Introduction
Acute myeloid leukaemia (AML) with karyotypic
discernible translocation (t)(8;21)(q22;q22) (RUNX1-RUNX1T1)
is observed in approximately 7% of adults (1), and is the most frequent
non-lymphoblastic leukaemia in children (2). This balanced translocation disrupts
two functionally distinct genes, RUNX1 (previously termed
AML1) and RUNX1T1 (commonly known as ETO or
MTG8) (3). Physiologically,
RUNX1 is a transcription factor that encodes to the alpha
subunit of the core biding factor (CBFα) which forms a heterodimer
with the CBFβ subunit and regulates differentiation of
haematopoietic stem cells into committed mature cells. The
translocated RUNX1T1 moiety gets activated as a result of
the fusion with RUNX1 (4,5).
Together, the hybridised RUNX1-RUNX1T1 (RR) oncoprotein functions
as a dominant negative form of RUNX1. As empirically shown, RR
exerts a repressive effect on the promoters of genes normally
activated by the RUNX1 transcription factor through interactions of
its RUX1T1 moiety with nuclear corepressors such as NCOR1
and Sin3A (6,7). Several studies have indicated that
this altered transcription pattern impairs myeloid precursor cell
differentiation, proliferation and apoptosis, and is therefore
thought to be the underlying pathogenesis of myeloid leukaemia
(8–13). However, this translocation itself is
not necessarily sufficient for the development of AML. It appears
that additional secondary mutations are required to impair
haematopoietic differentiation to recapitulate the phenotypic
features of AML. Based on recent studies there appears to be a
fairly consistent association between the t(8;21) translocation and
the classical MAPK signalling pathway (9,14).
One of many signalling pathways triggered by
haematopoietic cytokines is the ERK (Ras/Raf/MEK/ERK) pathway. The
MAP kinase ERK2 (MAPK1), encoded by MAPK1, plays an integral
role in the ERK cascade in relaying extracellular biological
signals from cell membrane to the nucleus. This mechanism modulates
the normal haematopoietic cell proliferation, differentiation and
prevention of apoptosis (15). The
ERK pathway, most extensively studied of all MAPK pathways, is
deregulated in a third of all human cancers, a frequent observation
in the pathogenesis of haematological malignancies such as AML
(16–18). In wide varieties of AMLs,
constitutive activation that centres on the MEK/ERK node in the
pathway has been attributed to the tumourigenic effects such as
independence of proliferation and evasion of apoptosis (16–19).
Previous study revealed that AML with t(8;21) encoded RR induces a
microRNA-dependent mechanism that utilises the MAPK cascade to
signal hyper-proliferation of myeloid progenitors and block their
differentiation (20). Another
group reported that Kasumi-1 cells that were refractory to
apoptosis following RR silencing activate an array of
signalling pathways involved in cell survival and proliferation,
prominently the ERK2 pathway (21).
These findings underscore the influence of RR on the MAPK pathway
towards the pathobiology of AML.
Although the role of constitutive activation of the
MEK/ERK pathway in the development of myeloid leukaemia is well
established, RR- and MAPK-driven intricate molecular and cellular
pathways that maintain the pathobiology of AML remain elusive. In
the present study we used siRNA-mediated gene silencing approach to
elucidate the transcription profiles of RR and MAPK1
suppressed AML cell lines and further analysed these expression
signatures to explore the molecular mechanisms involved in cell
proliferation, cell cycle distribution, apoptosis and
differentiation. In the present study, we report differential
regulation of gene expression by RR and MAPK1 as determined by
genome-wide expression analysis responsible for the phenotypic
features of AML.
Materials and methods
Cell culture and siRNA
transfection
Two suspension leukaemic cell lines with t(8;21),
Kasumi-1 and SKNO-1, were used in the present study. The former was
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA) while the latter was a kind gift from Professor Olaf
Heidenreich (Northern Institute for Cancer Research, Newcastle
University, UK). Kasumi-1 and SKNO-1 cells were cultured in
RPMI-1640 medium containing 10 and 20% foetal bovine serum (FBS)
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
respectively. SKNO-1 was also supplemented with 10 ng/ml of
granulocyte-macrophage colony stimulating factor (GM-CSF). Cell
lines were incubated at 37°C in a humidified incubator with 5%
CO2.
Kasum-1 and SKNO-1 cells cultivated to late log
growth phase, concentrated to 107 cells/ml in culture
medium without serum, were electroporated with siRNA using a
Bio-Rad Gene Pulser Xcell™ at 330 V for 10 m. The sequences of
siRNAs used are as follows: siRNA-MAPK1
(siMAPK1) sense, 5′-GUUCGAGUAGCUAUCAAGATT-3′ and antisense,
5′-UCUUGAUAGCUACUCGAACTT-3′; siRNA-RUNX1-RUNX1T1
(siRR) sense, 5′-CCUCGAAAUCGUACUGAGAAG-3′ and antisense,
5′-UCUCAGUACGAUUUCGAGGUU-3′; and scrambled mismatch siRNA
(siMM) from Qiagen (Hilden, Germany).
For isolated downregulation of RR and
MAPK1 gene expression, both cell types were
electro-transfected with 200 nM of siRR and 100 nM of
annealed siMAPK1 respectively; and for co-transfection a
mixture of both siRR and siMAPK1 were used. These
concentrations were derived from dose-dependent experiments
performed in our laboratory. In experiments requiring extended
transfection, cells were sequentially transfected on days 0, 4 and
7. Immediately after electro-transfection, cells were diluted
20-fold in complete culture medium and returned to the incubator.
All cell groups were electroporated in triplicates.
RNA isolation and integrity
evaluation
Quantitative RT-PCR total RNA was isolated using
RNeasy Mini kit (Qiagen) according to the manufacturer's protocol.
RNA integrity was analysed on an Agilent 2100 Bioanalyser RNA 6000
NanoLabChip (Agilent Technologies, Palo Alto, CA, USA) to produce
an electrophoresis trace of 18S and 28S peaks using 2100 Expert
Software (Agilent Technologies). The RNA integrity number (RIN) was
calculated directly from the peak areas, a value of 10 corresponded
to intact RNA while 1 indicates a total degradation. Our optimised
methods showed a 28S:18S ratio of 1.9–2.1:1 and an RIN of 9–10.
Quantitative real-time polymerase
chain reaction (q-RT-PCR)
Quantitative real-time-polymerase chain reaction
(qRT-PCR) was performed on RNA isolated at various time points
post-transfection to quantify the mRNA expression levels of
MAPK1 and RR genes in cells treated with respective
siRNAs.
Gene-specific primers for MAPK1 (forward,
5′-CTGCTGCTCAACACCACCT-3′ and reverse,
5′-GCCACATATTCTGTCAGGAACC-3′); and B2M (forward,
5′-GGCATTCCTGAAGCTGACAG-3′ and reverse,
5′-TCTGCTGGATGACGTGAGTAA-3′) were designed using PrimerPlex
software (Premier Biosoft, Palo Alto, CA, USA). Primers to quantify
RR expression were: Forward, 5′-AATCACAGTGGATGGGCCC-3′ and
reverse, 5′-TGCGTCTTCACATCCACAGG-3′ as described by Heidenreich
et al (5).
A mixture of 50 ng of total RNA together with 0.5 µM
of MAPK1 and B2M, and 0.2 µM of RR primers was
reverse transcribed and amplified simultaneously in the same
reaction tube using the QuantiFast SYBR®-Green RT-PCR
One-Step PCR Master Mix (Qiagen) in a final volume of 20 µl on a
StepOnePlus Real-Rime thermocycler (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). The thermocycler was
programmed at 50°C for 10 min for RNA reverse transcription into
complementary cDNA followed by 95°C for 5 min for the initial DNA
polymerase activation step. Quantitative amplification was carried
out in 35 cycles of denaturation at 95°C for 10 sec, and annealing
and extension at 60°C for 30 sec. All reactions were performed in
triplicates.
Relative quantity of MAPK1 and RR mRNA
expression was computed using the comparative cycle threshold
(2−∆∆Ct) method normalised with the β2
microglobulin B2M expression as endogenous reference gene.
Mock cells were used as a reference sample for normalisation and
AllStars negative siRNA (Qiagen) as a mismatch (siMM) control for
validation purposes.
Cellular differentiation assay
Differentiation ability of RR- and
MAPK1-suppressed Kasumi-1 cells was monitored by CD34
expression. Kasumi-1 cells sequentially transfected on days 0, 4
and 7 with either siRR, siMAPK1 or a combination of
siRR + siMAPK1 were double stained on day 9 with
allophycocyanin conjugated anti-CD34:APC and peridinin-chlorophyll
A protein conjugated anti-CD45:PerCP (Becton-Dickinson, Franklin
Lakes, NJ, USA).
Briefly, ~105 transfected Kasumi-1 cells
were treated with Fc blocking reagent [phosphate-buffered saline
(PBS) containing 0.02% sodium azide and 1% BSA] and incubated on
ice for 30 min to block non-specific antibody binding. After
incubation, the cells were washed and re-suspended in 100 µl of
PBS. Cells were subsequently stained with 0.5 µl of allophycocyanin
conjugated anti-CD34:APC and 2 µl of peridinin-chlorophyll A
protein conjugated anti-CD45:PerCP (Becton-Dickinson). Cells were
sorted on a FACSCanto II equipped with BD FACSCanto Clinical
Software for acquisition and analyses. Mock cells and
siMM-transfected Kasumi-1 cells were used as controls.
Cell apoptosis assay
To determine the suppressive effects of MAPK1
and RUNX1-RUNX1T1 genes on apoptosis induction, Kasumi-1
cells were sequentially transfected on days 0, 4 and 7 with
siMAPK1 and siRR singly, and in combination.
