Introduction
Prostate cancer is one of the most common
malignancies diagnosed in males (1).
The pathogenesis of prostate cancer has not been clearly elucidated
to date, and requires further exploration to facilitate early
diagnosis and effective treatment (2). Prostate is a human organ that is easily
susceptible to infections and inflammations. Chronic inflammation
plays a significant role in the development of various types of
cancer including prostate cancer (3).
Approximately 15–20% of cancer patients are
associated with infections or inflammation and prostate cancer is
one such cancer type (4). In a
self-reported prospective cohort study of 5,821 men over 65 years
of age, chronic prostatitis was shown to be a significant factor in
the occurrence of prostate cancer (5). Chronic inflammation triggers
proliferative inflammatory atrophy (PIA) of the prostate, which in
turn acts as a potential precursor lesion to prostatic
intraepithelial neoplasia (PIN) and carcinoma (6).
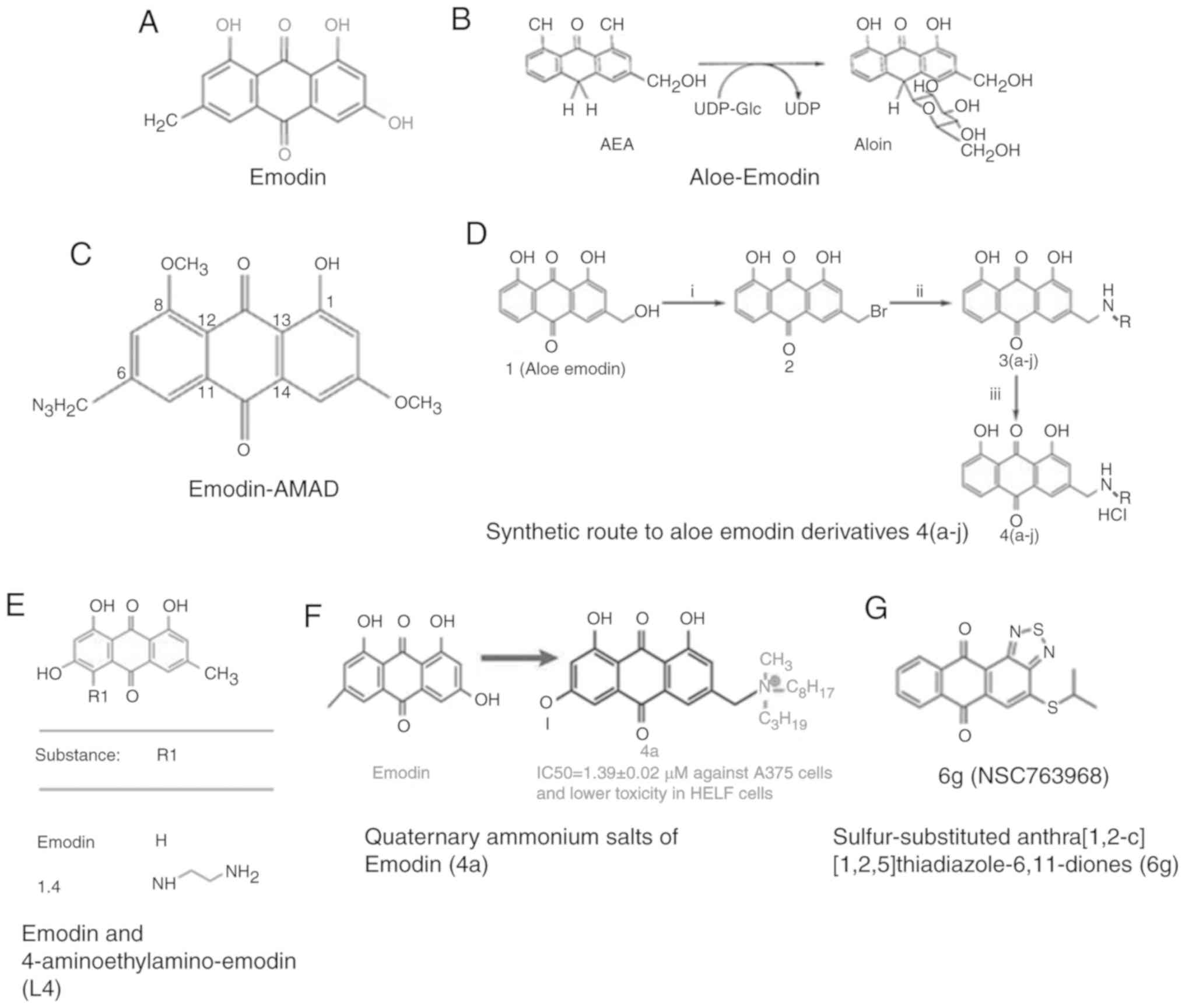
Emodin (1,3,8-3 hydroxy-6-methyl anthraquinone),
with a molecular formula of
C15H10O5, (Fig. 2A) is an active ingredient of the plant
rhubarb, and is mostly used for relieving abdominal distension,
constipation and other gastrointestinal symptoms (7). Recent studies show that emodin exhibits
anti-inflammatory and anticancer effects on prostate cancer
(8). In this review, we aim to
clarify the correlation between inflammation and prostate cancer,
as well as the mechanism of action of emodin in prostate cancer to
explore its role in the inhibition of prostate cancer through
anti-inflammatory pathways.
