Introduction
Gastric cancer is a leading cause of cancer-related
deaths worldwide (1). Recent
studies have revealed that immune-targeting therapy improves the
survival of patients with gastric cancer. Numerous chemotherapy
regimens have been clinically examined for treating gastric cancer;
however, there is an urgent need for novel therapeutic agents for
the treatment of gastric cancer.
Imatinib mesylate (imatinib) is a powerful tyrosine
kinase inhibitor that specifically targets BCR-ABL, KIT, and
platelet-derived growth factor receptor (PDGFR) kinases and is used
for treating chronic myelogenous leukemia (CML), gastrointestinal
stromal tumors (GISTs), and other types of cancers (2,3).
However, the cellular and molecular mechanisms underlying the
antitumor effects of imatinib are unknown.
PDGFR-α and PDGFR-β are transmembrane tyrosine
kinase receptors whose ligands play critical roles in cancer cell
migration and proliferation (4–6). The
PI3K/AKT pathway is an important downstream signaling pathway of
PDGFR that plays important roles in promoting cell proliferation,
suppressing cell motility, and inhibiting cell apoptosis. Increased
PDGFR expression is also detected in various solid cancers
(7).
The endoplasmic reticulum (ER) plays a major role in
protein synthesis and maturation, lipid synthesis, calcium
homeostasis, and protein folding (8). Misfolded and unfolded proteins,
particularly secretory and transmembrane proteins produced in the
ER, induce an evolutionarily conserved unfolded protein response
(UPR), an ER stress pathway, which in turn induces various
pathological events such as inflammation, aging, and
neurodegenerative disorders. The UPR, which improves folding, is a
survival response triggered by cells to restore ER homeostasis.
Chronic accumulation of unfolded proteins in the ER may lead to
death receptor-independent and mitochondria-mediated apoptotic
pathways (9–12).
At low concentrations, intracellular oxidants
function as signal transducers to induce growth factors, hypoxia,
and other receptor-ligand systems (13,14).
However, at concentrations above threshold levels, these oxidants
induce damage of lipids, proteins, RNA, and DNA, thus triggering
cell death through apoptosis and/or necrosis (15–17).
Reactive oxygen species (ROS) such as oxygen ions and peroxides are
chemically reactive molecules containing oxygen that are formed as
natural by-products of normal oxygen metabolism and play important
roles in cell signaling and homeostasis (18). Biological functions of ROS and their
potential roles in cancer development and disease progression have
been investigated over the past several decades. ROS mediate cancer
cell apoptosis induced by various anticancer agents and other
stimuli (19). However, various
studies have also shown that anticancer agents increase apoptosis
of malignant cells by decreasing ROS production (20).
However, the role of ROS, and mechanisms underlying
their antitumorigenic function in solid tumors, particularly
gastric cancer, are unknown. In the present study, we evaluated
whether imatinib exerted antitumorigenic effects on gastric cancer
cells by inducing apoptosis. Our results indicated that IRE1α-c-Jun
NH2-terminal kinase (JNK)- and C/EBP homologous protein
(CHOP)-associated ER stress was involved in imatinib-induced
apoptosis of gastric cancer cells, suggesting that imatinib is a
potential chemotherapeutic agent for treating gastric cancer.
Materials and methods
Reagents and antibodies
Imatinib was purchased from Novartis International
AG (Basel, Switzerland). JNK inhibitor SP600125 was obtained from
EMD/Merck KGaA (Darmstadt, Germany). Rabbit antibodies against PARP
(1:1,000 dilution; cat. no. 9542), caspase-3 (1:1,000 dilution;
cat. no. 9662), Bid (1:1,000 dilution; cat. no. 2002), Bim (1:1,000
dilution; cat. no. 2819), Puma (1:1,000 dilution; cat. no. 4976),
Noxa (1:1,000 dilution; cat. no. 14766), p-JNK (1:1,000 dilution;
cat. no. 9251), JNK (1:1,000 dilution; cat. no. 9252), PDGFR-α
(1:1,000 dilution; cat. no. 3164), PDGFR-β (1:1,000 dilution; cat.
no. 3169), p-PDGFR-α (1:1,000 dilution; cat. no. 4547), p- PDGFR-β
(1:1,000 dilution; cat. no. 3161), AKT (1:1,000 dilution; cat. no.
9272), p-AKT (1:1,000 dilution; cat. no. 4060), 4EBP1 (1:1,000
dilution; cat. no. 9644), p-4EBP1 (1:1,000 dilution; cat. no.
2855), p70 S6 kinase (1:1,000 dilution; cat. no. 2708), p-p70 S6
kinase (Ser371) (1:1,000 dilution; cat. no. 9208), p-p70 S6 kinase
(Thr 389) (1:1,000 dilution; cat. no. 9205), mTOR (1:1,000
dilution; cat. no. 2983), p38 MAPK (1:1,000 dilution; cat. no.
9212), p-p38 MAPK (1:1,000 dilution; cat. no. 9211), p44/42 MAPK
(ERK1/2) (137F5) (1:1,000 dilution; cat. no. 4695), p-p44/42 MAPK
(Erk1/2) (Thr202/Tyr204) (1:1,000 dilution; cat. no. 4370), eIF2α
(D7D3) (1:1,000 dilution; cat. no. 5324), p-eIF2α (Ser51) (1:1,000
dilution; cat. no. 3597) and GRP94 (1:1,000 dilution; cat. no.
2104) and IRE1α (1:1,000 dilution; cat. no. 3294) were obtained
from Cell Signaling Technology, Inc. (Beverly, MA, USA). Rabbit
antibody against p-IRE1 α (1:1,000 dilution; cat. no. ab48187) were
obtained from Abcam (Cambridge, UK). Mouse antibodies against
BCL2-associated X protein (Bax) ATF6 (1:1,000 dilution; cat. no.
sc-20067), Bcl-2 (1:1,000 dilution; cat. no. sc-509), Survivin
(1:1,000 dilution; cat. no. sc-17779) and CHOP (GADD 153) (1:1,000
dilution; cat. no. sc-7351) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Mouse antibodies against
ATF6 (1:1,000 dilution; cat. no. NBP1-40256) were obtained from
Novus Biologicals, LLC (Littleton, CO, USA). Anti-actin (1:2,000
dilution; cat. no. A2228) and DCFH-DA (cat. no. D6883) were
purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Cell culture
Human gastric cancer cell line AGS was obtained from
the American Type Culture Collection (ATCC; Manassas, VA, USA), and
SNU-638 and MKN45 cell lines were purchased from the Korean Cell
Line Bank (Seoul, Korea). CHOP−/− and corresponding
wild-type MEF cell lines were provided by Dr Randal J. Kaufman
(Sanford Burnham Medical Research Institute, CA, USA). Gastric
cancer cells were cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS) and 1% antibiotic-antimycotic solution
(Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified
atmosphere of 5% CO2.
Cell viability assay
Cell growth rate and viability were determined by
performing 3-(4,5-dimethylthiazol-2-ly)-2,5-diphenyl tetrazolium
bromide (MTT)-based colorimetric assays (Sigma-Aldrich; Merck
KGaA). Cells were grown in 96-well plates containing medium
supplemented with 10% fetal bovine serum at a density of
1×104 cells/well. The medium in each well contained
different concentrations of imatinib. After 48 h, 50 µl MTT
solution was added to each well, and the cells were incubated at
37°C for 4 h. Next, the supernatant was aspirated, and the dye was
solubilized using 200 µl dimethyl sulfoxide (DMSO). Cell viability
was determined according to the manufacturer's instructions by
measuring the absorbance in individual wells at 595 nm using a
microplate reader.
Cell cycle and apoptosis analyses
Cell cycle was analyzed by performing propidium
iodide (PI) staining. Adherent cells were harvested by treatment
with trypsin and were fixed with 5 mM EDTA and 85% ethanol. Fixed
cells were incubated with 50 µg/ml PI and 20 µg/ml RNase at 37°C
for 30 min and were analyzed by performing flow cytometry.
