Introduction
Liver cancer is commonly diagnosed, and the 5-year
relative survival rate is only 18% (1,2).
Current treatments applicable for liver cancer, include surgery,
transcatheter arterial chemoembolization and chemotherapy (3). In recent years, the efficacy of agents
that selectively target critical signaling pathways has been
assessed in several clinical trials, however, no relevant
improvement has been achieved to date (3,4).
Therefore, it is urgent to identify therapeutic strategies for more
effective therapy of liver cancer.
Recently, the activation of sonic Hedgehog (SHH)
signaling has been implicated in liver cancer (5). It was revealed to play a crucial role
in the initiation and maintenance of liver cancer and contribute to
chemotherapeutic resistance (6).
The SHH signaling pathway is initiated by the binding of the
patched (Ptch) receptor, which in turn relieves smoothened (Smo)
receptor from inhibition. Smo then triggers a series of
intracellular events, resulting in the activation of downstream
target genes including the glioma-associated oncogene homolog
(Gli), which are the early transcriptional targets of SHH signaling
(7,8). It has been determined that blocking
SMO can inhibit the activity of the SHH signaling pathway, and SMO
has been used as a target to develop related drugs for cancer
treatment (7). It was reported that
Smo antagonists including cyclopamine and vismodegib could inhibit
the growth of tumors (9).
Vismodegib (GDC0449) has been approved by the U.S. Food and Drug
Administration (FDA) for the treatment of metastatic or locally
advanced unresectable basal cell carcinoma (10).
Several studies have revealed that SHH regulates
sustained activation of histone deacetylases (HDACs) which is
required for cell growth (11,12).
Liver cancer patients with overexpression of HDAC1 exhibited higher
incidence of cancer cell invasion, poorer histological
differentiation, and a low survival rate (13,14).
Aberrant regulation of HDAC2 may play a pivotal role in the
development of liver cancer rendering HDAC2 a relevant target for
liver cancer therapy (15). In
numerous previous studies, SHH signaling regulated histone
acetylation and chromatin (16) and
led to carcinogenesis (17).
Further studies are still required to ascertain the use of dual
inhibition of SHH signaling and HDAC treat liver cancer. In the
present study, it was demonstrated that the combined use of SHH and
HDAC inhibitors effectively treated liver cancer cells in
vitro and ex vivo. These studies indicated that SHH
inhibition may be a reasonable strategy to extend the utility of a
HDAC inhibitor in liver cancer.
Materials and methods
Cell culture
The HepG2 cell line was obtained from the Cell Bank,
Chinese Academy of Sciences (Shanghai, China). This is a common
liver cancer cell line. The HepG2 cell line was derived from a
15-year-old white male with well-differentiated liver cancer. HepG2
cells were cultured in Dulbecco's modified Eagle's medium (DMEM)
(HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The HepG2 cells were cultured in a
humidified incubator containing 5% CO2 at 37°C.
Cell viability assay and drug
combination analysis
Cell viability was assessed by Cell Counting Kit-8
assay (CCK-8; Dojindo Molecular Technologies, Inc., Rockville, MD,
USA). Vismodegib and entinostat were purchased from Shanghai
Biochempartner Co., Ltd. (Shanghai, China). According to the
instructions, HepG2 cells cultured in 96-well plates were treated
with an indicated concentration of vismodegib (0, 1, 2, 4, 8 and 16
µM) or entinostat (0, 0.5, 1, 2, 4 and 8 µM) or a combination of
the two, or DMSO for 72 h. The half-maximal inhibitory
concentration (IC50) values were obtained from
dose-response curves with GraphPad Prism software (version 7.00;
GraphPad Software, Inc., La Jolla, CA, USA). The synergistic effect
was determined by calculating the combination index (CI) using the
Calcusyn software program (version 2.1; Biosoft, Great Shelford,
UK). Data from cell viability assays were expressed as the fraction
of growth inhibition by the single drugs or the combination.
Synergism was expressed as a CI value <1 and antagonism by a CI
value >1 at 0.5 fraction affected (FA).
Cell proliferation assay
The effects of the inhibitors on cell growth were
evaluated as previously described (18). Briefly, the cells were cultured in
6-well plates at 1,500 cells/well densities and then treated with
vismodegib (4 µM) or entinostat (2 µM) or combination (vismodegib
and entinostat at the respective concentrations). The cells were
treated for ~14 days. Then the cells were fixed, stained with
crystal violet, and extracted with glacial acetic acid. The optical
density (OD) was assessed at 570 nm by the microplate reader
(iMark™ Microplate Absorbance Reader; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Soft agar colony formation assay
The colony formation of the liver cancer cells in
soft agar medium was performed as previously described (19). Briefly, the cell suspension
(2×104 cells/well) was mixed with 0.4% soft agar (BD
Biosciences, Franklin Lakes, NJ, USA) prepared with DMEM containing
10% FBS and layered onto 0.6% soft agar prepared with DMEM
containing 10% FBS. In addition, liver cancer cells were treated
with media supplemented with the indicated drugs. The medium was
changed twice a week. The colonies were photographed and the area
of these colonies was assessed by Image-Pro Plus software (version
6.0; Media Cybernetics, Inc., Rockville, MD, USA) at the end of the
experiment.
Apoptosis analysis
HepG2 cells were grown in 6-well plates and treated
with vismodegib (4 µM), entinostat (2 µM), combination or DMSO
control for 48 h. Following treatment, apoptotic cells were
assessed by nuclear morphology and apoptotic cell numbers were
detected. Cells exposed to different treatments for 48 h were
stained with acridine orange (AO) and ethidium bromide (EB) as
previously described (20), and
examined with a fluorescence microscope (Leica Microsystems GmbH,
Wetzlar, Germany). Apoptosis in liver cancer cells was analyzed
with an Annexin V-FITC Apoptosis Detection Kit (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan). The cultured cells were
trypsinized, resuspended in Annexin V binding buffer, and incubated
with Annexin V and propidium iodide (PI) in the dark. Flow
cytometric analysis was performed using a BD Accuri™
flow cytometer (BD Biosciences, San Jose, CA, USA) to subject the
stained cells.