Inflammation in prostate cancer
development
Correlation between inflammation and
prostate cancer
Inflammation is regarded as a hallmark of the
occurrence and development of cancer (9). In 1863, Rudolf Virchow found leukocytes
in neoplastic tissues and hypothesized ‘lymphatic infiltration’ as
the first step in cancer progression in chronic inflammatory
regions. In the 1990s, the study of an inflammatory
microenvironment in tumor tissues was the main research focus in
the field of cancer research (4). A
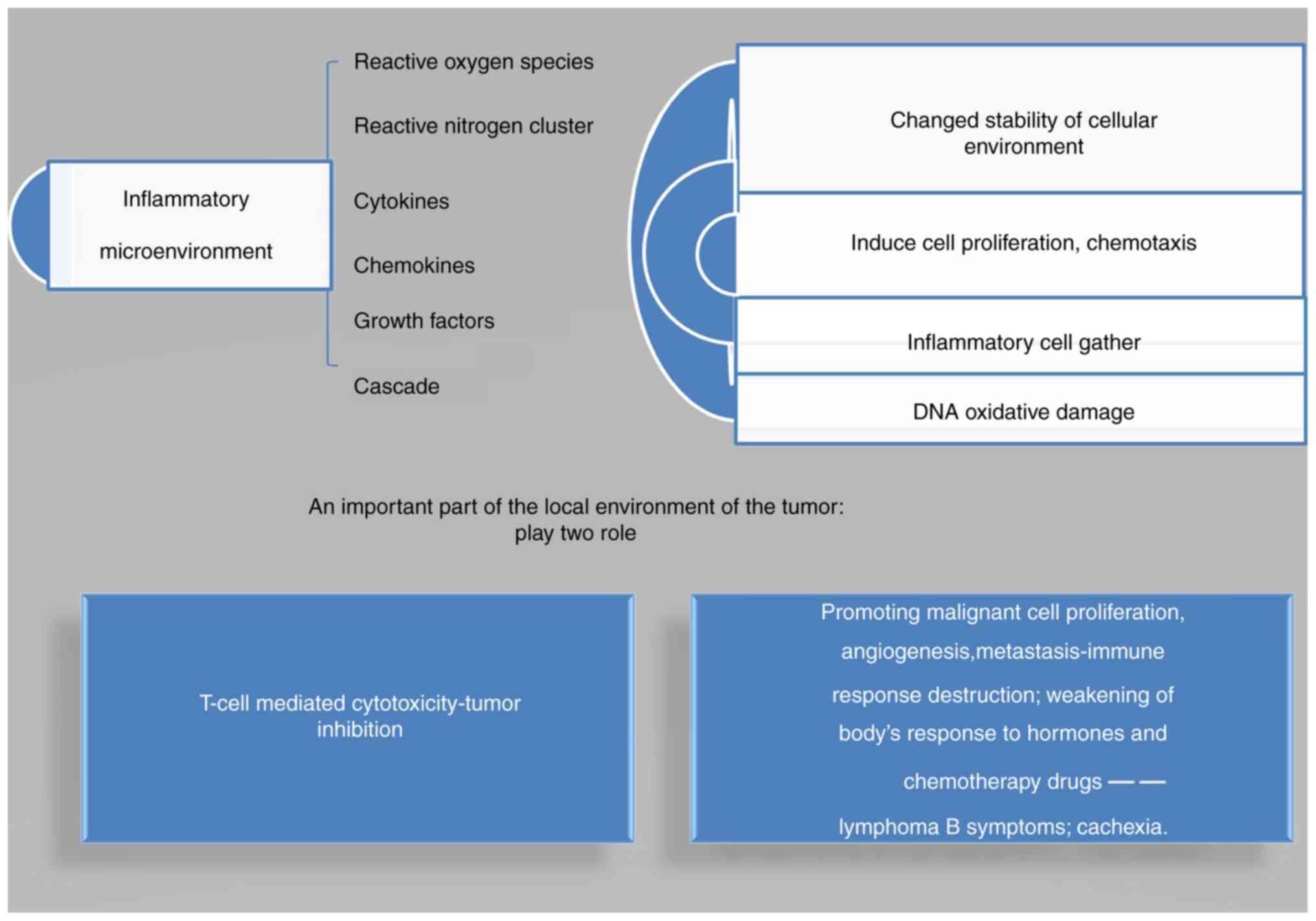
sustained inflammatory microenvironment induces several reactive
oxygen species (ROS), reactive nitrogen species (RNS), cytokines
and chemokines, growth factors and other inflammatory mediators,
changing the stability of the cellular environment. This in turn
induces cell proliferation, chemotaxis, and inflammatory cells,
leading to DNA oxidative damage. This indicates that the
proliferation of these cells loose their control in the
inflammatory microenvironment (10).
This finally induces tumors, causing inflammation in the local
environment of the tumor. Inflammation plays two roles in the
turmor microenvironment. Firstly, it shows benign local effects,
such as T-cell mediated cytotoxicity, leading to tumor inhibition;
and secondly, the inflammation is characterized by persistent,
adverse effects, such as promotion of malignant cell proliferation,
angiogenesis, and metastasis, which subsequently destroys the
immune response and weakens the body's response to hormones and
chemotherapy drugs. This results in the occurrence of lymphoma B
symptoms (i.e., fever, sweating, weight loss) and cachexia. At the
same time, oncogenes can induce the formation of an inflammatory
microenvironment (9) (Fig. 1).
The main signaling pathways,
inflammatory mediators, inflammatory cells, and cytokines involved
in the inflammatory immune system of prostate cancer
The prostate consists of three different regions:
the central zone (CZ), the transition zone (TZ) and the peripheral
zone (PZ). Benign prostatic hyperplasia (BPH) occurs in the TZ
area, whereas prostatitis and cancer occur mainly in the PZ area
(11). Several studies have reported
that inflammatory mediators such as inflammatory pathways,
inflammatory cytokines, and inflammatory cells promote the
development and progression of prostate cancer (9). Both external inflammatory pathways and
inherent genetic pathways can lead to activation of inflammatory
signaling pathways, mainly nuclear factor (NF)-κB signaling,
JAK/Stat, hypoxia-inducible factor 1 (HIF-1) signaling and
mechanistic target of rapamycin (mTOR) signaling pathways (9). NF-κB is a crucial transcription factor
in the inflammatory reaction, which increases the expression of
tumor-promoting factors, such as interleukin (IL)-6 and tumor
necrosis factor (TNF)-α. The crosstalk between NF-κB and multiple
pathways, including Stat3, AP1, interferon regulatory factor, Nrf2,
Notch, Wnt/β-catenin affects cancer cell behaviors in regards to
metabolism, invasion, metastasis, angiogenesis and resistance to
treatment. The JAK/Stat pathway is an inflammatory pathway that is
activated under moderate stress conditions. It is closely related
with the occurrence and development of prostate cancer (12). Stat1 regulates immune suppression
induced by macrophages and bone marrow-derived suppressor cells
(BMSCs) through the expression of nitric oxide synthase. Stat3 is
upregulated in cancer and immune cells, and is associated with
increased synthesis of key inflammatory mediators, cytokines and
chemokines. HIF1 signal transduction pathways are activated by
oncogenes, which in turn can assist in identifying the hypoxic
state of the tumor cells and adjust tumor metabolism by increasing
glycolysis and reducing mitochondrial function to promote the
survival of tumor cells (13). One
reason for age-related cancers such as prostate cancer could be an
inflammatory milieu driven by mTOR in senescent cells (14). The main function of TORC2 is
phosphorylation of Akt at Ser473 and TORC2 was found to play an
important role in the proliferation and anchorage-independent
growth of PC-3 prostate cells. Akt was fount to be highly activated
by phosphorylation at Ser473 in prostate cancer cell lines (PC-3
and LN-CaP). Knockdown of Rictor (a component of TORC2) reduced Akt
Ser473, delaying prostate cancer cell proliferation (15).
All inflammatory cytokines (IL-1, IL-6, IL-11,
IL-18, IL-7, TNF-α and mic-1/GDF-15) are involved in the occurrence
of prostate cancer (11). Both IL-1β
and TNF-α enhance the recruitment of immunosuppressive cells,
increase the expression of chemokine factors on the tumor cell
surface, promote the ability of tumor cell invasion and metastasis
ability and finally promote tumor formation (16). IL-6 is an important inflammatory
cytokine in the JAK/Stat pathway and a key regulatory factor in
tumor formation (17). Previous
studies revealed the existence of IL-6 in the epithelial cells of
prostate cancer patients (18). A
variety of inflammatory lesions stimulate IL-6, and together with
its gp130 (GP130) subunit activates JAK/STAT, ERK1/2, MARK, PI3K,
AKT and mTORC1 signaling pathways to regulate prostate cancer cell
proliferation and apoptosis (18).