Apoptotic cells were stained using an Annexin V-fluorescein
isothiocyanate (FITC) kit (Beckman Coulter, Inc., Brea, CA, USA),
according to the manufacturer's instructions, and were then
analyzed.
Western blot analysis
Cells harvested 48 h after imatinib treatment were
centrifuged at 14,000 × g and 4°C for 5 min and were washed twice
with phosphate-buffered saline (PBS). Next, the cells were lysed in
30 µl ice-cold RIPA buffer and were centrifuged again at 14,000 × g
and 4°C for 30 min. The obtained pellet was stored at −70°C for
further analysis. Protein concentrations were determined using the
Bradford method (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Lysates corresponding to equal protein amounts were boiled in
sample buffer for 5 min. Next, the samples were resolved by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10
or 12% gels, with 50 µg total protein loaded in each lane of the
gel. The resolved proteins were transferred onto a 0.45-µm
nitrocellulose membranes. The membranes were blocked with 5%
non-fat dry milk in Tris-buffered saline with Tween-20 (TBST)
buffer for 2 h to prevent non-specific binding, and was then washed
three times with TBST for 10 min each. Next, the membrane was
incubated with specific primary antibodies, followed by incubation
at 4°C overnight with horseradish peroxidase-conjugated secondary
antibody and then incubated with anti-mouse (1:2,500 dilution; cat.
no. 170-6516; Bio-Rad Laboratories) or anti-rabbit (1:2,500
dilution; cat. no. 7074; Cell Signaling Technology, Inc.) specific
polyclonal secondary antibodies for 2 h at 4°C. Then, the membrane
was washed three times with TBST for 10 min each. Signals were
detected by exposure to X-ray film using an EZ-Western Lumi Pico
kit (DoGEN, Seoul, Korea; cat. no. DG-WP-250).
ROS generation
ROS generation was assessed in cells treated with
imatinib for 30 or 60 min. Intracellular ROS levels were determined
using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), which
oxidizes within cells to fluorescent dichlorofluorescein. Control
and imatinib-treated cells were incubated with 100 µM DCFH-DA for
30 min at 37°C.
Next, the cells were fixed in 3.7% paraformaldehyde
for 10 min at room temperature. Subsequently, the cells were
co-stained with 2 µM 4′,6-diamidino-2-phenylindole (Molecular
Probes; Thermo Fisher Scientific, Inc.) at 37°C. After washing
three times with PBS, the cells were mounted using VECTASHIELD
mounting medium (Vector Laboratories, Inc., Burlingame, CA, USA),
and immunofluorescence was detected using a confocal microscope
(Carl Zeiss AG, Oberkochen, Germany). Then, MEF of MEF
CHOP−/− cells were stained with 100 µM DCFH-DA for 30
min at 37°C. The cells were then transferred to FACS tubes and
analyzed using the FL1 channel on a FACScan cytometer (BD
Biosciences, Franklin Lakes, NJ, USA).
Statistical analysis
Statistical analysis was performed using GraphPad
InStat 5 software (GraphPad Software, Inc., San Diego, CA, USA).
Statistical significance was determined by one-way analysis of
variance (ANOVA) followed by Bonferroni post hoc test for multiple
comparisons or Student's t-test. For all tests, P<0.05 or
P<0.01 were considered to indicate statistically significant
differences.
Results
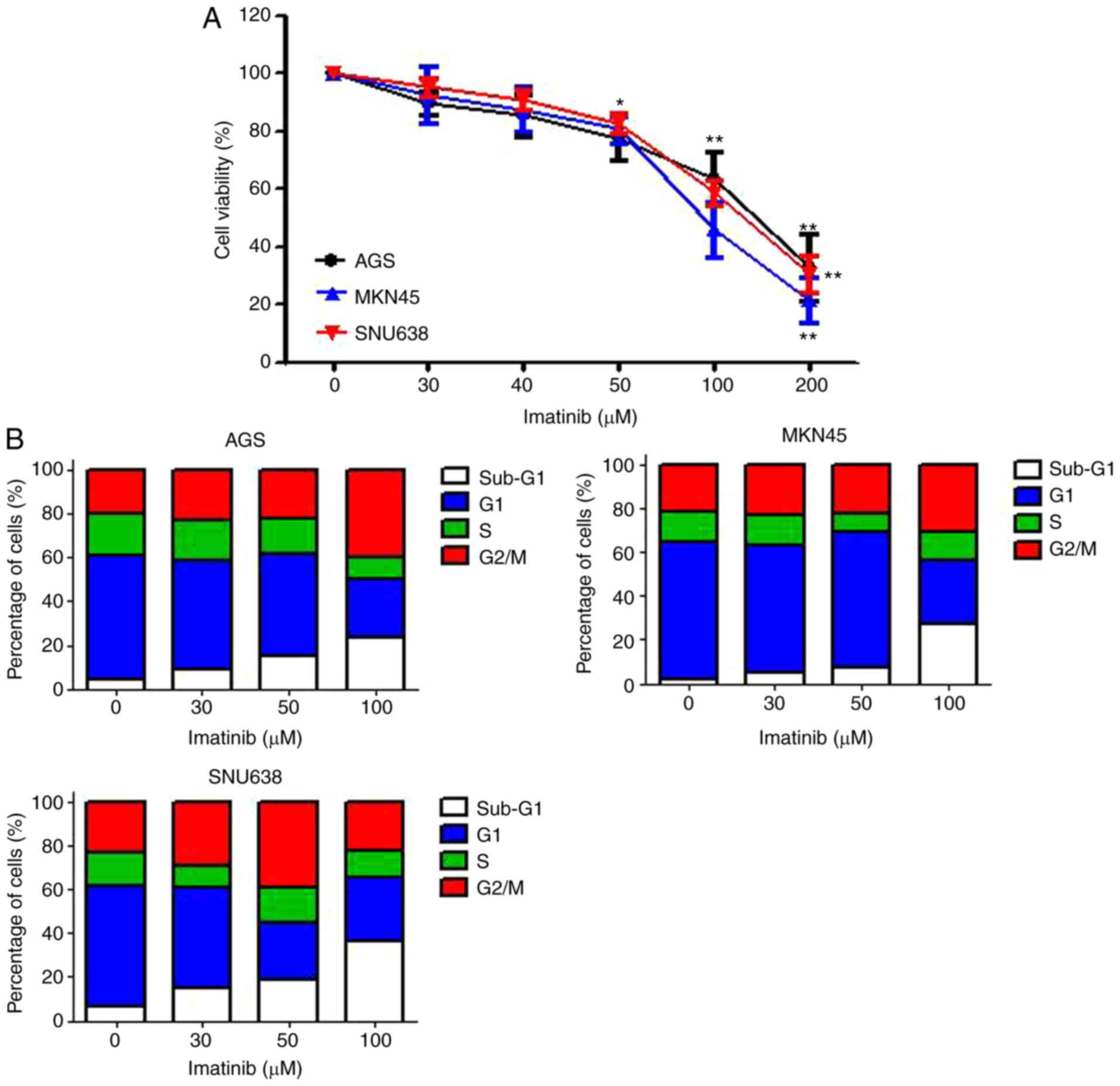
Imatinib decreases the viability of
gastric cancer cells
To investigate the antitumor activity of imatinib
against gastric cancer cells (AGS, MKN45 and SNU638 cells), the
cells were incubated at different concentrations of imatinib for 48
h and their viability was determined by performing MTT assays.
Results of the MTT assays revealed that imatinib treatment
significantly decreased cell viability in a dose-dependent manner.
Statistically significant cytotoxic effects were observed after
treatment with >50 µM imatinib (Fig.