Apoptosis was quantified on day 9 using an Annexin V/fluorescein
isothiocyanate (FITC) assay kit (AbD Serotec®; Bio-Rad
Laboratories, Raleigh, NC, USA) using a FACSCanto™ II (BD
Biosciences; Becton-Dickinson and Company, Franklin Lakes, NJ, USA)
flow cytometer. In brief, cells in logarithmic growth phase after
treatment with siRNA were washed once in cold 1X PBS and once in
cold 1X binding buffer. Cells were suspended to a density of
1×105 cells in 100 µl of 1X binding buffer and treated
with 5 µl of Annexin V:FITC for 10 min in the dark at room
temperature. On completion, the cells were washed again in 1X
binding buffer and re-suspended in 190 µl of 1X binding buffer with
10 µl of propidium iodide (PI) and directly analysed on a flow
cytometer. siMM-transfected cells were used as a negative
control.
Cell cycle analysis
Distribution of Kasumi-1 cells in different phases
of the cell cycle were examined using EZCell™ cell cycle analysis
kit (BioVision, Milpitas, CA, USA) according to the manufacturer's
protocol. Three aliquots of Kasumi-1 cells were eletrotransfected
on days 0 and 4 with siRR and siMAPK1, and in
combination respectively. Kasumi-1 cells electroporated with
siMM and mock-transfected cells were used as controls. Cell
cycle analysis was performed on day 6 by FACS analysis. Briefly,
cells were washed in PBS, adjusted to a cell density of
1×106 and fixed in 100% ice-cold ethanol for 24 h. On
the next day, PBS washed cells were digested with 100 µl of RNase A
for 30 min at 37°C followed by staining with 400 µl of PI and
subsequently analysed using FACSCanto II flow cytometer at 488 nm
wavelength using FL3 filters with wavelength of 670 nm for the
proportion cell cycle distribution in the
G0/G1, S and G2/M phase.
Cell viability and proliferation
assays
The rate of viable cells was determined using trypan
blue (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) staining
method that selectively stains dead cells. Cell counting was
carried out using a haemocytometer.
To quantitatively determine the number of viable
cells in proliferation on days 0, 4 and 8 following
electrotransfection, the siRR, siMAK1 and combined
siRR/siMAPK1-treated Kasumi-1 cells were seeded
separately, on the day of the experiment, into a 96-well plate at a
density of 5×104/well in 100 µl complete growth medium
in quadruples. Twenty microliters of
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
MTS/PMS solution (as instructed in the Cell Titer 96®
AQueousNon-Radioactive Cell Proliferation Assay kit literature;
Promega Corp., Madison, WI, USA) was added and the plate was
incubated for 2 h at 37°C in a humidified 5% CO2
incubator. The absorbance was measured at 490 nm using an ELISA
plate reader. Data are represented as mean ± SD of four independent
experiments.
Microarray experiments
Post electrotransfection, genome wide expression
(GWE) levels were determined in a series of four experimental
conditions set up for each cell line, Kasumi-1 and SNKO-1. They
include cells; co-transfected with siRR/siMAPK1;
singly transfected with siRR; siMM; and mock-transfected
cells. A total of 24 arrays, three replicates for each of the
transfection experiment, were processed using Illumina HumanHT-12
v3 Expression BeadChip™ array (Illumina, San Diego, CA, USA)
targeting 48,804 human transcripts per array with an average of
15-fold transcript redundancy.
Pre-hybridisation sample preparation used a total of
400–500 ng of RNA from each experiment, which was reverse
transcribed to cDNA followed by amplification and in vitro
transcription of cDNA to bio-11-dUTP-labelled complementary RNA
(cRNA). Quantified (500 ng) and labelled cRNA was then hybridised
to BeadChip arrays at 55°C overnight. BeadChips were then
wash-cleaned with Illumina high-stringent wash buffer. The
hybridisations were scanned after staining with 1 µg/ml
streptavidin-Cy3 (Amersham Biosciences, Piscatawy, NJ, USA) on the
Illumina BeadArray Reader confocal scanner and BeadScan software to
produce bead level data.
Background subtracted bead summary data were
exported to GeneSpring™ GX 11.0 software (Agilent Technologies,
Santa Clara, CA, USA) for transformation and normalisation, and
differential expression analyses. Microarray expression data were
normalised to the most stable three reference genes (ACTB,
B2M and UBC). Probe sets returning a P-value <0.05
(unpaired t-test) in comparison to the mock cells and experiment
classes were considered to be differentially expressed. The
differentially expressed genes were then filtered for gene sets
that had fold-changes >1.1, and >1.5 together with false
discovery rate (FDR) adjusted P-value ≤0.05.
The processed data were used for GO annotation and
KEGG pathway enrichment analyses using the Database for Annotation,
Visualization and Integrated Discovery [DAVID; (22)].
Statistical analysis
All statistical analysis was performed using SPSS
software version 17 (SPSS, Inc., Chicago, IL, USA). Paired t-test
was used to measure mean differences between two variables whereas
one-way ANOVA was used to measure mean differences between three
variables.
Results
siRNA induces the silencing of
RUNX1-RUNX1T1 and MAPK1 in Kasumi-1 cells
To identify the silencing efficacies of
siMAPK1 and siRR, we transfected Kasumi-1 cells with
100 and 200 nM of the respective siRNAs separately in triplicates.
Time course suppression of RR and MAPK1 genes in the
experimental cells were compared with mock-transfected cells. The
results of qRT-PCR showed that siMAPK1 inhibited the
expression of MAPK1 by 85% on day one, and lasted for at
least three days with 2% recovery (83%) on day two and 6% recovery
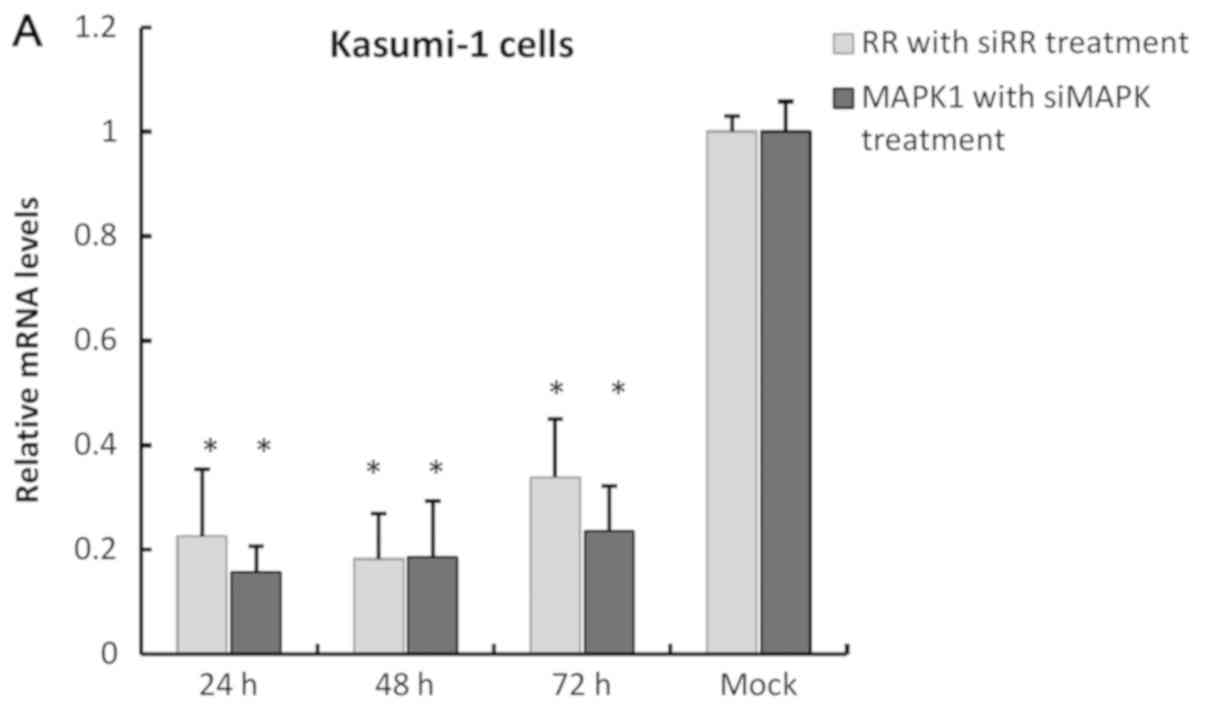
(77%) on day three (Fig. 1A).
siRR-treated Kasumi-1 cells exhibited up to 78% suppression
of RR expression at 24 h and continued to further suppress
to 82% at 48 h before 16% recovery was observed at 72 h. These
observations as measured by qRT-PCR validates that significant
knockdown (>80%) of RR and MAPK1 was achieved with
respective doses of 200 nM of siRR and 100 nM of
siMAPK1 at 48 h post-transfection. Melting curves for
RUNX1-RUNX1T1 and MAPK1 are included in Fig. 1B and C, respectively.
Whole genome expression profile
We conducted gene expression analysis on mRNA
extracts obtained at 48 h post-transfection using Illumina's
HumanHT-12 v3 Expression BeadChips. A total of 24 arrays, three
replicates for each of the four experimental conditions for both
Kasumi-1 and SKNO-1 cells (as described above) were profiled for at
least 48,804 transcripts to characterise the genes modulated when
RR and MAPK1 were downregulated.
Microarray data for Kasumi-1 cells co-transfected
with siMARK1 and siRR showed a total of 4,788
differentially expressed genes (DEG) as compared to the mock cells,
accounting for 9.8% of the transcriptome probed in our array.
However, when the 4,788 DEGs were conditioned to a fold change (FC)
≥1.5 and P<0.05 (t-test), the gene set condensed to 466
representing 262 upregulations and 204 downregulations. Relative to
Kasumi-1 cells, co-knockdown of RR and MAPK1 genes in
SKNO-1 cells revealed a more pronounced gene modulation presenting
with 6,465 DEGs accounting for 13.2% of the transcriptome studied.
Of these 6,465 DEGs, 732 sequences had a FC of ≥1.5 (at P<0.05)
representing 415 upregulations and 317 downregulations.