Cell cycle analysis
The treated cells were fixed in ice-cold 70% ethanol
and stored at 4°C overnight for cell cycle analysis. After the
fixed cells were washed with PBS, they were treated with 100 µg/ml
RNase A at 37°C for 20 min, and stained with 50 µg/ml PI (Dojindo
Molecular Technologies, Inc.). Then, the cells were subjected to
fluorescence-activated cell sorting. The cell populations that were
found in G0/G1, S and G2/M phases were quantified using the ModFit
software (version 4.0; Verity Software House, Inc., Topsham, ME,
USA).
Protein isolation and western
blotting
Cells exposed to vismodegib (4 µM) and/or entinostat
(2 µM) for 48 h were lysed by RIPA buffer (Nanjing KeyGen Biotech,
Co., Ltd., Nanjing, China) containing protease and phosphatase
inhibitors. The BCA method was used to quantify the protein of the
lysate. The whole cell extracts (30 µg) was separated by 8%
SDS-PAGE and the polyvinylidene fluoride (PVDF) membranes were
blocking by 5% skim milk for 2 h at 4°C, then were incubated with
the following primary antibodies staying overnight at 4°C: vinculin
(dilution 1:2,000; cat. no. V4139; Sigma-Aldrich, Merck KGaA,
Darmstadt, Germany), CDK1 (dilution 1:500; cat. no. 10762-1-AP),
cyclin B1 (dilution 1:500; cat. no. 55004-1-AP), Gli1 (dilution
1:500; cat. no. 25733-1-AP), Gli2 (dilution 1:500; cat. no.
18989-1-AP), Gli3 (dilution 1:500; cat. no. 19949-1-AP) (all from
ProteinTech Group, Inc., Chicago, IL, USA), cleaved-PARP (dilution
1:1,000; cat. no. 9541), pS6RPs255/256 (dilution
1:1,000; cat. no. 4858), pAKTs473 (dilution 1:1,000;
cat. no. 4060) and acetyl-histone H3 (dilution 1:1,000; cat. no.
9677) (all from Cell Signaling Technology, Inc., Danvers, MA, USA).
Subsequently, they were incubated with horseradish
peroxidase-conjugated secondary antibody (dilution 1:1,000; cat.
no. A0208; Beyotime Institute of Biotechnology, Shanghai, China),
which was followed by Thermo Scientific™ chemiluminescence (cat.
no. 32106; Thermo Fisher Scientific Co., Ltd., Shanghai, China)
detection.
Ex vivo culture of patient tumor
tissue
The case of a 59-year-old patient with diagnosed
primary moderately differentiated liver cancer without treatment
history was employed. The acquisition of tumor tissue was executed
with an Institutional Review Board protocol approved by Binzhou
Medical University (Yantai, China) and written informed consent was
provided by this patient. The primary liver cancer specimens from
this patient who underwent a hepatic segmentectomy were obtained
and subjected to ex vivo culture experiments.
For the ex vivo culture, 1 cm2
hemostatic gelatin dental sponges were soaked in the medium, in
which DMEM base was supplemented with 10% FBS, hydrocortisone (1
mg/100 ml; Sigma-Aldrich; Merck KGaA), antibiotic/antimycotic
solution (Gibco; Thermo Fisher Scientific, Inc.), and insulin (1
mg/100 ml) for 1 h. Then, liver cancer tissues were cut into 1
mm3 blocks, and transferred to the top surface of
hemostatic gelatin dental sponges. Tissue blocks were treated with
SHH pathway inhibitor vismodegib and HDAC inhibitor entinostat as
single-agents or in combination for 48 h, and then fixed with
paraformaldehyde. The tissue blocks were embedded in paraffin,
sectioned with microtome, and stained with hematoxylin and eosin
(H&E). Immunohistochemistry (IHC) was implemented using
antibodies Bcl-2 (dilution 1:200; cat. no. 12789-1-AP), Bax
(dilution 1:200; cat. no. 50599-2-Ig) and Ki-67 (dilution 1:400;
cat. no. 27309-1-AP) (all from ProteinTech Group, Inc.). The
results of the staining were quantified by Image-Pro Plus
software.
Statistical analysis
All numerical data were presented as averages and
the standard deviation (SD) of the mean. The data was analyzed by
one-way analysis of variance (ANOVA) followed by Dunnett's post hoc
test. Statistical significance was regarded at P<0.05. All
statistical analyses were performed using GraphPad Prism 5.0
(GraphPad Software, Inc., San Diego, CA, USA).
Results
Combined use of vismodegib and
entinostat inhibits the growth of liver cancer cells
Given that the SHH signaling pathway regulated
histone acetylation and led to cancinogenesis (20), the liver cancer cell line, HepG2,
was chosen for examination. Cell proliferation assay using CCK-8
revealed that the IC50 value of vismodegib for HepG2
cells was 4.012 µM (Fig. 1A), and
the IC50 value of entinostat for these cells was 1.992
µM (Fig. 1A). Then, the HepG2 cells
were treated with different concentrations of vismodegib and
entinostat, each alone and in combination for 72 h. The
median-effect analysis was used to assess the effect of the drug
combination on proliferation inhibition. Combined treatment with
vismodegib and entinostat resulted in a synergistic increase in
proliferation inhibition and synergistic CI value of <1 at 0.5
FA in HepG2 cells (Fig. 1B).
