Introduction
Traditional anticancer therapies target both cancer
and normal cells indiscriminately, and are often ineffective in the
treatment of relapsed tumors. Consequently, a major aim of cancer
drug development is to target cancer-specific genetic alterations
or signaling pathways. This strategy has lead to the identification
and development of some highly effective and less toxic drugs,
including small molecules and antibodies (1). However, such targeted drugs are
limited by the diversity of cancer genotypes and intratumor
heterogeneity, as well as the rapid development of drug resistance
(2,3). Thus, there is still an ongoing and
urgent need for the design of novel agents that can effectively
target cancer cells with broad-spectrum efficacy, while leaving
normal cells less affected. To this end, targeting cancer-specific
biochemical pathways that are crucial for the survival of malignant
cells and dispensable for normal cells are likely to be some of the
most promising anticancer strategies (4–6).
Increased production of reactive oxygen species
(ROS) is a distinct feature of various types of cancer (7). Intracellular ROS have been linked to a
myriad of physiological activities and pathological changes.
Although ROS can activate signaling pathways that regulate normal
and cancer cellular proliferation and growth, elevated ROS levels
denote oxidative stress that can damage cellular components and
jeopardize cell survival (8). In
order to survive, cancer cells rewire biochemical pathways to
antagonize the toxicity of increased ROS and enhance the function
of repair systems to withstand the resultant oxidative damage
(9). However, adaptation to the
intense oxidative pressure by cancer cells creates cancer-specific
conditions that are vulnerable to disruptions of the rewired
biochemical pathways (10). Thus,
further ROS insult, reductions in the antioxidants levels, and/or
blockage of damage repair pathways during the malignant
transformation of cancer genotypes may result in artificial
lethality and selective killing of cancer cells (4).
Icaritin is a natural compound derived from the
medicinal plants of the Epimedium family (11). Crude extracts of Epimedium
plants have been used in traditional Chinese herbal formulations to
treat bone, neural, cardiovascular and immune malfunctions and/or
are administered as aphrodisiacs for their sexual
performance-enhancing properties (12). The major bioactive herbal ingredient
of Epimedium plants has been identified to be icariin,
together with smaller amounts of icaritin, desmethylicaritin,
icariside I and icariside II (13).
In addition to the natural constituents of Epimedium plants,
icaritin, desmethylicaritin, icariside I and icariside II are also
generated from icariin through deglycosylation and demethylation by
intestinal microflora (13). These
Epimedium prenylflavonoids are structurally similar and
functionally related to estrogen and are, hence, called
phytoestrogens (14).
Depending on the working compound concentration and
cellular context, icaritin has demonstrated both agonistic and
antagonistic activities towards the various types of estrogen
receptors (ERs). By acting as an agonist of the canonical ERs (ERα
and ERβ), icaritin promotes repair of bone and cardiovascular
damage by inducing osteogenic and cardiomyogenic differentiation
(12,15). Similarly, icaritin stimulates
mammary epithelial cell proliferation (14) and stem cell self-renewal (16), while it inhibits neuronal apoptosis
and hence acts in a neuroprotective manner in certain
neurodegenerative models (17). In
addition to the canonical ERs, icaritin may also activate the
membrane-bound G-protein ER 1 to promote proliferation of some
ER-negative breast cancers (18).
However, most ER-negative breast cancers, as well as some BCR,
RhoGEF and GTPase activating protein (BCR)-ABL proto-oncogene 1,
non-receptor tyrosine kinase (ABL)+ leukemic cells,
overexpress the ERα variant ERα-36, and are therefore suppressed by
icaritin, whose action blocks ERα-36-mediated epidermal growth
factor receptor-Src-extracellular signal-regulated kinase (ERK)
and/or BCR-ABL-mediated growth factor receptor-bound protein 2-Ras
signaling (19–23). Moreover, icaritin binds to the aryl
hydrocarbon receptor in order to promote degradation of ERα and/or
androgen receptor (AR); whereas, it further suppresses ERα-positive
breast cancer and AR-positive prostate cancer (24,25).
In addition to the phytoestrogen-associated
cytotoxicity against breast and prostate cancer, icaritin has also
demonstrated potent toxicity against broader types of cancer, which
is independent of the expression of ER and AR (11,26).
The majority of the studies indicated that icaritin induces cell
cycle arrest and apoptosis or autophagic cell death in various
types of cancer, by distinct mechanisms of action, including
suppression of interleukin (IL)-6/Janus kinase 2 (Jak2)/signal
transducer and activator of transcription 3 (STAT3) and/or
mitogen-activated protein kinase (MAPK) signaling (27–30),
sustained activation of ERK1/2 or c-Jun N-terminal kinase (JNK1)
(26,31,32),
inhibition of phosphatidylinositol 3-kinase (PI3K)/RAC-α
serine/threonine-protein kinase (Akt) pathway (33) and 5′-AMP-activated protein kinase
(AMPK)-dependent inhibition of serine/threonine-protein kinase mTOR
(34). However, the molecular
mechanisms that link icaritin to these signaling pathways remain
undiscovered. Icaritin has been shown to stimulate ROS generation
in certain types of cells (34–38).
However, it is not known whether ROS play a role in the anticancer
toxicity of icaritin. Although, cervical cancer is among the top 10
cancers in incidence and mortality globally (39), the effect of icaritin on cervical
cancer has not been examined. In the present study, it was
demonstrated that icaritin treatment caused a rapid increase in ROS
in the human HeLa and SiHa cervical cancer cell lines, which
subsequently resulted in extensive oxidative DNA damage and large
numbers of DNA breaks, and eventually caused activation of the
intrinsic apoptosis pathway. These results suggest that icaritin
can cause cancer cell death via the induction of the DNA damage
response (DDR)-triggered cell death. Thus, icaritin may be an
optimal drug candidate for the treatment of cervical cancer.
Materials and methods
Cells and reagents
The human HeLa and SiHa cervical cancer cell lines,
and the non-cancerous 293 and CCD-1095Sk cell lines were purchased
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). The cells were cultured in 37°C with 5% CO2
according to the instructions provided by ATCC. Icaritin was
purchased from Yuanye Biotechnology (Shanghai, China). The purity
was measured by high-performance liquid chromatography (15) and determined to be 99.6%. Stock
solutions of icaritin were prepared in 100% dimethyl sulfoxide
(DMSO; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and working
solutions were in complete cell culture medium. Vehicle control
samples included the same amount of DMSO in the absence of
icaritin. N-acetyl cysteine (NAC) was purchased from Calbiochem
(EMD Millipore, Billerica, MA, USA). The sources of additional
reagents were specified in the relevant sections.
MTT assay
The cells were seeded in 96-well plates at a density
of 2,000 cells per well for 12 h, and then treated with icaritin or
vehicle control for 24 or 48 h. A total of 20 µl MTT (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) solution (5 mg/ml
in PBS) was added to each well. The plate was incubated at 37°C for
an additional 4 h. Following removal of the culture medium, the
cells were incubated in 150 µl 100% DMSO on a plate shaker for 10
min. The absorbance was measured at 595 nm was measured by a
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
The data were analyzed using the GraphPad Prism software (GraphPad
Software, Inc., La Jolla, CA, USA).
Colony formation assay
The cells were seeded in 6-well plates at 2,000
cells/well for 12 h and then treated with different concentrations
of icaritin or vehicle control for 8 days. Following washing with
PBS, the cells were fixed in ice-cold methanol and briefly stained
with crystal violet solution (0.5% in 25% methanol; Sigma-Aldrich;
Merck KGaA). The plates were air-dried overnight and photographs
were captured with a Canon digital camera (PowerShot ELPH 180
20-Megapixel; Canon, Inc., Tokyo, Japan). Ethanol (70%) was added
to dissolve the violet crystals and the absorbance at 595 nm was
measured by a microplate reader (Bio-Rad Laboratories, Inc.). The
data were analyzed using the GraphPad Prism software.
Wound healing assay
The cells were seeded in 12-well plates at a density
of 3×105 cells/well until they reached 70–80% confluence
as a monolayer. A vertical straight scratch across the center of
each well was made by gently and slowly scratching the monolayer
with a 1 ml pipette tip in one direction. The wells were washed
twice with medium to remove the detached cells and fresh medium was
added that contained different concentrations of icaritin and/or
vehicle control. phase-contrast microscopy images were captured at
0, 24 and 48 h and analyzed using the ImageJ software (National
Institutes of Health, Bethesda, MA, USA).
ROS measurement
Total cellular ROS were measured by flow cytometry
using a cell-based ROS assay kit (Beyotime Institute of
Biotechnology, Haimen, China). Following drug treatment, the cells
were washed twice with PBS and subsequently incubated with 10 µM
dichlorofluorescin diacetate (DCFH-DA) at 37°C for 30 min in the
dark. The cells were collected following trypsin digestion (Thermo
Fisher Scientific, Inc.) and then analyzed by the FACSCaliber flow
cytometer (BD Biosciences, San Jose, CA, USA). Total cellular ROS
levels were expressed as the averaged DCF fluorescence intensity of
the cells. The data presented is average of three independent
experiments.