1A). Next, we determined the effect of imatinib on cell cycle
progression. For this, the cells were treated with various
concentrations of imatinib, and cell cycle progression was assessed
after 48 h by performing flow cytometry with PI staining. Imatinib
treatment for 48 h significantly increased the percentage of cells
in the sub-G1 and G2/M phases of the cell cycle, in a
dose-dependent manner. These data indicated that imatinib treatment
arrested gastric cancer cells in the sub-G1 phase of the cell cycle
(Fig. 1B), and also indicated that
imatinib exerted anti-proliferative effects on gastric cancer
cells.
Imatinib induces apoptosis of gastric
cancer cells
We performed Annexin V/PI double staining to
evaluate whether imatinib induced apoptosis of the three gastric
cancer cell lines. Results of Annexin V/PI double staining revealed
that the percentage of Annexin V/PI-positive cells significantly
increased, indicating that imatinib treatment increased the early
apoptosis of gastric cancer cells (Fig.
2A). Next, we performed western blot analysis to confirm
imatinib-induced apoptosis of gastric cancer cells, and to identify
the underlying mechanism. Imatinib treatment for 48 h increased
poly(ADP-ribose) polymerase (PARP) cleavage, and cleaved caspase-3
and −9 expression in AGS cells (Fig.
2B). Bcl-2 is an anti-apoptotic protein, and Bax is a
pro-apoptotic protein. We observed that Bax and Bcl-2 levels were
unaffected in imatinib-treated AGS cells. Pro-apoptotic Bcl-2
family proteins (Noxa and Bid) were induced in imatinib-treated
cells. Similar results were obtained for imatinib-treated MKN45 and
SNU638 gastric cancer cells (Fig. 2C
and D). These results indicated that imatinib treatment induced
apoptosis of gastric cancer cells.
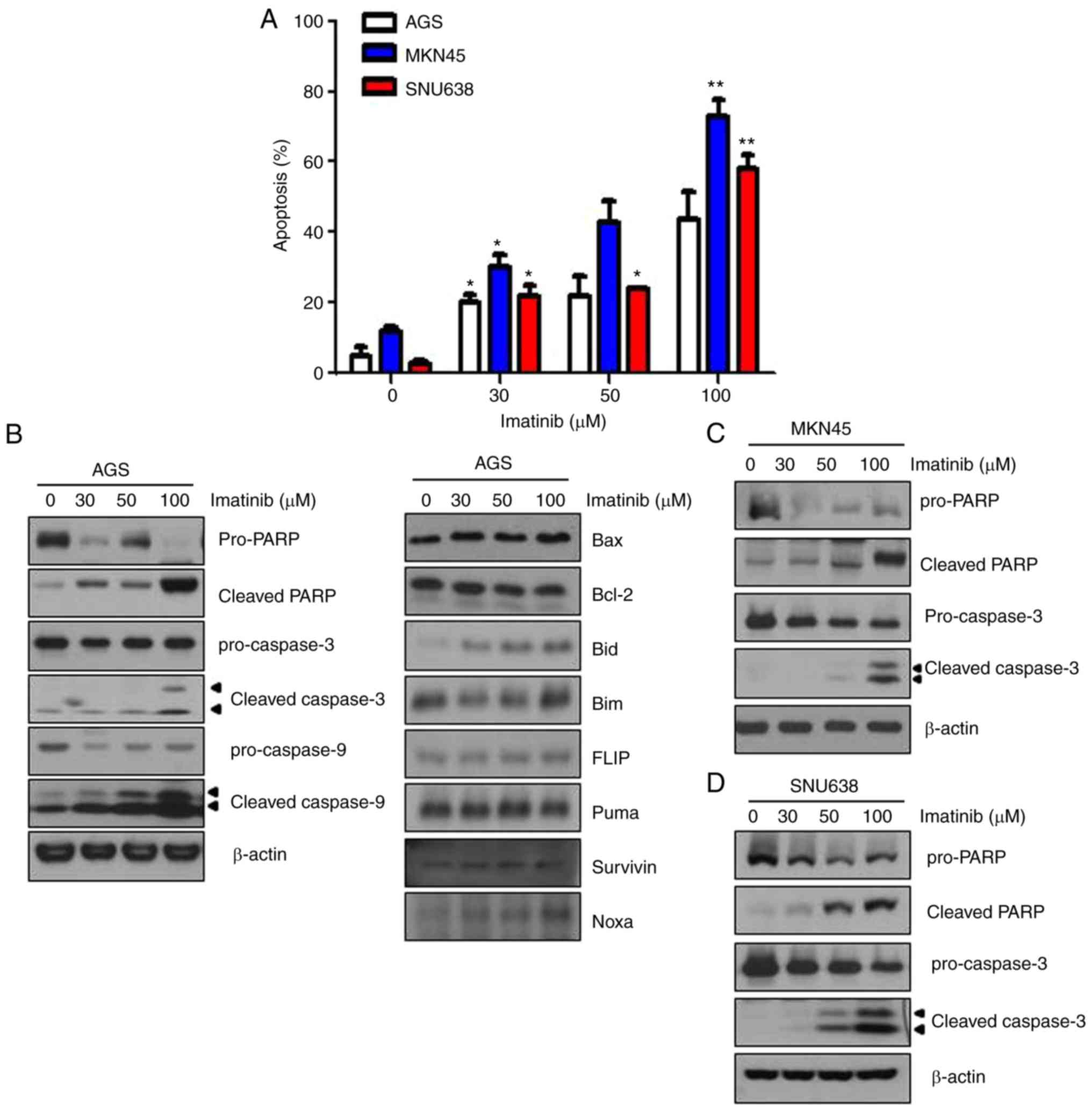 | Figure 2.Imatinib induces the apoptosis of
gastric cancer cells. (A) AGS, MKN45, and SNU638 cells were stained
with Annexin V-FITC and PI after incubation with the indicated
concentrations of imatinib for 48 h, and the number of apoptotic
cells was analyzed by performing flow cytometry. Statistical
analysis of the results of flow cytometry. Annexin V/PI-positive
cells were defined as apoptotic cells. Results are expressed as the
means ± standard error of three individual experiments. *P<0.05
and **P<0.01 compared with the control cells. (B-D) AGS, MKN45,
and SNU638 cells were treated with the indicated concentrations of
imatinib for 48 h. Levels of cleaved caspase-3, cleaved caspase-9,
cleaved PARP, Bax, and Bcl-2 were determined by performing
immunoblotting. β-actin was used as an internal standard. FITC,
fluorescein isothiocyanate; PI, propidium iodide; Bax,
BCL2-associated X protein; PARP, poly(ADP-ribose) polymerase. |
Imatinib decreases PDGFR signaling in
gastric cancer cells
Imatinib is a powerful tyrosine kinase inhibitor
that specifically targets BCR-ABL, KIT, and PDGFR kinases, and is
used for treating CML, GISTs, and other types of cancers (3,21–24).
In the present study, c-KIT and BCR-ABL expression was not detected
in the three gastric cancer cell lines (data not shown). Next, we
examined the effects of imatinib on PDGFR signaling in AGS cells
and found that imatinib efficiently suppressed PDGFR-induced
downstream signaling (Fig. 3A).
Imatinib inhibited the phosphorylation of PDGFR-α, PDGFR-β, AKT and
p70S6K (Ser371). Notably, the phosphorylation of PDGFR-β decreased
more markedly than that of PDGFR-α in imatinib-treated cells.
However, no significant changes were observed in total protein
levels in imatinib-treated cells.