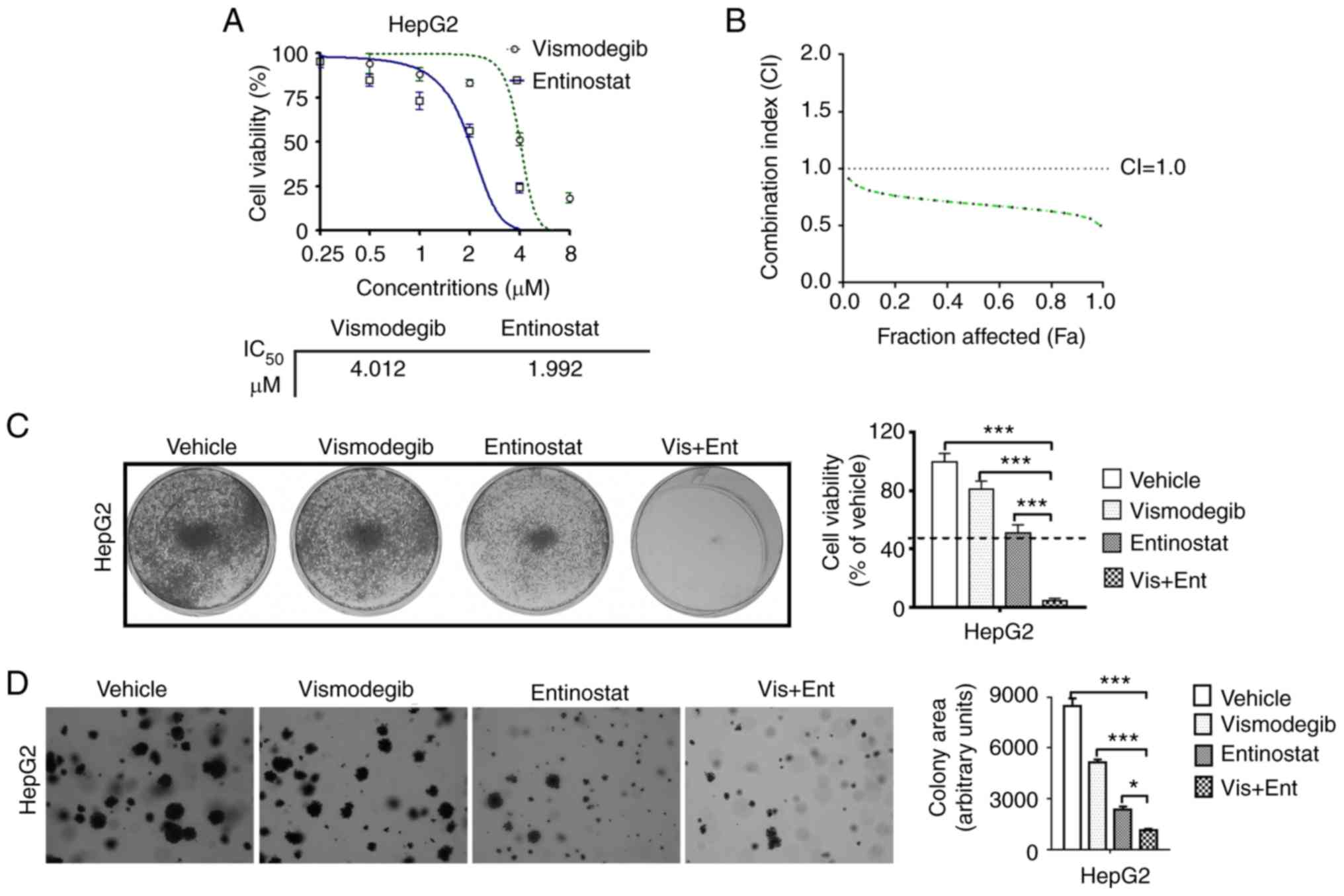 | Figure 1.The combined use of vismodegib and
entinostat synergistically inhibits the growth of liver cancer
cells. (A) The IC50 values of HepG2 cells treated with
vismodegib (0, 1, 2, 4, 8 and 16 µM) or entinostat (0, 0.5, 1, 2, 4
and 8 µM) for 72 h were assessed using a CCK-8 assay. (B) The HepG2
cells were treated with indicated drugs. The combination index (CI)
values were assessed using the results of the CCK-8 assay and
presented with FA combinations. (C) Liver cancer cells cultured in
the 6-well plates were treated with the inhibitors as indicated and
then stained with crystal violet. (D) Liver cancer cells were
cultured on soft agar and were treated with the indicated
inhibitors for ~4 weeks. The colonies were imaged and quantified
(magnification, ×200). The mean ± SD for 3 independent experiments
is presented. *P<0.05; ***P<0.001. CCK-8, Cell Counting
Kit-8; FA, fractions affected. |
We also observed that SHH signaling inhibitor
vismodegib as single-agent slightly reduced the viability of HepG2
cells (Fig. 1C). Conversely, the
HDAC inhibitor entinostat revealed a moderate inhibitory effect on
the viablity of these liver cancer cells (Fig. 1C). However, the combined use of
vismodegib and entinostat almost completely suppressed the growth
of HepG2 cells (Fig. 1C).
The observation that the combination treatment
inhibited cell proliferation in two-dimensional culture conditions
led us to further assess the effect of the drug combination in
three-dimensional conditions. Inhibition of the SHH pathway by the
use of vismodegib alone slightly inhibited colony formation
efficiently and inhibition of entinostat exhibited a moderate
inhibitory effect. As anticipated, the combined use of vismodegib
and entinostat inhibited the formation of colonies in HepG2 cells
almost completely (Fig. 1D). These
results indicated that dual use of SHH signaling inhibitor
vismodegib and HDAC inhibitor entinostat provided a synergistic
inhibitory effect on the proliferation of liver cancer cells.