Immunofluorescent staining
Following drug treatment, the cells were washed once
by PBS, fixed in methanol/acetone (v/v, 1/1) at 4°C for 15 min, and
washed 3 times with PBS. Fixed cells were blocked in 0.3% Triton
X-100 with 1.5% bovine serum albumin (BSA) at room temperature for
30 min, incubated with rabbit-anti-TP53-binding protein 1 (53BP1;
1:1,000; cat. no. A300-272A; Bethyl Laboratories, Inc., Montgomery,
TX, USA) and/or rabbit-anti-γH2AX histone (1:500; cat. no.
bs-3185R; BIOSS, Beijing, China) primary antibodies at 4°C
overnight, followed by incubation with Cy3-conjugated
goat-anti-rabbit (1:250; cat. no. 111-165-003; Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) secondary
antibody for 1 h at room temperature. The slides were washed three
times with PBS and sealed in VECTASHIELD Mounting Medium with DAPI
(Vector Laboratories, Inc., Burlingame, CA, USA).
Comet assay
The comet assay was performed using the OxiSelect
Comet assay kit (Cell Biolabs, Inc., San Diego, CA, USA) following
the manufacturer's instructions (40). The cells were collected by trypsin
digestion, washed twice with PBS, and then resuspended in 1.2%
low-melting point agarose maintained at 37°C. The density of the
cells used was 1×105 cells/ml. A total of 80 µl of
cell-containing agarose was layered on a frosted glass slide. The
slide was transferred to 4°C for 15 min and submerged in pre-cooled
lysis buffer (100 mM EDTA, 2.5 M NaCl, 10 mM Tris-HCl, 1% Triton
X-100 and 10% DMSO, pH 10.0) at 4°C for 1.5 h in the dark. The
slides were washed twice in enzyme buffer (40 mM HEPES, 0.1 M KCl,
0.5 mM EDTA and 0.2 mg/ml BSA, pH 8.0) and then incubated in enzyme
buffer with or without 8-oxoguanine glycosylase (OGG1; 1.0 µg/ml;
OriGene Technologies, Inc., Rockville, MD, USA) at 37°C for 45 min.
Following brief washing with enzyme buffer, the slides were
denatured in pre-cooled alkaline buffer (300 mM NaOH, 1 mM EDTA) in
an electrophoresis chamber for 30 min and then electrophoresis was
performed under 300 mA and 50 V for 30 min. The slides were
incubated in cooled neutralizing buffer (250 mM Tris-HCl, pH 7.5)
for 30 min, immersed in cold 70% ethanol for 5 min and then dried
in air. The cells on the slides were stained with Vista Green DNA
dye at room temperature for 15 min in the dark. The images were
captured using an Olympus fluorescent microscope (Olympus
Corporation, Tokyo, Japan). The tail moment was defined as
percentage of tail DNA × tail length, quantified using the CASP
software (Perceptive Instruments, Ltd., Edmunds, UK).
Western blot analysis
The cells were washed twice with PBS and removed
from the plate using a scraper and 100 µl radioimmunoprecipitation
buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate,
0.1% sodium dodecyl sulfate and 50 mM Tris, pH 8.0) containing 1 mM
phenylmethane sulfo-nylfluoride (Santa Cruz Technology, Inc.,
Dallas, TX, USA).
Following centrifugation at 12,000 × g for 20 min at
4°C, the protein concentration levels in the supernatants were
measured using a bicinchoninic acid kit (Beijing Dingguo Changsheng
Biotechnology Co., Ltd., Beijing, China). The samples were
denatured at 95°C for 10 min, separated on a 12% SDS-PAGE gel and
transferred to a polyvinylidene difluoride membrane (EMD
Millipore). The membranes were blocked in Tris-buffered saline with
Tween-20 [10 mM Tris (pH 7.5), 100 mM NaCl, 0.1% Tween-20; PBST)
containing 5% (w/v) nonfat milk for 2 h at room temperature,
followed by incubation with primary antibodies overnight at 4°C
[B-cell lymphoma 2 (Bcl-2; 1:500; cat. no. sc-7382) and apoptosis
regulator Bax (Bax; 1:500; cat. no. sc-7480) from Santa Cruz
Biotechnology, Inc.; cleaved caspase 3 (1:500; cat. no. ab2302),
cleaved caspase 9 (1:200; cat. no. ab2324) and X-linked inhibitor
of apoptosis protein (XIAP; 1:1,000; cat. no. ab28151) from Abcam
(Cambridge, UK); cyclin-dependent kinase 1 (CDK1)-pT14 (1:1,000;
cat. no. bs-3091R), cell division cycle 25C (CDC25C)-pS216
(1:1,000; cat. no. bs-3096R), β-actin (1:5,000; cat. no. bs-0061R)
and matrix metalloproteinase 9 (MMP9; 1:1,000; cat. no. bs-4593R)
from BIOSS]. Subsequently, the membranes were incubated with a
horseradish peroxidase-conjugated goat-anti-rabbit (1:10,000; cat.
no. 111-035-003; Jackson ImmunoResearch Laboratories, Inc.)
secondary antibody for 1 h at room temperature. The signals were
visualized with an ECL kit (EasySee Western Blot kit; Beijing
Transgen Biotech Co., Ltd., Beijing, China).
Cell cycle analysis
Following drug treatment, the cells were collected
by trypsin digestion, washed twice with PBS, and fixed in 70%
alcohol at −20°C for 1 h. Fixed cells were resuspended in 300 ml of
freshly prepared PBS with 0.1% Triton X-100, 0.2 mg/ml DNase-free
RNase A (Sigma-Aldrich; Merck KGaA), 10 µg/ml propidium iodide
(Roche Diagnostics, Basel, Switzerland), and incubated at 37°C for
20 min in the dark. The cells were filtered through a Filcon nylon
mesh (BD Biosciences) and analyzed using a FACScalibur flow
cytometer and ModFit software (BD Biosciences).
Apoptosis analysis by flow
cytometry
Following drug treatment, cells were collected by
trypsin digestion, washed twice with PBS and stained in working
solution using an Annexin V-fluorescein isothiocyanate/PI dual
staining kit (BestBio Science, Shanghai, China) according to the
manufacturer's instructions. The stained cells were analyzed by the
FACScalibur flow cytometer and the CellQuest software (BD
Biosciences).
Mitochondrial membrane potential
measurement
The mitochondrial membrane potential was measured
using the JC-1 dye (Beyotime Institute of Biotechnology) according
to the manufacturer's instructions. The cells were washed twice
with PBS, incubated with 1 ml JC-1 working solution for 20 min at
37°C, and sealed in VECTASHIELD medium (Vector Laboratories). Cell
images were imaged with an Olympus fluorescent microscope.
Statistical analysis
The data are expressed as the mean ± standard
deviation and the results were representative of three independent
experiments. All statistical analyses were performed using the
GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA,
USA). The statistical comparisons between two groups were performed
by unpaired two-tailed Student's t-test and multigroup comparisons
were conducted by using two-way ANOVA followed by the Dunnett or
Tukey test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Cytotoxicity of icaritin against HeLa
and SiHa cervical cancer cells
It has been reported that icaritin (Fig. 1A) induces considerable cytotoxicity
against several different types of cancer cells by mechanisms that
are not yet fully understood (26–34).
To examine the effects of icaritin on cervical cancer, the
cytotoxicity of icaritin against human HeLa and SiHa cells was
analyzed. HeLa and SiHa cancer cells were treated with a
concentration range of icaritin for 24, 48 and/or 72 h. The results
of the MTT assay indicated that the proliferation rates of HeLa and
SiHa cancer cells were significantly suppressed by icaritin
(Fig. 1B-D). Following 48 h of
treatment, the half maximal inhibitory concentration
(IC50) values of icaritin for HeLa and SiHa cells were
12.5 and 17 µM, respectively (Fig.
1B). The viabilities of HeLa and SiHa cells were reduced to
47.5 and 60%, respectively, following treatment by 12.5 µM icaritin
for 48 h, while those of the non-cancerous CCD-1095Sk fibroblasts
and H293 epithelial cells were not changed (Fig. 1C). These results indicated that
icaritin was selectively more toxic to cancer cells. Overall, the
inhibition of cancer cell proliferation by icaritin exhibited
optimal dose- and time-dependency (Fig.
1D).