The high expression of IL-17 in PIA lesions is the direct evidence
for the involvement of the inflammatory microenvironment in the
development of prostate cancer. The IL-17-MMP7 signaling pathway
has been found to be involved in the transition of prostate
epithelial neoplasia to prostate cancer (19). IL-18 helps in maintaining the
inflammatory microenvironment of prostate cancer by damaging the
function of natural killer (NK) cells and allowing tumor cells to
escape the host immune response (13). The expression of IL-32 in the prostate
gland helps reduce the occurrence of prostate cancer, and thus has
become a ‘hot’ research topic in the treatment of prostate cancer
(20). MIC-1/GDF-15 is regulated by
inflammatory cytokines, and the combination of MIC-1/GDF-15 and
prostate-specific antigen (PSA) in the serum can improve the
specificity of prostate cancer detection (11). Tumor cells and inflammatory cells
secrete TGF-β, which enhances the expression of CXCL12/CXCR4 and
CXC5/CXCR2 in tumor cells, suggesting the initiation of tumor
metastasis. INF-γ plays a positive role in inhibiting tumor
occurrence. Macrophage migration inhibitory factor (MIF) represents
a link between inflammation and cancer. This promotes macrophages
present in inflammation, local invasion, proliferation, activation
and secretion of TNF-α, IL-1 and IL-8 cytokines; and induces
macrophages to produce NO, increasing DNA damage. Moreover, MIF
with the help of Erk1/Erk2 phosphorylation events can activate
NF-κB, COX-2 and increase NOS2, with or without dependence on P53
apoptosis inhibition (21). A new
inflammatory factor (composed of NLR proteins) has recently been
reported to be associated with prostate cancer cell apoptosis and
can lead to the secretion and maturity of IL-1 and IL-18 cytokines
(20).
In the tumor tissues of prostate cancer,
distribution of tumor-associated macrophages (TAMs) can have an
effect on tumor stage and progression (21). The proportion of CD4+ T
cells in prostate cancer tissues was found to be much higher than
that in peripheral blood, especially TH1,
TH17 and Treg cells (22,23).
Infiltrating macrophages, mast cells, neutrophils, T cells, B cells
and their related subgroups in the tumor release TNF-α, epidermal
growth factor (EGF), vascular endothelial growth factor (VEGF),
fibroblast growth factor 2 (FGF2) and other cytokines. Infiltration
of these inflammatory mediators could also mediate
epithelial-mesenchymal transition (EMT), promoting tumor growth and
metastasis. Furthermore, inflammation could induce differentiation
of BMSCs and tumor stem cells into epithelial and interstitial
fibroblast cells, thus promoting tumor growth and angiogenesis.
Meanwhile, the inhibitory effects of BMSCs on cytotoxic T
lymphocytes (CTLs) and NK cells also maintain tumor cell viability
in the immune mechanism (24).
Other factors associated with
inflammation and prostate cancer
Mitochondria, complement activation, ROS and RNS,
DNA methylation, chemokines, innate immune genes, estrogen and
oxidative stress are all also involved in the immunoregulatory
function of prostate cancer. Mitochondria are known as the heart of
immunity, and can activate main innate immune signaling pathways
such as NF-κB (25). In addition to
the components of innate immunity, complement activation also
participates in the adaptive immune response and inflammation.
Complement activation end product and its receptor reduces cell
apoptosis, promotes cell differentiation, proliferation, and
migration by regulating immune response in the tumor
microenvironment (26). Continuous
generation of ROS (OH−, O2) and RNS (NO,
OONO−) in chronic inflammation induces damage of the
cellular macromolecules, especially DNA chain rupture and base
mutation. This subsequently leads to tumor-suppressor gene mutation
and protein modification after translation that is related to basic
processes such as cell apoptosis, DNA repair, and cell cycle
checkpoint, increasing the risk of chronic inflammation. In
addition, DNA methylation is one of the major epigenetic changes,
in which significant hypermethylation occurs in various tumors. The
hypermethylation of the promoter can induce transcriptional
silencing of APC, p16, BRCA1, Rb, MDM2, and other tumor-suppressor
genes. The methylated CpG site aids in easily removing ammonia,
leading to missense mutation of the tumor-related genes. Research
has revealed that hypermethylation is often triggered by chronic
inflammation in microbial infections, and thus DNA methylation also
reflects the association between inflammation and cancer (10). Chemokines induce inflammatory cells
towards the inflammatory site and surrounding lesions, regulating
the occurrence, development, adhesion, and spread of prostate
cancer cells (27). The migration and
invasion ability of PC-3 cells were significantly improved after
treatment with exogenous CXCL16, suggesting that CXCL16/CXCR6 might
act as an independent chemokine axis in prostate bone metastasis
(27). The expression of CXCL1/GROα
was found to be increased in prostate cancer after castration,
promoting prostate cancer cell proliferation, migration and
invasion by decreasing fibulin-1 expression through NF-κB/HDAC1
epigenetic regulation (28). CXCR4
interacts with matrix proteins (such as laminin, fibronectin,
collagen) in prostate cancer cells and acts on tumor cells through
its ligand CXCL12, thus regulating the expression of synthase, FAK
phosphorylation, p38MARK and ROCK kinase (27,29).
Two innate immune-related genes, the inactivated
mutants E265X and M1I in L- ribonuclease (RNaseL), have been
associated with prostate cancer susceptibility. RNase L, which is
activated and expressed after a novel γ-retrovirus infection, is
prone to cause prostatitis. RNase L when activated by its
cognate-induced ligand induces 2′,5′-related oligoadenylation,
which is encoded by the MIC1 gene. Locus is defined as the site of
susceptibility to prostate cancer. Single nucleotide polymorphisms
(SNPs) in RNase L are implicated in inflammation and prostate
cancer risk (30,31).
Androgen receptors (ARs) are ligand-activated
transcription factors of the nuclear receptor superfamily that
mediate the biological effects of androgens in the prostate. AR
signaling regulates inflammation, which in turn affects the
progression of BPH and plays an important role in prostate cancer
development and progression (32).
Studies on immune inflammation conducted in 105 BPH specimens
showed that patients with stronger immune inflammation have larger
prostate size, higher AR expression levels and higher serum PSA
levels (33). Immunohistochemical
analysis showed that BPH patients had more infiltrated macrophages
in the prostate and higher expression of CCL3 than those with
normal prostate. Stromal AR could increase the expression of CCL3
by recruiting infiltrating macrophages, which thereby promotes the
development of BPH (34).
Interestingly, the opposite effect of androgen/AR signaling showed
that dihydrotestosterone can regulate the immune system in BPH by
inhibiting inflammatory cytokines in stromal cells (35).
The overexpression of aromatase (AROM+)
can increase mast cells in the prostatic tissues of mice affected
by endogenous estrogen, and a large number of infiltrated
inflammatory cells in the matrix and cavity of the prostate.
According to PCR array, CCL20, CCL8, CCR6, CCR5, and CCR2 showed
significant expression as bridge factors in the association of
inflammation and cancer. The atypical epithelial cells and
micropapillary growth pattern then appeared in the inflammatory
infiltration area. Additionally, several atypical nuclei with
prominent nucleoli appeared, and these changes were associated with
high levels of stromal tumor cell proliferation in the surrounding
tissues, suggesting chronic inflammation and precancerous lesions.
Therefore, it is believed that estrogen acts as a connection
linking chronic prostatitis and prostatic intraepithelial neoplasia
(PIN) (36). Another potential
mechanism in which inflammation is related with cancer is that the
oxidative stress induced by inflammatory cytokines can lead to
epigenetic recruitment in the sites of DNA injury, producing DNA
methyltransferase, chromatin remodeling, and inhibiting factor
complex. Therefore, a wide abnormal DNA methylation and
transcriptional silencing of gene promoters occurred in prostate
cancer progression and metastasis (22,37).
