhibits the expression of
invasion- and EMT-related molecules in HepG2 cells
Epithelial-mesenchymal transition (EMT), which
endows tumor cells with enhanced motility and invasion capacities,
is a prerequisite for tumor infiltration and metastasis (21). In order to investigate the effect of
GA on the expression of invasion-related genes (MMP-2 and MMP-9)
and EMT-related genes (E-cadherin, ZO-1, vimentin and N-cadherin),
HepG2 cells were treated with GA (25 and 50 µM) for 24 h and then
the total cell RNA was extracted and subjected to RT-qPCR analysis.
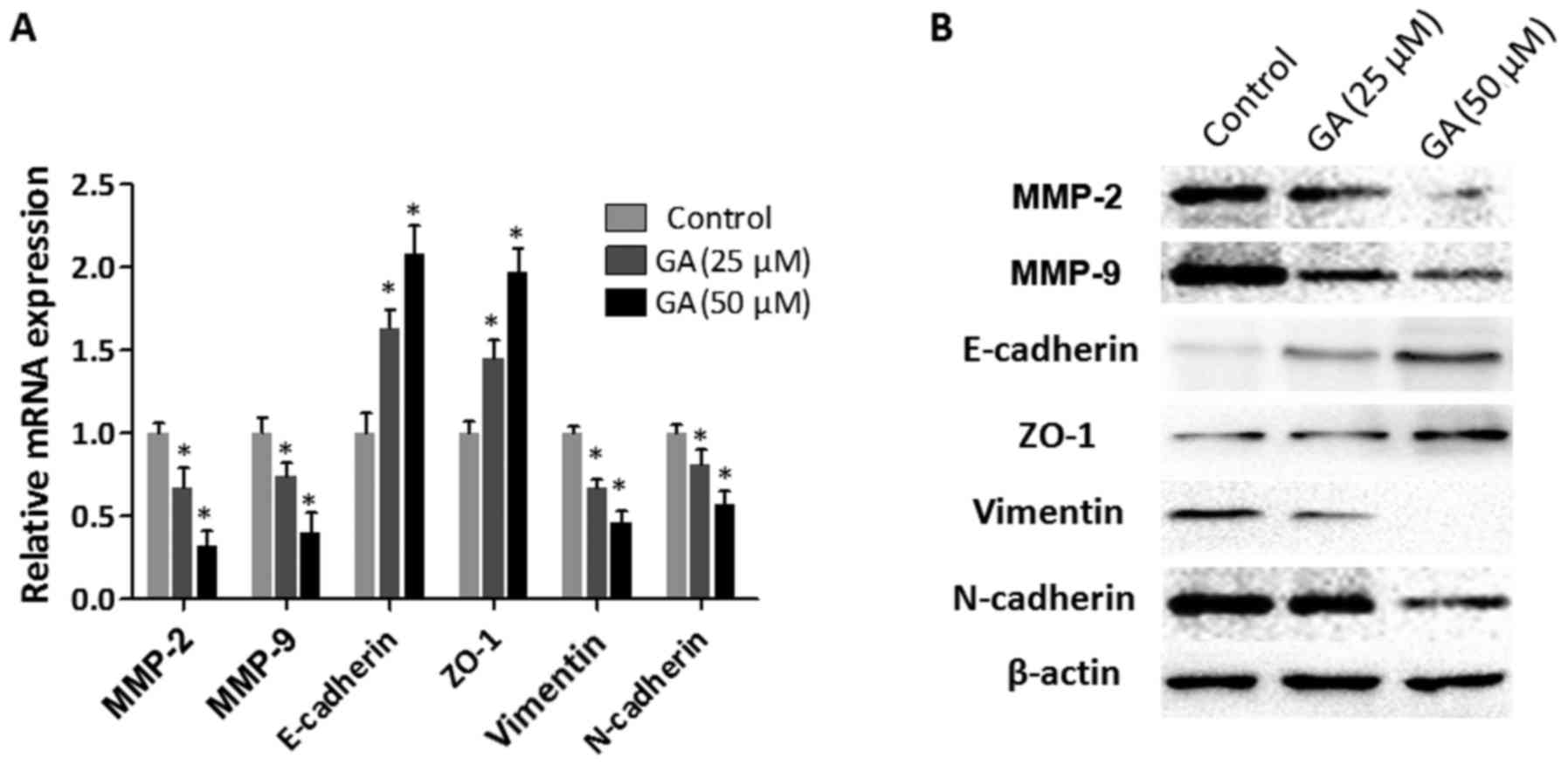
As revealed in Fig. 3A, the mRNA
expression levels of MMP-2 and MMP-9 were significantly reduced in
the HepG2 cells after treatment with GA. In addition, increased
expression levels of epithelial markers (E-cadherin and ZO-1) and
decreased expression levels of mesenchymal markers (vimentin and
N-cadherin) were detected in the HepG2 cells with GA intervention.