We then examined the 466 and 732 gene sets with FC
>1.5 for shared common genes modulated in both Kasumi-1 and
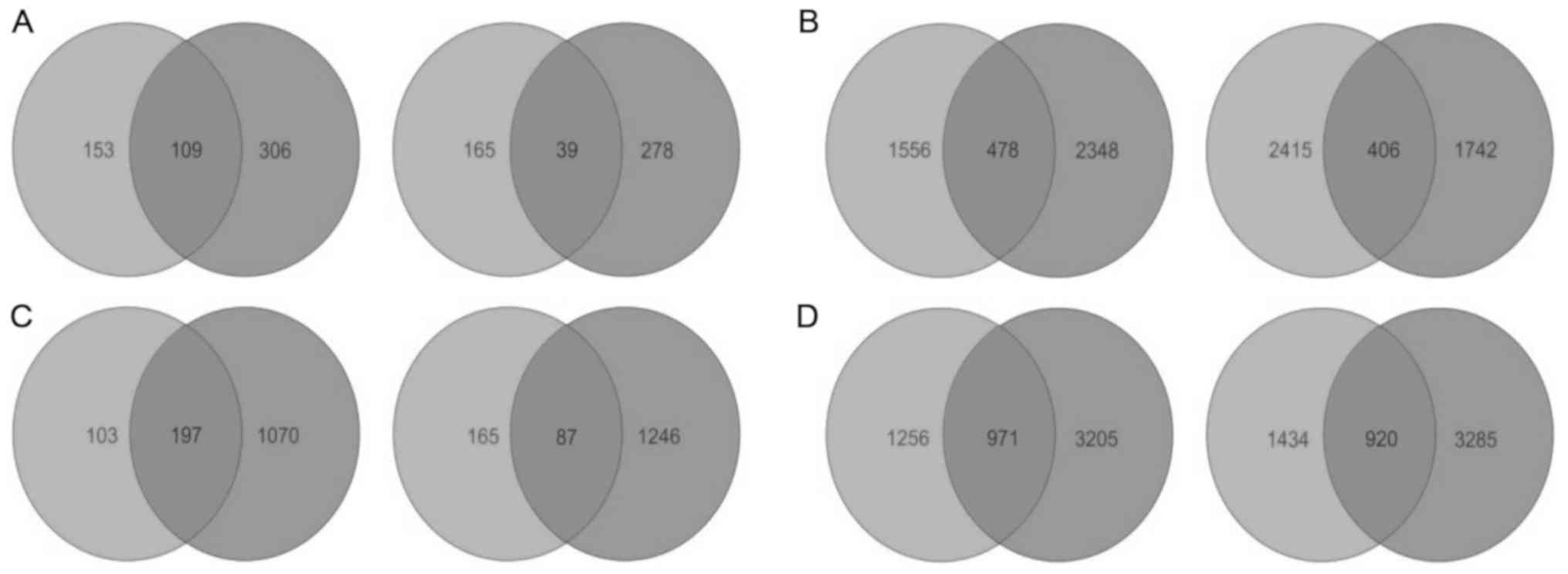
SKNO-1 cells using the two-way Venn diagram analysis. In total,
there were 148 overlapping genes, which included 109 upregulations
and 39 downregulations (Fig. 2A).
We also looked at the shared genes between the two cell lines when
the fold changes were relaxed to 1.1. At this fold change, we
observed 4,855 and 4,974 genes that were modulated in their
respective co-transfected Kasumi-1 and SKNO-1 cell lines when
compared to their respective mock-transfected cells. When used on
two-way Venn diagram, these two enriched gene sets presented with
884 overlapping genes, which included 478 upregulations and 406
downregulations (Fig. 2B).
Whole genome expression studies from isolated
RR-depleted Kasumi-1 cells presented with 5,178 DEGs
relative to mock-transfected cells, accounting for 10.6% of the
transcriptome probed. One-tenth of these, 552 genes, had a fold
change expression ≥1.5 including 300 upregulations and 252
downregulations. In turn, RR-depleted SKNO-1 altered the
expression of 9,049 genes. Of these, expression of 2,600 genes was
significantly changed at a ≥1.5-fold cut-off including 1,267
upregulations and 1,333 downregulations. Two-way Venn diagram
analysis enrolling the gene sets observed at fold changes ≥1.5 from
Kasumi-1 cells (552) and SKNO-1 cells (2,600) revealed that a
subset of 284 DEGs (including 197 upregulations and 87
downregulations) were common between the two cell lines (Fig. 2C), and support their association
with the RR knockdown. Moreover, when the absolute fold
change cut-off was adjusted to ≥1.1, we observed the number of
overlapping genes from the two cell lines rose to 1,891, with 971
upregulations and 920 downregulations (Fig. 2D).
Gene ontology analysis
To reveal the potential function of differentially
expressed genes common for both Kasumi-1 and SKNO-1 cell lines in
terms of their associated biological processes, cellular components
and molecular functions, we used the online data-mining tools of
DAVID software (23,24). Uploading the 148 overlapping gene
set, derived from the siRR + siMAPK experiment as depicted
on the Venn diagram Fig. 2A, into
DAVID with the default gene ontology (GO) setting (P<0.01)
indicated association to 267 biological processes (BP), 49 cellular
components (CC) and 52 molecular functions (MF). GO enrichment
analysis for the 284 overlapping gene set identified from the Venn
diagram analysis (Fig. 2C) from
siRR experiments on Kasumi-1 and SKNO-1 cell lines
identified 269 BP, 85 CC and 69 MF.
qRT-PCR validation of microarray
data
In order to validate the altered gene expression
results of the BeadChip experiments, we performed quantitative PCR
following reverse transcription (qRT-PCR) on the same RNA isolates
used on BeadChips. We chose to quantify the expression levels of
MAPK1 and RR, and compared the fold changes.
qRT-PCR assessment of RR and MAPK1
gene expression levels from the co-transfection experiment showed
comparable downregulated fold changes validating the microarray
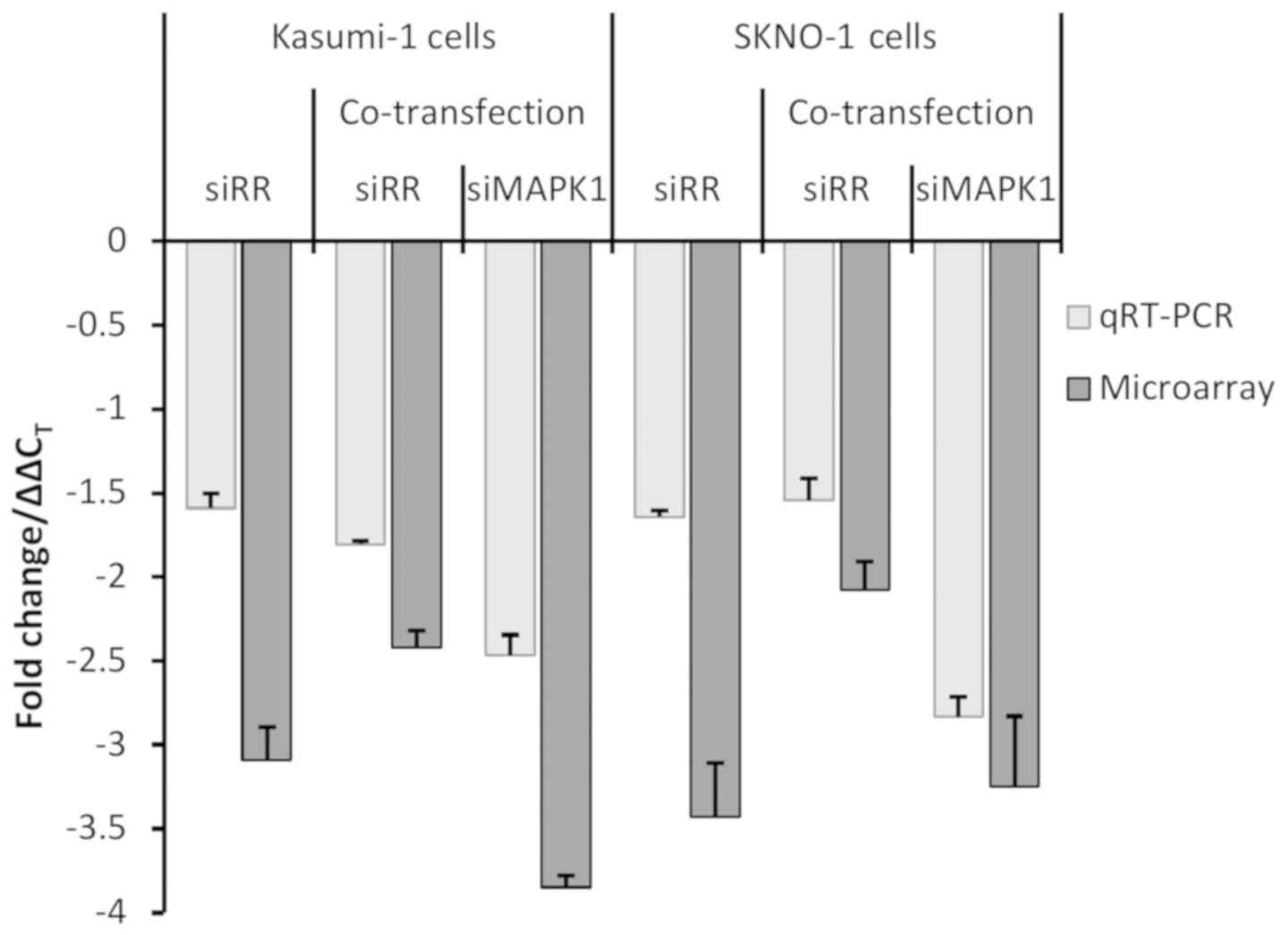
data. As illustrated in Fig. 3,
RR expression in co-transfected Kasumi-1 and SKNO-1 cells
revealed a fold change between 1.75–1.85 and 1.4–1.6, respectively
on qRT-PCR, while the same on the BeadChip had fold change
downregulations between 2.4–2.6 and 1.9–2.3, respectively. Similar
agreements were observed for MAPK1 in the co-transfected
experiments. MAPK1 had a downregulated fold changes between
2.4–2.6 and 2.6–3.0 by qRT-PCR and 3.8–3.9 and 2.8–3.6 by BeadChip
in Kasumi-1 and SKNO-1 cells, respectively.