Oxidative stress can also make inflammatory cytokines, such as TNF
release signals, leading to DNA cracking with proximity induction
of AR signals, making prostate epithelial cells and TMPRSS2-ERG
gene fusion, and promoting prostate cancer (38) (Table
I).
 | Table I.Role of the main signaling pathways,
inflammatory cells and cytokines in the inflammatory immune system
of prostate cancer. |
Table I.
Role of the main signaling pathways,
inflammatory cells and cytokines in the inflammatory immune system
of prostate cancer.
| Type of
inflammatory mediator | Name of
inflammatory mediator | Activity/mechanisms
of action on prostate cancer | Impact on prostate
cancer |
|---|
| Inflammatory
signaling pathways | NF-κB | Acts as a crucial
transcription factor in inflammatory reaction, increases the
expression of tumor-promoting factor IL-6 and TNF-α, crosstalk with
multiple pathways, including Stat3, AP1, IRF, Nrf2, Notch,
Wnt/β-catenin and JAK/Stat pathway an inflammatory pathway in
moderating stress. | Affects cancer cell
behaviors in regards to metabolism, invasion, metastasis,
angiogenesis and treatment resistance |
|
| JAK/Stat | An inflammatory
pathway in moderating stress. Stat1 regulates immune suppression
induced by macrophages and bone marrow-derived suppressor cells
through the expression of nitric oxide synthase. Stat3 enhances the
synthesis of key inflammatory mediators, cytokines and
chemokines | Is closely related
to the occurrence and development of prostate cancer |
|
| HIF1 | Increases
glycolysis and reduces mitochondrial function | Promotes the
survival of tumor cells |
|
| mTOR | Drives inflammatory
milieu in senescent cells. TORC2 is used for the phosphorylation of
Akt at Ser473 | Is associated with
age-related cancers such as prostate cancer. Also plays an
important part in proliferation and anchorage-independent growth of
PC-3 prostate cells |
|
| Erk1/Erk2 | Phosphorylation of
Erk1/Erk2 activates NF-κB, COX-2 and increases NOS2, with or
without dependence on P53 inhibition of apoptosis |
|
| Inflammatory
cytokines | IL-1β;TNF-α | Enhances the
recruitment of immunosuppressive cells, and increases the
expression of chemokine factors in the tumor cell surface | Promotes tumor cell
invasion and metastatic ability and promotes tumor formation |
|
| IL-6 | An important
inflammatory cytokine in the JAK/Stat pathway; a key regulatory
factor for tumor formation; activating JAK/Stat, Erk1/2, Mark,
PI3K, Akt and mTORC1 | Regulates prostate
cancer cell proliferation and apoptosis and promotes
tumorigenesis |
|
| IL-17 | IL-17-MMP7 signal
axis is considered as a signaling pathway for the transition of
prostate epithelial neoplasia to prostate cancer | Directly produces
evidence of prostate cancer caused by inflammatory
microenvironment. |
|
| IL-18 | Damages the
function of NK cells allowing tumor cells to escape the host immune
response | Aids in maintaining
the inflammatory microenvironment of prostate cancer |
|
| IL-32 | Regulates cell
growth, metabolism and immune function and is therefore involved in
the pathologic regulator or | Aids in reducing
the occurrence of prostate cancer protectant of inflammatory
diseases |
|
| Mic-1/GDF-15 | Combination of
Mic-1/GDF-15 and prostate-specific antigen (PSA) in serum | Improves the
specificity of prostate cancer detection |
|
| TGF-β | Enhances the
expression of CXCL12/ CXCR4 and CXC5/CXCR 2 in tumor cell
chemokines | Suggests the
beginning of tumor metastasis |
|
| INF-γ | Limits
proliferation and retards the invasive potential of prostate cancer
cells | Plays an essential
role in inhibiting tumor occurrence |
|
| MIF | Promotes
macrophages in inflammation, local invasion, proliferation,
activation and secretion of TNF-α, IL–1 and IL-8 cytokines; induces
macrophages to produce NO and increase DNA damage | One of the most
important medium that links inflammation and cancer |
|
| IL-1; IL-18 | Secreted and
matured by a new inflammatory factor (composed of NLR
proteins) | Associated with
prostate cancer cell apoptosis |
| Inflammatory
cells | TAMs | Distribution of
these cells can affect tumor stage and progression |
|
| CD4+ T
cells | Proportion of
CD4+ T cells (especially TH1, TH17 and Treg cells) is
much higher in prostate cancer tissues than that in peripheral
blood |
|
| Infiltrating
macrophages, mast cells, neutrophils, T cells, B cells | Release TNF-α, EGF,
VEGF, FGF2 and other cytokines. mediate EMT | Promotes tumor
growth and metastasis. |
|
| BMSCs, tumor stem
cells | Transfer into
epithelial and interstitial fibroblast cells; inhibit CTLs and NK
cells | Promote tumor
growth and angiogenesis; maintain tumor cell viability in the
immune mechanism |
| Other factors
associated with inflammation and prostate cancer | Mitochondria | Activate the main
innate immune signaling pathway such as NF-κB, known as heart of
immunity |
|
|
| Complement
activation end product and its receptor mediating | Regulates immune
response | Reduces cell
apoptosis, promote cell differentiation, proliferation, migration
in the tumor microenvironment. |
|
| ROS
(OH−, O2) and RNS (NO, OONO) | Induce damage of
cellular macromolecules, especially DNA chain rupture and base
mutation | Tumor-suppressor
gene mutation and protein modification after translation related to
the basic process such as cell apoptosis, DNA repair, cell cycle
checkpoint cell, and increase the risk of chronic
inflammation. |
|
| Hypermethylation of
promoters | Induces
transcriptional silencing of APC, p16, BRCA1, Rb, MDM2, and other
tumor suppressor genes | Induces the
transcriptional silencing of APC, p16, BRCA1, Rb, MDM2, and other
tumor-suppressor genes |
|
| Chemokines | Induce inflammatory
cells to gather and move toward inflammation tissue and surrounding
lesions tissue | Play an important
role in the process of occurrence, development, adhesion, and
spread of prostate cancer |
|
| RNaseL | When activated by
its cognate-induced ligand in a cell, 2′,5′-related
oligoadenylation is induced. The locus is the site of
susceptibility to prostate cancer | Prone to
prostatitis following infection with a novel γ-retrovirus. When
activated by its cognate-induced ligand in a cell, 2′,5′-related
oligoadenylation is induced, one of which is encoded by MIC1 gene.
The locus is the site of susceptibility to prostate cancer. |
|
| AR signaling | Regulates
inflammation; higher AR expression levels and higher serum PSA
levels indicative of stronger immune inflammation | Affects the
progression of BPH and plays an significant role in prostate cancer
development and progression |
|
| Endogenous
estrogen | Increases mast
cells in the prostatic tissues | Connection linking
chronic prostatitis and PIN |
Antitumor effect of emodin in prostate
cancer
PIM1, a proto-oncogene, is responsible for encoding
serine/threonine PIM1 kinase, and plays a vital role in regulating
prostate cancer cell cycle and apoptosis. As a kinase inhibitor,
emodin was found to selectively inhibit PIM kinase and inhibit the
growth of DU-145 prostate cancer cells, which were isolated from
the brain metastases of prostate cancer. DU145 cells are
androgen-independent prostate cancer cells with low differentiation
degree, and lack the expression of endogenous AR (39). PC3 cells, isolated from the bone
metastases of human prostate cancer, are low differentiated
androgen-independent prostate cancer cells, without endogenous AR.