These observations were confirmed at the protein level by the
results of the western blot assays (Fig. 3B). Collectively, these results
indicated that GA inhibited the expression of invasion-related
molecules and prevented the EMT process in HepG2 cells.
GA downregulates HGF/c-Met signaling
activity in HepG2 cells
A previous study has demonstrated that HGF/c-Met
signaling plays critical roles in the promotion of cell invasion
and the EMT process (8). To
determine whether GA has an influence on HGF/c-Met signaling
activation in liver cancer cells, HepG2 cells were treated with GA
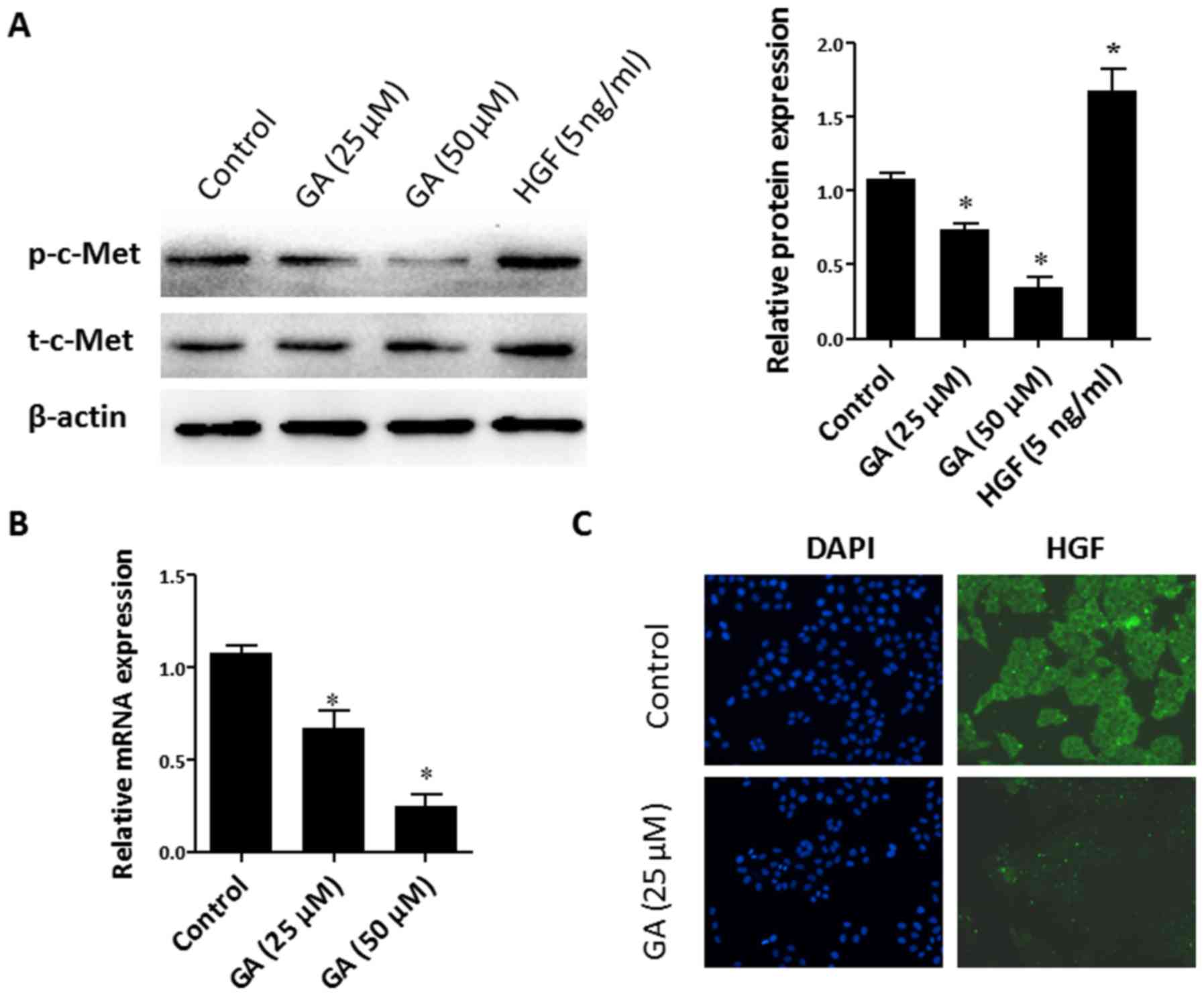
(25 and 50 µM) for 48 h, and then the phosphorylation level of
c-Met (p-c-Met) was determined by immunoblotting. Exogenous
recombinant HGF (5 ng/ml) was used as a positive control. As
observed in Fig. 4A, the total
c-Met (t-c-Met) remained unchanged in HepG2 cells after GA
treatment. However, treatment with GA resulted in a dose-dependent
decrease of p-c-Met expression in HepG2 cells. The binding of HGF
to its corresponding RTK c-Met is necessary for c-Met
phosphorylation and triggering of downstream events. To further
confirm that GA-prevented c-Met phosphorylation is mediated by a
reduction in HGF production, the expression of HGF after GA
intervention was detected. As revealed in Fig. 4B, the RT-qPCR results demonstrated
that the mRNA expression of HGF was suppressed by GA treatment.