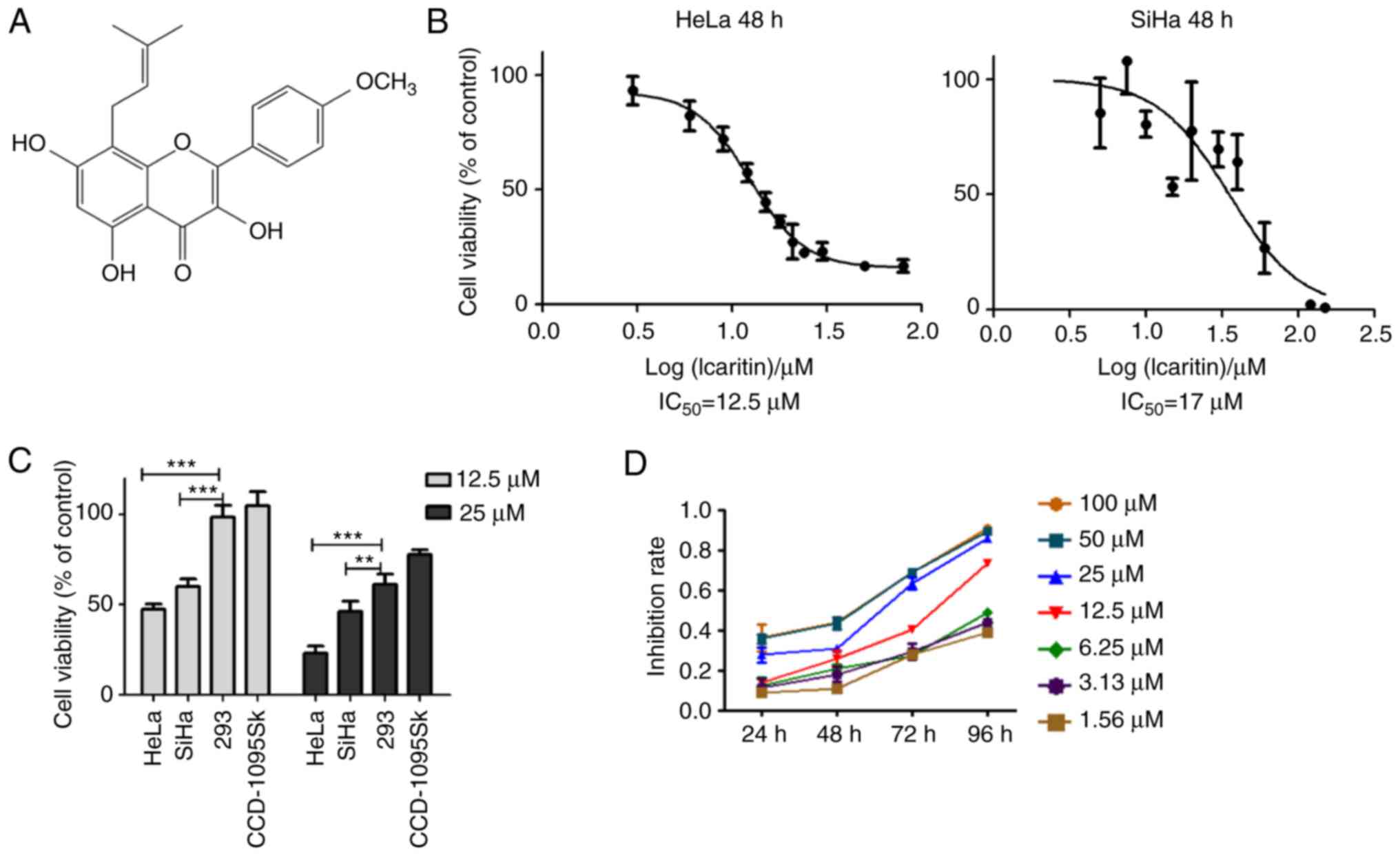 | Figure 1.Cytotoxicity of icaritin against
cervical cancer cells. (A) Chemical structure of icaritin. (B) HeLa
and SiHa cervical cancer cells treated with 0, 3, 6, 9, 12, 15, 18,
21, 24, 30, 50 or 80 µM of icaritin for 48 h and cell viability
determined by MTT assay. The results were analyzed using the
GraphPad Prism software by nonlinear regression (curve fit). (C)
Sensitivity of different types of cells: HeLa and SiHa cancer
cells, and CCD-1095Sk and 293 non-cancerous cells, were treated
with 12.5 µM or 25 µM of icaritin for 48 h and cell viability was
determined by the MTT assay (n=3, mean ± standard deviation;
**P<0.01; ***P<0.001). (D) Time- and dose-dependency;
representative results of HeLa cells. IC50, half maximal
inhibitory concentration. |
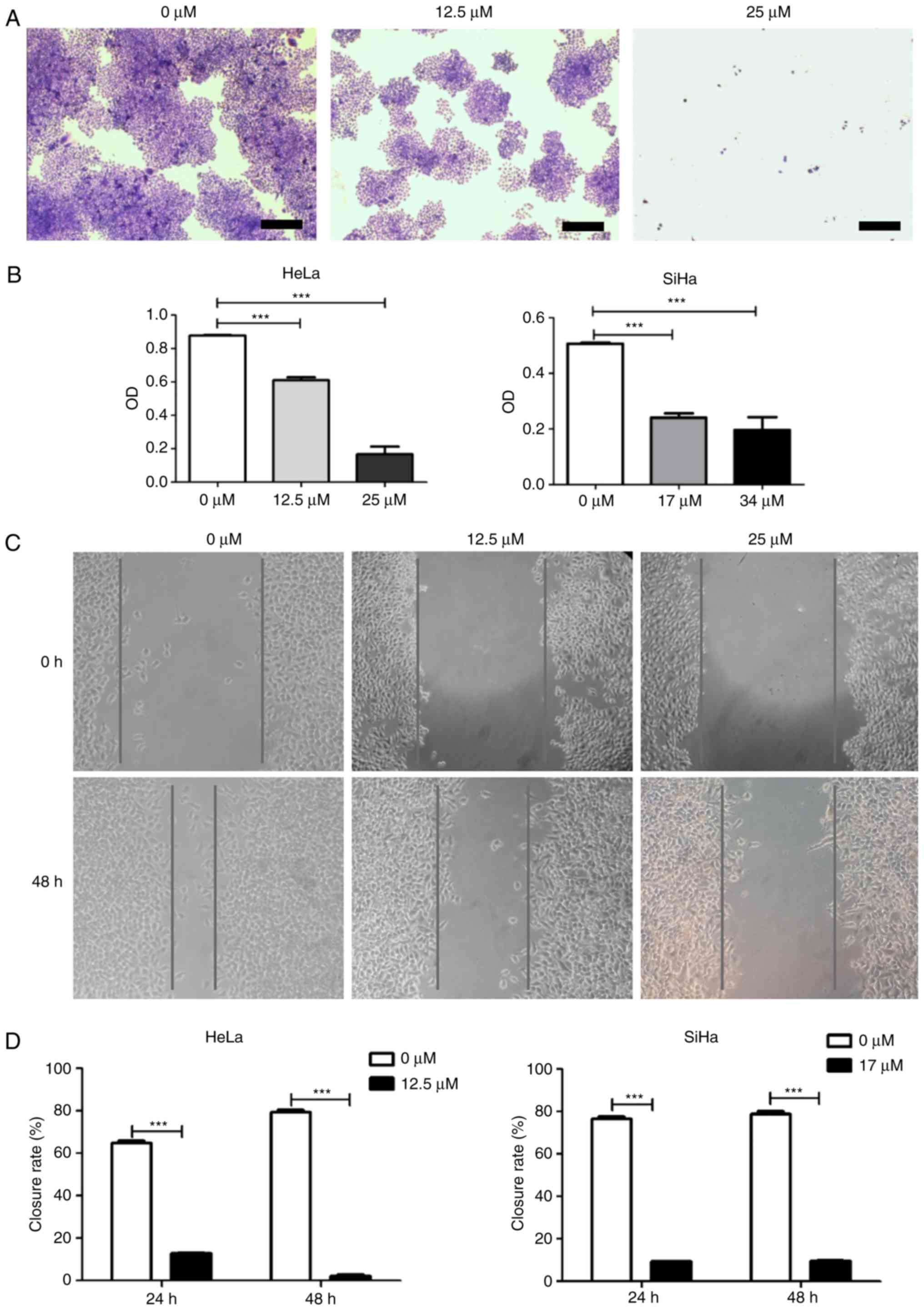
Subsequently, a colony formation assay was used to
confirm the inhibitory activity of icaritin against HeLa and SiHa
cervical cancer cells. After 8 days of treatment with icaritin at
the IC50 determined in the cytotoxicity assay, fewer and
smaller colonies were observed for SiHa and HeLa cells compared
with 0 µM treatment (Fig. 2A and
B). In contrast to this dose treatment, higher concentration
levels of icaritin (equivalent to 2× IC50) resulted in
further inhibition of cellular proliferation (Fig. 2A and B). However, the growth curves
of the non-cancerous CCD-1095Sk fibroblasts and 293 epithelial
cells that were treated with the same concentration points were
much less affected (data not shown). Taken collectively, these
results suggested that icaritin dose-dependently and selectively
suppressed the growth of HeLa and SiHa cervical cancer cells.
In addition to these proliferation and survival
assays, the impact of icaritin on the growth and/or migration of
HeLa and SiHa cervical cancer cells was further analyzed in wound
healing assays. The growth and/or migration of both HeLa and SiHa
cells were significantly decreased in the presence of icaritin
(Fig. 2C and D). Given the effects
revealed by the MTT and colony formation assays above, the
decreased wound healing was likely a result of an inhibitory effect
on proliferation, rather than on migration.
