Introduction
As one of the most frequently diagnosed cancers in
women worldwide, it is estimated that cervical cancer accounts for
more than 500,000 new cases and 250,000 deaths each year (1). Human papillomavirus (HPV) infection is
recognized as the most significant risk factor that presents in
most cervical cancers (2,3). Integration of HPV into the cellular
genome causes genome instability, transcriptional variations and
epigenetic alterations (4,5). However, HPV alone is not sufficient to
induce malignant transformation (6). Therefore, additional cancer-causing
genetic variations may underlie the development and progression of
cervical cancer.
MicroRNAs (miRNAs) are small, non-coding RNA
molecules of ~22 nucleotides in length, functioning by targeting
mRNAs to regulate gene expression at the post-transcriptional level
(7,8). miRNAs are involved in diverse
biological processes, including the cell cycle, differentiation and
metabolism (9). Increasing evidence
highlights the involvement of altered miRNA expression in cervical
cancer (10). Examples include
hsa-miR-21 (11), hsa-miR-196a
(12) and hsa-miR-497 (13). hsa-miR-21 is an oncogene
overexpressed in cervical cancer, the inhibition of which
upregulates the tumor suppressor, programmed cell death 4 (PDCD4)
and suppresses cell proliferation (11). Upregulation of hsa-miR-196a has also
been detected in cervical cancer tissues, in which it promotes
cancer cell proliferation (12).
Unlike hsa-miR-21 and hsa-miR-196a, hsa-miR-497 is a tumor
suppressor for cervical cancer and suppresses cancer cell migration
and invasion by targeting insulin-like growth factor 1 (IGF-1)
receptor (13).
Potential prognostic miRNA signatures of cervical
cancer have been identified. A miRNA signature for clinical
response consisting of hsa-miR-200a and hsa-miR-9 has been
previously identified by the expression analysis of candidate
miRNAs (14). According to the
expression levels of hsa-miR-200a and hsa-miR-9, cervical cancer
samples could be divided into low- and high-risk groups. Functional
analysis indicated that both hsa-miR-200a and hsa-miR-9 are likely
to play important roles in cervical cancer metastasis. However,
only a limited number of miRNAs were analyzed in the present study
(14), and miRNA expression
differences between pathologic stages have not yet been
examined.
To identify novel outcome predictors of cervical
cancer, we comprehensively analyzed the expression levels of miRNAs
using data from The Cancer Genome Atlas (TCGA). miRNAs
significantly differentially expressed between early (I and II) and
advanced (III and IV) pathologic stages were identified, followed
by the identification of an optimal subset of signature miRNAs. The
subset of signature miRNAs revealed good performance in progression
prediction and may serve as a promising prognostic predictor of
cervical cancer in clinical practice.
Materials and methods
Data source
mRNA and miRNA expression profiles (Illumina HiSeq
2000 RNA sequencing data) of cervical cancer were downloaded from
TCGA (https://gdc-portal.nci.nih.gov/)
database. In addition, the level-3 data in TCGA-CESC was acquired
on an online platform (http://gdac.broadinstitute.org/runs/stddata__2016_01_28/data/CESC/20160128/).
Samples for which both mRNA and miRNA data as well as information
on pathologic stage and survival were selected for the present
study. In total, data of 285 samples were collected, which were
randomly divided into training (143 samples) and validation (142
samples) datasets. Clinical characteristics of these samples are
summarized in Table I.
 | Table I.Summary of clinical characteristics
of the training, validation and entire datasets. |
Table I.
Summary of clinical characteristics
of the training, validation and entire datasets.
| Clinical
characteristics | Training dataset
(N=143) | Validation dataset
(N=142) | Entire dataset
(N=285) |
|---|
| Age (years, mean ±
SD) | 46.65±13.56 | 48.92±13.53 | 47.78±13.57 |
| Pathologic_M
(M0/M1/-) | 55/4/84 | 50/7/85 | 105/11/169 |
| Pathologic_N
(N0/N1/-) | 62/24/57 | 67/28/47 | 129/52/104 |
| Pathologic_T
(T1/T2/T3/T4/-) | 69/28/9/3/34 | 65/40/9/7/21 |
134/68/18/10/55 |
| Pathologic_stage
(I/II/III/IV) | 83/7/43/10 | 72/18/40/12 | 155/25/83/22 |
| Smoking
(Yes/No/-) | 69/56/18 | 56/71/15 | 125/127/33 |
| New tumors
(Yes/No/-) | 24/101/18 | 19/102/21 | 43/203/39 |
| Radiation therapy
(Yes/No/-) | 88/30/25 | 83/33/26 | 171/63/51 |
| Targeted molecular
therapy (Yes/No/-) | 69/33/41 | 59/30/53 | 128/63/94 |
| Status
(Deceased/Alive) | 35/108 | 35/107 | 70/215 |
| Overall survival
months (mean ± SD) | 32.07±39.29 | 32.25±38.57 | 32.16±38.86 |
Screening of significantly
differentially expressed miRNAs
Samples in the training dataset were divided into
early (I and II) and advanced (III and IV) pathologic-stage groups.
miRNA expression profiles were compared between the two groups
using the t-test (http://127.0.0.1:26738/library/stats/html/t.test.html)
and Wilcoxon rank-sum test (http://127.0.0.1:26738/library/stats/html/wilcox.test.html)
under R3.1.0. False discovery rate (FDR) <0.05 and |logFC (fold
change)|>0.263 were set as thresholds for the selection of
significantly differentially expressed miRNAs for both methods.
Significantly differentially expressed miRNAs (Table SI) in the two stages were further
compared using the t-test in excel. Two-way hierarchical clustering
analysis based on centered Pearson correlation (15) was performed using the pheatmap
package (https://cran.r-project.org/package=pheatmap) in R.
Correlation between clusters and pathologic stages were analyzed by
the Chi-square test using the chisq.test function (http://127.0.0.1:21869/library/stats/html/chisq.test.html)
in R. Kaplan-Meier survival analysis was conducted for different
clusters, using the survival package (version 2.40-1; http://cran.r-project.org/package=survival) under
R.
Selection and validation of an optimal
subset of a 7-miRNA signature
Signature miRNAs were selected from differentially
expressed miRNAs using the bootstrap algorithm (16) of the random forest package
(https://cran.r-project.org/package=randomForest)
(17) in R. The optimal subset of a
7-miRNA signature was the one yielding the minimum out-of-bag (OOB)
error.
Based on the expression values of the optimal subset
of miRNAs, two-way hierarchical clustering analysis was conducted
for both the training and the validation dataset, followed by the
Kaplan-Meier survival analysis of different clusters.
A cervical cancer-specific support vector machine
(SVM) classifier was constructed based on the expression values of
the optimal subset of miRNAs, using the SVM function (core
function: Sigmoid kernel; cross: 10-fold cross validation) in the
e1701 package in R (18). The SVM
classifier was used to predict the pathologic stages of samples.
Based on the predictions, samples in both the training and the
validation dataset were classified into either an early-stage-like
or an advanced-stage-like group. The prognosis of different groups
was analyzed using the Kaplan-Meier survival curve analysis.
Independence analysis of an
SVM-predicted group as a prognostic factor
Clinical information on age, pathologic M,
pathologic N, pathologic T, smoking, new tumor, radiation therapy,
and targeted molecular therapy was extracted from both the training
and the validation dataset. Correlations between the clinical
variables and prognoses were analyzed by univariate and
multivariate Cox regressions, using the survival package under
R3.1.0. Clinical variables with P<0.05 were considered to be
significant and independent prognostic factors.
Samples in both the training and the validation
dataset were further stratified according to different clinical
variables. The relation between the SVM-predicted group and the
prognosis of cervical cancer was analyzed using univariate Cox
regression and Kaplan-Meier survival curve analysis for each
stratum, with P<0.05 considered to be statistically
significant.