This was further confirmed by immunofluorescence against HGF
(Fig. 4C). These findings indicated
that GA suppressed the activity of HGF/c-Met in HepG2 cells via
reduction of HGF production.
Exogenous HGF supplementation improves
GA-impaired cell invasion ability
To further confirm that the GA-mediated
migratory/invasive response in HepG2 cells was due to HGF
suppression, recombinant HGF was used to treat HepG2 cells. As
revealed in Fig. 5A, consistent
with the previous data, 50 µM GA treatment resulted in reduced
expression of p-c-Met, HGF, MMP-2, MMP-9, vimentin and N-cadherin
and increased expression of E-cadherin and ZO-1 in HepG2 cells.
However, exogenous HGF (5 ng/ml) supplementation reversed these
effects that were mediated by GA. The inhibited expression of
p-c-Met, MMP-2, MMP-9, vimentin and N-cadherin by GA was improved
in the presence of HGF. Furthermore, the enhanced expression of
E-cadherin and ZO-1 by GA was markedly suppressed by HGF
supplementation. In addition, the Matrigel invasion assay results
revealed that exogenous HGF supplementation restored the inhibitory
effect of GA on the invasiveness of HepG2 cells (Fig. 5B). These results indicated that
reduced HGF expression was responsible for the GA-suppressed
invasion response in HepG2 cells.
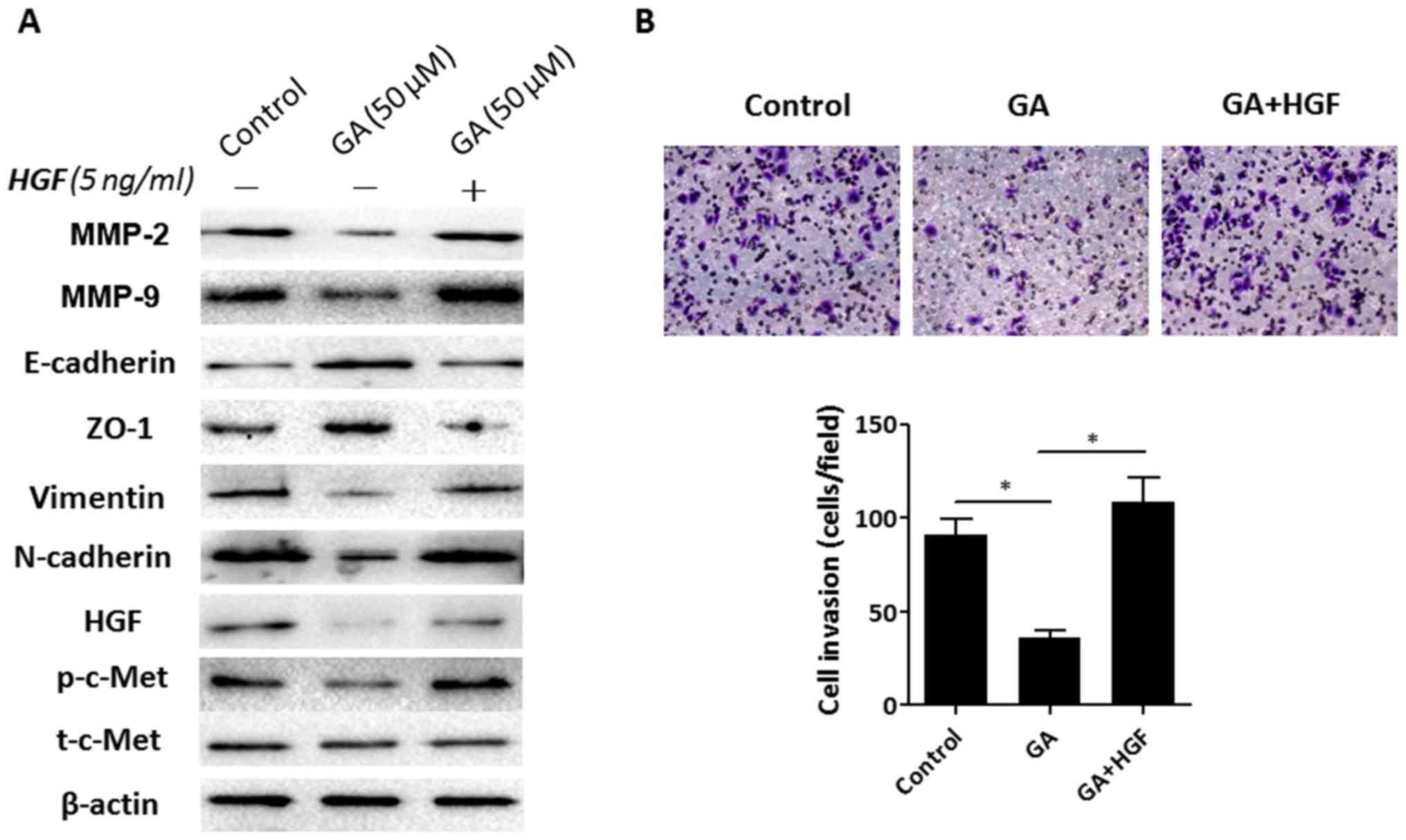 | Figure 5.Recombinant HGF improves
GA-suppressed invasion and EMT changes in HepG2 cells. (A) HepG2