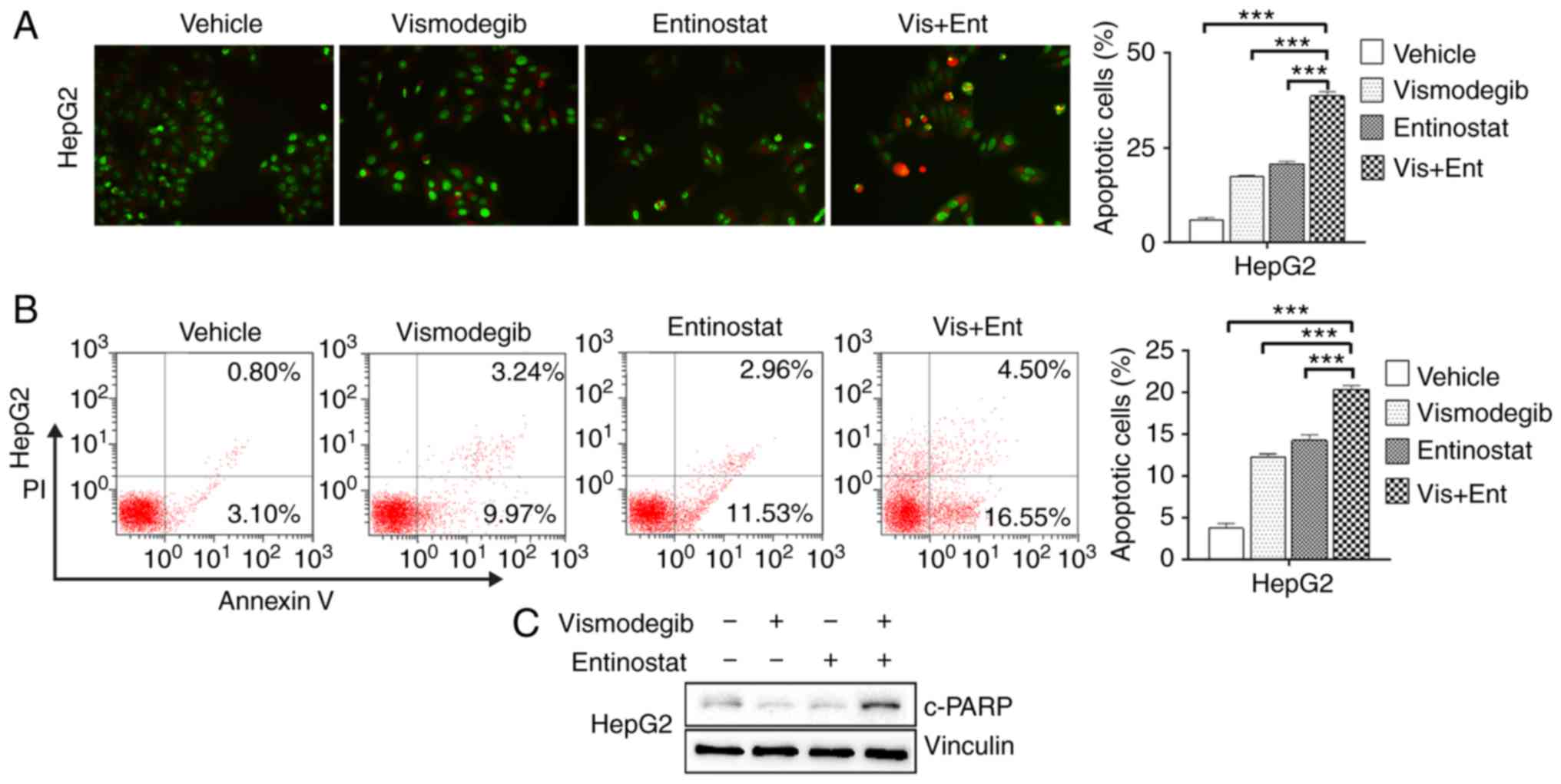
Combined use of vismodegib and
entinostat synergistically induces apoptosis in liver cancer
cells
To investigate whether apoptosis was the pathway of
cell death after treatment, cell morphological changes were
observed and the proportion of apoptotic cells was evaluated by
AO/EB staining and flow cytometric assays, respectively. The
results indicated that only sporadic apoptotic cells were observed
in the vehicle group, however, more apoptotic cells were observed
in the vismodegib and entinostat single-agent treated groups. The
number of apoptotic cells significantly increased in the combined
treatment group of HepG2 cells (Fig.
2A). While vismodegib or entinostat alone produced a mild
increase in Annexin V-positive cells, combined treatment resulted
in significantly increased Annexin V-positive cells in HepG2 cells
(Fig. 2B). Consistent with this
result, the combination treatment also enhanced the abundance of
cleaved-PARP, a marker for active apoptosis, in HepG2 cells.
(Fig. 2C).
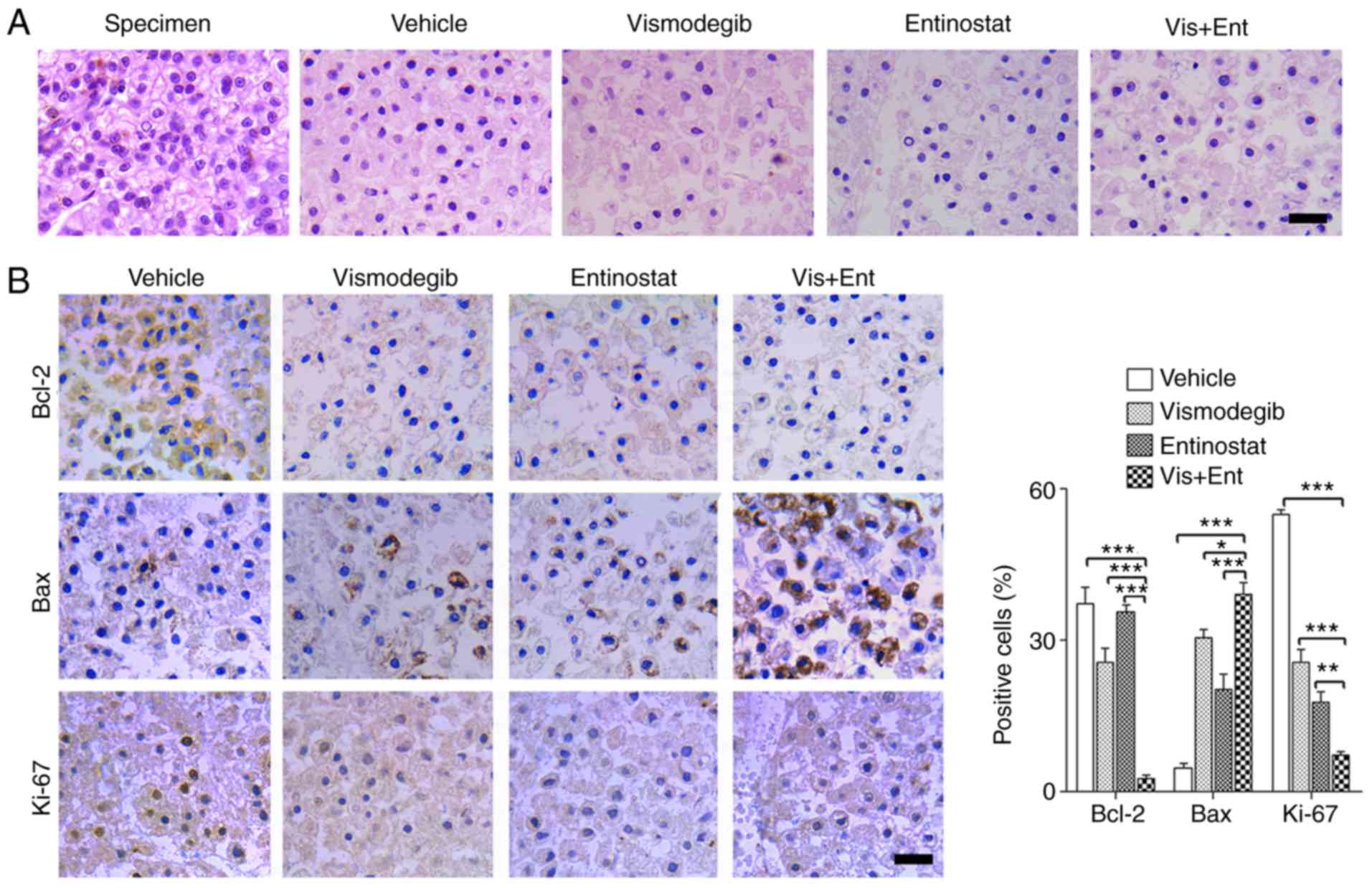
Combination treatment using vismodegib
and entinostat has potent inhibition activity in liver cancer
specimens
An ex vivo culture model of liver cancer was
employed to assess the therapeutic effect of vismodegib combined
with entinostat. Briefly, we dissected fresh surgical specimens of
primary liver cancer into ~1 mm3 blocks. These blocks
were exposed to vismodegib and entinostat as single-agents or in
combination for 48 h on the absorbable gelatin sponge. The results
of histological examination revealed that the tissue structure of
vehicle-treated explants was similar to that of the primary tumor