Functional analysis of targets of a
7-miRNA signature
mRNAs significantly differentially expressed between
early and advanced stages were screened as aforementioned for
miRNAs. mRNAs targeted by the optimal subset of a 7-miRNA signature
were predicted using miRWalk. The selected miRNA-mRNA pairs could
be retrieved in four databases including miRWalk (http://mirwalk.umm.uni-heidelberg.de/),
miRanda (19), miRDB (http://www.mirdb.org/), miRMap (https://mirmap.ezlab.org/app/), miRNAMap (http://mirnamap.mbc.nctu.edu.tw/), RNA22
(https://cm.jefferson.edu/rna22/) and
TargertScan (http://www.targetscan.org/mamm_31/), which were
further intersected with the significantly differentially expressed
mRNAs. Thus, the selected mRNAs were used for the construction of a
regulatory network of the optimal subset of a 7-miRNA signature.
mRNAs in the regulatory network were functionally annotated using
DAVID (https://david.ncifcrf.gov/) (20) and significantly enriched Gene
Ontology (GO) biological processes, and Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathways were retrieved, with P<0.05 set as a
threshold for significantly enriched terms.
Results
Differential miRNA expression between
the early- and advanced-stage groups
Among the 143 samples in the training dataset, 90
were early-stage and 53 were advanced-stage samples. miRNAs with
low expression (median expression value <1.0) were removed and
the remaining 318 miRNAs were used for further analysis. In total,
51 miRNAs were identified to be significantly differentially
expressed between early- and advanced-stage groups by t-test,
whereas 49 miRNAs were identified by Wilcoxon rank-sum test
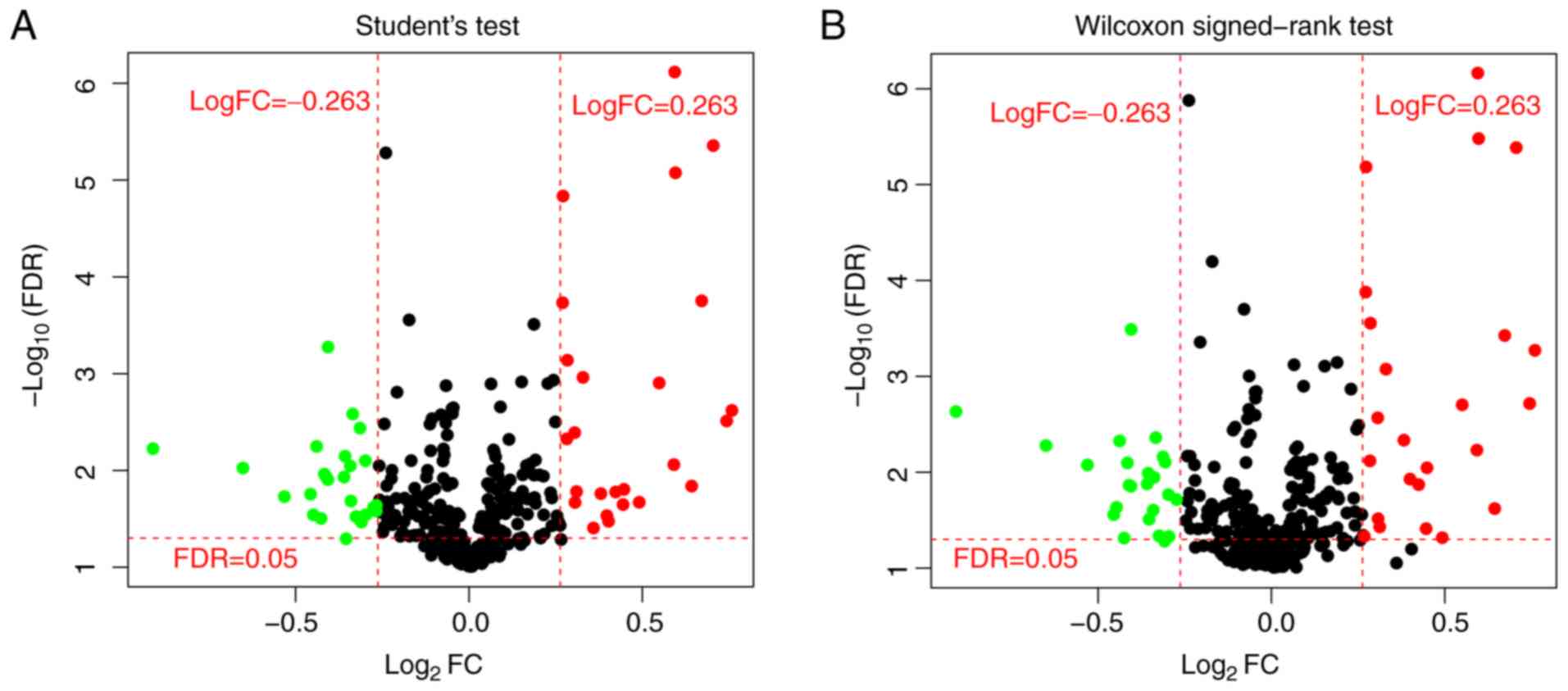
(Fig. 1). In total, 44 miRNAs were
identified to be significantly differentially expressed by both
t-test and Wilcoxon rank-sum test.
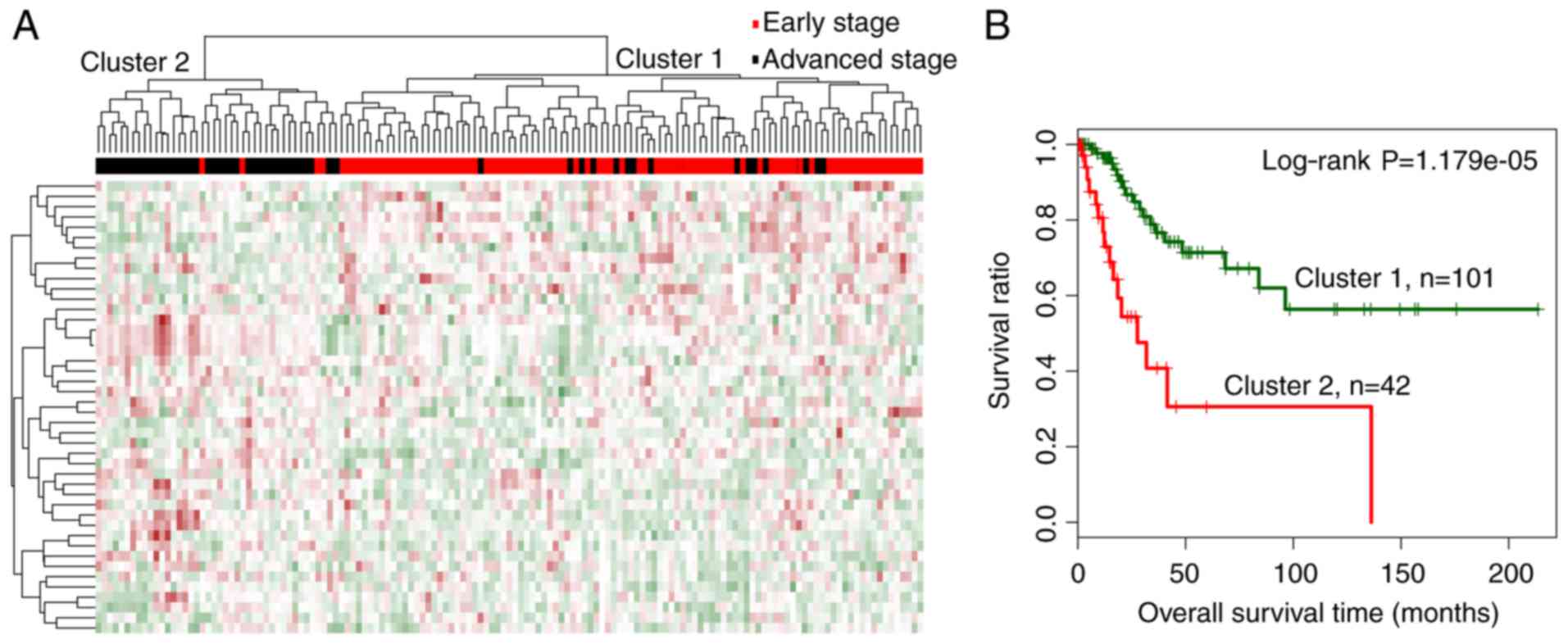
Two-way hierarchical clustering analysis was carried
out based on the expression levels of the 44 miRNAs and separated
the training dataset into 2 distinct clusters, designated as
clusters 1 and 2 (Fig. 2A). Cluster
1 mainly consisted of early-stage samples, including 86 early- and
15 advanced-stage samples. Cluster 2 mainly consisted of
advanced-stage samples, including 38 advanced- and 4 early-stage
samples. The overall accuracy of hierarchical clustering in
classifying samples was 86.71% (124 out of 143 samples). Moreover,
the 2 clusters were correlated significantly with cancer
progression status (χ2=69.5245, P<2.2e-16).