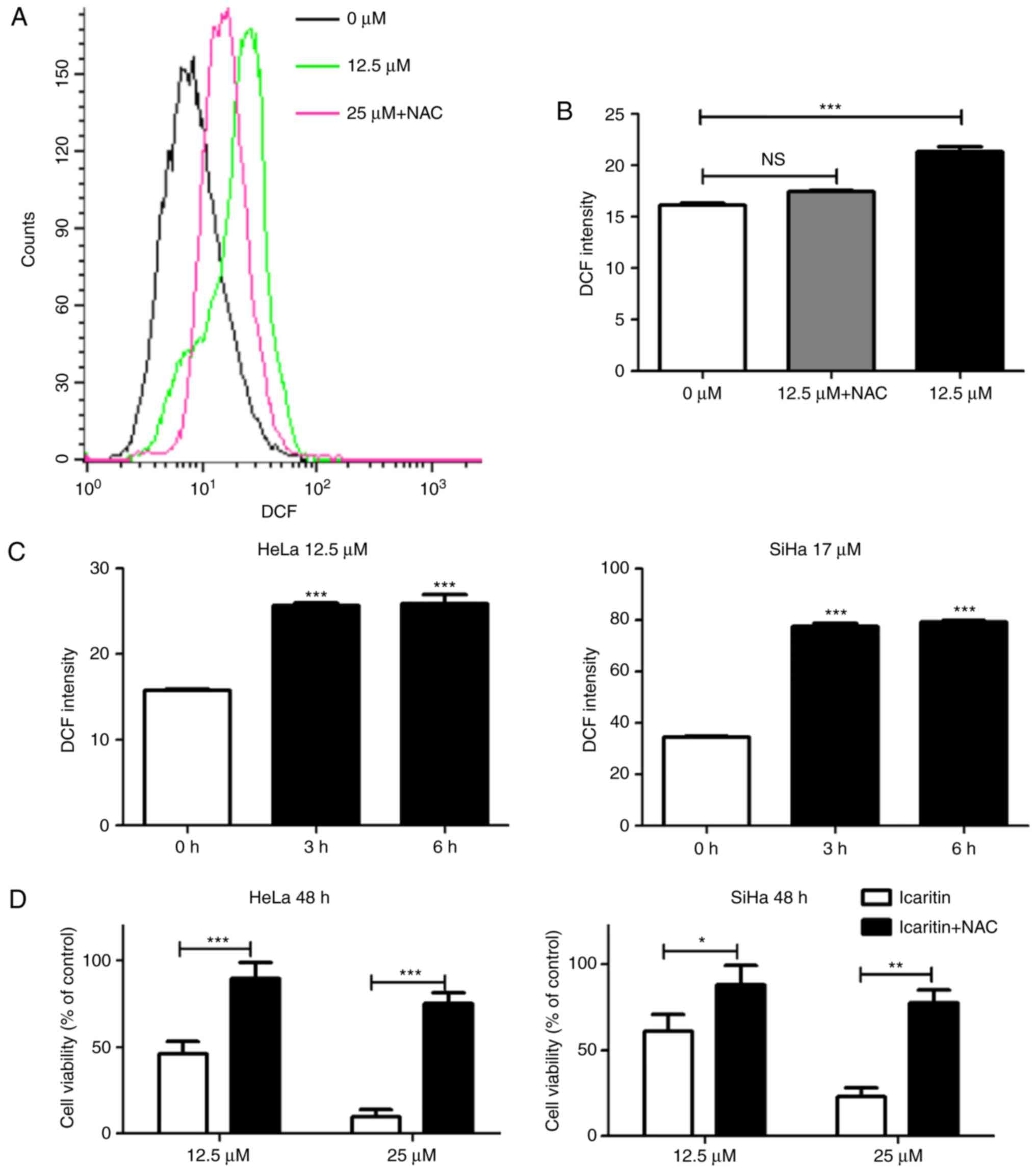
Icaritin increases ROS levels in
cervical cancer cells
Icaritin has been reported to stimulate ROS
generation in specific cell types (34–38).
To determine whether icaritin could induce ROS generation in HeLa
and SiHa cervical cancer cells, the fluorescent probe DCFH-DA was
used to measure total cellular ROS. Treatment with icaritin at
concentrations equivalent to the IC50 values for 12 h
caused a significant increase in ROS levels in HeLa and SiHa cancer
cells, which was blocked by pre-treatment with 5 mM NAC (Fig. 3A and B). A significant increase in
ROS levels was noted as early as 3 h after initiation of icaritin
treatment (Fig. 3C), whereas a
longer treatment did not result in an additional increase in ROS
levels. Notably, the inhibition of icaritin-induced ROS increase by
NAC significantly reduced the cytotoxicity of icaritin against HeLa
and SiHa cells (Fig. 3D). These
results suggested that treatment of cervical cancer cells with
icaritin caused a rapid and robust increase in ROS levels, which
contributed to the suppression of cancer cell proliferation by
icaritin.
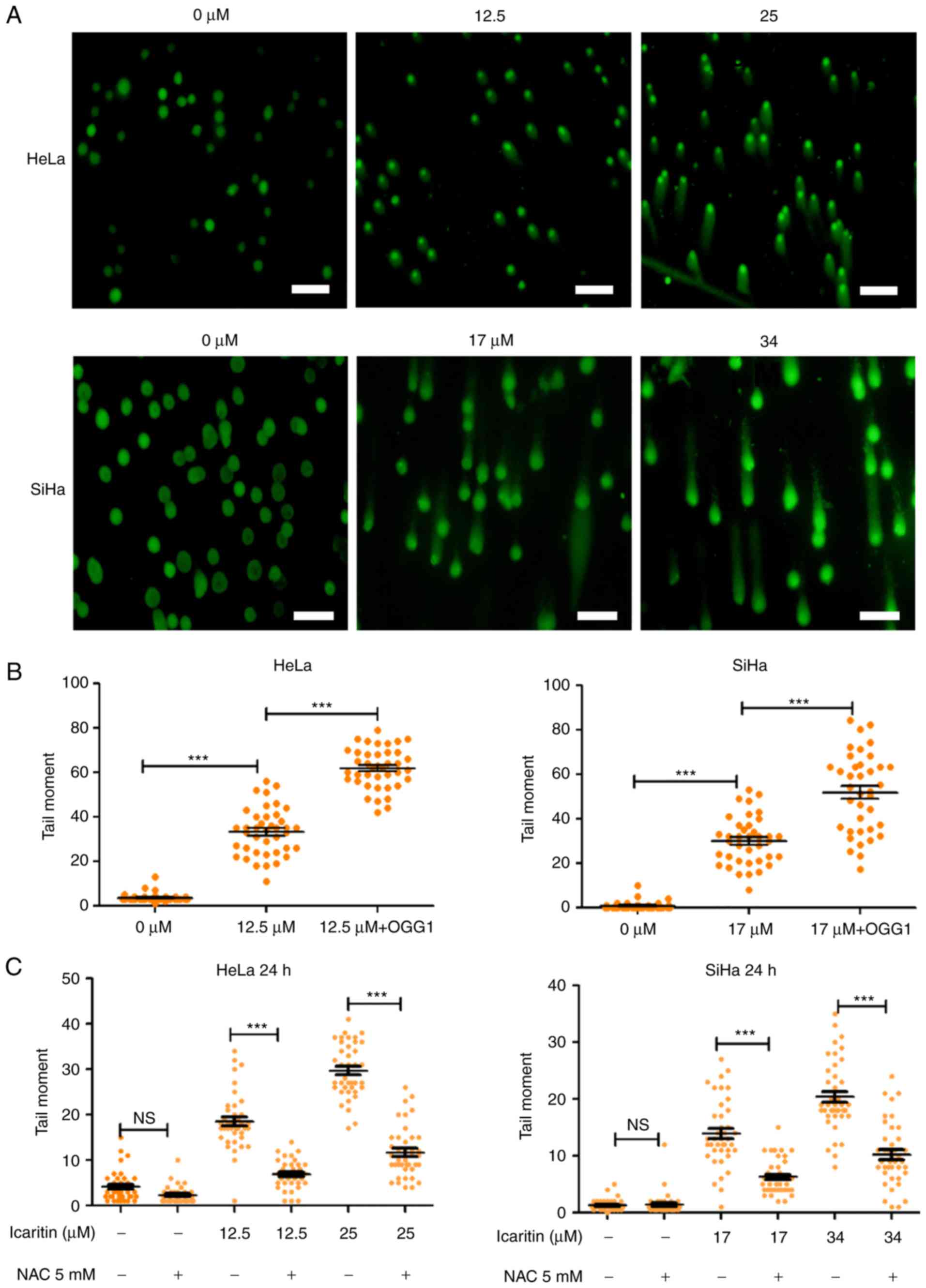
Icaritin-induced ROS overload results
in extensive DNA breaks
Elevated ROS levels results in oxidative stress,
which can damage cellular components and affect cell survival. For
example, repair of ROS-oxidized deoxynucleotides in DNA, such as
8-oxo-2′-deoxyguanosine (8-oxo-dG), may result in accumulation of
repair-associated DNA strand breaks, and subsequent induction of
cell cycle arrest and programmed cell death (41). To gain insight into the
physiological implications of the icaritin-induced ROS overload in
cancer cells, the number of DNA strand breaks prior to and
following treatment with icaritin was compared using the comet
assay (single-cell gel electrophoresis). Alkaline conditions were
used to convert all single-strand DNA breaks (SSB) to double-strand
breaks (DSB), suggesting that the size and intensity of the comet
tail (tail moment) was proportional to the number of total DNA
strand breaks (SSBs + DSBs). The results indicated that after 24 h
incubation of HeLa and SiHa cancer cells with 1× IC50
icaritin (12.5 µM for HeLa, 17 µM for SiHa), the number of cancer
cells with a comet tail, and the size and intensity of the comet
tail (tail moment), were increased (Fig. 4A and B). Treatment with 2×
IC50 of icaritin (25 µM for HeLa, 34 µM for SiHa)
further increased the number of comet tail-containing cells and
their corresponding tail size (Fig.
4A). These effects were blocked by pre-treatment with the ROS
inhibitor NAC, indicating that the DNA strand breaks occurred via
ROS production (Fig. 4C).
OGG1 is a glycosylase that specifically removes the
oxidized guanine base of 8-oxo-dG in DNA, leaving an apurinic site,
which can be converted into a DSB during an alkaline comet assay.