tissue (Fig. 3A). The blocks
treated with vismodegib and entinostat displayed marked disrupted
cellular integrity compared to that in the single-agent treatment
groups (Fig. 3A). Consistently, the
combination treatment significantly reduced proliferation as
determined by Ki-67, and significantly enhanced apoptosis as
determined by downregulated Bcl-2 and upregulated Bax (Fig. 3B). Collectively, these data
ascertained the potential use of vismodegib and entinostat to treat
liver cancer.
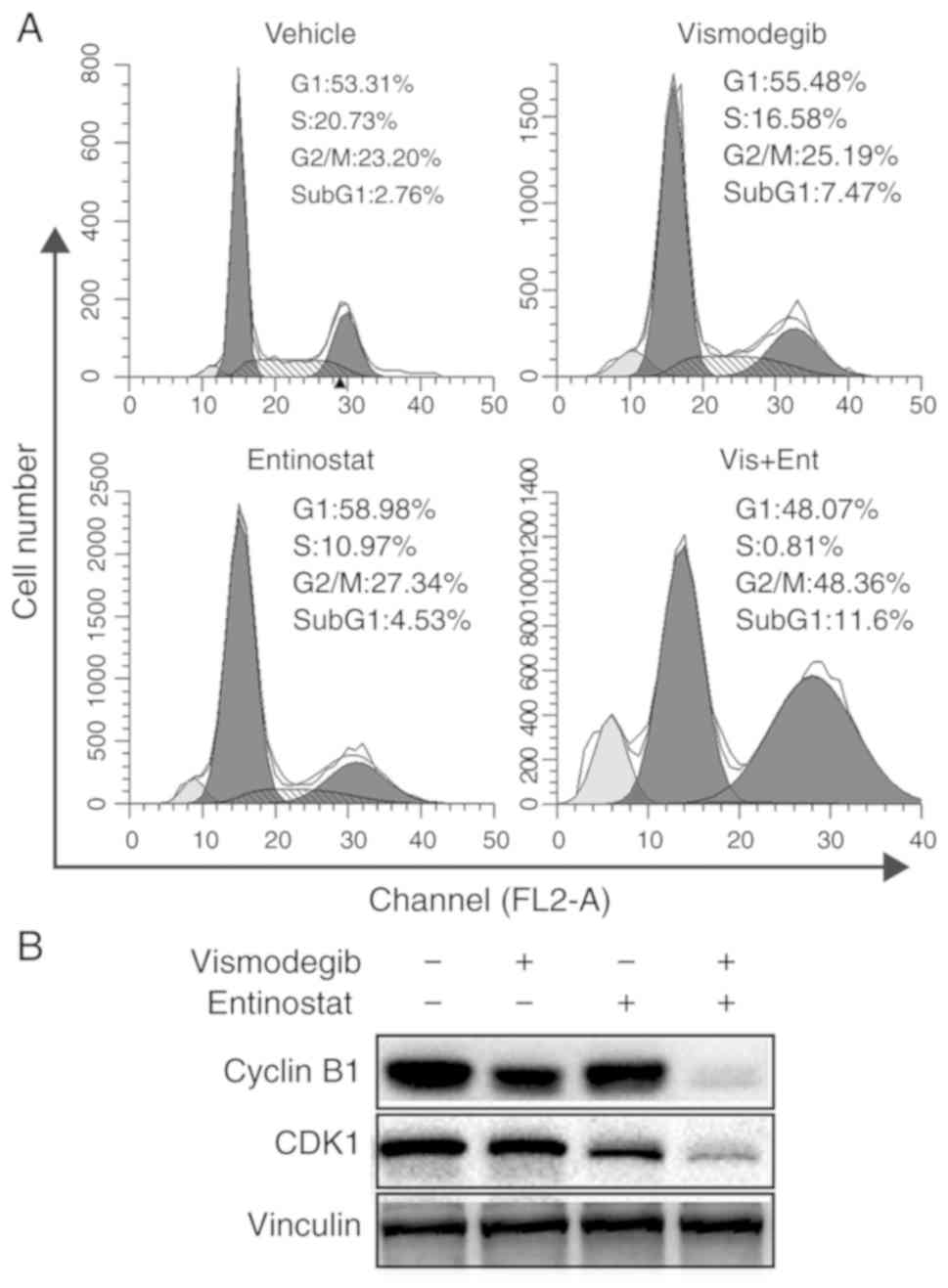
Combined use of vismodegib and
entinostat synergistically induces G2/M arrest of liver cancer
cells
To assess whether the effects of combination
treatment on liver cancer cells involved changes in the cell cycle,
HepG2 cells were treated with vismodegib and entinostat alone or
combined treatment for 48 h. The results revealed that combination
treatment significantly increased the number of cells at the G2/M
phase and significantly decreased the number of cells at the S
phase (Fig. 4A). However, an
increase of sub-G1 populations was also observed. While vismodegib
and entinostat as single-agents led to a slight increase of sub-G1
populations, dual treatment with vismodegib and entinostat resulted
in a marked increase of sub-G1 in HepG2 cells (Fig. 4A). The present study determined that
combination therapy induced G2/M arrest and triggered apoptosis of
HepG2 cells. With regards to the biomarkers of the cell cycle,
vismodegib combined with entinostat reduced the expression of
cyclin B1 and cyclin dependent kinase 1 (CDK1) (Fig. 4B), which further indicated that the
combination treatment resulted in G2/M phase arrest.