Kaplan-Meier analysis revealed that samples in cluster 1 were
related with a significantly better prognosis than those in cluster
2 (Fig. 2B; log-rank P=1.179e-05),
which was consistent with the dominant sample stages in the
respective clusters.
Cervical cancer-related optimal subset
of a 7-miRNA signature
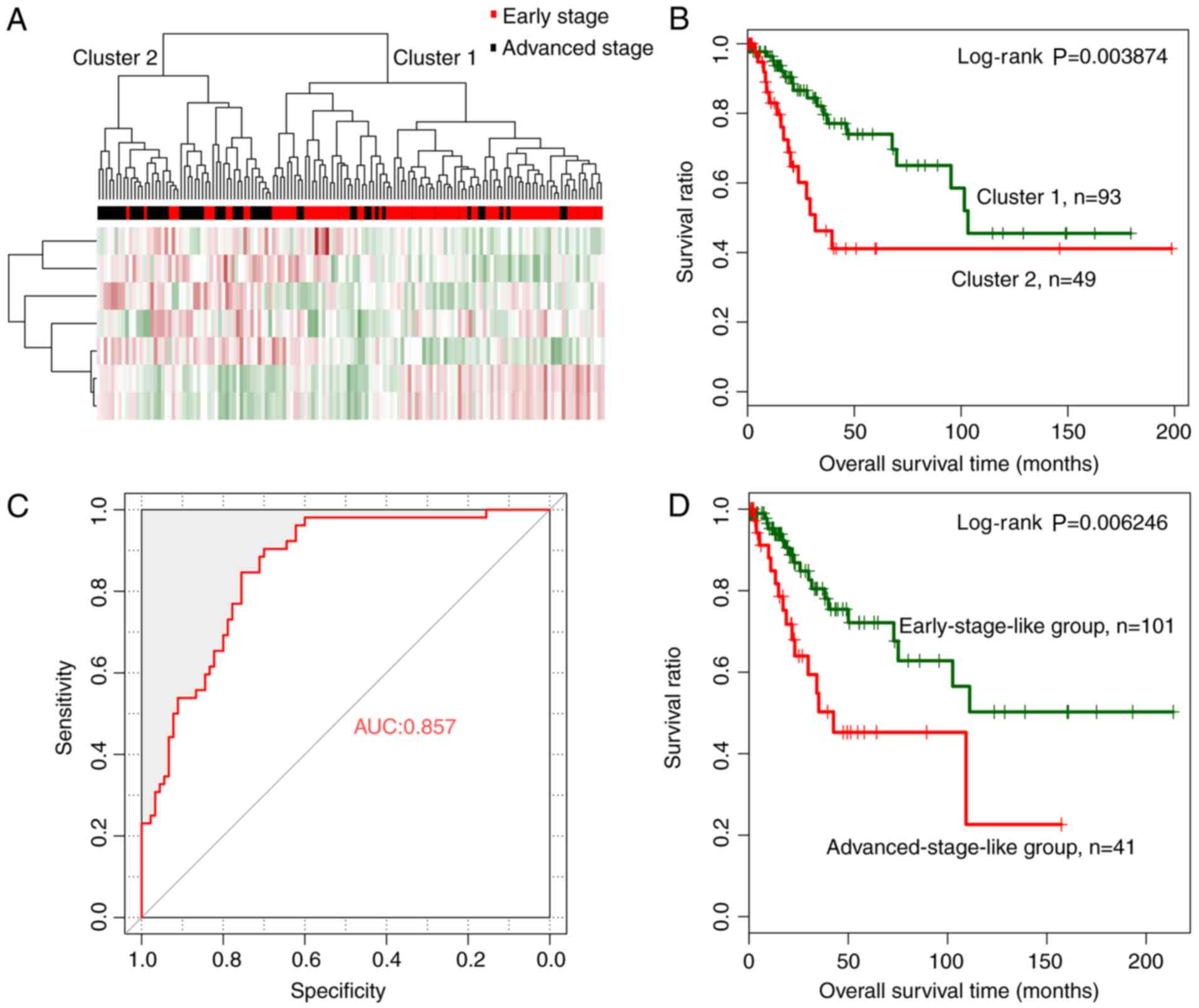
The random forest algorithm was used to identify an
optimal subset of a 7-miRNA signature from the 44 significantly
differentially expressed miRNAs, using the training dataset. The
results revealed that the OOB error reached a minimum (0.181) when
7 miRNAs were used for fitting (Fig.
3A). These 7 miRNAs, including miRNAs hsa-miR-144, hsa-miR-147,
hsa-miR-218, hsa-miR-425, hsa-miR-451, hsa-miR-483 and hsa-miR-486,
were selected as optimal miRNAs and are summarized in Table II. According to the expression
levels of these 7 miRNAs in the training dataset, hsa-miR-147 and
hsa-miR-218 exhibited a significantly higher expression in the
early than in the advanced stage, whereas the remaining 5 miRNAs
revealed a significantly lower expression level in the early stage
(Fig. 3B).