cells were treated with 50 µM GA or GA plus 5 ng/ml HGF for 48 h,
then the protein expression levels of invasion-related molecules
(MMP-2 and MMP-9), HGF, t-c-Met, p-c-Met and EMT markers
(E-cadherin, ZO-1, N-cadherin and vimentin) were assessed using
western blot assays. (B) Matrigel invasion assays were conducted in
order to detect the invasion ability of HepG2 cells after 50 µM GA
or GA plus 5 ng/ml HGF intervention. Images are representative of
three independent experiments (magnification, ×200). *P<0.05 vs.
the control group (0 µM GA). HGF, hepatocyte growth factor; GA,
ginkgolic acid; EMT, epithelial to mesenchymal transition. |
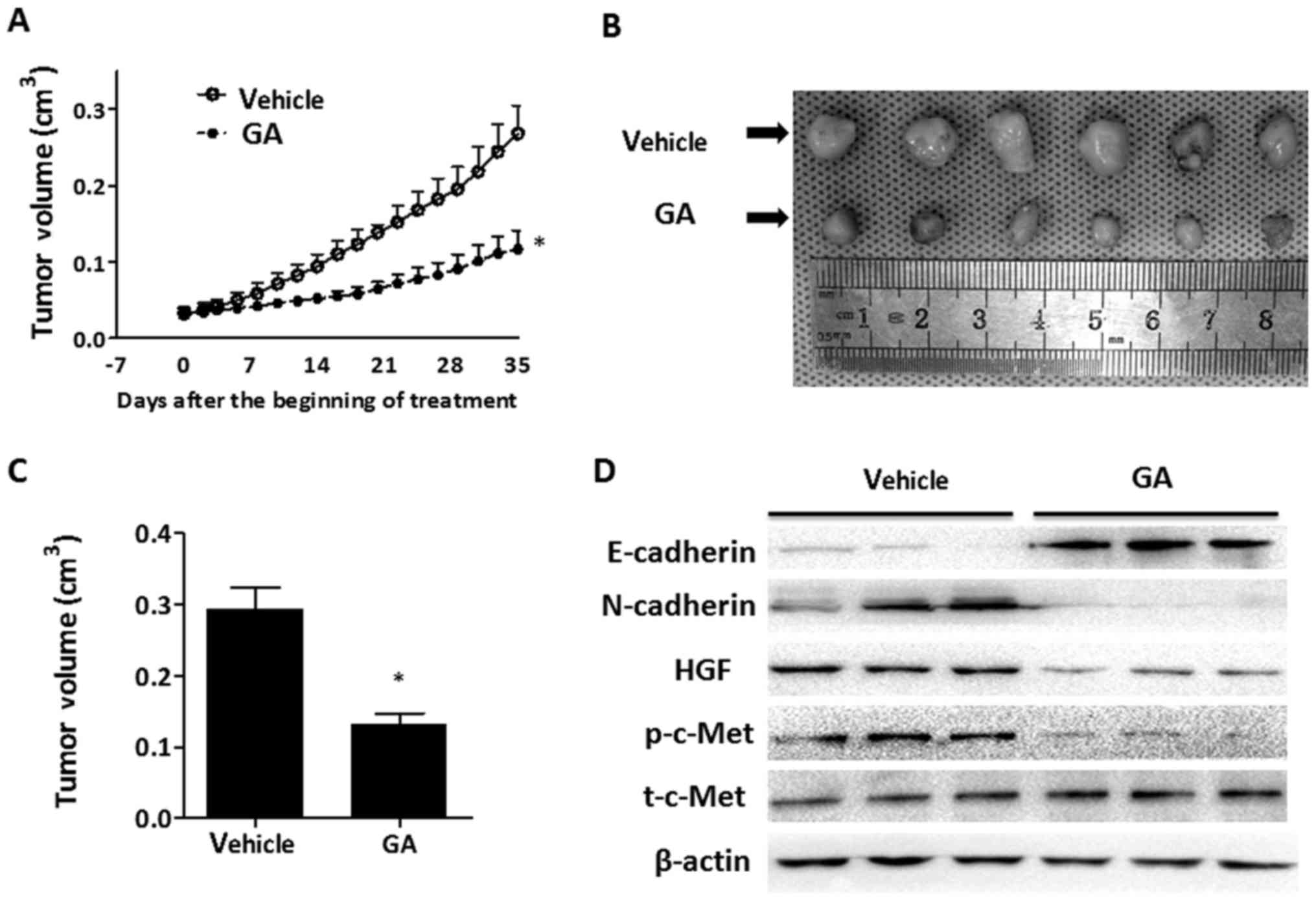
GA suppresses tumor growth in
vivo
Animal experiments were conducted in order to assess
the effect of GA on the growth of HepG2 cells in vivo. A
single-cell suspension (30 µl) of HepG2 cells (containing
1×106 cells) was inoculated subcutaneously into the back
of BALb/c nude mice. One week after inoculation, mice were randomly
divided into two cohorts, one of which received the vehicle and the
other was orally administrated with GA. Through monitoring the
tumor volume, tumor growth was found to be significantly suppressed
by GA intervention as compared to the mice in the vehicle group
(Fig. 6A). The volumes of the
tumors from the GA-treated mice were reduced by >50% compared
with the control mice at the end of the experiment (Fig. 6B and C). Western blotting was
performed in order to detect the expression of E-cadherin,
N-cadherin and p-c-Met in the tumor tissue. As shown in Fig. 6D, treatment with GA decreased the
expression of HGF and the phosphorylation level of c-Met. Levels of
E-cadherin were markedly increased, while levels of N-cadherin were
markedly decreased in tumor tissue from the GA-treated mice. We did
not observe any metastases both in the vehicle and GA-treated group
at the end of the experiment. These in vivo findings
demonstrated that GA effectively suppressed tumor growth, and
inhibition of the activation of c-Met in tumor cells and prevention
of EMT may be the underlying mechanism of GA.
Discussion
Liver cancer is one of the most devastating cancers,
with the majority of patients being diagnosed at late stages where
intrahepatic or extrahepatic metastasis is present and curative
surgical treatments are not possible. Tumor relapse and metastasis
remain the main obstacles for long-term survival even after
curative partial hepatic resection or orthotopic liver
transplantation (22,23). Thus, inhibition of cancer invasion
and metastasis may be of great clinical significance in improving
the survival of patients with liver cancer. The present study
revealed that GA suppressed migration, invasion and EMT-related
gene expression of HepG2 cells, and targeting of HGF/c-Met
signaling may be the underlying mechanism.