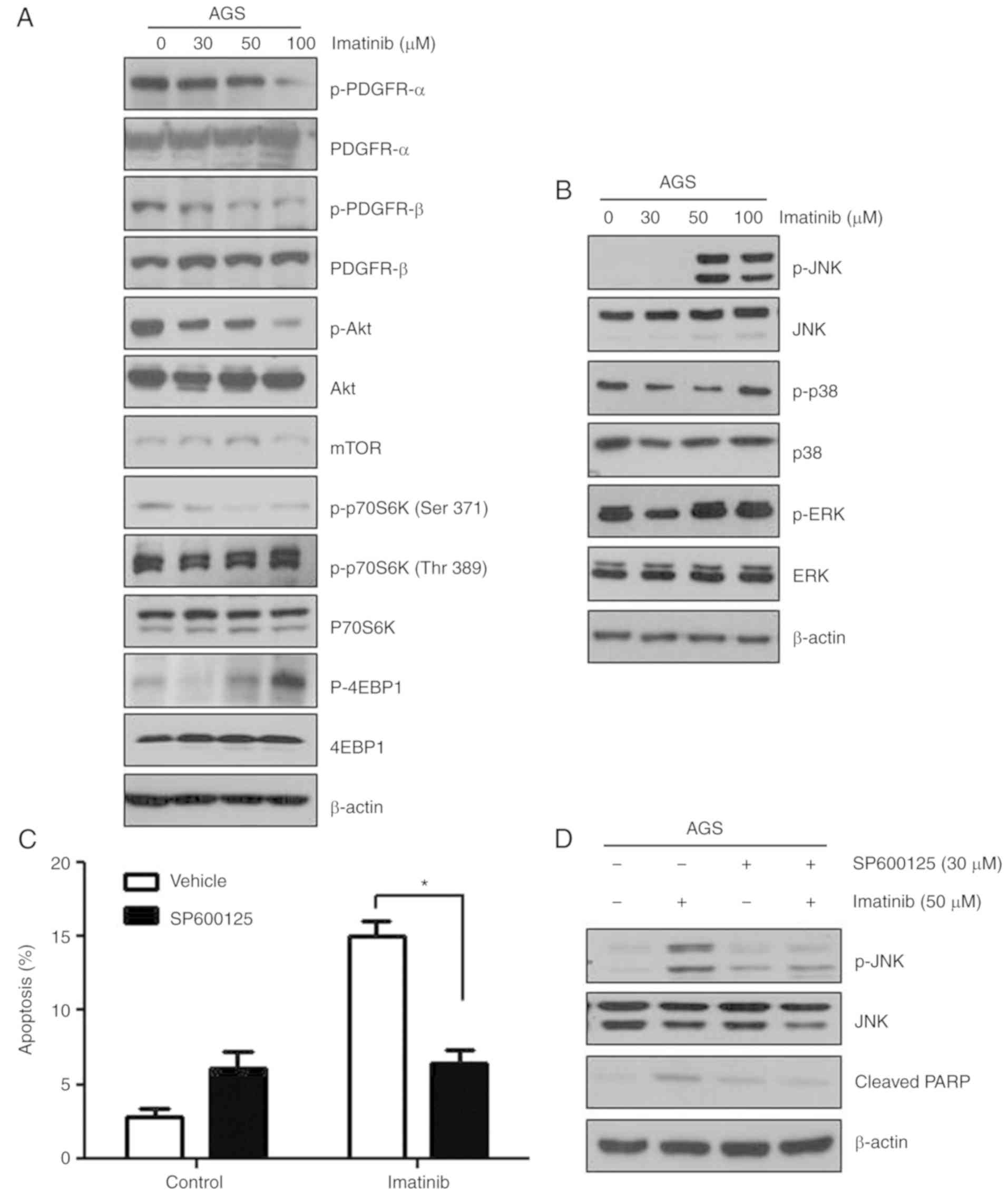 | Figure 3.Imatinib induces apoptosis by
phosphorylating JNK in gastric cancer cells. (A) AGS cells were
treated with the indicated concentrations of imatinib for 48 h.
Expression levels of proteins associated with the PDGFR pathway
were assessed by performing immunoblotting with the corresponding
antibodies. β-actin was used as an internal standard. (B) AGS cells
were treated with the indicated concentrations of imatinib for 48
h, and the levels of JNK, p-JNK, p38, p-p38, ERK, p-ERK and β-actin
were assessed by performing immunoblotting with the corresponding
antibodies. (C) Blocking of the JNK pathway effectively suppressed
imatinib-induced apoptosis of AGS cells. *P<0.05 compared to the
vehicle. (D) AGS cells were pretreated with 20 µM JNK inhibitor
(SP600125) for 1 h, followed by treatment with 50 µM imatinib for
48 h. Levels of cleaved PARP, JNK and p-JNK were determined by
performing immunoblotting. β-actin was used as an internal
standard. JNK, c-Jun NH2-terminal kinase; PDGFR, platelet-derived
growth factor receptor. |
Imatinib induces apoptosis by
phosphorylating JNK in gastric cancer cells
Various cellular stresses and stimuli induce
mitogen-activated protein kinase (MAPK) signaling that contributes
to apoptotic induction (25). To
determine the effect of imatinib on the expression and activity of
molecules involved in MAPK signaling, we treated AGS cells with 30,
50 and 100 µM imatinib for 48 h and performed western blot
analysis. Imatinib treatment increased JNK phosphorylation in a
dose-dependent manner, without affecting total JNK levels, but did
not affect p38 and ERK phosphorylation (Fig. 3B). To verify the involvement of JNK
in imatinib-induced apoptosis, the cells were co-treated with a JNK
pathway inhibitor (SP600125) and imatinib (50 µM). Treatment of
gastric cancer cells with SP600125 significantly decreased their
imatinib-induced apoptosis due to JNK inhibition (Fig. 3C). Moreover, treatment with SP600125
completely abolished imatinib-induced PARP activation (Fig. 3D). These results indicated that
imatinib-induced apoptosis was mediated by the JNK-MAPK
pathway.
Imatinib-induced apoptosis of gastric
cancer cells is mediated by ER stress induction and ROS
generation
AGS and SNU638 gastric cancer cells were used to
investigate mechanisms underlying imatinib-induced apoptosis.
Recent research has indicated that ER stress plays a crucial role
in the regulation of apoptosis. To confirm that ER stress was
involved in imatinib-induced apoptosis, we examined the expression
of ER stress-associated proteins, namely, activating transcription
factor 6 (ATF6), glucose-regulated protein 94 (GRP-74), eIF2α,
p-eIF2α, IRE1α, p-IRE1α, and CHOP in imatinib-treated AGS and
SNU638 cells. Notably, imatinib induced the expression of GRP94,
p-eIF2α, p-IRE1 α and CHOP in a concentration- and time-dependent
manner in both AGS and SNU638 cells (Fig. 4A-C), indicating that
imatinib-induced apoptosis was mediated by the ER stress pathway. A
recent study indicated that several anticancer agents induced
oxidative stress by promoting ROS generation, thus inducing
cytotoxicity and apoptosis in cancer cells (19). To investigate the potential role of
ROS in imatinib-induced apoptosis, we determined ROS production in
imatinib-treated gastric cancer cells using DCFH-DA.
Imatinib-treated cells produced significant fluorescence signals.
In addition, imatinib induced ROS production in a time-dependent
manner (Fig. 4D).
CHOP−/− MEFs were resistant to the effects of PARP-1
cleavage compared with wild-type CHOP MEFs (Fig. 4E). Imatinib reduced the elevated ROS
levels in CHOP−/− MEFs, providing evidence that imatinib
reduced endogenous ROS levels in a CHOP-dependent manner (Fig. 4F). To examine the relationship
between JNK and ER-stress activation during imatinib exposure, AGS
cells were pretreated with an inhibitor of JNK (SP600125), then
treated with imatinib. Pretreatment with SP600125 significantly
decreased imatinib-induced CHOP (Fig.
4G). These results collectively indicated that imatinib-induced
apoptosis of gastric cancer cells was mediated by ROS generation
and ER stress.