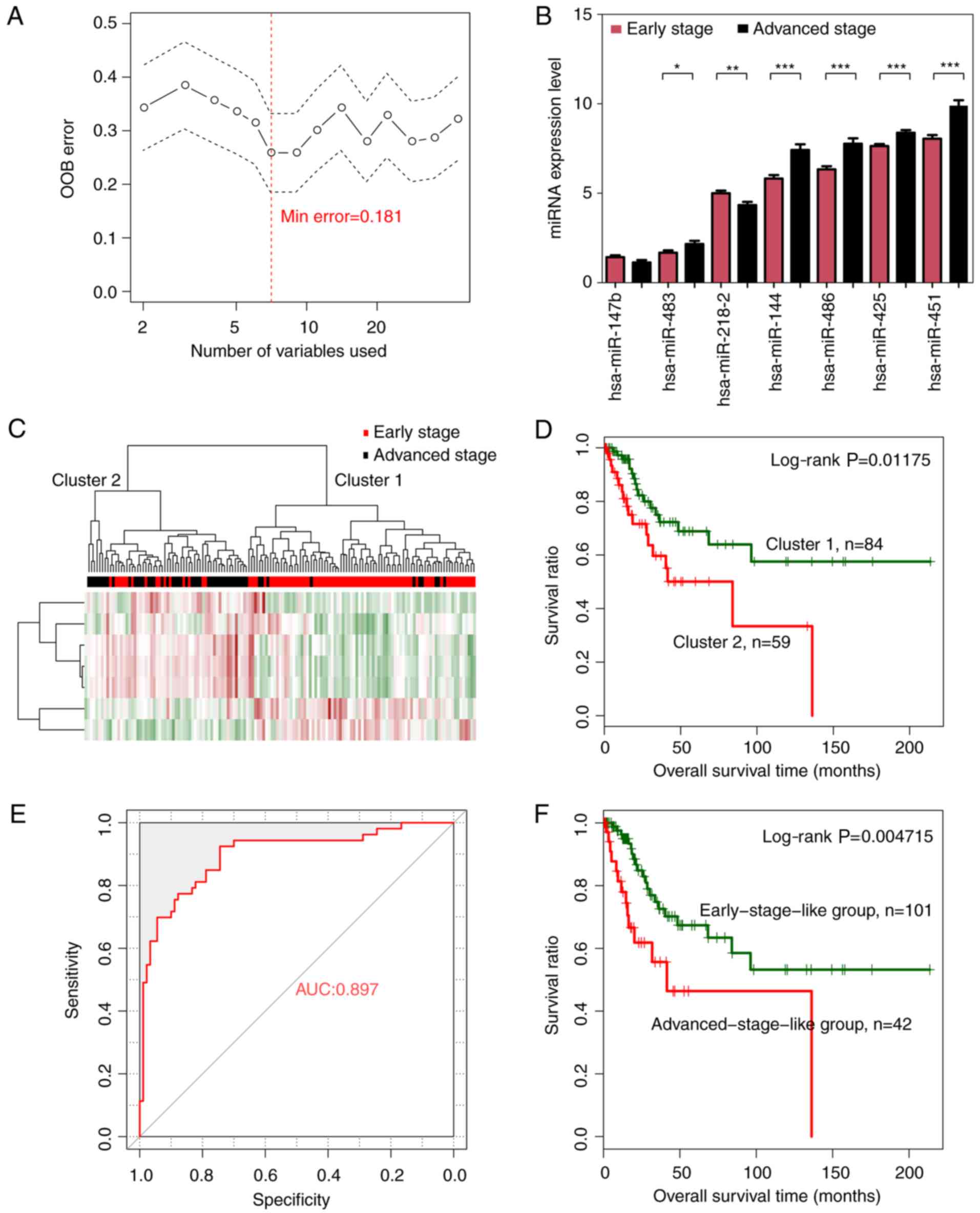 | Figure 3.Optimal subset of signature miRNAs
and their performance in sample classification. (A) OOB error curve
for signature miRNA selection using the training dataset. The OOB
error is plotted against the number of signature miRNAs used. The
red vertical dashed line indicates the minimum OOB error (0.181),
where the number of selected signature miRNAs is 7. (B) Expression
levels of the signature miRNAs of the optimal subset in early-
(red) and advanced (black)-stage groups using a t-test. *P<0.05,
**P<0.01, ***P<0.005. (C) Two-way hierarchical clustering of
samples in the training dataset based on the expression levels of
the 7 signature miRNAs yielded 2 clusters, which are labeled as
cluster 1 and cluster 2. Early- and advanced-stage samples are
indicated in red and black in the horizontal bar, respectively. (D)
Kaplan-Meier survival curves of the 2 clusters in C. Survival
curves for clusters 1 and 2 are indicated in green and red,
respectively. (E) ROC curves of the training dataset generated
using the SVM classifier. The AUC was calculated to be 0.897. (F)
Kaplan-Meier survival curves for early-stage-like (green curve) and
advanced-stage-like (red curve) groups as predicted by the SVM
classifier. OOB, out-of-bag; ROC, receiver operating
characteristic; SVM, specific support vector machine; AUC, area
under the curve. |
| Table II.Progression-related signature miRNAs
of cervical cancer. |
Table II.
Progression-related signature miRNAs
of cervical cancer.
|
| Wilcoxon test | t-test |
|---|
|
|
|
|
|---|
| miRNAs | logFC | FDR | P-value | logFC | FDR | P-value |
|---|
| hsa-miR-144 | 0.701563604 | 4.16E-06 | 1.78E-07 | 0.699709 | 4.46E-06 | 1.95E-07 |
| hsa-miR-147b | −0.654720572 | 0.005331 | 0.000228 | −0.65658 | 0.009557 | 0.000419 |
| hsa-miR-218-2 | −0.40945555 | 0.000328 | 1.40E-05 | −0.41131 | 0.000538 | 2.36E-05 |
| hsa-miR-425 | 0.268232408 | 6.62E-06 | 2.83E-07 | 0.266381 | 1.48E-05 | 6.49E-07 |
| hsa-miR-451 | 0.590364706 | 6.95E-07 | 2.97E-08 | 0.588512 | 7.74E-07 | 3.39E-08 |
| hsa-miR-483 | 0.740086852 | 0.001946 | 8.33E-05 | 0.738239 | 0.00312 | 0.000137 |
| hsa-miR-486 | 0.592724644 | 3.35E-06 | 1.43E-07 | 0.590872 | 8.53E-06 | 3.74E-07 |
Hierarchical clustering analysis based on the
expression levels of the 7-miRNA signature revealed that samples in
the training dataset could be separated into 2 clusters, (Fig. 3C). Similar to the hierarchical
clustering results based on the expression levels of all 44
significantly differentially expressed miRNAs, cluster 1 mainly
consisted of early-stage samples (74 early- vs. 10 advanced-stage
samples) whereas cluster 2 mainly consisted of advanced-stage
samples (16 early- vs. 43 advanced-stage samples). The overall
classification accuracy was 81.82% (117 out of 143 samples).
Additionally, cluster 1 was related with significantly better
prognosis than cluster 2 (Fig. 3D;
log-rank P=0.01175).
A cervical cancer-specific SVM
classifier for pathologic stage prediction
A cervical cancer-specific SVM classifier was
constructed based on the expression levels of a 7-miRNA signature
in the optimal subset. Samples in the training dataset were
classified as early-stage-like or advanced-stage-like using the SVM
classifier. The results revealed that the SVM classifier could
classify samples in the training dataset with an overall accuracy
of 85.31% (122 out of 143 samples; sensitivity, 79.81%,
specificity, 94.44%, positive prediction value (PPV), 88.095%,
negative prediction value (NPV), 84.16%, and area under the
receiver operating characteristic (ROC) curve (AUC), 0.897)
(Fig. 3E). Kaplan-Meier survival
analysis revealed that the predicted early-stage-like group had a
significantly better prognosis than the advanced-stage-like group
(Fig. 3F; log-rank P=0.004715).
Validation of the performance of a
7-miRNA signature in pathologic stage prediction
The validation dataset was used to validate the
performance of a 7-miRNA signature in predicting pathologic stage.
The validation samples were first classified into cluster 1 and
cluster 2 by hierarchical clustering (Fig. 4A). Similar to the results for the
training dataset, cluster 1 mainly consisted of early-stage samples
(78 early- vs. 15 advanced-stage samples) and cluster 2 mainly
consisted of advanced-stage samples (12 early- vs. 37
advanced-stage samples). The overall accuracy was 80.99% (115 out
of 142 samples). Prognosis in the 2 clusters was analyzed using
Kaplan-Meier survival curve analysis. Consistent with the findings
based on the training dataset (Fig.
4D), classification in cluster 1 indicated significantly better
prognosis (Fig. 4B).
Then, we classified the validation samples into
early-stage-like and advanced-stage-like groups using the SVM
classifier. Consistent with the training dataset findings, the
validation samples were classified with an accuracy of 80.98% (115
out of 142 samples; sensitivity, 73.46%, specificity, 91.11%, PPV,
80.49%, NPV, 81.19% and AUC, 0.857) (Fig. 4C). Kaplan-Meier survival curve
analysis revealed that the early-stage-like samples corresponded
with significantly better prognosis than the advanced-stage-like
group (Fig. 4D; log-rank
P=0.006246).