The results from the isolated RR knockdown
experiment were also consitent with those of BeadChip. On a log2
scale, qRT-PCR revealed that RR expression was downregulated
by 1.5–1.7- and 1.6–1.7-fold, while the same on microarray was
2.9–3.3 and 3.2–3.7 in Kasumi-1 and SKNO-1 cells, respectively.
RUNX1-RUNX1T1 and MAPK1 suppression
reduces the proliferation rate of t(8;21) cells
To determine the effect of RUNX1-RUNX1T1 and
MAPK1 suppression and their combined influence on the
proliferation rate, we transfected Kasumi-1 cells three successive
times in a gap of three days with their respective siRNAs
individually and in combination. MTS assays were performed on days
0, 4 and 8 to determine the proliferation activity. siMM and
mock-transfected Kasumi-1 cells served as controls.
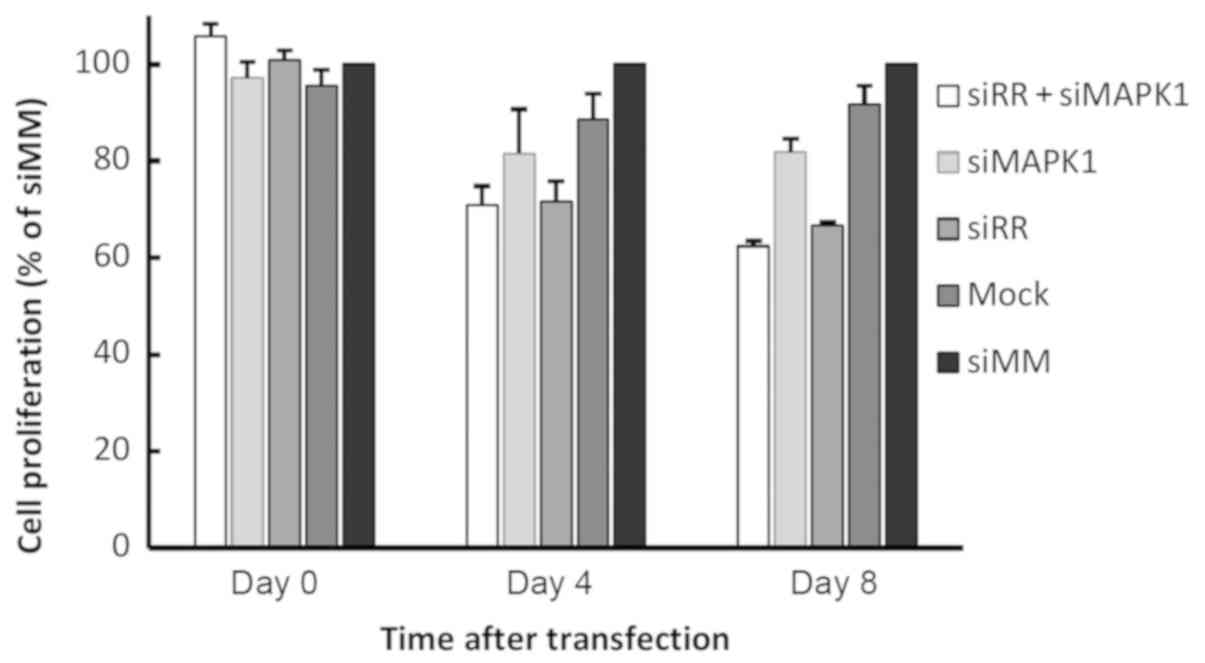
We observed that a single co-transfection with
siRR and siMAPK1 or isolated transfection of
siRR was not sufficient to inhibit the proliferation of
Kasumi-1 cells when compared with siMM (Fig. 4). However, sequential
electroporations on every third or fourth day attenuated the
proliferation viabilities on days 4 and 8 in both the experiments.
Similarly, two successive transfections of siMAPK1 were
required to inhibit Kasumi-1 cell proliferation by 18.5% on day 4.
The third successive transfection was only sufficient to sustain
the proliferation at the same level on day 8. These data suggest
that the fusion protein RR plays a more substantial role than MAPK1
in supporting the proliferation of leukaemic cells with
t(8;21).
Our GO enrichment analysis revealed that the
observed growth suppression upon RR knockdown was
accompanied by deregulated expression of classical positive
regulator genes that otherwise are known to enhance cell
proliferation. They include significant downregulation of FLT3,
HIF1A, KIT, MYCN, LYN, UBE2A, TBRG4, RPS9, RPS15A, RPA1, PRDX3,
PDF, ODC1, IL13RA1, ITGB1, GRN, FABP1, CDK6, F2R, CDC123, CAPNS1,
MARCKSL1, CD40, BCL2L1, AGPAT1, MXI1, CAV2, ENG, EMP3, IL8,
MFN2 and TRIM24 genes, and upregulation of CEBPA,
S100A11, ING4 and SSTR2 genes. On comparison, GO
enrichment analysis for category of proliferation from RR
knockdown and RR/MAPK1 co-knockdown experiments revealed
several shared differentially expressed genes. On exclusion of the
simultaneously altered genes, we observed that downregulation of
JUN, NRAS, and STAT1 genes and upregulation of
VHL, ZBTB16 and BTG1 genes were unique to
MAPK1 depletion. The results of the GO enrichment analysis
are summarised in Table I.
 | Table I.Summary of Gene Ontology Enrichment
Analysis according to pathways indicated when genes were silenced
using the respective siRNAs. |
Table I.
Summary of Gene Ontology Enrichment
Analysis according to pathways indicated when genes were silenced
using the respective siRNAs.
| Pathways |
RUNX1-RUNX1T1 (siRR) | MAPK1
(siMAPK1) |
|---|
| Cell | Upregulated | Upregulated |
| proliferation | genes | genes |
|
| CEBPA | VHL |
|
| S100A11 | ZBTB16 |
|
| ING4 | BTG1 |
|
| SSTR2 |
|
|
| Downregulated | Downregulated |
|
| genes | genes |
|
| FLT3 | FLT3 |
|
| HIF1A | NRAS |
|
| KIT | STAT1 |
|
| MYCN |
|
|
| LYN |
|
|
| UBE2A |
|
|
| TBRG4 |
|
|
| PRDX3 |
|
|
| CD40 |
|
|
| IL8 |
|
|
| BCL2L1 |
|
| Apoptosis | Upregulated
genes | Upregulated
genes |
|
| BCL2 | TP53 |
|
| SPHK2 | TNFSF10 |
|
| CFLAR | ADA |
|
| CD24 |
|
|
| NOTCH1 |
|
|
| ADAM17 |
|
|
| BIRC3 |
|
|
| HMGB1 |
|
|
| Downregulated
genes | Downregulated
genes |
|
| IP6K2 | JUN |
|
| BECN1 | NRAS |
|
Differentiation | Upregulated
genes | Upregulated
genes |
|
| CD24 | ADA |
|
| NOTCH1 |
|
|
| DNMT38 |
|
|
| CEBPA |
|
|
| CEBPE |
|
|
| ID2 |
|
|
| JMJD6 |
|
|
| IK2F1 |
|
|
| Downregulated
genes | Downregulated
genes |
|
| RHOA | JUN |
|
| CBFB |
|
|
| KIT |
|
|
| CDK6 |
|
|
|
FLT3 |
|
| Cell cycle | Upregulated
genes | Upregulated
genes |
|
| FOXO4 | RASSF1 |
|
| ING4 | GADD45A |
|
|
| p53 |
|
|
| FBX06 |
|
| Downregulated
genes | Downregulated
genes |
|
| TPX2 | MAPK11 |
|
| CDC2 |
|
|
| CKAP5 |
|
|
| IL8 |
|
|
| MAD21L |
|
|
| CCNE2 |
|
|
| CCNG2 |
|
|
| CDK6 |
|
|
| TBRG4 |
|
RUNX1-RUNX1T1 suppression exerts an
anti-apoptotic effect while MAPK1 suppression induces apopotosis in
t(8;21) cells
Next, we examined the consequences of isolated and
co-depletions of RR and MAPK1 on cell apoptosis.
Kasumi-1 cells harvested on day 9, after three consecutive
transfections, were double stained using Annexin V/FITC and PI.