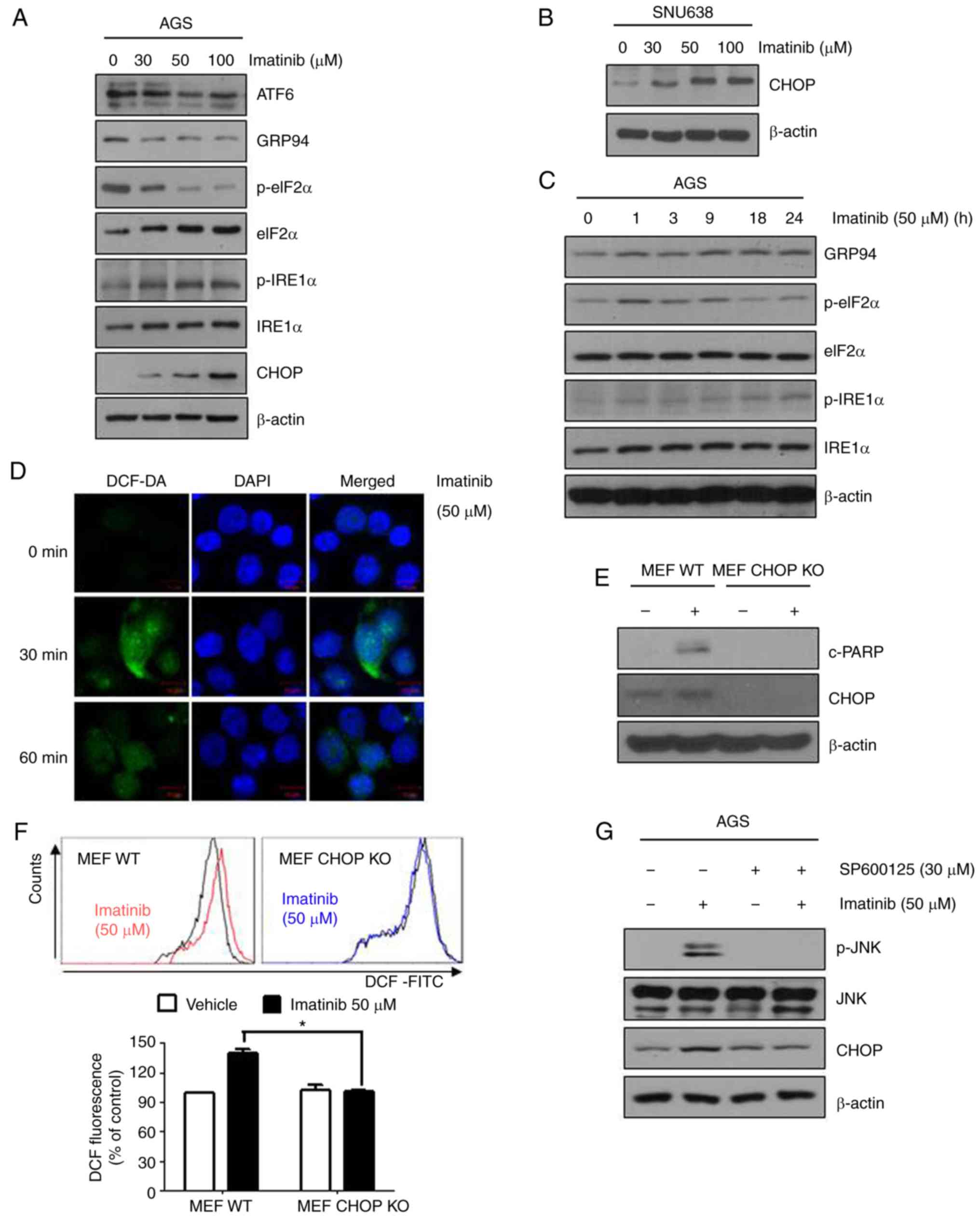 | Figure 4.Imatinib-induced apoptosis is
mediated by the ER stress pathway. (A-C) AGS and SNU638 cells were
treated with the indicated concentrations of imatinib for 48 h (A
and B) or with 50 µM imatinib for the indicated time periods (C).
Levels of ER stress pathway-associated proteins were assessed by
performing immunoblotting with the corresponding antibodies (D).
AGS cells were treated with 50 µM imatinib for 30 or 60 min,
stained with the FITC probe DCFH-DA (10 µM) for 30 min at 37°C, and
were visualized under a confocal microscope. (E) Wild-type and
CHOP−/− MEFs were treated with 50 µM imatinib for 24 h,
and levels of cleaved PARP and CHOP were determined by performing
immunoblotting. β-actin was used as an internal standard. (F) ROS
generation of wild-type and CHOP−/− MEFs was detected
using DCFDA staining 1 h after the indicated treatments. Graphs
represent the means ± SEM. *P<0.05 compared with treatment of
imatinib in MEF WT cells. (G) AGS cells were pretreated with 20 µM
JNK inhibitor (SP600125) for 1 h, followed by treatment with 50 µM
imatinib for 48 h. Levels of cleaved CHOP, JNK, and p-JNK were
determined by immunoblotting. β-actin was used as an internal
standard. ER, endoplasmic reticulum; fluorescein isothiocyanate;
PARP, poly(ADP-ribose) polymerase; CHOP, C/EBP-homologous protein;
ROS, reactive oxygen species; JNK, c-Jun NH2-terminal kinase. |
Imatinib and chemotherapy agents
synergistically decrease growth of gastric cancer cells
We examined the effect of combination treatment with
imatinib and chemotherapy agents on the viability of gastric cancer
cells. Treatment with imatinib or irinotecan alone resulted in
limited suppression of the viability of gastric cancer cells
(<15%) at 24 h (Fig. 5A and C).
In contrast, combination treatment with a fixed concentration of
irinotecan and varying concentrations of imatinib significantly
decreased the viability of gastric cancer cells. We used 50 µM
imatinib for performing subsequent experiments since this
concentration of imatinib significantly reduced cell viability
during combination treatment with irinotecan, with only a slight
decrease in the percentage of viable cells. Next, we examined
whether the combined effect of irinotecan and imatinib treatment on
the viability of gastric cancer cells was mediated by apoptotic
induction. Co-treatment of gastric cancer cells with irinotecan and
imatinib significantly increased the percentage of apoptotic cells,
whereas treatment with irinotecan or imatinib alone only slightly
increased the percentage of apoptotic cells (Fig. 5D) compared with that among untreated
cells. Next, we evaluated the effect of combination treatment with
0.5–2 µM 5-Fu and 30–50 µM imatinib on the viability of AGS cells
by performing MTT assays. Combination treatment with 50 µM imatinib
and 2 µM 5-Fu significantly inhibited the growth of AGS cells
compared with treatment with imatinib or 5-Fu alone (Fig. 5B and E). Similarly, imatinib also
increased irinotecan or 5-Fu-mediated inhibition of MKN45 and
SNU638 cell growth (Fig. 5F-I).
These findings indicated that combined treatment with imatinib and
chemotherapy agents (irinotecan or 5-Fu) effectively induced the
apoptosis of gastric cancer cells.