Independence of the SVM-predicted
group in progression prediction
Correlations between clinical variables and
prognosis were analyzed using Cox regression. Both univariate and
multivariate Cox regression revealed that the SVM-predicted group,
pathologic T, and new tumors were independent prognostic factors,
since they were significantly correlated with prognosis (P<0.05)
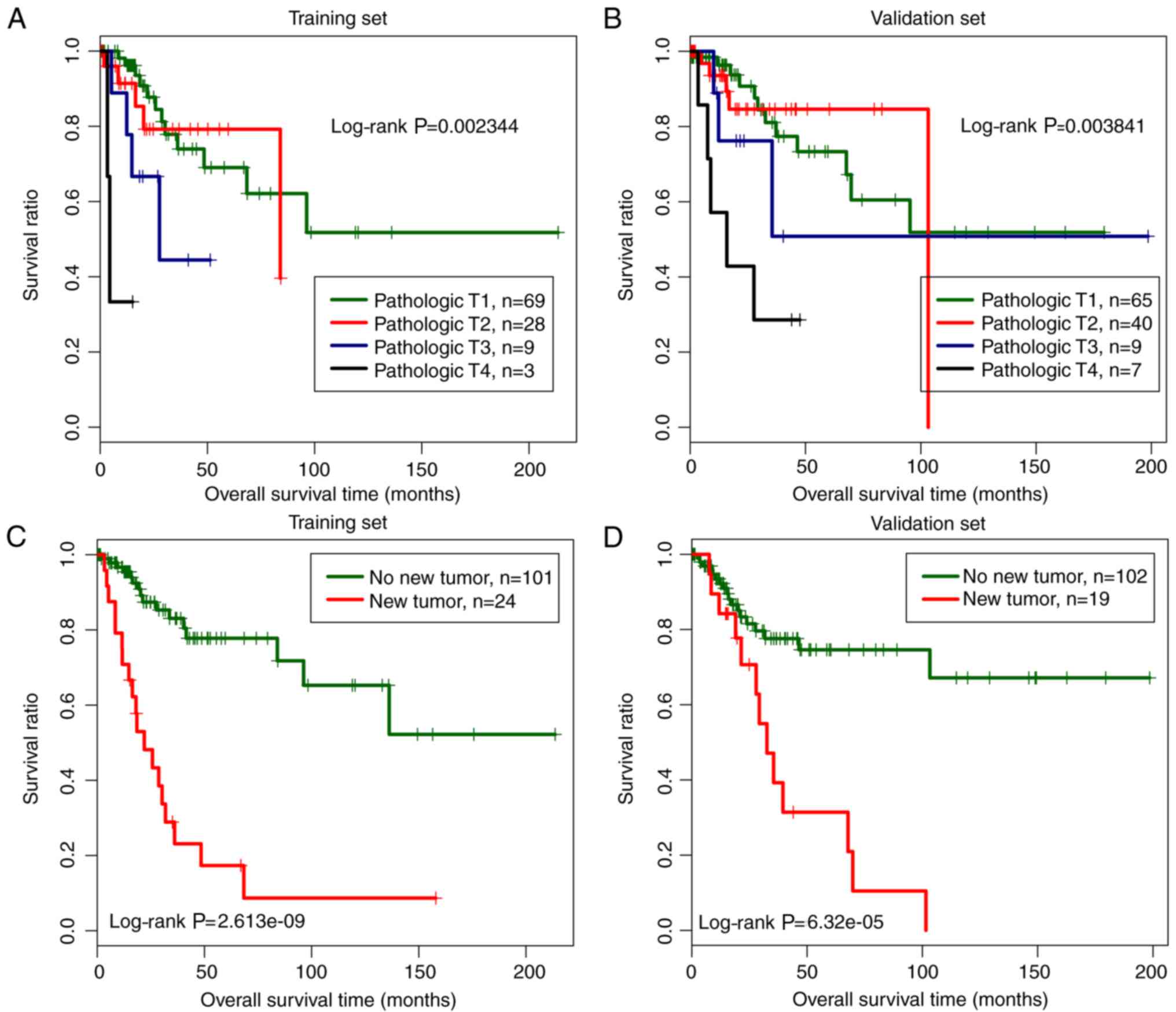
in both the training and the validation dataset (Table III). Kaplan-Meier survival curve
analysis for both the training and the validation dataset revealed
that samples under pathologic T1 and T2 categories were related to
a significantly better prognosis than those under pathologic T3 and
T4 categories, and samples without new tumors were correlated with
a better prognosis than those with new tumors (Fig. 5).
| Table III.Cox regression analysis of the
correlations between the clinical variables and prognosis for both
the training and the validation datasets. |
Table III.
Cox regression analysis of the
correlations between the clinical variables and prognosis for both
the training and the validation datasets.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variables | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Training dataset
(N=143) |
| SVM predicted
group | 2.601 | 1.308–5.172 | 0.00472 | 2.07 | 0.341–2.587 | 0.0272 |
|
(Early/Advanced stage) |
| Age (years) | 1.239 | 0.633–2.426 | 0.531 | – | – | – |
|
(≤45/>45) |
| Pathologic M | – | – | – | – | – | – |
|
(M0/M1) |
| Pathologic N | 4.861 | 1.583–14.93 | 0.00227 | 3.037 | 0.902–10.22 | 0.0729 |
|
(N0/N1) |
| Pathologic T | 2.076 | 1.27–3.395 | 0.00234 | 2.916 | 1.209–7.032 | 0.0172 |
|
(T1/T2/T3/T4) |
| Smoking | 1.461 | 0.726–2.944 | 0.286 | – | – | – |
|
(Yes/No) |
| New tumors | 6.036 | 3.083–11.82 | 2.60E-09 | 6.101 | 1.964–14.24 | 0.00053 |
|
(Yes/No) |
| Radiation
therapy | 0.9697 | 0.4204–2.237 | 0.943 | – | – | – |
|
(Yes/No) |
| Targeted molecular
therapy | 0.891 | 0.4168–1.904 | 0.765 | – | – | – |
|
(Yes/No) |
| Validation dataset
(N=142) |
| SVM predicted
group | 2.473 | 1.265–4.836 | 0.00625 | 1.233 | 0.422–3.601 | 0.00702 |
|
(Early/Advanced stage) |
| Age (years) | 1.248 | 0.628–2.48 | 0.526 | – | – | – |
|
(≤45/>45) |
| Pathologic M | – | – | – | – | – | – |
|
(M0/M1) |
| Pathologic N | 1.81 | 0.732–4.474 | 0.193 | – | – | – |
|
(N0/N1) |
| Pathologic T | 1.71 | 1.174–2.491 | 0.00384 | 1.739 | 1.119–2.701 | 0.01388 |
|
(T1/T2/T3/T4) |
| Smoking | 1.39 | 0.683–2.827 | 0.362 | – | – | – |
|
(Yes/No) |
| New tumors | 3.94 | 1.908–8.136 | 6.32E-05 | 5.229 | 2.117–12.919 | 0.000337 |
|
(Yes/No) |
| Radiation
therapy | 1.155 | 0.517–2.578 | 0.725 | – | – | – |
|
(Yes/No) |
| Targeted molecular
therapy | 1.074 | 0.472–2.446 | 0.865 | – | – | – |
|
(Yes/No) |
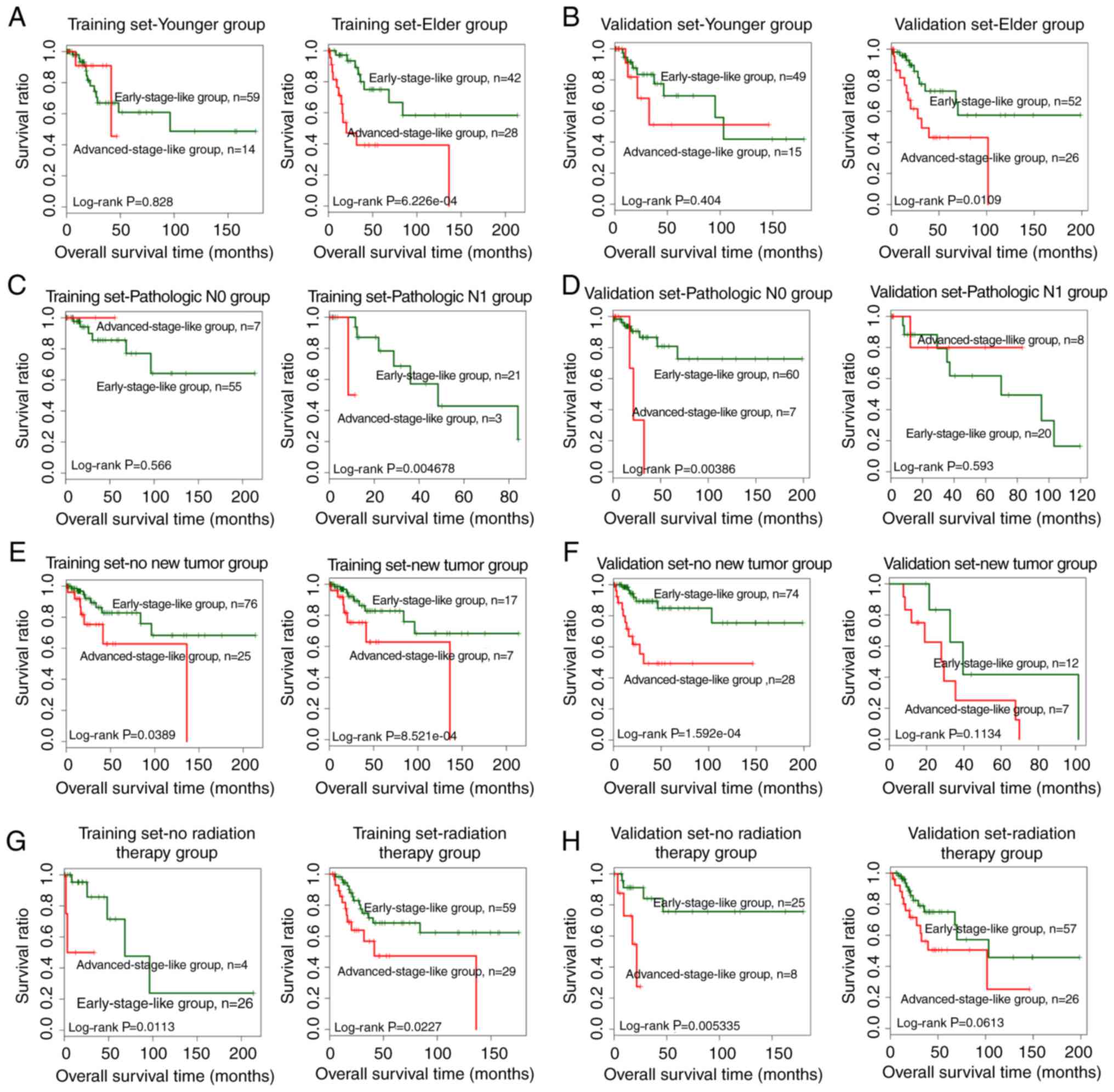
Stratified analysis was performed to evaluate the
independence of the SVM-predicted group as a prognostic factor.