PC3 prostate cancer cell experiments have shown that emodin
inhibited the growth of PC3 cells by activating the Notch signaling
pathway, inducing cell apoptosis, and blocking the cells in the
G2/M phase. In addition, emodin inhibited the role of VEGF in
anticancer mechanisms (40). Emodin
was found to act as a strong growth inhibitor in LNCaP cells
isolated from the left supraclavicular lymph node of a male patient
in 1977, which is sensitive to androgen, and the cytotoxic
mechanism was related to the production of ROS. In oxygen and
low-oxygen environment, emodin can reduce the expression of AR in
LNCaP cells (41).
The chemokine receptor CXCR4-CXCL12 axis promotes
invasion and metastasis of protate tumor cells. Emodin was found to
inhibit the activation of NF-κB and to lower CXCR4 and HER2 at the
transcription level, thereby inhibiting the invasion and metastasis
of DU145 prostate cancer cells (42).
Compared with PC3 cells, emodin significantly induced the apoptosis
of LNCaP cells and inhibited their proliferation. These findings
suggested that emodin inhibits prostate LNCaP cell proliferation by
regulating the activity of AR and p53-p21 pathways, increasing
caspase-3 and −9, increasing the ratio of Bax/Bcl-2, and inducing
LNCaP cell apoptosis through mitochondrial signaling pathway
(43). Emodin enhanced the
cytotoxicity of chemotherapeutic drugs in prostate cancer cells,
and its mechanism was found to be related to the inhibition of
multidrug resistance (MDR) and hypoxia-inducing factors. Emodin is
proven to be an ROS generator and a novel small inhibitor of HIF-1.
It has been shown that co-treatment with emodin plus cisplatin in
DU145 cells and mouse xenografts dramatically elevated ROS levels,
downregulated MDR1 expression and HIF-1 transactivation. HIF1 acts
as an upstream controller of MDR1 and is redox-sensitive (44).
Emodin was found to downregulate the transcriptional
activity of AR by preventing AR nuclear translocation, disrupting
the association between AR and heat shock protein 90 (HSP90),
increasing the interaction and ubiquitination of AR with E3 ligase
MDM2 (murine double minute 2), leading to AR degradation through a
proteasome-mediated pathway in a ligand-independent manner
(8). In in vivo studies,
emodin demonstrated low drug toxicity and maintained physical
activity in prostate cancer-induced C3(1)/SV40 transgenic mice
(8).
The activation of p53 can induce the expression of
p21. Emodin increased the expression of p53 and p21 in LNCaP cells,
thus inducing significant apoptosis. LNCaP is an AR- and
LRP1-positive prostate cancer cell line, and has been found to be
more susceptible to emodin than PC-3 cells, which are AR-negative
LRP-positive prostate cancer cells. LRP1 and AR are expressed in
prostate cancer and are upregulated in hypoxic conditions. In
AR-positive LNCaP cells, AR was markedly upregulated under
CoCl2-induced hypoxia-like conditions, decreased by
emodin and CoCl2 co-treatment. These data indicate that
emodin induces ROS-mediated growth inhibition (41).
Antitumor effects of emodin derivatives in
prostate cancers
Aloe vera is the most commercialized
Aloe species belonging to the Xanthorrhoeaceae family
(45). Aloe-Emodin (AE) (Fig. 2B) is an ahydroxyanthraquinone obtained
from Aloe vera leaves, which is structurally very similar to
emodin. AE was found to suppress the proliferation and
anchorage-independent growth in PC-3 cells in a dose-dependent
manner with a peak concentration at 15 mM. The phosphorylation of
Akt at Ser473 was strongly inhibited by AE (15). In an ex vivo pull-down assay,
AE-conjugated Sepharose 4B beads pulled down endogenous Rictor
together with mTOR and Akt (15).
Emodin-derived aloin can combine with mTORC2 in the
cells to inhibit kinase activity by inhibiting mTORC2, Akt and
PKC-α substrate downstream activity. This in turn was found to
inhibit the proliferation and nondependent growth of PC3 cells
(15). This suggests that aloin can
inhibit the development of prostate cancer through the mTOR
signaling pathway (15) (Table II). An experiment that investigated
the effect of AE in rat C6 glioma cells showed that AE led to the
formation of intracytoplasmic acidic vesicles indicating autophagic
cell death, blockage of the cell cycle and caspase-dependent
apoptosis. AE had no affects on the activation of MAPK p38,
Jun-N-terminal kinase, or transcription factor NF-κB. But according
to the results of cell-based ELISA, AE markedly inhibited the
activation of extracellular signal-regulated kinases 1 and 2
(ERK1/2) in C6 cells. These results indicated that anti-glioma
action of AE involved ERK-independent induction of both apoptosis
and autophagy (46). However, whether
AE also shows these effects in prostate cancer warrants further
investigation.
| Table II.Mechanisms of emodin in prostate
cancer. |
Table II.
Mechanisms of emodin in prostate
cancer.
| Natural drug
name | Mechanism in
prostate cancer | Impact on prostate
cancer |
|---|
| Emodin | Inhibits PIM
kinase | Inhibits the growth
of DU-145 prostate cancer cells |
|
| Activates Notch
signaling pathway; inhibits VEGF | Inhibited the
growth of PC3 prostate cancer cells, induced cell apoptosis; and
blocked the cell cycle in G2/M phase; anti-cancer mechanisms |
|
| Reduces the
expression of AR in oxygen and low-oxygen environment | Effect in LNCaP
prostate cancer cells |
| Emodin derivative
aloin | Inhibits mTOR
signaling pathway through inhibition of mTORC2, Akt and PKC-α
substrate downstream activity | Inhibits the
proliferation and nondependent growth of PC3 cells in the
development of prostate cancer |
| Emodin | Inhibits the
activation of NF-κB and lowers CXCR4 and HER2 | Inhibit the
invasion and metastasis of prostate cancer cells |
| Emodin | Regulates the
activity of AR and p53-p21 pathways, increases caspase-3 and −9,
increases the ratio of Bax/Bcl-2; and induces LNCaP cell apoptosis
through the mitochondrial pathway | Inhibits prostate
LNCaP cell proliferation |
| Emodin | Inhibits multidrug
resistance and hypoxia-inducing factors | Enhances the
cytotoxicity of chemotherapy drugs in prostate cancer cells |
| Co-treatment with
emodin plus cisplatin | Dramatically
elevates ROS levels, downregulates MDR1 expression and HIF-1
transactivation | Inhibit the tumor
growth in DU145 cells and mouse xenografts, owing to oxidative
stress and MDR1 down-regulation within tumors |
| Emodin | Downregulates AR
transcriptional activity by preventing AR nuclear translocation,
disrupts the association between AR and heat shock protein 90,
increasing the interaction and ubiquitination of AR with E3 ligase
MDM2, leading to AR degradation through proteasome-mediated pathway
in a ligand-independent manner | Low drug toxicity;
maintains physical activity in prostate cancer-induced C3(1)/SV40
transgenic mice |
| Emodin | Increases the
expression of p53 and p21; thus, inhibits ROS-mediated growth in
AR- and LRP1-positive prostate cell line LNCaP | Induces significant
apoptosis in LNCaP cells |
| Aloe-Emodin | Inhibits
phosphorylation of Akt at Ser473 | Suppresses
proliferation and anchorage-independent growth in PC-3 cells |
AMAD (Fig. 2C), an
emodin azide methyl anthraquinone derivative extracted from the
nature knotweed rhizome, has potent cytotoxic effects on human
breast cancer cell line MDA-MB-453 and human lung adenocarcinoma
Calu-3 cells. Moreover, AMAD was found to induce apoptosis via a
mitochondrial pathway involving caspase-8/Bid activation in both
cell lines (47). Overexpression of
HER2/neu is well-known to predict a poor prognosis in cancer.