Three consecutive siRR treatments
significantly (P=0.0001) lowered the live cells by 24% when
compared with the siMM control indicating that RUNX1-RUNX1T1
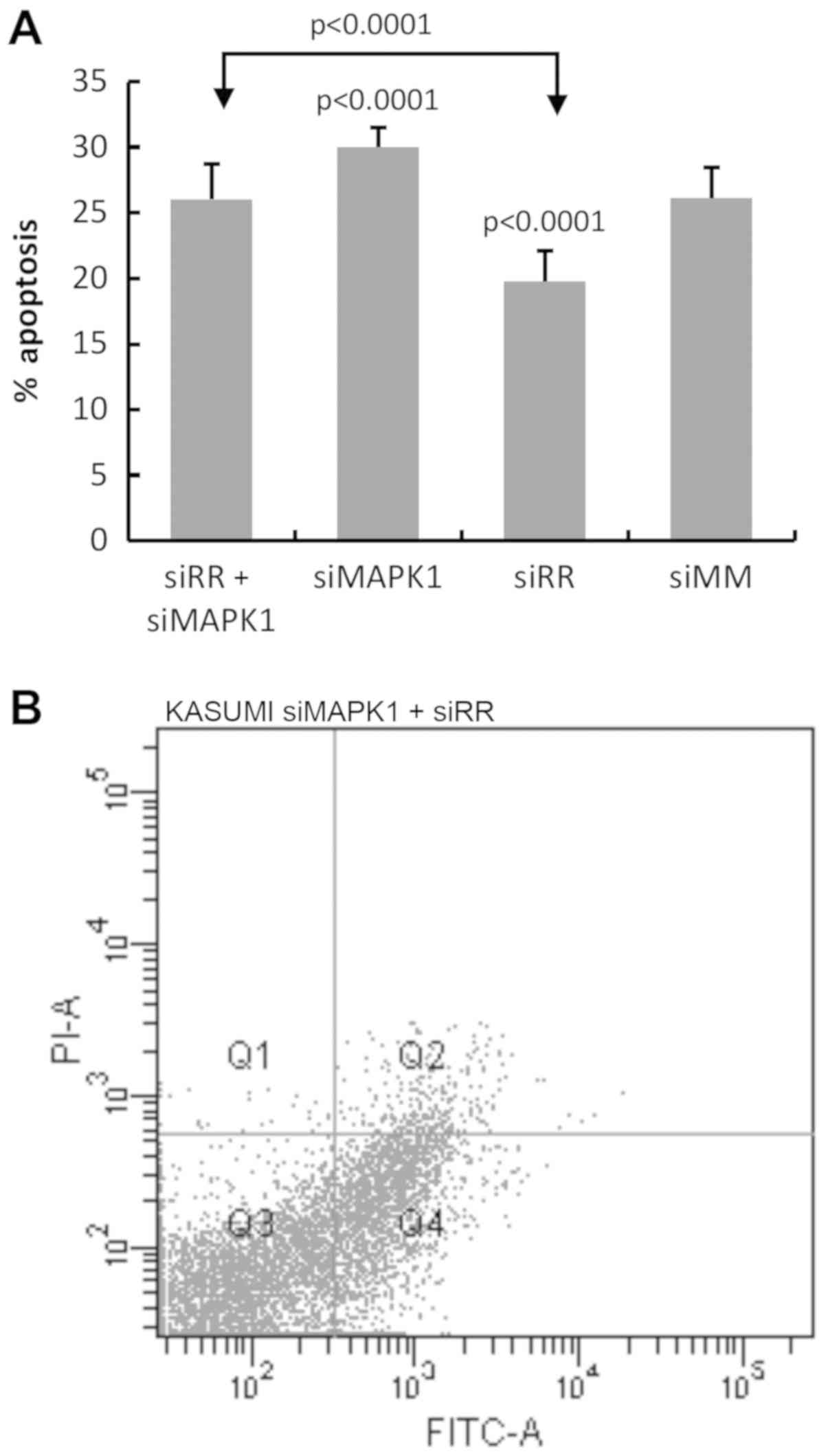
suppression has an apoptotic effect on Kasumi-1 cells (Fig. 5). Notably, pronounced apoptosis
(30±1.6%) was observed upon isolated MAPK1 deletion, and
revealed a significant difference (P<0.0001) when compared with
control cells. However, co-depeletion demonstrated no difference
when compared with the siMM control cells. FACS results for the
apoptosis analysis are shown in Fig.
5B-E.
GO enrichment analysis on shared DEGs from both
Kasumi-1 and SKNO-1 cells with fold change of ≥1.5 in response to
RR depletion exhibited several changes to genes involved in
negative regulation of apoptosis. They included significant
upregulation of BCL2, SPHK2, CFLAR, CD24, NOTCH1, ADAM17,
BIRC3 and HMGB1, and downregulation of IP6K2 and
BECN1 genes. Exclusion of overlapping differentially
expressed genes involved in apoptosis between the isolated RR
knockdown and the co-knockdown experiments revealed five genes
specific for MAPK1 depletion. They include upregulated
expression of TP53, TNFSF10 and ADA genes as well as
downregulated expression of JUN and NRAS genes. The
results of the GO enrichment analysis are summarised in Table I.
RUNX1-RUNX1T1 suppression supports,
while MAPK1 depletion inhibits the differentiation of t(8;21)
cells
To investigate the effect of RUNX1-RUNX1T1
and MAPK1 depletion on the progenitor status of Kasumi-1
cells, we analysed the CD34 expression levels as an indicator of
differentiation. Compared with the cells, RR suppression
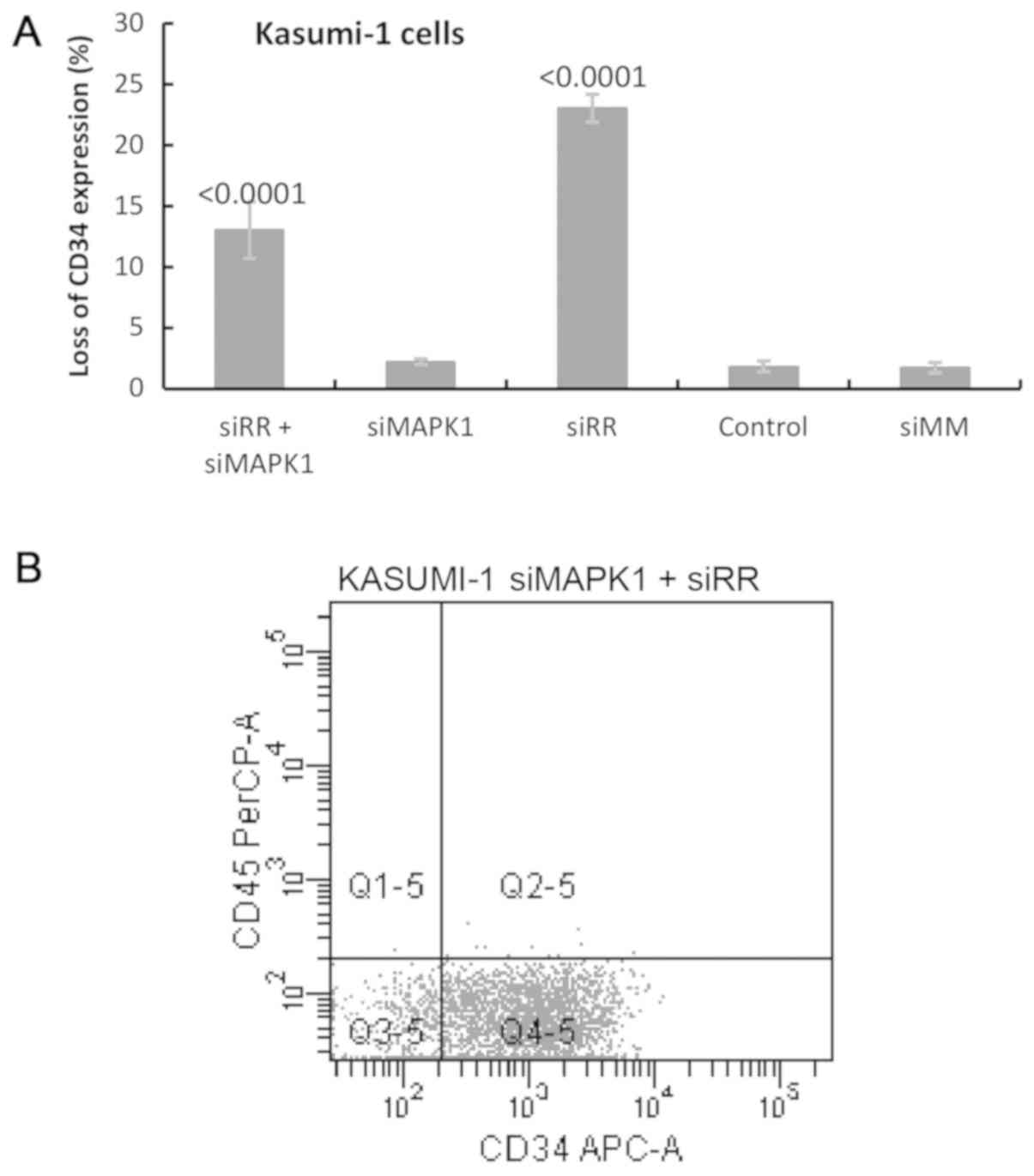
showed statistically significant (P=0.0001) loss of CD34-expressing
cells by ~23±1.1% 9 days after the first of the three successive
electroporations indicating that the cells were undergoing apparent
differentiation (Fig. 6A). The loss
of CD34-expressing cells corresponded with an increase in cells
expressing CD45 which indicated differentiation. The FACS results
are shown in Fig. 6B-E,
respectively. Based on the gene ontology analysis on the
RR-suppressed microarray data, we identified several genes involved
in cell differentiation to be activated or repressed. Upregulated
genes included CD24, NOTCH1, DNMT3B, CEBPA, CEBPE, ID2,
JMJD6 and IKZF1, while RHOA, CBFB, KIT, CDK6 and
FLT3 were downregulated.
When both MAPK1 and RR were
co-suppressed, the expression of CD34 in Kasumi-1 cells were
relatively less pronounced (13±2.3%) (Fig. 6A). However, CD34 suppression
revealed a significant difference (23±1.1%), when compared with the
control cell expression in which only RR was suppressed. This
implies that MAPK1 abrogated the effect of RR by ~40%
suggesting that tandem MAPK1 suppression exerted an
anti-differentiation effect. Due to this antagonist effect of
MAPK1 and RR co-suppression, we used the same
strategy mentioned in the apoptosis section to identify the genes
more specific for MAPK1 knockdown. After exclusion,
MAPK1 knockdown revealed significant downregulation of
c-JUN and upregulation of ADA, both identified to
have anti-differentiation effect.
FACS experiment on isolated MAPK1 suppression
showed no significant loss of CD34 expression when compared with
mock and AllStar siRNA-treated control cells (Fig. 6A). The results of the GO enrichment
analysis are summarised in Table
I.