Samples in both the training and the validation dataset were
stratified according to age, pathologic M, pathologic N, pathologic
T, smoking, new tumors, radiation therapy and targeted molecular
therapy. Univariate Cox regression revealed that the SVM-predicted
group was significantly correlated with the prognosis for elder
patients (>45 years of age) in both the training and the
validation dataset (Table IV).
Similar results were obtained for patients without new tumors and
radiation therapy (Table IV).
Additionally, the SVM-predicted group was significantly correlated
with pathologic N1 patients in the training dataset and pathologic
N0 patients in the validation dataset (Table IV). Kaplan-Meier survival curves
for samples stratified by age, pathologic N, new tumors and
radiation therapy are presented in Fig.
6.
| Table IV.Stratified prognosis analysis of
patients in both the training and the validation datasets. |
Table IV.
Stratified prognosis analysis of
patients in both the training and the validation datasets.
|
| Training
dataset |
| Validation
dataset |
|---|
|
|
|
|
|
|---|
| Variables | HR | 95% CI | P-value | Variables | HR | 95% CI | P-value |
|---|
| Age (years) |
|
|
| Age (years) |
|
|
|
| ≤45
(N=73) | 0.846 | 0.187–3.825 | 0.828 | ≤45
(N=64) | 1.647 |
0.504–5.388 | 0.404 |
| >45
(N=70) | 4.379 | 1.751–10.95 | 0.000623 | >45
(N=78) | 2.865 | 1.229–6.684 | 0.0109 |
| Pathologic N |
|
|
| Pathologic N |
|
|
|
| N0
(N=62) | 1.34 | 0.813–2.506 | 0.566 | N0
(N=67) | 8.396 | 1.983–35.55 | 0.00386 |
| N1
(N=24) | 1.321 | 0.571–2.285 | 0.004678 | N1
(N=28) | 0.565 | 0.0677–4.715 | 0.593 |
| Pathologic T |
|
|
| Pathologic T |
|
|
|
| T1+T2
(N=97) | 3.153 | 0.976–10.18 | 0.04289 | T1+T2
(N=105) | 2.135 | 0.673–6.769 | 0.187 |
| T3+T4
(N=12) | 1.062 | 0.178–6.326 | 0.948 | T3+T4
(N=16) | 1.359 | 0.320–5.77 | 0.676 |
| Smoking |
|
|
| Smoking |
|
|
|
| Yes
(N=56) | 1.697 | 0.635–4.537 | 0.286 | Yes
(N=56) | 1.873 | 0.719–4.871 | 0.191 |
| No
(N=69) | 6.558 | 2.119–20.3 | 0.000186 | No
(N=71) | 2.138 | 0.741–6.174 | 0.1503 |
| New tumors |
|
|
| New tumor |
|
|
|
| Yes
(N=24) | 4.868 | 1.759–13.47 | 0.000852 | Yes
(N=19) | 0.3594 | 0.0961–1.345 | 0.1134 |
| No
(N=101) | 2.769 | 1.012–7.576 | 0.0389 | No
(N=102) | 5.213 | 2.006–13.55 | 0.000159 |
| Radiation
therapy |
|
|
| Radiation
therapy |
|
|
|
| Yes
(N=88) | 2.407 | 1.104–5.246 | 0.0227 | Yes
(N=83) | 2.12 | 0.948–4.74 | 0.0613 |
| No
(N=30) | 1.843 | 1.173–6.056 | 0.0113 | No
(N=33) | 1.884 | 1.422–4.372 | 0.005335 |
| Targeted molecular
therapy |
|
|
| Targeted molecular
therapy |
|
|
|
| Yes
(N=69) | 2.405 | 0.995–5.812 | 0.0512 | Yes
(N=59) | 1.643 | 1.389–2.292 | 0.006816 |
| No
(N=33) | 3.257 | 0.622–7.05 | 0.139 | No
(N=30) | 2.33 | 0.829–6.541 | 0.0988 |
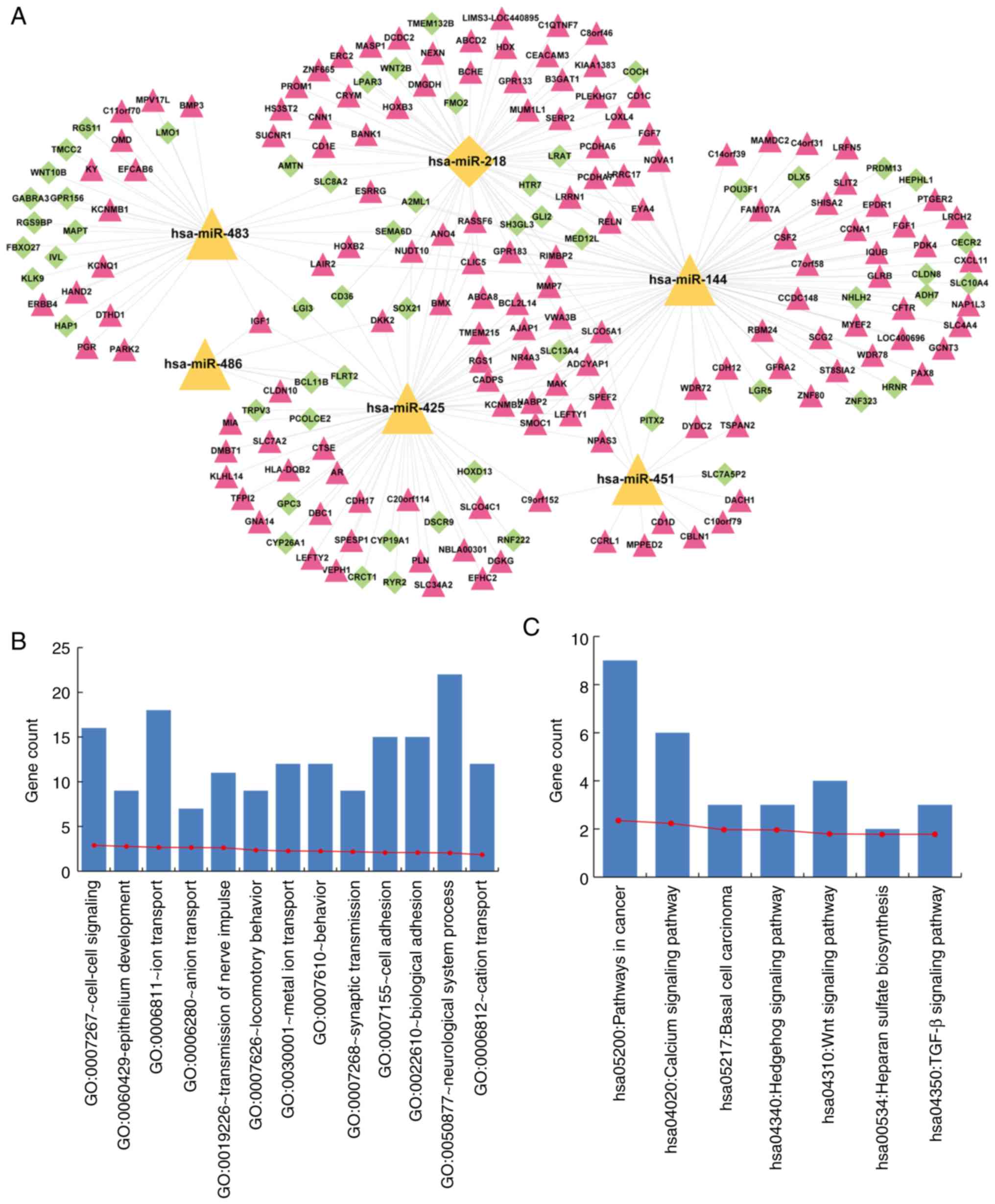
Functional annotation of a 7-miRNA
signature
mRNA expression profiles of early- and
advanced-stage samples were compared, and 535 significantly
differentially expressed mRNAs were identified. Target mRNAs of a
7-miRNA signature were predicted using miRwalk. As a result, we
found that 56, 47, 34, 14, 30 and 31 significantly differentially
expressed mRNAs were targeted by hsa-miR-218, hsa-miR-144,
hsa-miR-425, hsa-miR-483, hsa-miR-486 and hsa-miR-451,
respectively, while none were targeted by hsa-miR-147. We
constructed a cervical cancer-related miRNA-mRNA regulation
network, consisting of a 7-miRNA signature and significantly
differentially expressed mRNAs targeted by this 7-miRNA signature
(Fig. 7A). The network consisted of
207 nodes, containing a 6-miRNA signature and 201 significantly
differentially expressed target mRNAs.
To interpret the potential roles of the miRNA
signature in cervical cancer, functional enrichment analysis was
conducted using DAVID (20). The
results revealed that mRNAs in the network were significantly
enriched for cancer-related GO biological processes and KEGG
pathways (Fig. 7B and C). The GO
terms included cell-cell signaling, epithelium development, ion
transport and adhesion (Fig. 7B).
The KEGG terms included pathways in cancer, calcium signaling
pathway, basal cell carcinoma, Hedgehog signaling pathway, Wnt
signaling pathway, heparan sulfate biosynthesis and TGF-β signaling
pathway (Fig. 7C).
Discussion
Although overwhelming evidence has demonstrated that
numerous microRNAs (miRNAs) are probably correlated with cervical
cancer (10–14), few studies focused on whether these
miRNAs were responsible for the progression of cervical cancer. In
the present study, we comprehensively analyzed cervical
cancer-related miRNA expression profiles from TCGA database and
identified 44 miRNAs significantly differentially expressed between