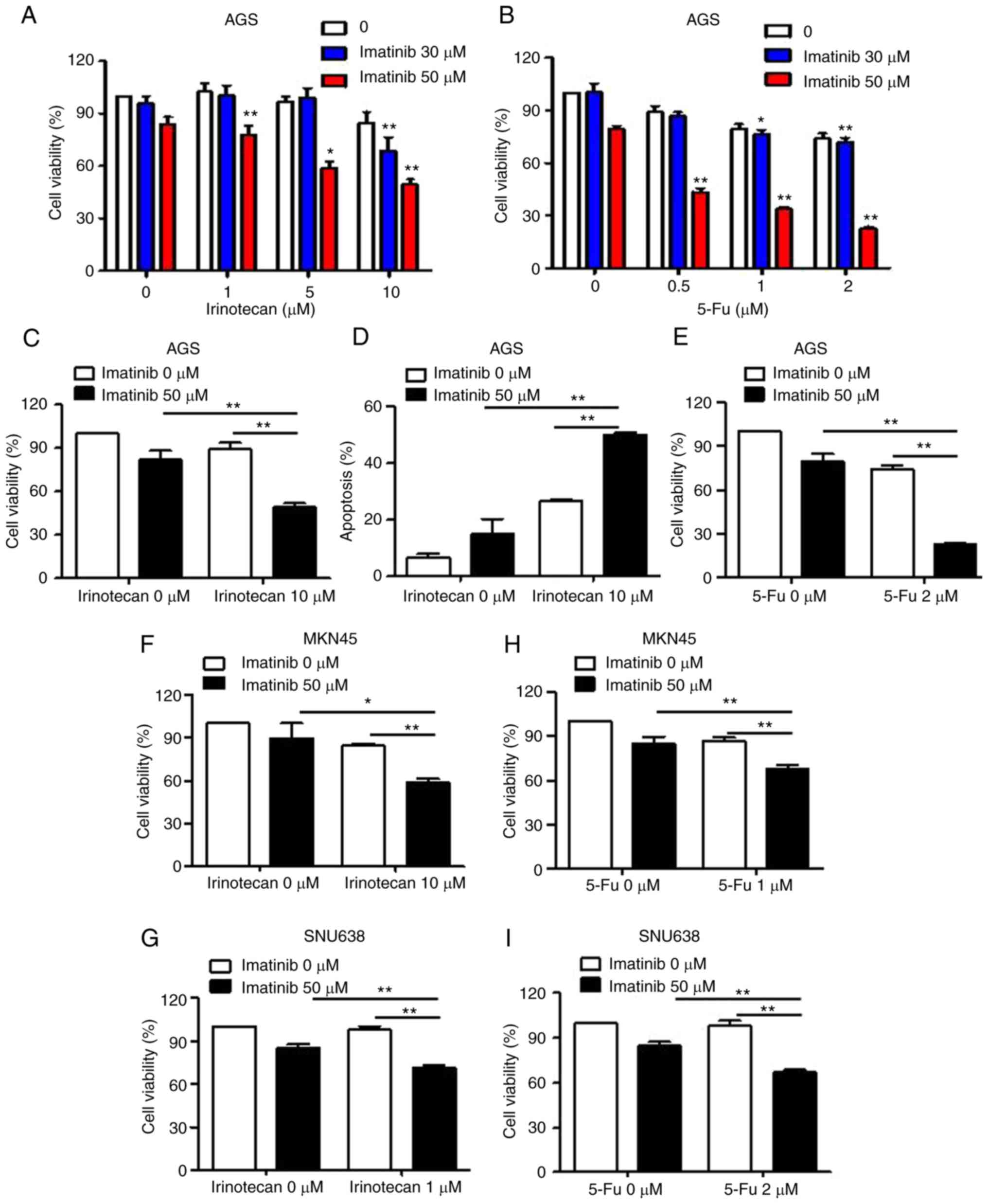 | Figure 5.Effects of combination treatment with
imatinib and chemotherapy agents on the growth of gastric cancer
cells. (A and C) MTT assays were performed to determine the effects
of treatment with imatinib or irinotecan alone, or in combination,
on the growth of AGS cells. AGS cells were treated with various
concentrations of imatinib and irinotecan for 48 h. Data are
expressed as the percentage of apoptotic cells compared with that
in control cells. *P<0.05 and **P<0.01, indicate significant
difference from values obtained for co-treatment group as compared
to the other groups. (D) Cells were stained with Annexin V and PI,
and flow cytometry was performed. Data are expressed as the means ±
SD of three independent experiments. (B and E) AGS cells were
treated with various concentrations of imatinib and 5-Fu for 48 h.
(F and G) MTT assays were performed to determine the effects of
treatment with imatinib or irinotecan alone, or in combination, on
the growth of MKN45 and SNU638 cells. Cells were treated with
various concentrations of imatinib and irinotecan for 48 h. Each
point represents the mean ± SD of at least three independent
experiments. (H and I) MKN45 and SNU638 cells were treated with
various concentrations of imatinib and 5-Fu for 48 h. MTT,
3-(4,5-dimethylthiazol-2-ly)-2,5-diphenyl tetrazolium bromide; PI,
propidium iodide. |
Discussion
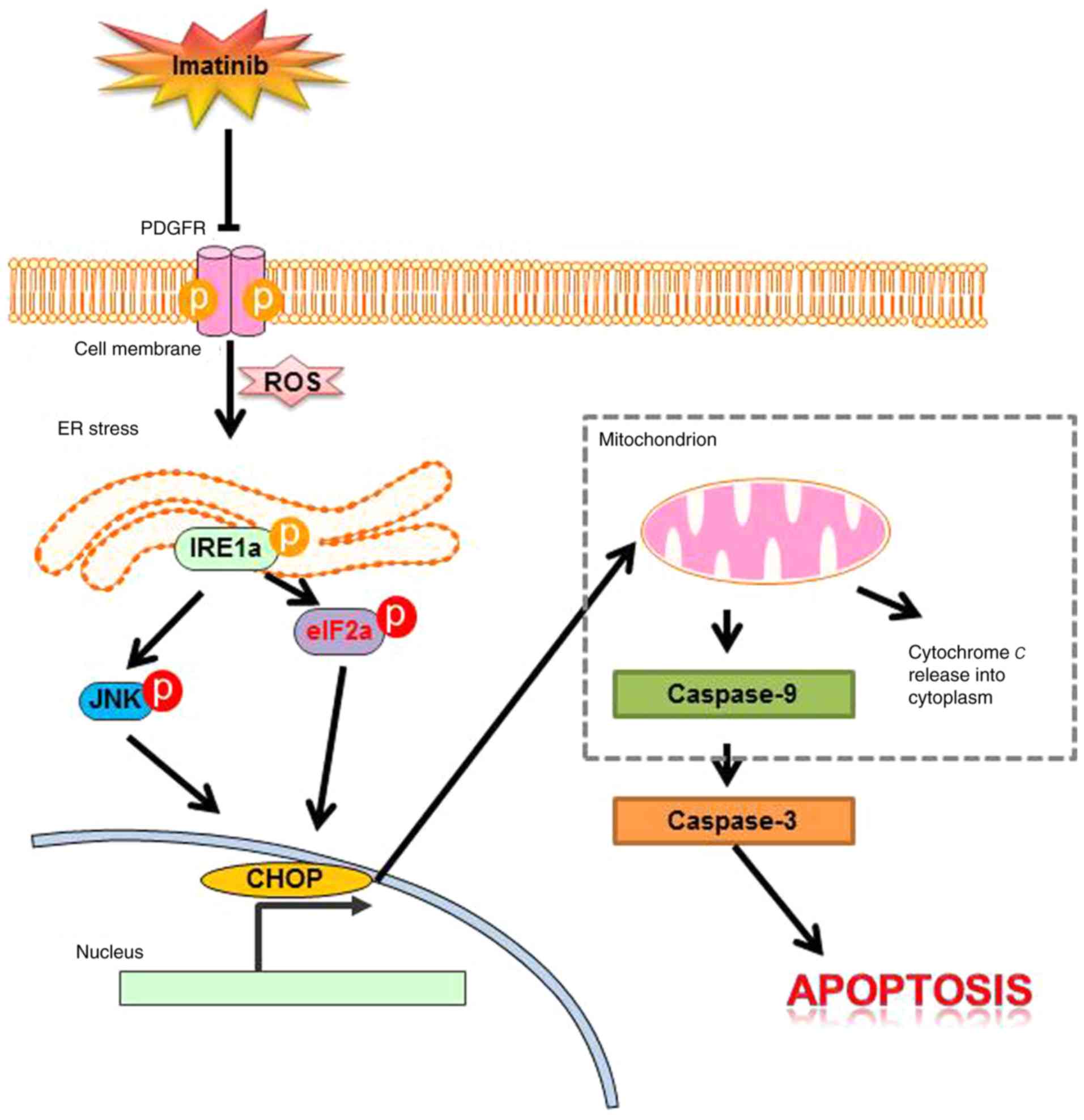
This is the first study, to the best of our
knowledge, to show that imatinib induces the apoptosis of gastric
cancer cells through the JNK/ROS/ER stress pathway (Fig. 6). It is important to determine
mechanisms underlying imatinib-induced death of gastric cancer
cells. Imatinib is a tyrosine kinase inhibitor with activity
against the BCR-ABL fusion oncoprotein, PDGFR, and c-KIT in GISTs,
prostate cancer, malignant gliomas, and ovarian cancer (26–29).