RUNX1-RUNX1T1 and MAPK1 depletion
promotes cell cycle arrest
Finally, we examined the effect of different siRNAs
on cell cycle progression. Kasumi-1 cells were electro-transfected
twice o a gap of three days with either siRR, siMAPK1 or
co-transfected with siRR/siMAPK1. Cell phase distributions
were analysed on day 6 by flow cytometry. In comparison with the
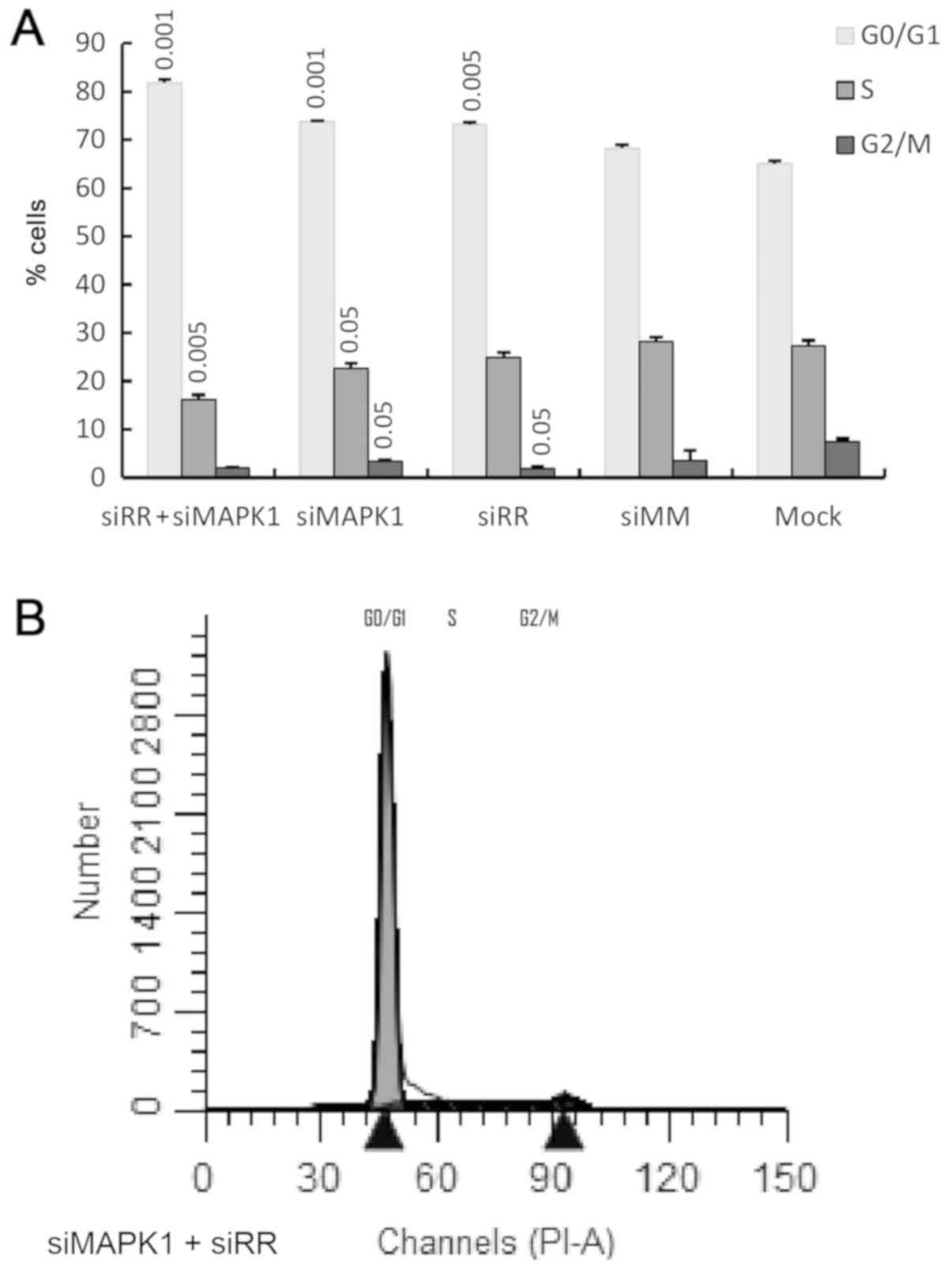
mock-transfected cells, RR suppression showed a reduction in
the percentage of cells in the S and G2M phases (Fig. 7A). This was correlated with
increased cells in the G0/G1 phase by ~13%.
Gene expression data following RR depletion in the Kasumi-1
and SKNO-1 cell lines showed that several genes involved in
positive and negative regulation of the cell cycle were variably
expressed. Upregulated genes included FOXO4 and ING4,
and downregulated genes were TPX2, CDC2, CKAP5, IL8, MAD2L1,
CCNE2, CCNG2, CDK6, TBRG4 and KPNA2. Isolated
MAPK1 suppression also caused an increased growth arrest.
This was accompanied with reduction in G2M and S phases, while
cells in the G0/G1 phase were increased
(Fig. 7A). FACS results for the
cell cycle analysis are included in Fig. 7B-F, respectively.
Combined targeting of RR and MAPK1
yielded further reduction of cells in S and in G2M phase by ~40 and
70%, respectively, when compared to the mock cells. This also led
to an increased number of cells in the G0/G1
phase by ~25%. Gene ontology for double knockdown of RR and
MAPK1, after exclusion of common target genes, demonstrated
that several protein coding genes were uniquely responsive to the
depletion of MAPK1; they included upregulation of RASSF1,
GADD45A, P53 and FBXO6, and downregulation of
MAPK11. The results of the GO enrichment analysis are
summarised in Table I.
Discussion
The consequence of RR translocation in
haematopoietic precursors is characterised by clonal proliferation
with reduced capacity to differentiate into committed progenitors
and therefore functional myeloid cells. Although the primary
genetic aberration t(8;21) found in self-renewing malignant
haematopoietic stem cells significantly alters the regulatory
processes controlling the growth and differentiation programmes, an
associated secondary lesion is often essential for the leukaemic
transformation and maintenance. Taken together, both primary and
secondary lesions confer a proliferation advantage and impair
haematopoietic differentiation in AML. Deregulated signalling in
the RAS/RAF/MEK/ERK pathway has often been implicated with
leukaemia, particularly in the RAS-RAF node of the pathway
(25,26). In this study, our focus was to
identify the patterns and networks of genes that were either
upregulated or downregulated driving the tumourigenic processes
including proliferation, apoptosis, cell cycle and differentiation
when RR and MAPK1 were suppressed. Gene expression
profiling has played a key role in providing us with important
clues to these processes.
We showed that continuous suppression of RR
is important for inhibiting cell proliferation, a finding
extensively validated and characterised by many researchers
(21,27). Our GO enrichment analysis showed
that this dampened proliferation was largely due to the
downregulation of several genes that are otherwise known to enhance
cell proliferation. Most important among them were KIT and
FLT3 genes that encode to class III transmembrane receptor
tyrosine kinases (RTK), which play a crucial role in
haematopoiesis. While the expression of FLT3 in
haematopoietic stem cells and progenitors are important to maintain
cell survival (28), c-Kit
expression by myeloblasts observed in ~80% of patients with AML
typically enhances proliferation (29). A negative correlation between the
level of c-Kit expression intensity on myeloblasts and the
number of leukocytes in the blood of AML patients has been reported
suggesting its role in myeloblast egression into peripheral
circulation (30). As these RTKs
are immediately upstream of the Ras/RAF/MAPK/ERK pathway, their
constitutive activation may lead to aberrant signalling,
proliferation and differentiation. Leveraging on this abnormal
signalling mechanism, several small-molecule tyrosine kinase
inhibitors have been developed and investigated as potential FLT3-
and c-KIT-targeted therapies for RTKs expressed on myeloblasts in
AML. It appears that patients with AML benefit from a synergistic
approach of kinase inhibitors used in combination with conventional
chemotherapy in controlling proliferation (31). On further dissection, the same set
of genes involved in inhibiting proliferation is also involved in
other biological processes. Based on the functional and biological
connectivity analysis on shared DEGs between cell lines in response
to RR suppression, at least eight (HIF1A, ITGB1, BCL2L1, CDK6,
CEBPA, IL8, FLT3 and KIT) DEGs were involved in cancer
pathways, five genes (IL13RA1, CD40, KIT, FLT3 and
IL8) in cytokine-cytokine receptor interactions and three
genes (CEBPA, KIT and FLT3) in the AML pathway.
We also showed that the antiproliferative
consequence of the co-suppression of MAPK1 and RR was
biologically no different from the effect exerted by isolated
repression of RR in Kasumi-1 cells. This failure of
MAPK1 depletion to augment the level of the
antiproliferation effect resulting from RR suppression
suggests the lack of coordinative synergy between the DEG sets.
Thus delivery of MAPK1 with RR siRNA may not serve as an attractive
gene therapy modality to control abnormal proliferation in t(8;21)
AML cells. GO enrichment analysis of the BeadChip data from the
MAPK1 suppressed Kasumi-1 and SKNO-1 cell lines demonstrated
six (6) DEGs with the biological
process of regulating cell proliferation. Among them, we found that
the expression of the NRAS oncogene (an upstream GTPase in
the classical ERK pathway) was suppressed in a
MAPK-1-dependent manner illustrating a possible feedback
loop within the ERK pathway. A previous study reported that ERK
exerts a negative feedback by interfering with RAS activation by
phosphorylating son of sevenless (SOS), an immediate early
gene in the ERK cascade (32).
Another interesting target of MAPK1 associated with the
proliferation and apoptosis in Kasumi-1 cells was the transcription
factor JUN, a subunit of the activator protein-1 (AP-1)
important for transactivation of CDKN2D. Downregulated
expression of JUN, in the absence of MAPK1 suggests a
cross-talk between the ERK and JNK pathways. A similar observation
has been made where ERK and Jun kinases underlie the
vascular endothelial cell growth factor-induced proliferation in
endothelial cells (33). These
observations suggest that MAPK1, the terminal kinase in the
ERK signalling pathway, may play a less significant role in the
progression of AML cells, and could be less effective as a
druggable target than RR in terms of impeding cell
proliferation. Another target of MAPK1 was the downregulation of
ZBTB16, a transcription factor that regulates the expression
of genes involved in cell growth and apoptosis, and may have
complemented the reduced proliferation observed in our study.