early- and advanced-stage samples. Subsequently, an optimal subset
of a 7-miRNA signature was extracted, including hsa-miR-144,
hsa-miR-147, hsa-miR-218, hsa-miR-425, hsa-miR-451, hsa-miR-483 and
hsa-miR-486. Moreover, both hierarchical clustering and support
vector machine (SVM) prediction results demonstrated that a 7-miRNA
signature played essential roles in the prediction of cervical
cancer progression. Subsequently, the high performance of this
subset of 7 miRNAs was validated in hierarchical clustering and SVM
prediction using a validation dataset. Additionally, Kaplan-Meier
survival analysis revealed that the SVM-predicted early-stage-like
group exhibited significant better prognosis than the
advanced-stage-like group for both the training and the validation
dataset. We also examined the independence of SVM prediction as a
prognostic factor. Both univariate and multivariate Cox regression
analyses confirmed the independence of the SVM-predicted group in
outcome prediction for the training and validation datasets. The
prognostic power of SVM prediction was additionally evaluated using
stratified analysis. Elder patients (>45 years of age) in both
datasets could be classified into early-stage-like and
advanced-stage-like groups with significant differences in
prognosis. Similar results were acquired for patients with no new
tumors and patients that had received no radiation therapy in both
datasets. Collectively, the results demonstrated that SVM
prediction was a prognostic factor independent of other clinical
factors, including age, new tumors and radiation therapy.
miRNAs act by regulating the expression of target
mRNAs (8,9). To interpret the functions of the
7-miRNA signature in cervical cancer, potential target mRNAs were
identified and subjected to GO and KEGG functional enrichment
analyses. Cancer-related GO biological processes and KEGG pathways
were overrepresented among these mRNAs, which supported the
importance of the 7-miRNA signature in cervical cancer. Two KEGG
terms, Hedgehog signaling pathway and Wnt signaling pathway,
deserve special attention. Both pathways are essential for cancer
cell proliferation, migration, and invasion in various types of
cancers, and thus, represent promising targets for cancer therapy
(21,22).
Accumulating evidence has suggested that the 7-miRNA
signature may be associated with the initiation of cervical cancer
and other cancers. hsa-miR-218, as a tumor suppressor, is encoded
by slit guidance ligand 2 (SLIT2) (23). It has been reported that the
expression of hsa-miR-218 was markedly decreased in the sera from
cervical patients compared to the age-matched normal healthy women,
and its decreased expression was associated with later stages,
cervical adenocarcinoma and lymphatic node metastasis (24). In vitro, miR-218
overexpression inhibited cervical cancer progression by regulating
clonogenicity, migration, invasion and metastasis by targeting
surviving (25). Consistent with
the tumor-suppressive role of hsa-miR-218, we revealed that
hsa-miR-218 expression was reduced in advanced-stage patients. With
regards to the aberrant expression of miR-218, previous studies
indicated that the presence of human papillomavirus (HPV)-16 and
HPV-1 increased E6 protein expression, which play an important role
in the rapid ubiquitin-dependent degradation of p53, resulting in
reduced expression of miR-218 by suppressing the transcription of
SLIT2 (26,27). In addition, hsa-miR-486, hsa-miR-425
and hsa-miR-144 have also been reported to be involved in cervical
cancer (28–30). It has been reported that
hsa-miR-486-3p acted as a tumor suppressor by inhibiting cell
growth and metastasis by targeting ECM1 (28). Sun et al revealed that
hsa-miR-425 was significantly upregulated in cervical cancer
compared with benign cervical disease patients and healthy
controls, and may serve as a prognostic indicator related to high
TNM stage and positive lymph node metastasis (29). hsa-miR-144 was significantly
downregulated in metastatic cervical cancer (30), although its role remains elusive.
The remaining 3 miRNAs have not been reported to be associated with
cervical cancer. However, multiple studies have suggested that they
play crucial roles in other types of cancer. Dysregulation of
hsa-miR-483 has been found in various types of cancer, including
adrenocortical (31), prostate
cancer (32) and lung
adenocarcinoma (33). hsa-miR-451
has been reported to be a tumor suppressor in different cancers
(34,35) and a protective effect of hsa-miR-147
has been found in ovarian cancer (36). Collectively, we inferred that the
7-miRNA signature may play important roles in the development and
progression of cervical cancer and may serve as a potential
biomarker of this disease.