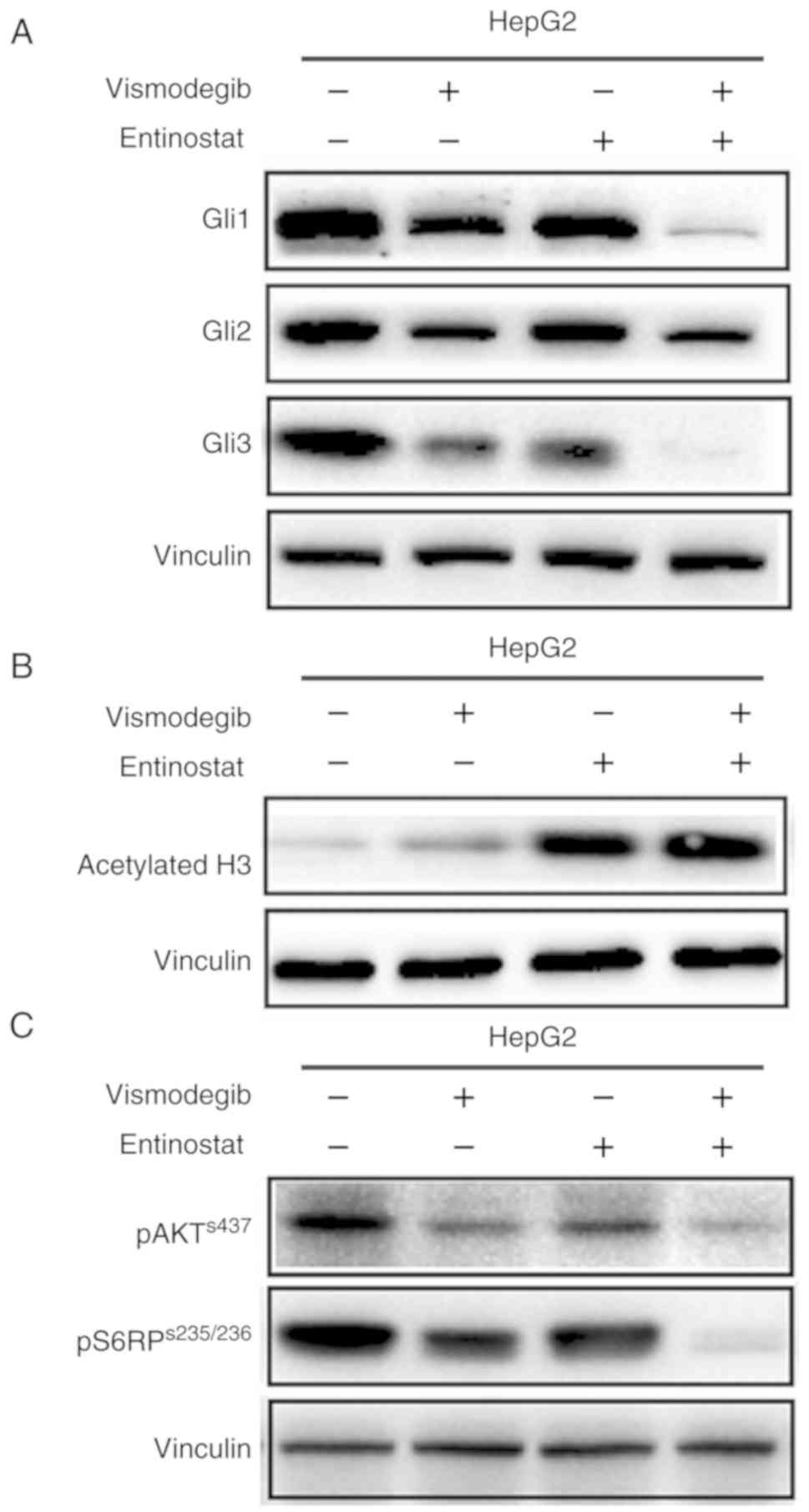
Vismodegib as a single agent or in
combination with entinostat attenuates PI3K/mTOR signaling
Western blot analysis revealed that vismodegib as a
single agent or in combination with entinostat resulted in a marked
reduction of the expression of Gli1, Gli2 and Gli3 effectors of SHH
signaling in HepG2 cells (Fig. 5A).
Notably, a slight effect was observed with SHH signaling in liver
cancer cells treated with entinostat as a single-agent. In
addition, the results revealed that entinostat as a single agent or
in combination with vismodegib led to a marked increase in
acetylated histone 3 in HepG2 cells (Fig. 5B).
It has been reported that inhibition of SHH
signaling regulated pancreatic tumorigenesis via inhibition of the
PI3K/AKT signaling pathway (21).
Thus, it was investigated whether the dual inhibition of the SHH
pathway and HDAC inhibited the growth of liver cancer cells via
inhibition of the PI3K/AKT signaling pathway. Liver cancer cells,
HepG2, were treated with vismodegib or entinostat as single-agents
or in combination. The results revealed that vismodegib as a
single-agent or in combination with entinostat caused markedly
reduced phosphorylated levels of AKT and S6RP, a downstream
effector of PI3K/mTOR signaling, in HepG2 cells (Fig. 5C). Collectively, the results of the
present study indicated that combined use of inhibitors vismodegib
and entinostat may downregulate the PI3K/mTOR signaling.
Discussion
Liver cancer is a complex and heterogeneous disease
associated with genomic aberrations. In order to solve the
difficult-to-treat characteristics of liver cancer, an SHH
signaling pathway inhibitor and a HDAC inhibitor were used to treat
liver cancer cells based on previous research. Our results revealed
that dual use of SHH and HDAC inhibitors effectively induced
apoptosis, inhibited cell proliferation in HepG2 cells, and
promoted cell death in liver cancer cells in an ex vivo
culture model of liver cancer.