Thus, incubation of the cells with OGG1 prior to electrophoresis
can aid the assessment of the amount of 8-oxo-dG in DNA during the
comet assay. The data indicated that incubation of icaritin-treated
cells with OGG1 further increased the tail moments (Fig. 4B), suggesting that icaritin-induced
ROS overload caused extensive oxidative DNA damage, which
subsequently resulted in large numbers of DNA breaks.
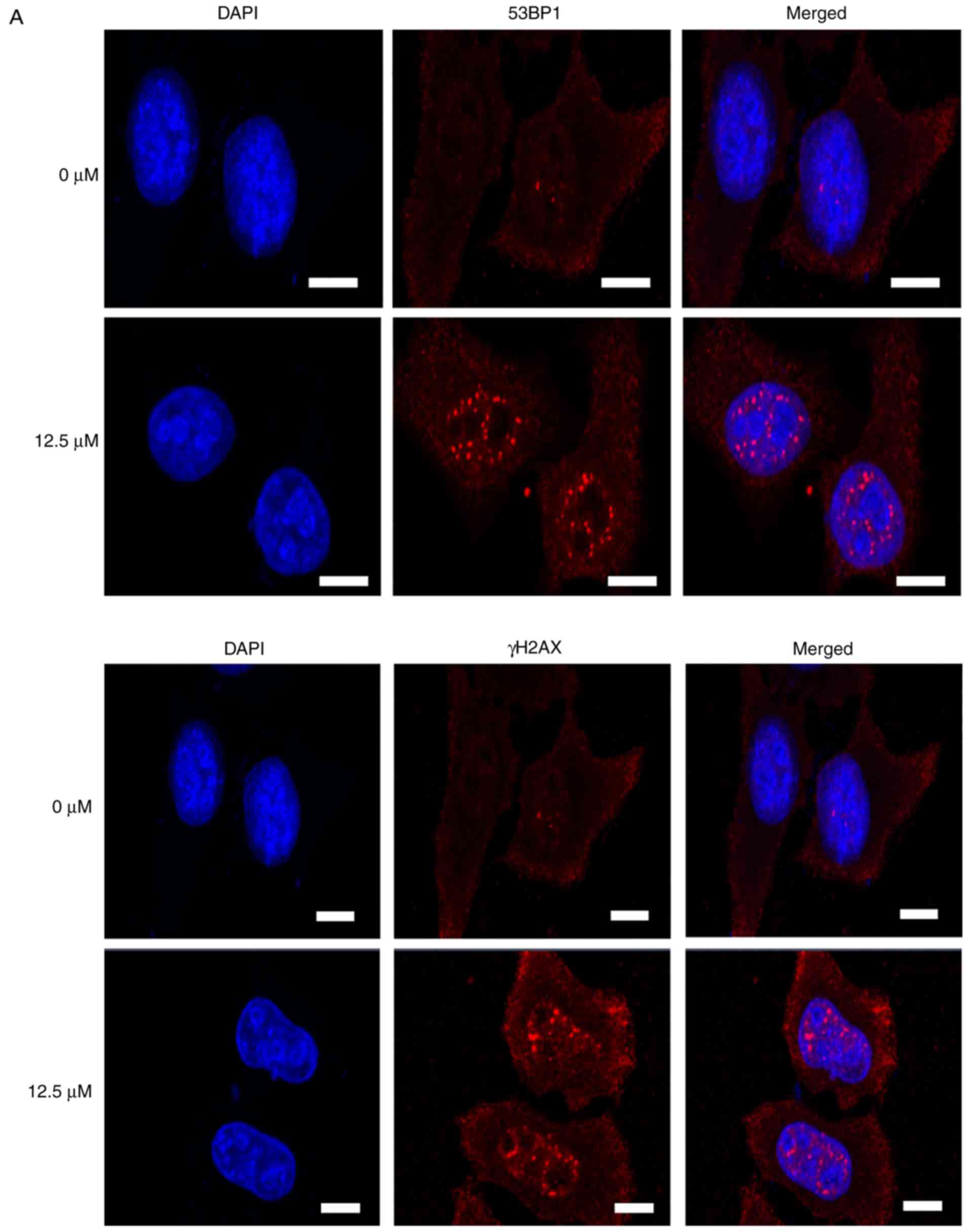
Subsequently, immunostaining was used to directly
visualize and quantify cellular DSBs by analyzing the DSB markers
γH2AX and 53BP1. Treatment with icaritin for 12 h caused a dramatic
increase in the number of cells with brightly stained nuclear 53BP1
and γH2AX foci (Fig. 5A-C). The
earliest increase in the foci of γH2AX was noted at 3 h
post-icaritin treatment, and for 53BP1 it was noted at 6 h
post-icaritin treatment in HeLa (Fig.
5B) and SiHa cancer cells (Fig.
5C). Higher concentration of icaritin resulted in a higher
number of positive cells and higher number of positive foci per
cell. Pre-incubation with 5 mM NAC completely blocked the
icaritin-induced increase in 53BP1 and γH2AX staining (data not
shown).
Icaritin treatment arrests cervical
cancer cell growth at the G2/M phases
To assess the consequences of the icaritin-induced
DNA damage, icaritin-induced changes in the cell cycle distribution
were analyzed by flow cytometry. The results revealed that icaritin
increased the percentage of cells at the G2/M phase,
while slightly decreasing the percentage of cells at the S phase
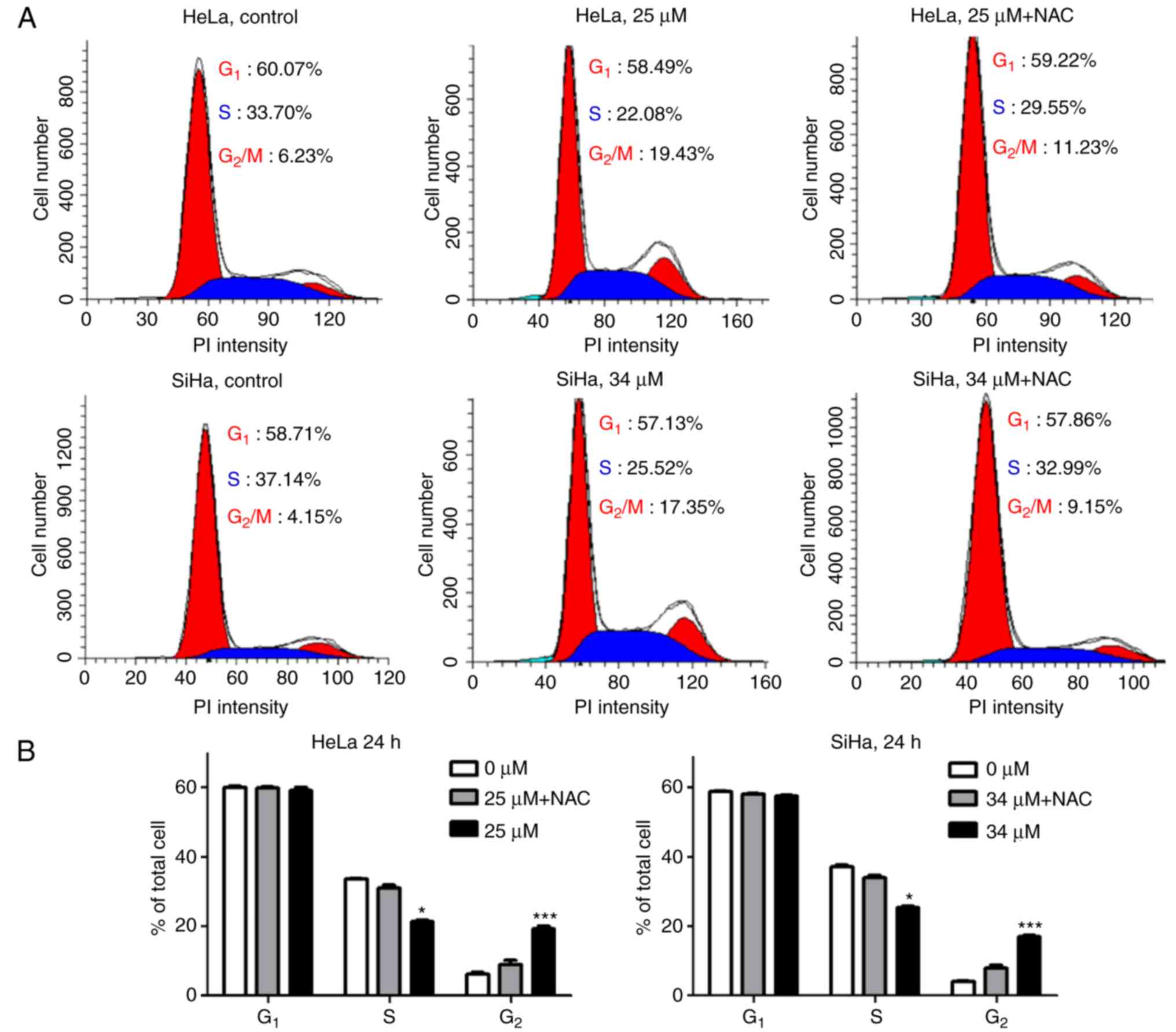
(Fig. 6). Following treatment with
25 µM icaritin for 24 h, the percentage of HeLa cells at the
G2/M phase was increased from 6.23 to 19.43%. A similar
effect was noted after treatment SiHa treatment with 34 µM icaritin
for 24 h (Fig. 6A). The percentage
of SiHa cells at the G2/M phases was increased from 4.15
to 17.35% (Fig. 6A). When the cells
were pre-incubated with NAC, the increase in the percentage of
G2/M cells was partially rescued (Fig. 6A and B). These results indicated
that icaritin-induced ROS caused cell cycle arrest and blocked the
progression of cervical cancer cells to the G2/M phases
of the cell cycle.