This study identified a panel of 10 genes whose
differential expression exerted an anti-apoptotic effect on
leukaemic myeloblast secondary to selective RR silencing. These
identified included upregulated BCL2, SPHK2, CFLAR, CD24,
NOTCH1, ADAM17, BIRC3 and HMGB1, and downregulated
IP6K2 and BECN1 genes. More important were the
overexpression of anti-apoptotic BCL2, BIRC3 and
CFLAR genes. Studies have shown that enhanced expression of
BCL2 in leukaemic myeloblasts offers them a survival
advantage by regulating the release of apoptogenic proteins from
mitochondria, notably cytochrome c, and resistance to
chemotherapy (34,35). This anti-apoptotic characteristic is
further strengthened by the increased activity of BIRC3, a member
of the IAP family of proteins, that acts as endogenous inhibitor of
caspases (36). Furthermore, the
loss of RR caused increased expression of CFLAR, a
critical anti-apoptotic regulator of apoptosis.
Although the selective RR silencing inflicts
an anti-apoptotic phenotype in t(8;21) myeloblasts, MAPK1
suppression reciprocally induces apoptosis while their
co-suppression ameliorates the effect (Fig. 5). This relative refractory survival
of t(8;21)-positive blasts upon enforced silencing of MAPK1
may be due to the apoptotic mitochondrial changes stimulated by the
activity of upregulated TP53 and TNFSF10, and
downregulated JUN gene, which regulates the release of
cytochrome c and drives the proteolytic caspases.
Additionally, NRAS and JUN, members of the classical
MAP kinase and JNK pathways, respectively, were
downregulated following the treatment of siMAPK1, demonstrating a
link between the two pathways. It was previously shown that
oncogenic RAS activates c-JUN via a separate pathway
from the activation of extracellular signal-regulated kinases
(37).
The significant loss of CD34 expression in the
RR-suppressed cells suggested that the fusion gene expression
exerted an anti-differentiation effect. Biological analysis
revealed that 13 genes, in response to RR suppression, were
involved in driving this process, and among them at least eight
genes (CEBPA, CEBPE, ID2, JMJD6 and IKZF1, CBFB, KIT
and CDK6) were directly responsible for myeloid progenitor
cell differentiation. Both CEBPA (CCAAT enhancer binding
protein α) and CEPBE are myeloid-specific transcription
factors that function as crucial regulators of granulopoiesis.
Their respective knockout mice have shown to block the
differentiation of common myeloid progenitor cells into
granulocyte-macrophage progenitor cells accompanied by
granulocytopaenia (38,39). Apart from its role in
differentiation, the consistent upregulation of myeloid
transcription factor CEBPA has been shown to arrests t(8;21) AML
cell proliferation through direct inhibition of CDK2 and CDK4
(40). A similar mechanism may be
partly responsible for reduced proliferation in our study as
RR knockdown resulted in upregulation of CEBPA and
downregulation of the G1 kinase CDK6, the homolog of
CDK4. The observed increased expression of ID2 subsequent to
RR suppression may function at later stages of differentiation of
t(8;21) to silence myeloblasts. This conclusion is proposed as
previous research has shown that ID2 mRNA is constitutively
expressed in more mature myeloid blast cells and the level markedly
increased towards terminal myeloid differentiation (41). Notably, we observed that
CBFB, which is required for myeloid and lymphoid
differentiation, was downregulated with the suppression of
RR suggesting that its deficiency does not block the
differentiation of myeloid progenitors. The observed loss of CD34
expression is further intensified by the downregulation of
CDK6 which otherwise is known to block myeloid
differentiation by interfering with RUNX1 DNA binding and
RUNX1-C/EBPα interaction (42).
Knockdown of MAPK1, but not RR,
exhibited elevated expression of ADA and downregulation of
JUN; both of which have been known to confer
anti-differentiation effects (43).
Therefore, it is not surprising that MAPK1-mediated
differential expression of ADA and JUN did not alter
the differentiation characteristics of the AML blasts (Fig. 6).
We showed that reduced proliferation upon isolated
RR and MAPK1 depletion was paralleled by cell cycle
arrest at the G0/G1 phase. A tightly
regulated molecular mechanism that controls the accurate transition
from G1 phase of the cell cycle to S phase is crucial
for eukaryotic cell proliferation, and its de-regulation promotes
oncogenesis. Upon siRNA-mediated RR depletion, we showed that eight
genes (TBRG4, CCNE2, FOXO4, CDK6, ING4, IL8, MAD2L1 and
CCNG2) under the GO category of negative regulation of the
cell cycle were downregulated except FOXO4. Noteworthy is
that the accumulation of Kasumi-1 cells in the G1 phase
of the cell cycle in response to RR suppression was important due
to the combined downregulated expression of CDK6 and
CCNE2 (cyclin E2). Regulatory protein CDK6 is a catalytic
subunit of the protein kinase complex that is important for cell
cycle G1 phase progression and G1/S
transition. GO enrichment analysis shows that the observed
G1 arrest in response to MAPK1 depletion in
t(8;21) myeloblasts was the result of increased expression of
negative regulators of the cell cycle: RASSF1, FBXO6,
DADD45A and P53. RASSF1A is an important human
tumour-suppressor protein that inhibits the accumulation of cyclin
D1 and induces cell cycle arrest acting at the level of
G1/S phase cell cycle progression by engaging the Rb
protein family (44).
GADD45α-induced cell cycle arrest leads to a reduced cell
proliferation rate. This is achieved by its interaction with
cyclin-dependent kinase inhibitor p21waf/cip/mda-6
leading to cell cycle arrest at G1/S transition due to
disruption in the formation of a CDK/cyclin complex (45). Most importantly, depletion of
MAPK1 significantly increased the expression of P53 and
leads to cell cycle arrest and apoptosis as observed in this
study.
The present study primarily utilised siRNAs to
silence target genes. Recently, more elaborate methods to induce
gene knockdown have been utilized. These include gene editing
techniques such as CRISPR (46–48)
which gives higher specificity and minimises off targets.
Nevertheless, gene silencing using small interfering RNA (siRNA)
should not be discounted altogether given its simplicity and
reproducibility. Other factors that should be considered in gene
silencing experiments for minimizing off targets include using more
than one target siRNA for each targeted gene or having a mismatch
control. Overexpression studies of MAPK1 merits further
investigation and could be carried out to ascertain the link
between RR and MAPK1. Furthermore, the expression of other partners
in the Ras/Raf/MEK/ERK pathways could be explored both at the gene
and protein levels to shed some mechanistic insight regarding the
interactions between MAPK1, RR and leukemogenesis.
In conclusion, in the present study, we showed that
MAPK1 simultaneously promotes myeloid cell proliferation and
differentiation by cell cycle progression and blocking apoptosis.
GO enrichment analysis showed that the processes involved in cell
cycle, cell proliferation, and cell differentiation were affected
by MAP1 downregulation.
Acknowledgements
Not applicable.
Funding
This study was supported by Research University
grants 1001/CIPPT/812095 and 1001/CIPPT/813064 from Universiti
Sains Malaysia.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SAASB, OH and NMY conceived and designed the study.
SAASB performed the experiments. MS and EJM wrote the paper and
analysed the data. EJM, NMY and OH reviewed and edited the
manuscript. NMY funded the project. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Grimwade D, Hills RK, Moorman AV, Walker
H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ and Burnett
AK; National Cancer Research Institute Adult Leukaemia Working
Group, : Refinement of cytogenetic classification in acute myeloid
leukemia: Determination of prognostic significance of rare
recurring chromosomal abnormalities among 5876 younger adult
patients treated in the United Kingdom Medical Research Council
trials. Blood. 116:354–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nucifora G and Rowley JD: AML1 and the
8;21 and 3;21 translocations in acute and chronic myeloid leukemia.
Blood. 86:1–14. 1995.PubMed/NCBI
|
|
3
|
Miyoshi H, Kozu T, Shimizu K, Enomoto K,
Maseki N, Kaneko Y, Kamada N and Ohki M: The t(8;21) translocation
in acute myeloid leukemia results in production of an AML1-MTG8
fusion transcript. EMBO J. 12:2715–2721. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chang KS, Fan YH, Stass SA, Estey EH, Wang
G, Trujillo JM, Erickson P and Drabkin H: Expression of AML1-ETO
fusion transcripts and detection of minimal residual disease in
t(8;21)-positive acute myeloid leukemia. Oncogene. 8:983–988.
1993.PubMed/NCBI
|
|
5
|
Heidenreich O, Krauter J, Riehle H,
Hadwiger P, John M, Heil G, Vornlocher HP and Nordheim A: AML1/MTG8
oncogene suppression by small interfering RNAs supports myeloid
differentiation of t(8;21)-positive leukemic cells. Blood.
101:3157–3163. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Trombly DJ, Whitfield TW, Padmanabhan S,
Gordon JA, Lian JB, van Wijnen AJ, Zaidi SK, Stein JL and Stein GS:
Genome-wide co-occupancy of AML1-ETO and N-CoR defines the t(8;21)
AML signature in leukemic cells. BMC Genomics. 16:3092015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hildebrand D, Tiefenbach J, Heinzel T,
Grez M and Maurer AB: Multiple regions of ETO cooperate in
transcriptional repression. J Biol Chem. 276:9889–9895. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan Y, Zhou L, Miyamoto T, Iwasaki H,
Harakawa N, Hetherington CJ, Burel SA, Lagasse E, Weissman IL,
Akashi K and Zhang DE: AML1-ETO expression is directly involved in
the development of acute myeloid leukemia in the presence of
additional mutations. Proc Natl Acad Sci USA. 98:10398–10403. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maiques-Diaz A, Chou FS, Wunderlich M,
Gómez-López G, Jacinto FV, Rodriguez-Perales S, Larrayoz MJ,
Calasanz MJ, Mulloy JC, Cigudosa JC and Alvarez S: Chromatin
modifications induced by the AML1-ETO fusion protein reversibly
silence its genomic targets through AML1 and Sp1 binding motifs.