Aberrant HGF/c-Met signaling has been implicated in
the acquisition of an aggressive phenotype with metastatic
potential in several types of malignancy by promoting tumor cell
migration, invasion, EMT and angiogenesis (9,24).
Upon binding with its high-affinity ligand HGF, c-Met undergoes
dimerization, auto-phosphorylation of its tyrosine residues and
formation of the multifunctional docking site for adaptor protein
binding, resulting in activation of downstream signal transduction
pathways (25). The role of the
HGF/c-Met axis in liver cancer has been systematically reviewed in
the past, and pharmacological c-Met inhibition is a promising
therapeutic strategy for liver cancer (10,19).
It has been reported that c-Met activation is significantly
associated with vascular invasion, neoangiogenesis and poor
outcomes in liver cancer (26). In
addition, overexpression of c-Met in liver cancer tissue has been
revealed to be correlated with early tumor recurrence or metastasis
after hepatectomy (27). Tivantinib
(ARQ 197), a selective oral inhibitor of c-Met, has exhibited
promising anticancer activity both in vitro and in
vivo (28). Results from a
phase II trial (29) revealed that
tivantinib statistically significantly improved the time to
progression and overall survival versus a placebo among patients
with unresectable liver cancer, indicating that inhibition of the
c-Met pathway by tivantinib may provide an effective and safe
second-line option for patients with advanced liver cancer,
particularly for those with c-Met overexpression. Collectively,
these results indicated that targeting c-Met may serve as a
promising therapeutic strategy for the treatment of liver
cancer.
EMT is a process by which epithelial cells lose
their cell polarity and intercellular adhesion, and acquire
mesenchymal features with migratory and invasive properties
(30). EMT has been revealed to
play critical roles in embryonic development, wound healing, tissue
regeneration, organ fibrosis and malignant transformation (31). Previous studies indicated that the
EMT process exerted pleiotropic functions in cancer progression
(31,32). Cancer cells undergoing EMT are
endowed with enhanced migratory, invasive and metastatic
properties. In addition, there is some evidence that EMT is
associated with chemoresistance and immunosuppressive tumor
microenvironment formation in cancer (33,34).
Loss of the epithelial adhesion protein E-cadherin and gain of
mesenchymal markers such as N-cadherin and/or vimentin are a
hallmark of EMT (35). HGF has been
shown to be an effective inducer of EMT in liver cancer (36). The present study revealed that the
expression of E-cadherin and ZO-1 was upregulated, while that of
N-cadherin and vimentin was downregulated in HepG2 cells after GA
intervention, accompanied by weakened invasion and migration
properties. However, these changes were reversed by exogenous HGF
supplementation.
The anticancer function of GA has attracted
interest. The inhibitory effects of GA on cancer cells have been
verified by a series of studies (15,17,18).
It was reported that GA can serve as a safe and potent anticancer
agent against pancreatic cancer by inducing AMPK activation and
inhibiting the signaling pathway and genes involved in lipogenesis
(15). In addition, GA inhibited
the migration of breast cancer cells without causing cytotoxicity
to the non-cancerous cell line (18). In a recently published study, the
anticancer effect of GA in colon cancer has been studied and both
in vitro and in vivo data revealed that GA suppressed
the proliferation, migration and invasion of colon cancer cells
without toxicity (16). Induction
of AMPK activation and inhibition of the expression of
invasion-associated proteins was found to be responsible for
GA-suppressed proliferation, migration and invasion of colon cancer
cells (17). In the present study,
we mainly focused on the effect of GA on the invasion ability and
EMT changes of HepG2 cells, and our results indicated that GA could
suppress the invasion ability and prevented EMT progression of
HepG2 cells efficiently. These results were consistent with
previous research on other cancer types (15,37).
In addition, reduction of the HGF/c-Met axis activity may be the
underlying mechanism. In this study, we did not investigate the
effect of GA on the apoptosis of HepG2 cells. However, a previous
study has shown that GA can induce apoptosis of human cancer cells
by decreasing the Bcl-2/Bax ratio (38). In conclusion, these results
indicated that GA may serve as a promising agent for liver cancer
treatment. Whether GA has a promoting effect on liver cancer cell
apoptosis and the underlying mechanisms warrants further study.