In the present study, c-KIT and BCR-ABL were not expressed in the
gastric cancer cell lines (data not shown). Mireskandari et
al reported that expression of c-KIT in gastric cancer appears
to be a very unlikely event (30).
Imatinib was revealed to induce apoptosis in, and may modulate the
metastasis of, gastric cancer cells by upregulating KAI1
expression (31). Biswas et
al reported that imatinib induced programmed cell death in
retinal ganglion cells by inhibiting PDGFR-mediated PI3K/AKT
signaling (32).
Another study suggested that the effect of imatinib
on the migration of medulloblastoma cells was not mediated by early
induction of apoptosis (33). A
recent study indicated that treatment with low and high
concentrations of imatinib induced cell growth arrest and
apoptosis, respectively, in glioblastoma cells. Consistently,
results of the present study revealed that imatinib induced
apoptosis at relatively high concentrations (20–100 µM), and
inhibited cell metastasis at lower concentrations (1–10 µM) (data
not shown). However, the mechanism underlying imatinib-induced cell
death is not completely understood. To clearly determine the
mechanism underlying imatinib-induced apoptosis, we identified the
possible involvement of a MAPK subfamily protein, since
accumulating evidence suggests important regulatory roles of MAPKs
in different physiological and pathological processes (34). It was observed that imatinib
treatment activated JNK in the late stage, but did not activate
ERK. Imatinib-induced activation of JNK/MAPK in the present study
indicated that these proteins perform distinct physiological
functions in determining the fate of gastric cancer cells.
Similarly, Chang et al reported that treatment with
high-dose imatinib induced JNK phosphorylation by elevating ROS
production in melanoma cells (34).
A study by Yu et al revealed that treatment with 5 mM STI571
interrupted cytoprotective 42/44 MAPK activation response in human
myeloid leukemia cells (35). These
results indicated that iron chelators activate different target
MAPKs in different cell types.
ER stress is suggested to be a significant
contributor to cell death. JNK activation plays a significant role
in UPR (36,37). Induction of the UPR in the ER, which
causes ER stress, induces several pathological and physiological
alterations such as glucose depletion, hypoxia, and oxidative
stress. Han et al reported that imatinib decreased JNK
activation and ER stress in the liver of a diabetic mouse model
(38). However, imatinib induced ER
stress in gastric cancer cells. Moreover, we found that imatinib
induced the apoptosis of gastric cancer cells by modulating ER
stress. This is the first study to report that imatinib induced
significant apoptosis of gastric cancer cells, which is mediated by
ER stress. Imatinib was also revealed to trigger ER stress in CML
cells expressing BCR-ABL (39). In
contrast, Zhang et al reported that imatinib did not induce
ER stress in Ph1-positive leukemia cells (40). These results indicated that imatinib
induced ER stress in a cell-specific manner. IRE1α-mediated JNK
activation in the ER induced apoptosis. Notably, we found that
imatinib-induced apoptosis of gastric cancer cells was mediated by
the JNK/ROS/ER stress pathway.
Generally, for patients with gastric cancer, therapy
is combined with cytotoxic chemotherapy and targeted therapy
(41). Therefore, it is very
important to find a target agent that has synergistic effects while
reducing toxicity of cytotoxic agents. Clinical studies on the
combination of imatinib, cisplatin and 5-fluoruracil or
capecitabine have been reported (42). In one of these clinical trials, the
safety and tolerability of combination of imatinib plus
5-fluoruracil was confirmed.
In summary, it was revealed that imatinib is a
potent antitumor agent that induces ER stress-mediated apoptosis of
gastric cancer cells. We observed that imatinib induced ER stress
by activating IRE1α, p-JNK, and CHOP. To the best of our knowledge,
this is the first study to determine mechanisms underlying
imatinib-induced apoptosis of gastric cancer cells. However,
further studies are required to determine the antitumorigenic
effects of imatinib in animal models. According to pathological
features, it is necessary to further study the anticancer effect of
imatinib in gastric cancer cells. Thus, our results indicated that
imatinib-induced activation of ER stress is a novel therapeutic
strategy for treating gastric cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Korea University Guro Hospital (O1600121) and supported by a grant
from the National Research Foundation (NRF) of Korea funded by the
Korean government (MSIP) (NRF-2017R1A2B2011684).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SCO and JLK conceived and designed the study and
critically revised the manuscript. JLK and DHL designed and
performed the experiments, analyzed the data and were major
contributors in writing the manuscript. SJ and BRK supervised the
western blot experiments and also performed the statistical
analysis. YJN, SHP, MJJ and YAJ performed the Annexin V/PI
apoptosis assay experiments. SJ, BRK, YJN, SHP, MJJ and YAJ
provided advice on the experiments and technical assistance. SCO
supervised the study. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BAX
|
BCL2-associated X protein
|
|
CHOP
|
C/EBP-homologous protein
|
|
eIF2α
|
eukaryotic translation initiation
factor 2α
|
|
ER
|
endoplasmic reticulum
|
|
FITC
|
fluorescein isothiocyanate
|
|
JNK
|
c-Jun NH2-terminal kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MTT
|
3-(4,5-dimethylthiazol-2-ly)-2,5-diphenyl tetrazolium bromide
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
PBS
|
phosphate-buffered saline
|
|
PI
|
propidium iodide
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakatani H, Araki K, Jin T, Kobayashi M,
Sugimoto T, Akimori T, Namikawa T, Okamoto K, Nakano T, Okabayashi
T, et al: STI571 (Glivec) induces cell death in the
gastrointestinal stromal tumor cell line, GIST-T1, via endoplasmic
reticulum stress response. Int J Mol Med. 17:893–897.
2006.PubMed/NCBI
|
|
3
|
Kadivar A, Kamalidehghan B, Akbari Javar
H, Karimi B, Sedghi R and Noordin MI: Antiproliferation effect of
imatinib mesylate on MCF7, T-47D tumorigenic and MCF 10A
nontumorigenic breast cell lines via PDGFR-β, PDGF-BB, c-Kit and
SCF genes. Drug Des Dev Ther. 11:469–481. 2017. View Article : Google Scholar
|
|
4
|
Donovan J, Abraham D and Norman J:
Platelet-derived growth factor signaling in mesenchymal cells.
Front Biosci. 18:106–119. 2013. View
Article : Google Scholar
|
|
5
|
Cumpănas AA, Cimpean AM, Ferician O,
Ceausu RA, Sarb S, Barbos V, Dema A and Raica M: The involvement of
PDGF-B/PDGFRβ axis in the resistance to antiangiogenic and
antivascular therapy in renal cancer. Anticancer Res. 36:2291–2295.
2016.PubMed/NCBI
|
|
6
|
Kazlauskas A: PDGFs and their receptors.
Gene. 614:1–7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pietras K, Sjöblom T, Rubin K, Heldin CH
and Ostman A: PDGF receptors as cancer drug targets. Cancer Cell.
3:439–443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Minamino T, Komuro I and Kitakaze M:
Endoplasmic reticulum stress as a therapeutic target in
cardiovascular disease. Circ Res. 107:1071–1082. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsai YC and Weissman AM: The unfolded
protein response, degradation from endoplasmic reticulum and
cancer. Genes Cancer. 1:764–778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Malhotra JD, Miao H, Zhang K, Wolfson A,
Pennathur S, Pipe SW and Kaufman RJ: Antioxidants reduce
endoplasmic reticulum stress and improve protein secretion. Proc
Natl Acad Sci USA. 105:18525–18530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thuerauf DJ, Marcinko M, Gude N, Rubio M,
Sussman MA and Glembotski CC: Activation of the unfolded protein
response in infarcted mouse heart and hypoxic cultured cardiac
myocytes. Circ Res. 99:275–282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: Cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mansfield KD, Simon MC and Keith B:
Hypoxic reduction in cellular glutathione levels requires
mitochondrial reactive oxygen species. J Appl Physiol.