Icaritin induces apoptosis in HeLa and
SiHa cervical cancer cells
Consistent with the observations of cell cycle
analysis, examination of the nuclear morphology of icaritin-treated
HeLa cells demonstrated that icaritin dose-dependently increased
the number of apoptosis hallmarks, namely pyknosis and condensed
chromatin (Fig. 7A). This suggested
that apoptosis was induced by icaritin treatment.
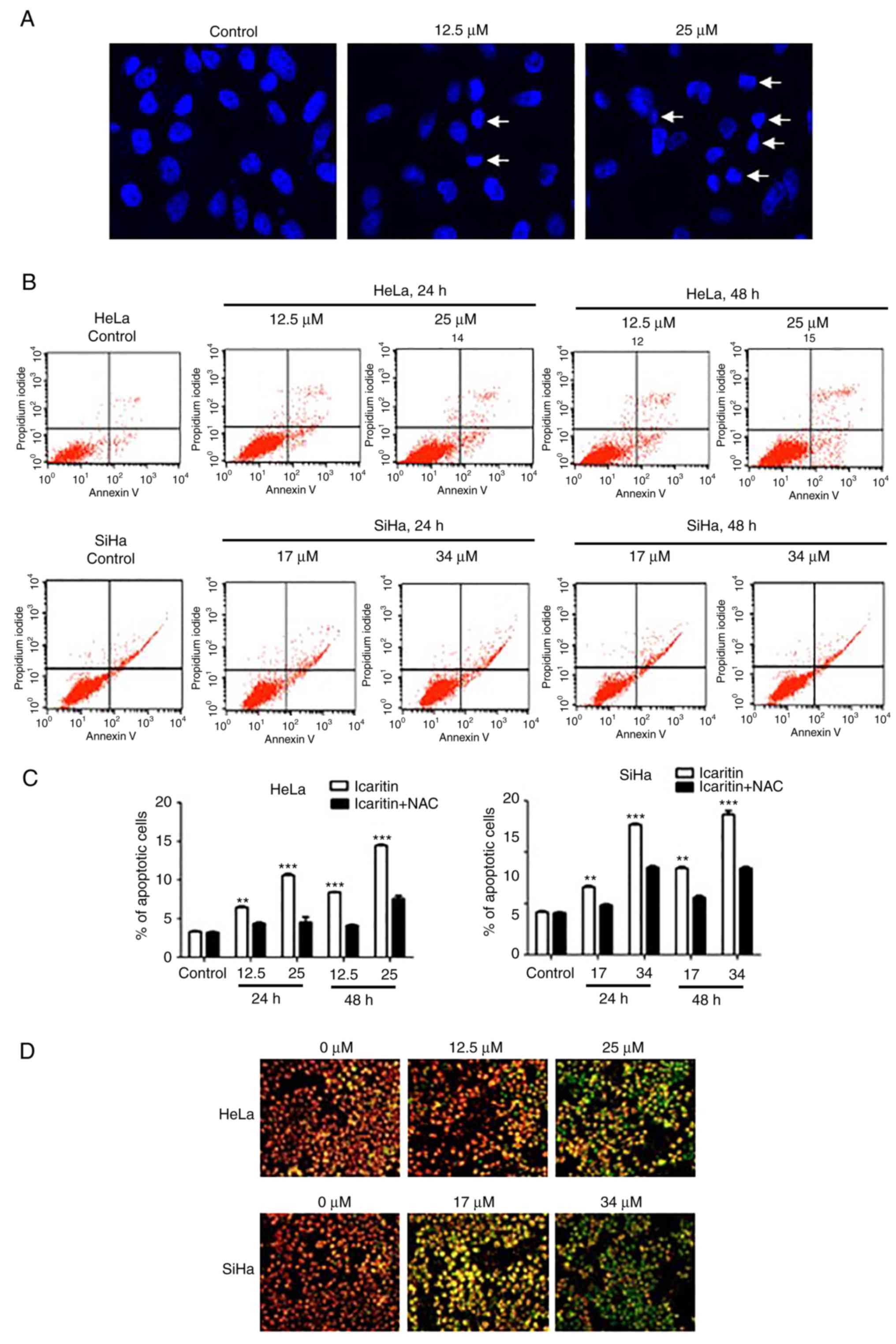
The percentage of apoptotic cells was analyzed by
Annexin V and propidium (PI) double-staining and flow cytometry.
The data demonstrated that icaritin dose- and time-dependently
induced apoptosis in HeLa and SiHa cells (Fig. 7B and C). Following treatment with
icaritin (1× IC50 and 2× IC50) for 24 h, the
percentage of apoptotic HeLa cells increased to 8.67 and 12.35%,
respectively, and the percentage of apoptotic SiHa cells increased
to 15.61 and 23.16%, respectively (Fig.
7B). Longer treatment (48 h) resulted in higher percentage of
apoptotic cells (11.11 and 17.36% for HeLa, 30.05 and 33.11% for
SiHa; Fig. 7B and C).
Pre-incubation with NAC significantly blocked icaritin-induced
apoptosis (Fig. 7C).
The loss of mitochondrial membrane potential is
characteristic of the activation of the intrinsic apoptotic
pathway. The mitochondria membrane potential probe, JC-1, was used
to assess the integrity of mitochondrial membranes in HeLa and SiHa
cells. The results demonstrated that 24 h treatment with icaritin
dose-dependently increased the intensity of green fluorescence,
while the intensity of the red fluorescence decreased (Fig. 7D), suggesting a prompt dissipation
of mitochondrial membrane potential and an activation of the
intrinsic apoptotic pathway caused by icaritin treatment.
To further explore the mechanism of action of
icaritin, western blot analysis was used to analyze the changes of
proteins associated with apoptosis. Treatment of the cells with the
icaritin IC50 for 24 h decreased MMP-9 levels in HeLa
and SiHa cells, whereas NAC blocked the icaritin-induced decrease
in MMP-9 (Fig. 8A). This indicated
that ROS have a role in the downregulation of MMP-9, which may be
involved in inhibited cell migration. The ATR serine/threonine
kinase (ATR)-checkpoint kinase 1 (Chk1) axis senses DSBs and
replication stress to regulate DNA repair, replication and cell
cycle progression and/or induce apoptosis (42). Treatment of the cells with icaritin
for 24 h resulted in reduced expression levels of CDK1-pT14 and an
increase in the expression levels of CDC25C-pS216 (Fig. 8B). Both changes were eliminated by
NAC pre-treatment, indicating that ROS-associated activation of the
ATR-Chk1 pathway occurs in icaritin-treated cancer cells, and that
inhibition of CDK1 activity was potentially responsible for
arresting the cell cycle at the G2/M phase. Finally,
treatment of the cells with icaritin for 24 h dose-dependently
increased the levels of the pro-apoptotic protein Bax, and the
levels of activated caspase 3 and 9 enzymes; whereas, it
concomitantly decreased the levels of the anti-apoptotic proteins,
Bcl-2 and XIAP (Fig. 8C and D).
These findings suggest that icaritin activated the intrinsic
apoptotic pathway, potentially as a result of the DDR signaling
pathway.