The aberrant activation of SHH signaling and the
mutation of its ligand are closely related to the progression of
cancer (8,22). Early clinical trial results have
revealed that vismodegib had good efficacy and safety in basal cell
carcinoma (23), however, it has
been recently revealed that Smo mutation conferred vismodegib
resistance to medulloblastoma (24). Recently studies have revealed that
vismodegib inhibited the SHH signaling pathway rendering tumor
cells sensitive to platinum-based chemotherapy in non-small cell
lung cancer (25) and was sensitive
to liver cancer radiation therapy (26). However, SHH inhibition in liver
cancer with vismodegib is challenging, due to the possible presence
of non-canonical Gli activation mechanisms. For the treatment of
tumors using SHH signaling inhibitors, a combination therapy is
used to increase the therapeutic effect of the inhibitor and
sensitivity to tumors. Therefore, in the present study a HDAC
inhibitor was used in combination with an SHH inhibitor to enhance
the efficacy of the SHH inhibitor and provide more possibilities
for the selection of drug treatment options for liver cancer.
Although high HDAC1 expression is associated with
activated SHH signaling in neural progenitors and medulloblastomas,
loss of HDAC activity was revealed to inhibit Hedgehog-dependent
growth of neural progenitors and tumor cells (11,16,27,28).
HDACs plays a role in regulating many proteins that are closely
related to the initiation and progression of cancer. Several HDACs
have been revealed to have aberrant expression in liver cancer
(15). In the present study
concerning gene expression characteristics as a predictor of
survival in liver cancer patients, it was determined that HDAC
overexpression was associated with lower survival rates in liver
cancer patients (4,29). It has been reported that Gli1 and
Gli2 are acetylated proteins, and their HDAC-mediated deacetylation
was revealed to promote transcriptional activation and positive
autoregulation of the loop by Hedgehog-induced upregulation of
HDAC1 (28,30). Consistent with this, our present
study revealed that the dual use of SHH and HDAC inhibitors could
effectively inhibit Gli expression and exert effective antitumor
activity in liver cancer cells. In the present study, an ex
vivo culture model of surgically resected fresh liver cancer
specimens was used to evaluate drug efficacy. Since the cultured
tissue explants retained tissue structure and cellularity, as
observed in primary tumors, this approach provided a suitable
platform to assess acute treatment response to liver cancer. Our
results indicated that the combination of vismodegib and entinostat
was effective in promoting the death of tumor explant cells.
The present results revealed that inhibition of SHH
signaling not only inhibited the growth of liver cancer cells and
promoted apoptosis, but also induced G2/M cell cycle arrest. In
addition, SHH signaling pro-apoptotic factors (cleaved PARP and
Bax) were upregulated, and the activity of anti-apoptotic factors
(including Bcl-2) were inhibited. SHH-related pathways do not
always work directly, but rather induce complex cascades of
networks that intersect with other pathways to function in
different biological processes. The PI3K/mTOR signaling pathway is
closely related to SHH (31).
Studies have indicated that the PI3K/mTOR signaling has a
synergistic effect on SHH signaling in embryonic development and
cancer (21,31). To further investigate the
relationship between the PI3K/mTOR and SHH pathways in liver
cancer, the results in the present study clearly revealed that
combined use of SHH and HDAC inhibitors blocked the activation of
the PI3K/mTOR pathway and played an antitumor role. Therefore, it
is suggested that the treatment of SHH signaling should consider
the effect of PI3K/mTOR pathway.
In summary, our data indicated that HDAC inhibition
contributed to the sensitivity of SHH inhibition, and inhibition of
PI3K/mTOR by inhibition of SHH and HDAC may play a key role in this
synergy. Although the number of liver cancer cell line models and
ex vivo models studied in the present study was limited, our
data inidcated that the combined use of SHH and HDAC inhibitors may
benefit patients with liver cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shandong
Provincial Natural Science Foundation (nos. ZR2013HM047 and
ZR201709250448), the Provincial Medicine and Health Science
Technology Development Program Shandong (nos. 2017WSB29030 and
2017WSB29031), and the Provincial Higher Education Science
Technology Development Program of Shandong (no. J18KB138).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
DW, JL and HC conceived the project and designed the
experiments. JL and HL performed the cell viability assay and the
drug combination analysis. HC contributed to the western blotting
tests and data analysis. HC and YSh were responsible for the cell
proliferation assay and the soft agar colony formation assay. HS
contributed to the apoptosis and cell cycle analysis experiments.
YL, YSu and YW contributed to the experiments about the clinical
specimen. HC and DW wrote the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The acquisition of tumor tissue was executed with an
Institutional Review Board protocol approved by Binzhou Medical
University (Yantai, China) and written informed consent was
provided by this patient.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heimbach JK, Kulik LM, Finn RS, Sirlin CB,
Abecassis MM, Roberts LR, Zhu AX, Murad MH and Marrero JA: Aasld
guidelines for the treatment of hepatocellular carcinoma.
Hepatology. 67:358–380. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi KJ, Baik IH, Ye SK and Lee YH:
Molecular targeted therapy for hepatocellular carcinoma: Present
status and future directions. Biol Pharm Bull. 38:986–991. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Y, Han C, Lu L, Magliato S and Wu T:
Hedgehog signaling pathway regulates autophagy in human
hepatocellular carcinoma cells. Hepatology. 58:995–1010. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Q, Huang S, Yang L, Zhao L, Yin Y,
Liu Z, Chen Z and Zhang H: Down-regulation of Sonic Hedgehog
signaling pathway activity is involved in 5-fluorouracil-induced
apoptosis and motility inhibition in Hep3B cells. Acta Biochim
Biophys Sin. 40:819–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li P, Lee EH, Du F, Gordon RE, Yuelling
LW, Liu Y, Ng JM, Zhang H, Wu J, Korshunov A, et al: Nestin
mediates hedgehog pathway tumorigenesis. Cancer Res. 76:5573–5583.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hanaoka J, Shimada M, Utsunomiya T, Morine
Y, Imura S, Ikemoto T and Mori H: Significance of sonic Hedgehog
signaling after massive hepatectomy in a rat. Surg Today.