Acknowledgements
We would like to thank Spandidos Publications
English Language Editing Service for English language editing.
Funding
The present study was supported by the Natural
Science Basic Research Project of Shaanxi Province (grant no.
2012JQ4016).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Author's contributions
HL and YL conceived and designed the study; XM, DZ
and XX acquired the data. DZ and SL analyzed and interpreted the
data; XX and HL wrote, reviewed, and revised the manuscript; XM and
XX provided administrative, technical and material support; YL
supervised the study. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The experimental protocols were authorized by the
Ethics Committee of The Second Affiliated Hospital of Medical
College, Xi'an Jiaotong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Forner A, Reig M and Bruix J:
Hepatocellular carcinoma. Lancet. 391:1301–1314. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Birchmeier C and Gherardi E: Developmental
roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends
Cell Biol. 8:404–410. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huh CG, Factor VM, Sánchez A, Uchida K,
Conner EA and Thorgeirsson SS: Hepatocyte growth factor/c-met
signaling pathway is required for efficient liver regeneration and
repair. Proc Natl Acad Sci USA. 101:4477–4482. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Suzuki K, Hayashi N, Yamada Y, Yoshihara
H, Miyamoto Y, Ito Y, Ito T, Katayama K, Sasaki Y, Ito A, et al:
Expression of the c-met protooncogene in human hepatocellular
carcinoma. Hepatology. 20:1231–1236. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kiss A, Wang NJ, Xie JP and Thorgeirsson
SS: Analysis of transforming growth factor (TGF)-alpha/epidermal
growth factor receptor, hepatocyte growth Factor/c-met, TGF-beta
receptor type II, and p53 expression in human hepatocellular
carcinomas. Clin Cancer Res. 3:1059–1066. 1997.PubMed/NCBI
|
|
8
|
Suárez-Causado A, Caballero-Díaz D,
Bertrán E, Roncero C, Addante A, García-Álvaro M, Fernández M,
Herrera B, Porras A, Fabregat I and Sánchez A: HGF/c-Met signaling
promotes liver progenitor cell migration and invasion by an
epithelial-mesenchymal transition-independent, phosphatidyl
inositol-3 kinase-dependent pathway in an in vitro model. Biochim
Biophys Acta. 1853:2453–2463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer: Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goyal L, Muzumdar MD and Zhu AX: Targeting
the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res.
19:2310–2318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hua Z, Wu C, Fan G, Tang Z and Cao F: The
antibacterial activity and mechanism of ginkgolic acid C15:1. BMC
Biotechnol. 17:52017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lü JM, Yan S, Jamaluddin S, Weakley SM,
Liang Z, Siwak EB, Yao Q and Chen C: Ginkgolic acid inhibits HIV
protease activity and HIV infection in vitro. Med Sci Monit.
18:BR293–BR298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li R, Shen Y, Zhang X, Ma M, Chen B and
van Beek TA: Efficient purification of ginkgolic acids from Ginkgo
biloba leaves by selective adsorption on Fe3O4 magnetic
nanoparticles. J Nat Prod. 77:571–575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li L, Yao QQ, Xu SY, Hu HH, Shen Q, Tian
Y, Pan LY, Zhou H, Jiang HD, Lu C, et al: Cyclosporin A affects the
bioavailability of ginkgolic acids via inhibition of P-gp and BCRP.
Eur J Pharm Biopharm. 88:759–767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma J, Duan W, Han S, Lei J, Xu Q, Chen X,
Jiang Z, Nan L, Li J, Chen K, et al: Ginkgolic acid suppresses the
development of pancreatic cancer by inhibiting pathways driving
lipogenesis. Oncotarget. 6:20993–21003. 2015.PubMed/NCBI
|
|
16
|
Liu Y, Yang B, Zhang L, Cong X, Liu Z, Hu
Y, Zhang J and Hu H: Ginkgolic acid induces interplay between
apoptosis and autophagy regulated by ROS generation in colon
cancer. Biochem Biophys Res Commun. 498:246–253. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiao L, Zheng J, Jin X, Wei G, Wang G, Sun
X and Li X: Ginkgolic acid inhibits the invasiveness of colon
cancer cells through AMPK activation. Oncol Lett. 14:5831–5838.