However, there were still multiple limitations in
the present study. Firstly, a sophisticated bioinformatics analysis
depending on a larger sample size would be required to examine the
potential regulatory role of the 7-miRNA signature in cervical
cancer. In addition, the corresponding experiments need to be
conducted to confirm our predictable results such as several
candidate RNA transcripts and critical signaling pathways.
Additionally, extensive clinical information is also required to
integrate into a comprehensive analysis to decipher the regulatory
mechanisms of cervical cancer.
In conclusion, we identified a 7-miRNA signature of
cervical cancer by a comprehensive miRNA expression analysis. The
7-miRNA signature was significantly associated with the progression
of cervical cancer and was used for the construction of a cervical
cancer-specific SVM classifier. The SVM is a promising predictor of
progression and outcome. Meanwhile, the 7-miRNA signature may be a
novel therapeutic target in future clinical practice. However,
further experimental and functional studies are required to reveal
the specific roles of these signature miRNAs in cervical
cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CS and YY conceived, designed the research and
drafted the manuscript; LZ and TZ were responsible for the
acquisition of data; LZ and JY analyzed and interpreted the data;
TZ, SQ and YG performed the statistical analysis; JY, SQ and YG
revised the manuscript for important intellectual content. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Walboomers JM, Jacobs MV, Manos MM, Bosch
FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ and Muñoz N:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Doorbar J: Molecular biology of human
papillomavirus infection and cervical cancer. Clin Sci.
110:525–541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiménez-Wences H, Peralta-Zaragoza O and
Fernández-Tilapa G: Human papilloma virus, DNA methylation and
microRNA expression in cervical cancer (Review). Oncol Rep.
31:2467–2476. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Akagi K, Li J, Broutian TR, Padilla-Nash
H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, et al:
Genome-wide analysis of HPV integration in human cancers reveals
recurrent, focal genomic instability. Genome Res. 24:185–199. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schiffman M, Wentzensen N, Wacholder S,
Kinney W, Gage JC and Castle PE: Human papillomavirus testing in
the prevention of cervical cancer. J Natl Cancer Inst. 103:368–383.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liz J and Esteller M: lncRNAs and
microRNAs with a role in cancer development. Biochim Biophys Acta.
1859:169–176. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Morris KV and Mattick JS: The rise of
regulatory RNA. Nat Rev Genet. 15:423–437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reshmi G and Pillai MR: Beyond HPV:
Oncomirs as new players in cervical cancer. FEBS Lett.
582:4113–4116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yao Q, Xu H, Zhang QQ, Zhou H and Qu LH:
MicroRNA-21 promotes cell proliferation and down-regulates the
expression of programmed cell death 4 (PDCD4) in HeLa cervical
carcinoma cells. Biochem Biophys Res Commun. 388:539–542. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hou T, Ou J, Zhao X, Huang X, Huang Y and
Zhang Y: MicroRNA-196a promotes cervical cancer proliferation
through the regulation of FOXO1 and p27Kip1. Br J
Cancer. 110:1260–1268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luo M, Shen D, Zhou X, Chen X and Wang W:
MicroRNA-497 is a potential prognostic marker in human cervical
cancer and functions as a tumor suppressor by targeting the
insulin-like growth factor 1 receptor. Surgery. 153:836–847. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu X, Schwarz JK, Lewis JS Jr, Huettner
PC, Rader JS, Deasy JO, Grigsby PW and Wang X: A microRNA
expression signature for cervical cancer prognosis. Cancer Res.
70:1441–1448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Xue W, Lv J, Han P, Liu Y and Cui B:
Identification of potential long non-coding RNA biomarkers
associated with the progression of colon cancer. Oncotarget.
8:75834–75843. 2017.PubMed/NCBI
|
|
16
|
Zapf A, Brunner E and Konietschke F: A
wild bootstrap approach for the selection of biomarkers in early
diagnostic trials. BMC Med Res Methodol. 15:432015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Toloşi L and Lengauer T: Classification
with correlated features: Unreliability of feature ranking and
solutions. Bioinformatics. 27:1986–1994. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Q and Liu X: Screening of feature
genes in distinguishing different types of breast cancer using
support vector machine. Onco Targets Ther. 8:2311–2317.
2015.PubMed/NCBI
|
|
19
|
John B, Enright AJ, Aravin A, Tuschl T,
Sander C and Marks DS: Human microRNA targets. PLoS Biol.
2:e3632004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takebe N, Miele L, Harris PJ, Jeong W,
Bando H, Kahn M, Yang SX and Ivy SP: Targeting Notch, Hedgehog, and
Wnt pathways in cancer stem cells: Clinical update. Nat Rev Clin
Oncol. 12:445–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nwabo Kamdje AH, Takam Kamga P, Tagne Simo
R, Vecchio L, Seke Etet PF, Muller JM, Bassi G, Lukong E, Kumar
Goel R, Mbo Amvene J, et al: Developmental pathways associated with
cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol Med.
14:109–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Culp TD, Budgeon LR, Marinkovich MP,
Meneguzzi G and Christensen ND: Keratinocyte-secreted laminin 5 can
function as a transient receptor for human papillomaviruses by
binding virions and transferring them to adjacent cells. J Virol.
80:8940–8950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu J, Wang Y, Dong R, Huang X, Ding S and
Qiu H: Circulating microRNA-218 was reduced in cervical cancer and
correlated with tumor invasion. J Cancer Res Clin Oncol.
138:671–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kogo R, How C, Chaudary N, Bruce J, Shi W,
Hill RP, Zahedi P, Yip KW and Liu FF: The microRNA-218~Survivin
axis regulates migration, invasion, and lymph node metastasis in
cervical cancer. Oncotarget. 6:1090–1100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Liu J, Yuan C, Cui B, Zou X and Qiao
Y: High-risk human papillomavirus reduces the expression of
microRNA-218 in women with cervical intraepithelial neoplasia. J
Int Med Res. 38:1730–1736. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scheffner M, Huibregtse JM, Vierstra RD
and Howley PM: The HPV-16 E6 and E6-AP complex functions as a
ubiquitin-protein ligase in the ubiquitination of p53. Cell.
75:495–505. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye H, Yu X, Xia J, Tang X, Tang L and Chen
F: MiR-486-3p targeting ECM1 represses cell proliferation and
metastasis in cervical cancer. Biomed Pharmacother. 80:109–114.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun L, Jiang R, Li J, Wang B, Ma C, Lv Y
and Mu N: MicroRNA-425-5p is a potential prognostic biomarker for
cervical cancer. Ann Clin Biochem. 54:127–133. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding H, Wu YL, Wang YX and Zhu FF:
Characterization of the microRNA expression profile of cervical
squamous cell carcinoma metastases. Asian Pac J Cancer Prev.
15:1675–1679. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Patterson EE, Holloway AK, Weng J, Fojo T
and Kebebew E: MicroRNA profiling of adrenocortical tumors reveals
miR-483 as a marker of malignancy. Cancer. 117:1630–1639. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang ZG, Ma XD, He ZH and Guo YX:
miR-483-5p promotes prostate cancer cell proliferation and invasion
by targeting RBM5. Int Braz J Urol. 43:1060–1067. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song Q, Xu Y, Yang C, Chen Z, Jia C, Chen
J, Zhang Y, Lai P, Fan X, Zhou X, et al: miR-483-5p promotes
invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1
and ALCAM. Cancer Res. 74:3031–3042. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pan X, Wang R and Wang ZX: The potential
role of miR-451 in cancer diagnosis, prognosis, and therapy. Mol
Cancer Ther. 12:1153–1162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu D, Liu C, Wang X, Ingvarsson S and
Chen H: MicroRNA-451 suppresses tumor cell growth by
down-regulating IL6R gene expression. Cancer Epidemiol. 38:85–92.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kleemann M, Bereuther J, Fischer S,
Marquart K, Hänle S, Unger K, Jendrossek V, Riedel CU, Handrick R
and Otte K: Investigation on tissue specific effects of
pro-apoptotic micro RNAs revealed miR-147b as a potential biomarker
in ovarian cancer prognosis. Oncotarget. 8:18773–18791. 2017.
View Article : Google Scholar : PubMed/NCBI
|