43:300–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lauth M, Bergstrom A, Shimokawa T and
Toftgard R: Inhibition of gli-mediated transcription and tumor cell
growth by small-molecule antagonists. Proc Natl Acad Sci USA.
104:8455–8460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Axelson M, Liu K, Jiang X, He K, Wang J,
Zhao H, Kufrin D, Palmby T, Dong Z, Russell AM, et al: U.S. Food
and drug administration approval: Vismodegib for recurrent, locally
advanced, or metastatic basal cell carcinoma. Clin Cancer Res.
19:2289–2293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee SJ, Lindsey S, Graves B, Yoo S, Olson
JM and Langhans SA: Sonic Hedgehog-induced histone deacetylase
activation is required for cerebellar granule precursor hyperplasia
in medulloblastoma. PLoS One. 8:e714552013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anestopoulos I, Voulgaridou GP,
Georgakilas AG, Franco R, Pappa A and Panayiotidis MI: Epigenetic
therapy as a novel approach in hepatocellular carcinoma. Pharmacol
Ther. 145:103–119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hardy T and Mann DA: Epigenetics in liver
disease: From biology to therapeutics. Gut. 65:1895–1905. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie HJ, Noh JH, Kim JK, Jung KH, Eun JW,
Bae HJ, Kim MG, Chang YG, Lee JY, Park H, et al: HDAC1 inactivation
induces mitotic defect and caspase-independent autophagic cell
death in liver cancer. PLoS One. 7:e342652012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Noh JH, Jung KH, Kim JK, Eun JW, Bae HJ,
Xie HJ, Chang YG, Kim MG, Park WS, Lee JY, et al: Aberrant
regulation of HDAC2 mediates proliferation of hepatocellular
carcinoma cells by deregulating expression of G1/S cell cycle
proteins. PLoS One. 6:e281032011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu M, Hernandez M, Shen S, Sabo JK, Kelkar
D, Wang J, O'Leary R, Phillips GR, Cate HS and Casaccia P:
Differential modulation of the oligodendrocyte transcriptome by
sonic hedgehog and bone morphogenetic protein 4 via opposing
effects on histone acetylation. J Neurosci. 32:6651–6664. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Valdora F, Banelli B, Stigliani S, Pfister
SM, Moretti S, Kool M, Remke M, Bai AH, Brigati C, Hielscher T, et
al: Epigenetic silencing of DKK3 in medulloblastoma. Int J
Mol Sci. 14:7492–7505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang D, Li C, Zhang Y, Wang M, Jiang N,
Xiang L, Li T, Roberts TM, Zhao JJ, Cheng H, et al: Combined
inhibition of pi3k and parp is effective in the treatment of
ovarian cancer cells with wild-type PIK3CA genes. Gynecol
Oncol. 142:548–556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Herynk MH, Stoeltzing O, Reinmuth N,
Parikh NU, Abounader R, Laterra J, Radinsky R, Ellis LM and Gallick
GE: Down-regulation of c-Met inhibits growth in the liver of human
colorectal carcinoma cells. Cancer Res. 63:2990–2996.
2003.PubMed/NCBI
|
|
20
|
Chao H, Wang L, Hao J, Ni J, Chang L,
Graham PH, Kearsley JH and Li Y: Low dose histone deacetylase
inhibitor, LBH589, potentiates anticancer effect of docetaxel in
epithelial ovarian cancer via PI3K/Akt pathway in vitro. Cancer
Lett. 329:17–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song Z, Du Y and Tao Y: Blockade of sonic
Hedgehog signaling decreases viability and induces apoptosis in
retinoblastoma cells: The key role of the pi3k/akt pathway. Oncol
Lett. 14:4099–4105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang S, He J, Zhang X, Bian Y, Yang L,
Xie G, Zhang K, Tang W, Stelter AA, Wang Q, et al: Activation of
the hedgehog pathway in human hepatocellular carcinomas.
Carcinogenesis. 27:1334–1340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rudin CM: Vismodegib. Clin Cancer Res.
18:3218–3222. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yauch RL, Dijkgraaf GJ, Alicke B, Januario
T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, et
al: Smoothened mutation confers resistance to a Hedgehog pathway
inhibitor in medulloblastoma. Science. 326:572–574. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Giroux Leprieur E, Vieira T, Antoine M,
Rozensztajn N, Rabbe N, Ruppert AM, Lavole A, Cadranel J and Wislez
M: Sonic Hedgehog pathway activation is associated with resistance
to platinum-based chemotherapy in advanced non-small-cell lung
carcinoma. Clin Lung Cancer. 17:301–308. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsai CL, Hsu FM, Tzen KY, Liu WL, Cheng AL
and Cheng JC: Sonic Hedgehog inhibition as a strategy to augment
radiosensitivity of hepatocellular carcinoma. J Gastroenterol
Hepatol. 30:1317–1324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chun SG, Zhou W and Yee NS: Combined
targeting of histone deacetylases and hedgehog signaling enhances
cytoxicity in pancreatic cancer. Cancer Biol Ther. 8:1328–1339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Canettieri G, Di Marcotullio L, Greco A,
Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E,
Ferretti E, Miele E, et al: Histone deacetylase and
Cullin3-RENKCTD11 ubiquitin ligase interplay regulates
Hedgehog signalling through Gli acetylation. Nat Cell Biol.
12:132–142. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z,
Roskams T, Durnez A, Demetris AJ and Thorgeirsson SS:
Classification and prediction of survival in hepatocellular
carcinoma by gene expression profiling. Hepatology. 40:667–676.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hay JF, Lappin K, Liberante F, Kettyle LM,
Matchett KB, Thompson A and Mills KI: Integrated analysis of the
molecular action of Vorinostat identifies epi-sensitised targets
for combination therapy. Oncotarget. 8:67891–67903. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen JS, Huang XH, Wang Q, Huang JQ, Zhang
LJ, Chen XL, Lei J and Cheng ZX: Sonic Hedgehog signaling pathway
induces cell migration and invasion through focal adhesion
kinase/AKT signaling-mediated activation of matrix
metalloproteinase (MMP)-2 and MMP-9 in liver cancer.
Carcinogenesis. 34:10–19. 2013. View Article : Google Scholar : PubMed/NCBI
|