Another study showed that AMAD has the capability of potently
decreasing Her2/neu protein and inhibiting the downstream MAPK and
PI3K-Akt signaling pathways in dose- and time-dependent manners
(48). Realistic data suggested that
blockage of Her2/neu binding to Hsp90 followed by proteasomal
degradation of Her2/neu were involved in emodin AMAD-induced
apoptosis in Her2/neu-overexpressing cancer cells (48). Further investigation is needed to
ascertain whether AMAD has a role in this respect in prostate
cancer.
Emodin analog development in prostate
cancer
Emodin has some drawbacks as a therapeutic agent,
such as low water solubility, low bioavailability after oral
admission and relatively high toxicity to the liver and kidney
(7). The structural modification of
emodin side chain, such as polymethyleneamine, sugar, or
minocycline, combined with methyl, hydroxyl and aryl ring sites
showed enhanced antitumor efficacy (49). The other achieved derivatives through
intercalating amino groups and glycosidic bonds displayed higher
levels of antitumor activities (50).
For example, addition of sugar chains in the emodin C3-hydroxyl
site not only increased its solubility but also significantly
improved its antitumor activity. Similarly, when the hydroxyl
groups of C-1 and C-8 position of emodin were methylated, its
anti-proliferative activity also showed improvement (51). EM-d-Rha is an anthracene
L-rhamnopyranoside derivative of emodin by connecting
L-rhamnopyranosides to a planar aromatic molecule, which has
10-fold stronger antitumor activity and growth inhibitory effects.
Introduction of one or two positively charged side chains in the
tricyclic coplanar structure of the tricyclic compounds, such as
anthranone compound anthracenedion, decreased the toxic effect on
tumor cells (51). The use of emodin
for tumor treatment may be one of the future research directions if
the side effects of emodin can be reduced without affecting its
antitumor effect.
Through the combination of amino acid esters and
substituted aromatic amines, water-soluble derivatives of AE have
been synthesized (Fig. 2D). These
compounds have demonstrated a more effective antitumor activity in
HepG2 and NCI-H460 cells. The structural activity relationship of
L-serine methyl ester, β-alanine ethyl ester and 3-(2-aminoethyl)
pyridine substituents showed improvement in their antitumor
activity (52). GSK-3β plays a vital
role in Wnt signal transduction by mediating degradation of
β-catenin. Revoking GSK-3β-dependent phosphorylation of β-catenin
by Wnt growth factors or inhibitors of GSK-3β led to the
accumulation of hypophosphorylated uncomplexed β-catenin in the
cytosol and nucleus, prompting the transcription of target gene. A
new emodin derivative 4-[N-(aminoethyl) 2-amino]-Emodin (L4
compound) (Fig. 2E) acts as a strong
GSK-3β inhibitor. It binds to the ATP binding site that is close to
the two key residues Asp133 and Val135 and prevents TCF/LEF
transactivation. Meanwhile, the L4 compound demonstrated low
cytotoxicity when compared with other GSK-3β inhibitors (53).
A series of novel quaternary ammonium salts of
emodin were synthesized in an experiment and their anticancer
activities have been investigated in vitro in A375, BGC-823,
HepG2 and HELF cells. The results revealed that compound 4a
(Fig. 2F) has the ability to induce
morphological changes and decrease cell viability. Compound 4a
induced apoptosis of A375 cells by dissipating mitochondrial
membrane potential (ΔΨm), resulting in the upregulation of P53 and
caspase-3. In addition, the results of this research group showed
no direct correlation between alkylating reactivities of emodin
derivatives and their anticancer activities and the presence of
only a weak interaction between emodin and DNA. This implied that
DNA might not be the main target of emodin derivatives. At the same
time, the compound's hydrophilicity was unfavorable
intracellularly. But a conclusion can be drawn that there is a
close relationship between the ability to generate ROS and
anticancer activity. Both compound 4d and 4a were found to be fat
soluble and contain one and two long carbon chains in N cations,
respectively. Compound 4a has the highest capability to generate
ROS (4a > 4d > emodin) (54). A
series of sulfur-substituted
anthra[1,2-c][1,2,5]thiadiazole-6,11-diones were synthesized. Among
the tested compounds, 6g (Fig. 2G)
appears to be the most active compound of this series that not only
induced apoptosis in DU-145 cancer cells but also attenuated ERK1/2
and p38 signaling pathways. Further development of these compounds
as potential anticancer agents is required (55).
Prospects of emodin in the inflammatory
immune microenvironment of prostate cancer
There is limited research regarding the mechanism of
emodin in the inflammatory immune microenvironment of prostate
cancer, and the affect on the immune microenvironment in a tumor by
this agent has raised wide concern (56,57). The
following suggestions have been proposed for future research.
The difference between beneficial inflammation that
stimulates the body's positive immunity and detrimental
inflammation that aggravates pathological damage must be explored.
For example, the M1 phenotype can promote the differentiation of
monocytes into macrophages and promote the sustained and effective
adaptive immune response to tumors via the TLR4-Myd88 signaling
pathway. In the pathogenesis of prostate cancer, how can benign
immunity be triggered with agents is still an unanswered question.
The exact mechanisms involved in transforming a tumor-promoted
microenvironment (TH2 cells and M2 macrophages) to a
tumor-inhibited microenvironment (TH1 cells and M1 macrophages)
warrants further elucidation.
Sex steroid hormones act as influential factors in
the occurrence, development, and outcomes of prostate cancer. One
reason for this might be due to the innate immune system or the
nonspecific immune system regulation of inflammation (58); macrophages in prostate cancer can
produce IL-1, in which the selective androgen receptor modulators
(SARMs) were used to activate the function of androgen
receptor-induced gene instead of inhibition. This process contains
TAB2 protein, which is a type of inflammatory signal sensor, and is
the component of TAB2/N-CoR/HDAC blocker complex. Inflammation
induces the phosphorylation of TAB2, triggering more gene
transcription, and effecting prostate cancer by regulating the
inflammatory microenvironment. The mechanism of emodin in hormone
intervention treatment of prostate cancer should be further
elucidated.
TAMs are specialized partners of tumor cell
migration, invasion, and metastasis, and their affect on tumor cell
metastasis is based on the macrophage environment. It has been
reported that macrophages have increased peritoneal proliferation
in ovarian cancer cells, and whether such a condition exists in
prostate cancer is still unclear. ANXA5 usually induces
anti-inflammatory and cell death activities. It has been found that
ANXA5 plays an inhibitory role on Cox-2 in prostate cancer and
could induce phosphorylation of NF-κBp65, but its role in cancer is
unclear (13,59,60).