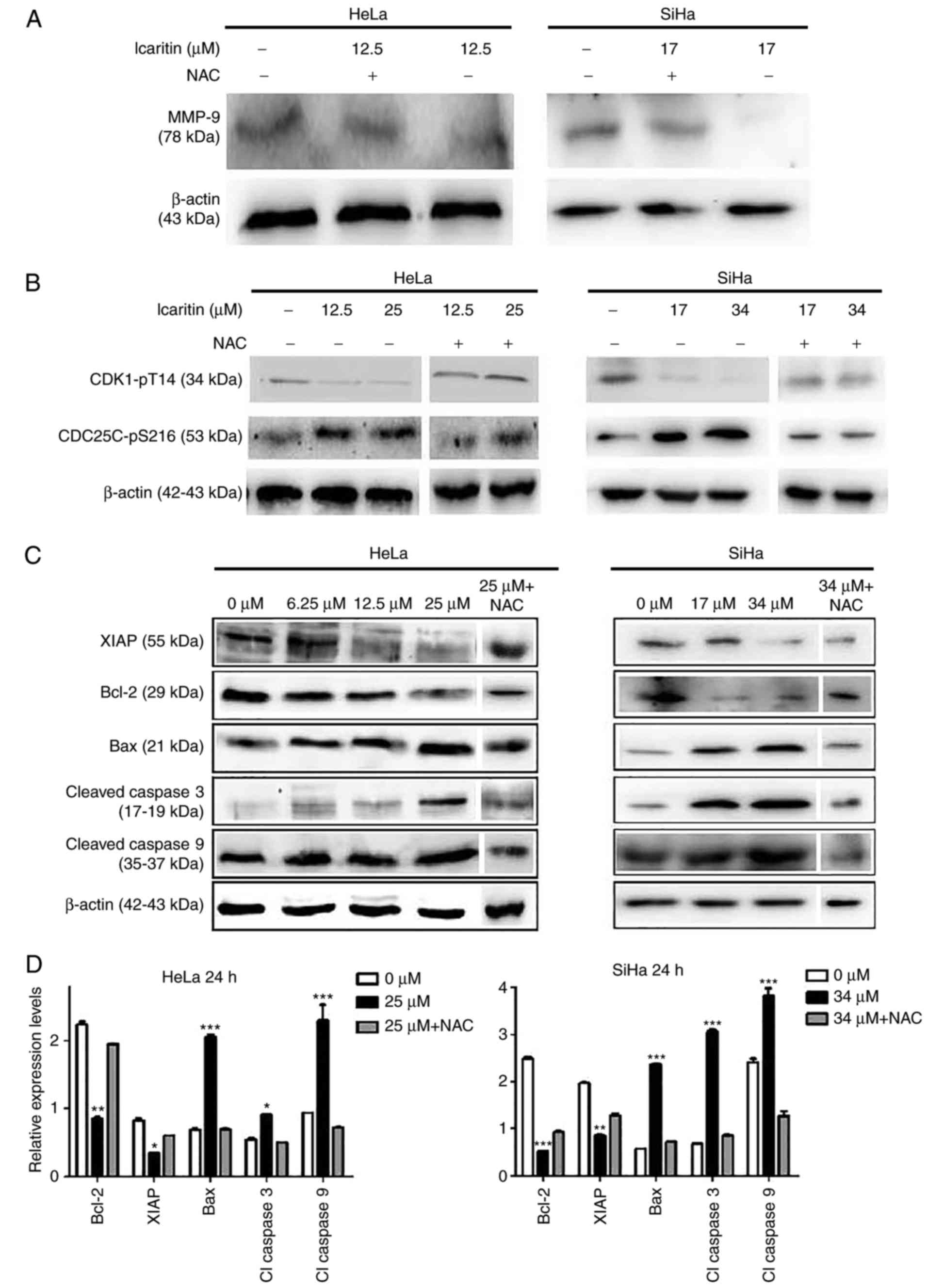 | Figure 8.Western blot analysis. HeLa and SiHa
cells with or without pre-incubation (1 h) with 5 µM NAC were
treated by icaritin for 24 h. Multiple SDS-PAGE gels were prepared
for each sample, and each gel was used for the detection of one
protein. Thus, one loading control, β-actin, was used for all
proteins of each sample. (A) Detection of MMP-9. (B) Detection of
cell cycle-associated proteins. (C) Detection of
apoptosis-associated proteins. (D) Quantification of
apoptosis-associated proteins in the 0 µM, 2× IC50 and
2× IC50 + NAC groups. Relative protein levels were
quantified from western blots using ImageJ and presented as ratios
of each protein band relative to the loading control. (*P<0.05,
**P<0.01, ***P<0.001 vs. 0 µM). NAC, N-acetyl cysteine,
MMP-9, matrix metalloproteinase-9; CDK1, cyclin-dependent kinase 1;
CDC25C, cell division cycle 25C; XIAP, X-linked inhibitor of
apoptosis protein; Bax, apoptosis regulator Bax; Bcl-2, B-cell
lymphoma-2. |
Discussion
In the present study, icaritin caused a potent
suppression of the growth of HeLa and SiHa cervical cancer cells,
although this effect was not observed for non-cancerous 293
epithelial and CCD-1095Sk fibroblast cells. Based on previous
reports in the literature that demonstrated the cytotoxicity of
this compound against a variety of cancer types (11,26–28,31),
the present study explored the hypothesis that icaritin possesses
broad-spectrum anticancer activity. Icaritin is one of the major
pharmaceutical components of the Epimedium family of plants
(13), extracts of which have long
been used in traditional Chinese herbal formulations (15). Studies have reported that icaritin
possesses a variety of biological functions in a wide range of cell
types. By inducing stem cell self-renewal (16) and osteogenic and cardiomyogenic
differentiation, icaritin promotes repair of bone and
cardiovascular injuries (12,15).
Icaritin has been shown to enhance osteoblastic differentiation and
mineralization, inhibit bone resorption and induce apoptosis of
osteoclasts (12). Animal studies
indicate that icariin can prevent bone loss in ovariectomized rats
and mice (12). A phase 1 clinical
trial to determine the potential of icaritin treatment for
osteoporosis and cardiovascular diseases is currently ongoing
(trial no. NCT02931305). By inhibiting neuronal apoptosis, icaritin
has neuroprotective effects against β-amyloid-induced neurotoxicity
(17); and by acting as a highly
selective ERa36 modulator, icaritin inhibited the growth of
multiple types of cancer cells both in vitro and in
vivo (11). Phase 1 and 1b
clinical trials to assess safety and efficacy of oral icaritin in
advanced solid tumors have been completed (trial nos. NCT01278810
and NCT02496949); although the results have not been published, the
fact that phase 2 and phase 3 clinical trials have been initiated
(trial nos. NCT01972672, NCT03236636 and NCT03236649) indicates
that the safety and efficacy of icaritin are encouraging. Thus, it
appears that this compound may be a good candidate and/or may serve
as a chemical scaffold for the development of novel anticancer
drugs.
Previous studies have reported that icaritin can
suppress various different types of cancer by induction of cell
cycle arrest, apoptosis and/or autophagic cell death (11,26,32,34).
Several different, and sometimes contradictory, mechanisms have
been proposed to explain its anticancer activity, including
suppression of IL-6/Jak2/STAT3 or MAPK signaling (27–30),
sustained activation of ERK1/2 and/or JNK1 (26,31,32),
inhibition of the PI3K/Akt pathway (33) and AMPK-dependent inhibition of mTOR
(34). However, the exact way by
which icaritin treatment affects these diverse signaling pathways
remains unclear. The data reported in the present study
demonstrated that treatment of HeLa and SiHa cervical cancer cells
with icaritin resulted in an immediate increase in cellular ROS
(within 3 h of icaritin treatment), which was immediately followed
by a dramatic increase in DNA 8-oxo-dG levels, in the number of
cells with numerous bright γH2AX and 53BP1 foci, and in the number
of DNA strand breaks, as determined by the comet assay. These
effects were not noted in the non-cancerous 293 and CCD-1095Sk
cells (data not shown). The blockage of ROS elevation using the ROS
inhibitor, NAC, decreased the expression of markers of oxidative
DNA damage. Cell cycle arrest and apoptosis were not noted when
cells were treated with icaritin for 12 h, but were apparent
following treatment with this compound for 24 h. Thus, these
studies suggested that an increase in ROS levels caused by icaritin
resulted in a rapid increase in oxidative DNA damage, which likely
stimulated base excision repair and generated a large number of
SSBs. Rapid and large-scale increases in SSB can saturate cellular
repair capacity, convert SSBs into numerous DSBs, and eventually
result in DDR signaling which induces cell cycle arrest and
apoptosis (7,10).
Notably, icaritin was recently reported to activate
the mitochondrial apoptotic pathway in human oral squamous cell
carcinoma cells by downregulating the expression of the
transcription factor Sp1 (43),
which was also noted to be induced by the human ribosomal protein
uL3 (rpL3) (44). rpL3
downregulates Sp1 to inhibit the expression and protein stability
of cystathionine-β-synthase (CBS). This in turn promotes
mitochondrial translocation of CBS to trigger the intrinsic
apoptotic pathway in p53 mutant cancer cells (44,45).
Furthermore, rpL3 increases cellular ROS levels by controlling the
cellular redox status (46). Thus,
it is likely that icaritin activates the mitochondrial apoptotic
pathway via multiple complex mechanisms.
Acknowledgements
Not applicable.
Funding
The present work was supported in part by grants
from National Natural Science Foundation of China (grant nos.
81320108025 and 81472662) and Science and Technology Development
Plan of Jilin Province (grant no. 20160204013YY).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XC performed the experiments, analyzed the results,
and wrote the manuscript. LS and YH performed the experiments. FL
conceived, designed and coordinated the study. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dancey JE, Bedard PL, Onetto N and Hudson
TJ: The genetic basis for cancer treatment decisions. Cell.
148:409–420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Block KI, Gyllenhaal C, Lowe L, Amedei A,
Amin ARMR, Amin A, Aquilano K, Arbiser J, Arreola A, Arzumanyan A,
et al: Designing a broad-spectrum integrative approach for cancer
prevention and treatment. Semin Cancer Biol. (35 Suppl):S276–S304.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu X and Zhang Z: Understanding the
genetic mechanisms of cancer drug resistance using genomic
approaches. Trends Genet. 32:127–137. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zecchini V and Frezza C: Metabolic
synthetic lethality in cancer therapy. Biochim Biophys Acta.
1858:723–731. 2017. View Article : Google Scholar
|
|
5
|
Fece de la Cruz F, Gapp BV and Nijman SM:
Synthetic lethal vulnerabilities of cancer. Annu Rev Pharmacol
Toxicol. 55:513–531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moloney JN and Cotter TG: ROS signalling
in the biology of cancer. Semin Cell Dev Biol. 80:50–64. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rudd SG, Valerie NCK and Helleday T:
Pathways controlling dNTP pools to maintain genome stability. DNA
Repair. 44:193–204. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rai P, Onder TT, Young JJ, McFaline JL,
Pang B, Dedon PC and Weinberg RA: Continuous elimination of
oxidized nucleotides is necessary to prevent rapid onset of
cellular senescence. Proc Natl Acad Sci USA. 106:169–174. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang X, Zhu D and Lou Y: A novel
anticancer agent, icaritin, induced cell growth inhibition,
G1 arrest and mitochondrial transmembrane potential drop
in human prostate carcinoma PC-3 cells. Eur J Pharmacol. 564:26–36.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Indran IR, Liang RL, Min TE and Yong EL:
Preclinical studies and clinical evaluation of compounds from the
genus Epimedium for osteoporosis and bone health. Pharmacol
Ther. 162:188–205. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tan HL, Chan KG, Pusparajah P, Saokaew S,
Duangjai A, Lee LH and Goh BH: Anti-cancer properties of the
naturally occurring aphrodisiacs: Icariin and its derivatives.