97:1358–1366. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Waypa GB, Guzy R, Mungai PT, Mack MM,
Marks JD, Roe MW and Schumacker PT: Increases in mitochondrial
reactive oxygen species trigger hypoxia-induced calcium responses
in pulmonary artery smooth muscle cells. Circ Res. 99:970–978.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ostadal P, Elmoselhi AB, Zdobnicka I,
Lukas A, Elimban V and Dhalla NS: Role of oxidative stress in
ischemia-reperfusion-induced changes in
Na+,K+-ATPase isoform expression in rat
heart. Antioxid Redox Signal. 6:914–923. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nojiri H, Shimizu T, Funakoshi M,
Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M, Tachibana T,
Ishikawa H, et al: Oxidative stress causes heart failure with
impaired mitochondrial respiration. J Biol Chem. 281:33789–33801.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morgan WA: DNA single-strand breakage in
mammalian cells induced by redox cycling quinones in the absence of
oxidative stress. J Biochem Toxicol. 10:227–232. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Devasagayam TP, Tilak JC, Boloor KK, Sane
KS, Ghaskadbi SS and Lele RD: Free radicals and antioxidants in
human health: Current status and future prospects. J Assoc
Physicians India. 52:794–804. 2004.PubMed/NCBI
|
|
19
|
Gong K, Xie J, Yi H and Li W: CS055
(Chidamide/HBI-8000), a novel histone deacetylase inhibitor,
induces G1 arrest, ROS-dependent apoptosis and
differentiation in human leukaemia cells. Biochem J. 443:735–746.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang YJ, Huang YP, Li ZL and Chen CH:
GRP78 knockdown enhances apoptosis via the down-regulation of
oxidative stress and Akt pathway after epirubicin treatment in
colon cancer DLD-1 cells. PLoS One. 7:e351232012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Atari-Hajipirloo S, Nikanfar S, Heydari A
and Kheradmand F: Imatinib and its combination with
2,5-dimethyl-celecoxibinduces apoptosis of human HT-29 colorectal
cancer cells. Res Pharm Sci. 12:67–73. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akram AM, Iqbal Z, Akhtar T, Khalid AM,
Sabar MF, Qazi MH, Aziz Z, Sajid N, Aleem A, Rasool M, et al:
Presence of novel compound BCR-ABL mutations in late chronic and
advanced phase imatinib sensitive CML patients indicates their
possible role in CML progression. Cancer Biol Ther. 18:214–221.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mahon FX: Discontinuation of TKI therapy
and ‘functional’ cure for CML. Best Prac Res Clin Haematol.
29:308–313. 2016. View Article : Google Scholar
|
|
24
|
Abu-Amna M, Awadie H and Bar-Sela G:
Imatinib-induced gastrointestinal vascular ectasia in a patient
with advanced gIST: Case report and literature review. Anticancer
Res. 36:6151–6154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Balachandran VP, Cavnar MJ, Zeng S,
Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C,
Rossi F, et al: Imatinib potentiates antitumor T cell responses in
gastrointestinal stromal tumor through the inhibition of Ido. Nat
Med. 17:1094–1100. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakatani H, Kobayashi M, Jin T, Taguchi T,
Sugimoto T, Nakano T, Hamada S and Araki K: STI571 (Glivec)
inhibits the interaction between c-KIT and heat shock protein 90 of
the gastrointestinal stromal tumor cell line, GIST-T1. Cancer Sci.
96:116–119. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matei D, Emerson RE, Lai YC, Baldridge LA,
Rao J, Yiannoutsos C and Donner DD: Autocrine activation of
PDGFRalpha promotes the progression of ovarian cancer. Oncogene.
25:2060–2069. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Kleeff J, Guo J, Fischer L, Giese N,
Büchler MW and Friess H: Effects of STI571 (gleevec) on pancreatic
cancer cell growth. Mol Cancer. 2:322003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mireskandari M, Shafaii AF, Kayser G and
Kayser K: Lack of CD117 and rare bcl-2 expression in stomach cancer
by immunohistochemistry. An immunohistochemical study with review
of the literature. Diagn Pathol. 1:72006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shandiz SA, Farasati S, Saeedi B,
Baghbani-Arani F, Asl EA, Keshavarz-Pakseresht B, Rahimi A, Assadi
A, Noorbazargan H, RahimpourHesari M, et al: Up regulation of KAI1
gene expression and apoptosis effect of imatinib mesylate in
gastric adenocarcinoma (AGS) cell line. Asian Pac J Trop Dis.
6:120–125. 2016. View Article : Google Scholar
|
|
32
|
Biswas SK, Zhao Y and Sandirasegarane L:
Imatinib induces apoptosis by inhibiting PDGF-but not
insulin-induced PI 3-kinase/Akt survival signaling in RGC-5 retinal
ganglion cells. Mol Vis. 15:1599–1610. 2009.PubMed/NCBI
|
|
33
|
Abouantoun TJ and MacDonald TJ: Imatinib
blocks migration and invasion of medulloblastoma cells by
concurrently inhibiting activation of platelet-derived growth
factor receptor and transactivation of epidermal growth factor
receptor. Mol Cancer Ther. 8:1137–1147. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang SP, Shen SC, Lee WR, Yang LL and
Chen YC: Imatinib mesylate induction of ROS-dependent apoptosis in
melanoma B16F0 cells. J Dermatol Sci. 62:183–191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu C, Krystal G, Varticovksi L, McKinstry
R, Rahmani M, Dent P and Grant S: Pharmacologic mitogen-activated
protein/extracellular signal-regulated kinase
kinase/mitogen-activated protein kinase inhibitors interact
synergistically with STI571 to induce apoptosis in
Bcr/Abl-expressing human leukemia cells. Cancer Res. 62:188–199.
2002.PubMed/NCBI
|
|
36
|
Lenna S, Han R and Trojanowska M:
Endoplasmic reticulum stress and endothelial dysfunction. IUBMB
Life. 66:530–537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim JL, Lee DH, Na YJ, Kim BR, Jeong YA,
Lee SI, Kang S, Joung SY, Lee SY, Oh SC and Min BW: Iron
chelator-induced apoptosis via the ER stress pathway in gastric
cancer cells. Tumour Biol. 37:9709–9719. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han MS, Chung KW, Cheon HG, Rhee SD, Yoon
CH, Lee MK, Kim KW and Lee MS: Imatinib mesylate reduces
endoplasmic reticulum stress and induces remission of diabetes in
db/db mice. Diabetes. 58:329–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pattacini L, Mancini M, Mazzacurati L,
Brusa G, Benvenuti M, Martinelli G, Baccarani M and Santucci MA:
Endoplasmic reticulum stress initiates apoptotic death induced by
STI571 inhibition of p210 bcr-abl tyrosine kinase. Leuk Res.
28:191–202. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang X, Inukai T, Akahane K, Hirose K,
Kuroda I, Honna H, Goi K, Kagami K, Tauchi T, Yagita H, et al:
Endoplasmic reticulum stress inducers, but not imatinib, sensitize
Philadelphia chromosome-positive leukemia cells to TRAIL-mediated
apoptosis. Leuk Res. 35:940–949. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lopez A, Harada K, Mizrak Kaya D and Ajani
JA: Current therapeutic landscape for advanced gastroesophageal
cancers. Ann Transl Med. 6:78–96. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mayr M, Becker K, Schulte N, Belle S,
Hofheinz R, Krause A, Schmid RM, Röcken C and Ebert MP: phase I
study of imatinib, cisplatin and 5-fluoruracil or capecitabine in
advanced esophageal and gastric adenocarcinoma. BMC Cancer.
12:587–593. 2012. View Article : Google Scholar : PubMed/NCBI
|