Programmed death ligand 1 (PD-L1) interacts with programmed death 1
(PD-1) receptor on T cells to induce an immune response, which
induces tumor cells to avoid immune monitoring. The role of
anti-PD-1 antibodies and drugs in tumor immunotherapy has attracted
much attention (61). It is
interesting to understand how emodin regulates and interferes with
PD-L1/PD-1, thereby affecting tumor immunity. Mesenchymal stem
cells (MSCs) are a major concern in studying cancer and the immune
microenvironment in recent years. MSCs are often recruited for
local inflammation and tumor microenvironment, promoting the
production of proinflammatory cytokines. The same is true for
prostate cancer, and its specific mechanisms and drug intervention
study should be further identified (3).
Abnormal expression of integrin induces tumor cell
migration and invasion abilities, which can change the
intracellular signal transduction to make them survive in the
microenvironement of other organs and does not trigger the internal
mechanisms of apoptosis (62). It can
also cause drug resistance in tumor cells (63). The abnormal expression of α6β1 in
prostate cancer can promote the metastasis of prostate cancer,
activate PI3K/AKT and NF-κB signaling pathways, and inhibit cell
apoptosis, thus maintaining the survival of tumor cells. The role
of emodin on this aspect should be further studied.
The acquisition and maintenance of type M1/M2
macrophage phenotypes depends on the regulation of transcription
and post-transcriptional levels. Epigenetic changes are the key
molecular mechanisms for controlling heterogeneity and plasticity
of macrophages. Of these, the study on microRNAs (miRNAs), DNA
methylation (DNAm), and epigenetic regulation of histone
modification has been extensively studied. miRs are important
regulatory factors in the proliferation, differentiation, and
apoptosis of macrophage cells, and regulation of phenotypic balance
of macrophages in miRs can alleviate inflammation and immune
function (64,65).
In conclusion, the inflammatory response should be
considered as an increased risk of prostate cancer. Emodin exhibits
anticancer action by targeting the inflammatory pathway such as
ROS, HIF-1, PIM1, AR, p53 and PI3K-Akt-mTOR, providing a
prospective area as an adjuvant therapy. Chemical modification may
improve the solubility and utilization ratio of emodin avoiding the
side effects; this issue deserves further investigation.
Acknowledgements
YT thanks Professor Benyi Li, who is from the
Department of Urology, the University of Kansas Medical Center,
Kansas City, USA. He helped to revise and correct the article. YT
thanks Professor Zhaoqin Fang and Professor Aidong Yang for their
continued encouragement and support. With their support, YT was
able to complete her thesis successfully.
Funding
The present study was supported partially by grants
from National Natural Science Foundation of China (grant no.
81673855), the Heritage Talent Project of Shanghai University of
Traditional Chinese Medicine of China (grant no. A1-GY010101), and
partially by the Cinical Basic Subject of Shanghai University of
Traditional Chinese Medicine of China (grant no. 12ZLJ08).
Availability of data and materials
Not applicable.
Authors' contributions
YT conceived the research design, collected the
literature, researched the information, constructed the figures and
table and wrote the paper. ZF, AY, BT and ZW carried out the
literature analysis, revised the paper and provided funding
support. All authors reviewed the results and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PIA
|
proliferative inflammatory atrophy
|
|
PIN
|
prostatic intraepithelial
neoplasia
|
|
CZ
|
central zone
|
|
TZ
|
transition zone
|
|
PZ
|
peripheral zone
|
|
BPH
|
benign prostatic hyperplasia
|
|
HIF1
|
hypoxia-inducible factor 1
|
|
PSA
|
prostate-specific antigen
|
|
TAMs
|
tumor-associated macrophages
|
|
EMT
|
epithelial-mesenchymal
transdifferentiation
|
|
CTL
|
cytotoxic T lymphocytes
|
|
NK
|
natural killer
|
|
ROS
|
reactive oxygen species
|
|
RNS
|
reactive nitrogen species
|
|
SNP
|
single nucleotide polymorphism
|
|
AR
|
androgen receptor
|
|
VEGF
|
vascular endothelial growth factor
|
|
MDR
|
multidrug resistance
|
|
HSP90
|
heat shock protein 90
|
|
SARMs
|
selective androgen receptor
modulators
|
|
PD-L1
|
programmed death ligand 1
|
|
PD-1
|
programmed death protein 1
|
|
MSCs
|
mesenchymal stem cells
|
|
miRNAs
|
microRNAs
|
|
DNAm
|
DNA methylation
|
|
mTORC
|
mammalian target of rapamycin
complex
|
|
PTEN
|
phosphatase and tensin homolog
|
References
|
1
|
Torre LA, Siegel RL, Ward EM and Jemal A:
Global cancer incidence and mortality rates and trends-an update.
Cancer Epidemiol Biomarkers Prev. 25:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pang C, Guan Y, Li H, Chen W and Zhu G:
Urologic cancer in China. Jpn J Clin Oncol. 46:497–501. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang KQ, Liu Y, Huang QH, Mo N, Zhang QY,
Meng QG and Cheng JW: Bone marrow-derived mesenchymal stem cells
induced by inflammatory cytokines produce angiogenetic factors and
promote prostate cancer growth. BMC Cancer. 17:8782017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Balkwill F and Mantovani A: Inflammation
and cancer: Back to virchow? Lancet. 357:539–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Daniels NA, Ewing SK, Zmuda JM, Wilt TJ
and Bauer DC; Osteoporotic Fractures in Men (MrOS) Research Group,
: Correlates and prevalence of prostatitis in a large
community-based cohort of older men. Urology. 66:964–970. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Marzo AM, Marchi VL, Epstein JI and
Nelson WG: Proliferative inflammatory atrophy of the prostate:
Implications for prostatic carcinogenesis. Am J Pathol.
155:1985–1992. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dong X, Fu J, Yin X, Cao S, Li X, Lin L
and Ni J; Huyiligeqi: Emodin: A review of its pharmacology,
toxicity and pharmacokinetics. Phytother Res. 30:1207–1218. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cha TL, Qiu L, Chen CT, Wen Y and Hung MC:
Emodin down-regulates androgen receptor and inhibits prostate
cancer cell growth. Cancer Res. 65:2287–2295. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Diakos CI, Charles KA, McMillan DC and
Clarke SJ: Cancer-related inflammation and treatment effectiveness.
Lancet Oncol. 15:e493–e503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Z, Xiao B, Mao XH and Zou QM: Research
progress on relationship between inflammationand tumor. Prog Mod
Biomed. 9:591–594. 2009.
|
|
11
|
Karan D and Dubey S: From inflammation to
prostate cancer: The role of inflammasomes. Adv Urol.
2016:31403722016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taniguchi K and Karin M: NF-κB,
inflammation, immunity and cancer: Coming of age. Nat Rev Immunol.
18:309–324. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Laberge RM, Sun Y, Orjalo AV, Patil CK,
Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et
al: MTOR regulates the pro-tumorigenic senescence-associated
secretory phenotype by promoting IL1A translation. Nat Cell Biol.
17:1049–1061. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu K, Park C, Li S, Lee KW, Liu H, He L,
Soung NK, Ahn JS, Bode AM, Dong Z, et al: Aloe-emodin suppresses
prostate cancer by targeting the mTOR complex 2. Carcinogenesis.