Front Pharmacol. 7:1912016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang ZQ and Lou YJ:
Proliferation-stimulating effects of icaritin and desmethylicaritin
in MCF-7 cells. Eur J Pharmacol. 504:147–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu DY and Lou YJ: Inducible effects of
icariin, icaritin, and desmethylicaritin on directional
differentiation of embryonic stem cells into cardiomyocytes in
vitro. Acta Pharmacol Sin. 26:477–485. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tsang WP, Zhang F, He Q, Cai W, Huang J,
Chan WY, Shen Z and Wan C: Icaritin enhances mESC self-renewal
through upregulating core pluripotency transcription factors
mediated by ERα. Sci Rep. 7:408942017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Zhang X, Wang H, Qi L and Lou Y:
Neuroprotective effects of icaritin against beta amyloid-induced
neurotoxicity in primary cultured rat neuronal cells via
estrogen-dependent pathway. Neuroscience. 145:911–922. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma HR, Wang J, Chen YF, Chen H, Wang WS
and Aisa HA: Icariin and icaritin stimulate the proliferation of
SKBr3 cells through the GPER1-mediated modulation of the EGFR-MAPK
signaling pathway. Int J Mol Med. 33:1627–1634. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang XT, Kang LG, Ding L, Vranic S,
Gatalica Z and Wang ZY: A positive feedback loop of ER-α36/EGFR
promotes malignant growth of ER-negative breast cancer cells.
Oncogene. 30:770–780. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Omarjee S, Jacquemetton J, Poulard C,
Rochel N, Dejaegere A, Chebaro Y, Treilleux I, Marangoni E, Corbo L
and Romancer ML: The molecular mechanisms underlying the
ERα-36-mediated signaling in breast cancer. Oncogene. 36:2503–2514.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thiebaut C, Chamard-Jovenin C, Chesnel A,
Morel C, Djermoune EH, Boukhobza T and Dumond H: Mammary epithelial
cell phenotype disruption in vitro and in vivo through ERalpha36
overexpression. PLoS One. 12:e01739312017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Zheng N, Dong J, Wang X, Liu L and
Huang J: Estrogen receptor-α36 is involved in icaritin induced
growth inhibition of triple-negative breast cancer cells. J Steroid
Biochem Mol Biol. 171:318–327. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen M, Turhan AG, Ding H, Lin Q, Meng K
and Jiang X: Targeting BCR-ABL+ stem/progenitor cells
and BCR-ABL-T315I mutant cells by effective inhibition of the
BCR-ABL-Tyr177-GRB2 complex. Oncotarget. 8:43662–43677.
2017.PubMed/NCBI
|
|
24
|
Tiong CT, Chen C, Zhang SJ, Li J, Soshilov
A, Denison MS, Lee LS, Tam VH, Wong SP, Xu HE, et al: A novel
prenylflavone restricts breast cancer cell growth through
AhR-mediated destabilization of ERα protein. Carcinogenesis.
33:1089–1097. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun F, Indran IR, Zhang ZW, Tan MH, Li Y,
Lim ZL, Hua R, Yang C, Soon FF, Li J, et al: A novel prostate
cancer therapeutic strategy using icaritin-activated
arylhydrocarbon-receptor to co-target androgen receptor and its
splice variants. Carcinogenesis. 36:757–768. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He J, Wang Y, Duan F, Jiang H, Chen MF and
Tang SY: Icaritin induces apoptosis of HepG2 cells via the JNK1
signaling pathway independent of the estrogen receptor. Planta Med.
76:1834–1839. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu JF, Li ZJ, Zhang GS, Meng K, Kuang WY,
Li J, Zhou XF, Li RJ, Peng HL, Dai CW, et al: Icaritin shows potent
anti-leukemia activity on chronic myeloid leukemia in vitro and in
vivo by regulating MAPK/ERK/JNK and JAK2/STAT3 /AKT signalings.
PLoS One. 6:e237202011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang JG, Lu R, Ye XJ, Zhang J, Tan YQ and
Zhou G: Icaritin reduces oral squamous cell carcinoma progression
via the inhibition of STAT3 signaling. Int J Mol Sci. 18:E1322017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li S, Priceman SJ, Xin H, Zhang W, Deng J,
Liu Y, Huang J, Zhu W, Chen M, Hu W, et al: Icaritin inhibits
JAK/STAT3 signaling and growth of renal cell carcinoma. PLoS One.
8:e816572013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu S, Wang Z, Li Z, Peng H, Luo Y, Deng
M, Li R, Dai C, Xu Y, Liu S, et al: Icaritin suppresses multiple
myeloma, by inhibiting IL-6/JAK2/STAT3. Oncotarget. 6:10460–10472.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guo Y, Zhang X, Meng J and Wang ZY: An
anticancer agent icaritin induces sustained activation of the
extracellular signal-regulated kinase (ERK) pathway and inhibits
growth of breast cancer cells. Eur J Pharmacol. 658:114–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tong JS, Zhang QH, Huang X, Fu XQ, Qi ST,
Wang YP, Hou Y, Sheng J and Sun QY: Icaritin causes sustained
ERK1/2 activation and induces apoptosis in human endometrial cancer
cells. PLoS One. 6:e167812011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Q, Huai L, Zhang C, Wang C, Jia Y, Chen
Y, Yu P, Wang H, Rao Q, Wang M, et al: Icaritin induces AML cell
apoptosis via the MAPK/ERK and PI3K/AKT signal pathways. Int J
Hematol. 97:617–623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Z, Meng X and Jin L: Icaritin induces
apoptotic and autophagic cell death in human glioblastoma cells. Am
J Transl Res. 8:4628–4643. 2016.PubMed/NCBI
|
|
35
|
Wo YB, Zhu DY, Hu Y, Wang ZQ, Liu J and
Lou YJ: Reactive oxygen species involved in prenylflavonoids,
icariin and icaritin, initiating cardiac differentiation of mouse
embryonic stem cells. J Cell Biochem. 103:1536–1550. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li C, Peng W, Song X, Wang Q and Wang W:
Anticancer effect of icaritin inhibits cell growth of colon cancer
through reactive oxygen species, Bcl-2 and cyclin D1/E signaling.
Oncol Lett. 12:3537–3542. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Song J, Shu L, Zhang Z, Tan X, Sun E, Jin
X, Chen Y and Jia X: Reactive oxygen species-mediated mitochondrial
pathway is involved in Baohuoside I-induced apoptosis in human
non-small cell lung cancer. Chem Biol Interact. 199:9–17. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zheng Q, Liu WW, Li B, Chen HJ, Zhu WS,
Yang GX, Chen MJ and He GY: Anticancer effect of icaritin on human
lung cancer cells through inducing S phase cell cycle arrest and
apoptosis. J Huazhong Univ Sci Technol Med Sci. 34:497–503. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Musa J, Achenbach CJ, O'Dwyer LC, Evans
CT, McHugh M, Hou L, Simon MA, Murphy RL and Jordan N: Effect of
cervical cancer education and provider recommendation for screening
on screening rates: A systematic review and meta-analysis. PLoS
One. 12:e01839242017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ding Y, Wang H, Niu J, Luo M, Gou Y, Miao
L, Zou Z and Cheng Y: Induction of ROS overload by alantolactone
prompts oxidative DNA damage and apoptosis in colorectal cancer
cells. Int J Mol Sci. 17:5582016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dong L, Wang H, Niu J, Zou M, Wu N, Yu D,
Wang Y and Zou Z: Echinacoside induces apoptotic cancer cell death
by inhibiting the nucleotide pool sanitizing enzyme MTH1. Onco
Targets Ther. 8:3649–3664. 2015.PubMed/NCBI
|
|
42
|
Yazinski SA and Zou L: Functions,
regulation, and therapeutic implications of the ATR checkpoint
pathway. Annu Rev Genet. 50:155–173. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jin L, Miao J, Liu Y, Li X, Jie Y, Niu Q
and Han X: Icaritin induces mitochondrial apoptosis by
up-regulating miR-124 in human oral squamous cell carcinoma cells.
Biomed Pharmacother. 85:287–295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pagliara V, Saide A, Mitidieri E,
d'Emmanuele di Villa Bianca R, Sorrentino R, Russo G and Russo A:
5-FU targets rpL3 to induce mitochondrial apoptosis via
cystathionine-β-synthase in colon cancer cells lacking p53.
Oncotarget. 7:50333–50348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Russo A, Saide A, Cagliani R, Cantile M,
Botti G and Russo G: rpL3 promotes the apoptosis of p53 mutated
lung cancer cells by down-regulating CBS and NFκB upon 5-FU
treatment. Sci Rep. 6:383692016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Russo A, Saide A, Smaldone S, Faraonio R
and Russo G: Role of uL3 in multidrug resistance in p53-mutated
lung cancer cells. Int J Mol Sci. 18:E5472017. View Article : Google Scholar : PubMed/NCBI
|