2017.PubMed/NCBI
|
|
18
|
Hamdoun S and Efferth T: Ginkgolic acids
inhibit migration in breast cancer cells by inhibition of NEMO
sumoylation and NF-κB activity. Oncotarget. 8:35103–35115. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Venepalli NK and Goff L: Targeting the
HGF-cMET axis in hepatocellular carcinoma. Int J Hepatol.
2013:3416362013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Illam SP, Narayanankutty A, Mathew SE,
Valsalakumari R, Jacob RM and Raghavamenon AC: Epithelial
mesenchymal transition in cancer progression: Prev entive
phytochemicals. Recent Pat Anticancer Drug Discov. 12:234–246.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cha C, Fong Y, Jarnagin WR, Blumgart LH
and DeMatteo RP: Predictors and patterns of recurrence after
resection of hepatocellular carcinoma. J Am Coll Surg. 197:753–758.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martínez Ares D, Suárez López FJ, Souto
Ruzo J, Otero Ferreiro A, Gómez Gutiérrez M, González Conde B,
Fernández Sellés C, Gala López B, Arnal Monreal F and Vázquez
Iglesias JL: Liver transplantation in patients with hepatocellular
carcinoma: Factors implicated in tumor relapse. Rev Esp Enferm Dig.
96:22–31. 2004.(In English, Spanish). PubMed/NCBI
|
|
24
|
Yap TA, Sandhu SK, Alam SM and de Bono JS:
HGF/c-MET targeted therapeutics: Novel strategies for cancer
medicine. Curr Drug Targets. 12:2045–2058. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Comoglio PM, Giordano S and Trusolino L:
Drug development of MET inhibitors: Targeting oncogene addiction
and expedience. Nat Rev Drug Discov. 7:504–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaposi-Novak P, Lee JS, Gòmez-Quiroz L,
Coulouarn C, Factor VM and Thorgeirsson SS: Met-regulated
expression signature defines a subset of human hepatocellular
carcinomas with poor prognosis and aggressive phenotype. J Clin
Invest. 116:1582–1595. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu FS, Zheng SS, Wu LJ, Ding W, Ma ZM,
Wang ZM, Teng LS and Zhao WH: Study on the prognostic value of
hepatocyte growth factor and c-met for patients with hepatocellular
carcinoma. Zhonghua Wai Ke Za Zhi. 44:603–608. 2006.(In Chinese).
PubMed/NCBI
|
|
28
|
Munshi N, Jeay S, Li Y, Chen CR, France
DS, Ashwell MA, Hill J, Moussa MM, Leggett DS and Li CJ: ARQ 197, a
novel and selective inhibitor of the human c-Met receptor tyrosine
kinase with antitumor activity. Mol Cancer Ther. 9:1544–1553. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Santoro A, Rimassa L, Borbath I, Daniele
B, Salvagni S, Van Laethem JL, Van Vlierberghe H, Trojan J, Kolligs
FT, Weiss A, et al: Tivantinib for second-line treatment of
advanced hepatocellular carcinoma: A randomised, placebo-controlled
phase 2 study. Lancet Oncol. 14:55–63. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
The importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia
XF, Sun X, Li GG, Hu QD, Fu QH and Liang TB: Hypoxia-induced
epithelial-to-mesenchymal transition in hepatocellular carcinoma
induces an immunosuppressive tumor microenvironment to promote
metastasis. Cancer Res. 76:818–830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ogunwobi OO and Liu C: Hepatocyte growth
factor upregulation promotes carcinogenesis and
epithelial-mesenchymal transition in hepatocellular carcinoma via
Akt and COX-2 pathways. Clin Exp Metastasis. 28:721–731. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baek SH, Ko JH, Lee JH, Kim C, Lee H, Nam
D, Lee J, Lee SG, Yang WM, Um JY, et al: Ginkgolic acid inhibits
invasion and migration and TGF-β-induced EMT of lung cancer cells
through PI3K/Akt/mTOR inactivation. J Cell Physiol. 232:346–354.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou C, Li X, Du W, Feng Y, Kong X, Li Y,
Xiao L and Zhang P: Antitumor effects of ginkgolic acid in human
cancer cell occur via cell cycle arrest and decrease the Bcl-2/Bax
ratio to induce apoptosis. Chemotherapy. 56:393–402. 2010.
View Article : Google Scholar : PubMed/NCBI
|