Leukemia. 26:1329–1337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klampfer L, Zhang J, Zelenetz AO, Uchida H
and Nimer SD: The AML1/ETO fusion protein activates transcription
of BCL-2. Proc Natl Acad Sci USA. 93:14059–14064. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Linggi B, Müller-Tidow C, van de Locht L,
Hu M, Nip J, Serve H, Berdel WE, van der Reijden B, Quelle DE,
Rowley JD, et al: The t(8;21) fusion protein, AML1 ETO,
specifically represses the transcription of the p14(ARF) tumor
suppressor in acute myeloid leukemia. Nat Med. 8:743–750. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vangala RK, Heiss-Neumann MS, Rangatia JS,
Singh SM, Schoch C, Tenen DG, Hiddemann W and Behre G: The myeloid
master regulator transcription factor PU.1 is inactivated by
AML1-ETO in t(8;21) myeloid leukemia. Blood. 101:270–277. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pabst T, Mueller BU, Harakawa N, Schoch C,
Haferlach T, Behre G, Hiddemann W, Zhang DE and Tenen DG: AML1-ETO
downregulates the granulocytic differentiation factor C/EBPalpha in
t(8;21) myeloid leukemia. Nat Med. 7:444–451. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rio-Machín A, Menezes J, Maiques-Diaz A,
Agirre X, Ferreira BI, Acquadro F, Rodriguez-Perales S, Juaristi
KA, Alvarez S and Cigudosa JC: Abrogation of RUNX1 gene expression
in de novo myelodysplastic syndrome with t(4;21)(q21;q22).
Haematologica. 97:534–537. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta
1773. 1263–1284. 2007.
|
|
16
|
Morgan MA, Dolp O and Reuter CW:
Cell-cycle-dependent activation of mitogen-activated protein kinase
kinase (MEK-1/2) in myeloid leukemia cell lines and induction of
growth inhibition and apoptosis by inhibitors of RAS signaling.
Blood. 97:1823–1834. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Towatari M, Iida H, Tanimoto M, Iwata H,
Hamaguchi M and Saito H: Constitutive activation of
mitogen-activated protein kinase pathway in acute leukemia cells.
Leukemia. 11:479–484. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Milella M, Kornblau SM, Estrov Z, Carter
BZ, Lapillonne H, Harris D, Konopleva M, Zhao S, Estey E and
Andreeff M: Therapeutic targeting of the MEK/MAPK signal
transduction module in acute myeloid leukemia. J Clin Invest.
108:851–859. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zaidi SK, Dowdy CR, van Wijnen AJ, Lian
JB, Raza A, Stein JL, Croce CM and Stein GS: Altered Runx1
subnuclear targeting enhances myeloid cell proliferation and blocks
differentiation by activating a miR-24/MKP-7/MAPK network. Cancer
Res. 69:8249–8255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Spirin PV, Lebedev TD, Orlova NN,
Gornostaeva AS, Prokofjeva MM, Nikitenko NA, Dmitriev SE, Buzdin
AA, Borisov NM, Aliper AM, et al: Silencing AML1-ETO gene
expression leads to simultaneous activation of both pro-apoptotic
and proliferation signaling. Leukemia. 28:2222–2228. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shih LY, Huang CF, Wang PN, Wu JH, Lin TL,
Dunn P and Kuo MC: Acquisition of FLT3 or N-ras mutations is
frequently associated with progression of myelodysplastic syndrome
to acute myeloid leukemia. Leukemia. 18:466–475. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bacher U, Haferlach T, Kern W, Haferlach C
and Schnittger S: A comparative study of molecular mutations in 381
patients with myelodysplastic syndrome and in 4130 patients with
acute myeloid leukemia. Haematologica. 92:744–752. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martinez N, Drescher B, Riehle H, Cullmann
C, Vornlocher HP, Ganser A, Heil G, Nordheim A, Krauter J and
Heidenreich O: The oncogenic fusion protein RUNX1-CBFA2T1 supports
proliferation and inhibits senescence in t(8;21)-positive leukaemic
cells. BMC Cancer. 4:442004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kikushige Y, Yoshimoto G, Miyamoto T, Iino
T, Mori Y, Iwasaki H, Niiro H, Takenaka K, Nagafuji K, Harada M, et
al: Human Flt3 is expressed at the hematopoietic stem cell and the
granulocyte/macrophage progenitor stages to maintain cell survival.
J Immunol. 180:7358–7367. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ikeda H, Kanakura Y, Tamaki T, Kuriu A,
Kitayama H, Ishikawa J, Kanayama Y, Yonezawa T, Tarui S and Griffin
JD: Expression and functional role of the proto-oncogene c-kit in
acute myeloblastic leukemia cells. Blood. 78:2962–2968.
1991.PubMed/NCBI
|
|
30
|
Woźniak J and Kopeć-Szlezak J: c-Kit
receptor (CD117) expression on myeloblasts and white blood cell
counts in acute myeloid leukemia. Cytometry Part B Clin Cytometry.
58:9–16. 2004. View Article : Google Scholar
|
|
31
|
Stone RM, Mandrekar SJ, Sanford BL,
Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Döhner K,
Marcucci G, et al: Midostaurin plus chemotherapy for acute myeloid
leukemia with a FLT3 mutation. N Engl J Med. 377:454–464.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dong Chen, Waters SB, Holt KH and Pessin
JE: SOS phosphorylation and disassociation of the Grb2-SOS complex
by the ERK and JNK signaling pathways. J Biol Chem. 271:6328–6332.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pedram A, Razandi M and Levin ER:
Extracellular signal-regulated protein kinase/Jun kinase cross-talk
underlies vascular endothelial cell growth factor-induced
endothelial cell proliferation. J Biol Chem. 273:26722–26728. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Andreeff M, Jiang S, Zhang X, Konopleva M,
Estrov Z, Snell VE, Xie Z, Okcu MF, Sanchez-Williams G, Dong J, et
al: Expression of Bcl-2-related genes in normal and AML
progenitors: Changes induced by chemotherapy and retinoic acid.
Leukemia. 13:1881–1892. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
36
|
Salvesen GS and Duckett CS: IAP proteins:
Blocking the road to death's door. Nat Rev Mol Cell Biol.
3:401–410. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Westwick JK, Cox AD, Der CJ, Cobb MH, Hibi
M, Karin M and Brenner DA: Oncogenic Ras activates c-Jun via a
separate pathway from the activation of extracellular
signal-regulated kinases. Proc Natl Acad Sci USA. 91:6030–6034.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus
ML, Dayaram T, Owens BM, Shigematsu H, Levantini E, Huettner CS,
Lekstrom-Himes JA, et al: Enhancement of hematopoietic stem cell
repopulating capacity and self-renewal in the absence of the
transcription factor C/EBP alpha. Immunity. 21:853–863. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yamanaka R, Barlow C, Lekstrom-Himes J,
Castilla LH, Liu PP, Eckhaus M, Decker T, Wynshaw-Boris A and
Xanthopoulos KG: Impaired granulopoiesis, myelodysplasia, and early
lethality in CCAAT/enhancer binding protein epsilon-deficient mice.
Proc Natl Acad Sci USA. 94:13187–13192. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang H, Iakova P, Wilde M, Welm A, Goode
T, Roesler WJ and Timchenko NA: C/EBPα arrests cell proliferation
through direct inhibition of Cdk2 and Cdk4. Mol Cell. 8:817–828.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ishiguro A, Spirin KS, Shiohara M, Tobler
A, Gombart AF, Israel MA, Norton JD and Koeffler HP: Id2 expression
increases with differentiation of human myeloid cells. Blood.
87:5225–5231. 1996.PubMed/NCBI
|
|
42
|
Fujimoto T, Anderson K, Jacobsen SE,
Nishikawa Si and Nerlov C: Cdk6 blocks myeloid differentiation by
interfering with Runx1 DNA binding and Runx1-C/EBPalpha
interaction. EMBO J. 26:2361–2370. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rangatia J, Vangala RK, Treiber N, Zhang
P, Radomska H, Tenen DG, Hiddemann W and Behre G: Downregulation of
c-Jun expression by transcription factor C/EBPα is critical for
granulocytic lineage commitment. Mol Cell Biol. 22:8681–8694. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shivakumar L, Minna J, Sakamaki T, Pestell
R and White MA: The RASSF1A tumor suppressor blocks cell cycle
progression and inhibits cyclin D1 accumulation. Mol Cell Biol.
22:4309–4318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kearsey JM, Coates PJ, Prescott AR,
Warbrick E and Hall PA: Gadd45 is a nuclear cell cycle regulated
protein which interacts with p21Cip1. Oncogene. 11:1675–1683.
1995.PubMed/NCBI
|
|
46
|
Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan
CA and Musunuru K: Enhanced efficiency of human pluripotent stem
cell genome editing through replacing TALENs with CRISPRs. Cell
Stem Cell. 12:393–394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shafie NH, Saleem M, Moses EJ, Razak SR
and Yusoff NM: The CRISPR-Cas9 system: A new dawn in gene editing.
J Bioanalys Biomed. 6:45–48. 2014.
|
|
48
|
Kleinstiver BP, Pattanayak V, Prew MS,
Tsai SQ, Nguyen NT, Zheng Z and Joung JK: High-fidelity CRISPR-Cas9
nucleases with no detectable genome-wide off-target effects.
Nature. 529:490–495. 2016. View Article : Google Scholar : PubMed/NCBI
|