33:1406–1411. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Denko NC: Hypoxia, HIF1 and glucose
metabolism in the solid tumour. Nat Rev Cancer. 8:705–713. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shalapour S and Karin M: Immunity,
inflammation, and cancer: An eternal fight between good and evil. J
Clin Invest. 125:3347–3355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Liang C and Chen X: Research
progress on the relationship between chronic prostatic inflammation
and prostate cancer. J Mod Urol. 20:207–210. 2015.
|
|
19
|
Zhang Q, Liu S, Ge D, Zhang Q, Xue Y,
Xiong Z, Abdel-Mageed AB, Myers L, Hill SM, Rowan BG, et al:
Interleukin-17 promotes formation and growth of prostate
adenocarcinoma in mouse models. Cancer Res. 72:2589–2599. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hong JT, Son DJ, Lee CK, Yoon DY, Lee DH
and Park MH: Interleukin 32, inflammation and cancer. Pharmacol
Ther. 174:127–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Z and Qi Y: Inflammation: Tumor
catalyst. World Latest Med Inf. 16:70–71. 2016.
|
|
22
|
Sfanos KS, Yegnasubramanian S, Nelson WG
and De Marzo AM: The inflammatory microenvironment and microbiome
in prostate cancer development. Nat Rev Urol. 15:11–24. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sfanos KS, Bruno TC, Maris CH, Xu L,
Thoburn CJ, DeMarzo AM, Meeker AK, Isaacs WB and Drake CG:
Phenotypic analysis of prostate-infiltrating lymphocytes reveals
TH17 and Treg skewing. Clin Cancer Res. 14:3254–3261. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Su huan and Chen ming: Research progress
on the mechanism of inflammatory response and tumor
microenvironment in prostate cancer. J Southeast Univ. 36:847–851.
2017.(Medical Science Edition).
|
|
25
|
Mills EL, Kelly B and O'Neill LAJ:
Mitochondria are the powerhouses of immunity. Nat Immunol.
18:488–498. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Afshar-Kharghan V: The role of the
complement system in cancer. J Clin Invest. 127:780–789. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou W, Hu W and Xu W: Effects of
CXCL16/CXCR6 axis on proliferation and invasion of human prostate
cancer cell line in vitro. Med J Wuhan University. 31:479–482.
2010.
|
|
28
|
Kuo PL, Shen KH, Hung SH and Hsu YL:
CXCL1/GROα increases cell migration and invasion of prostate cancer
by decreasing fibulin-1 expression through NF-κB/HDAC1 epigenetic
regulation. Carcinogenesis. 33:2477–2487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, et al: The relationship between
chemokines, inflammation, and prostate cancer. Mod Prev Med.
42:952–956. 2015.
|
|
30
|
Schoenfeld JD, Margalit DN, Kasperzyk JL,
Shui IM, Rider JR, Epstein MM, Meisner A, Kenfield SA, Martin NE,
Nguyen PL, et al: A single nucleotide polymorphism in inflammatory
gene RNASEL predicts outcome after radiation therapy for localized
prostate cancer. Clin Cancer Res. 19:1612–1619. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiklund F, Jonsson BA, Brookes AJ,
Strömqvist L, Adolfsson J, Emanuelsson M, Adami HO,
Augustsson-Bälter K and Grönberg H: Genetic analysis of the RNASEL
gene in hereditary, familial, and sporadic prostate cancer. Clin
Cancer Res. 10:7150–7156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Izumi K, Li L and Chang C: Androgen
receptor and immune inflammation in benign prostatic hyperplasia
and prostate cancer. Clin Investig (Lond). 4:935–950. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu ZL, Yuan Y, Geng H and Xia SJ:
Influence of immune inflammation on androgen receptor expression in
benign prostatic hyperplasia tissue. Asian J Androl. 14:316–319.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Lin WJ, Izumi K, Jiang Q, Lai KP,
Xu D, Fang LY, Lu T, Li L, Xia S and Chang C: Increased infiltrated
macrophages in benign prostatic hyperplasia (BPH): Role of stromal
androgen receptor in macrophage-induced prostate stromal cell
proliferation. J Biol Chem. 287:18376–18385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vignozzi L, Cellai I, Santi R, Lombardelli
L, Morelli A, Comeglio P, Filippi S, Logiodice F, Carini M, Nesi G,
et al: Antiinflammatory effect of androgen receptor activation in
human benign prostatic hyperplasia cells. J Endocrinol. 214:31–43.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ellem SJ, Wang H, Poutanen M and
Risbridger GP: Increased endogenous estrogen synthesis leads to the
sequential induction of prostatic inflammation (prostatitis) and
prostatic pre-malignancy. Am J Pathol. 175:1187–1199. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aryee MJ, Liu W, Engelmann JC, Nuhn P,
Gurel M, Haffner MC, Esopi D, Irizarry RA, Getzenberg RH, Nelson
WG, et al: DNA methylation alterations exhibit intraindividual
stability and interindividual heterogeneity in prostate cancer
metastases. Sci Transl Med. 5:169ra102013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mani RS, Amin MA, Li X, Kalyana-Sundaram
S, Veeneman BA, Wang L, Ghosh A, Aslam A, Ramanand SG, Rabquer BJ,
et al: Inflammation-induced oxidative stress mediates gene fusion
formation in prostate cancer. Cell Rep. 17:2620–2631. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giraud F, Akué-Gédu R, Nauton L, Candelon
N, Debiton E, Théry V, Anizon F and Moreau P: Synthesis and
biological activities of 4-substituted pyrrolo[2,3-a]carbazole Pim
kinase inhibitors. Eur J Med Chem. 56:225–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Deng G, Ju X, Meng Q, Yu ZJ and Ma LB:
Emodin inhibits the proliferation of PC3 prostate cancer cells in
vitro via the Notch signaling pathway. Mol Med Rep. 12:4427–4433.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Masaldan S and Iyer VV: Exploration of
effects of emodin in selected cancer cell lines: Enhanced growth
inhibition by ascorbic acid and regulation of LRP1 and AR under
hypoxia-like conditions. J Appl Toxicol. 34:95–104. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ok S, Kim SM, Kim C, Nam D, Shim BS, Kim
SH and Ahn KS, Choi SH and Ahn KS: Emodin inhibits invasion and
migration of prostate and lung cancer cells by downregulating the
expression of chemokine receptor CXCR4. Immunopharmacol
Immunotoxicol. 34:768–778. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yu CX, Zhang XQ, Kang LD, Zhang PJ, Chen
WW, Liu WW, Liu QW and Zhang JY: Emodin induces apoptosis in human
prostate cancer cell LNCaP. Asian J Androl. 10:625–634. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang XZ, Wang J, Huang C, Chen YY, Shi
GY, Hu QS and Yi J: Emodin enhances cytotoxicity of
chemotherapeutic drugs in prostate cancer cells: The mechanisms
involve ROS-mediated suppression of multidrug resistance and
hypoxia inducible factor-1. Cancer Biol Ther. 7:468–475. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kumar S, Yadav M, Yadav A, Rohilla P and
Yadav JP: Antiplasmodial potential and quantification of aloin and
aloe-emodin in Aloe vera collected from different climatic regions
of India. BMC Complement Altern Med. 17:3692017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mijatovic S, Maksimovic-Ivanic D, Radovic
J, Miljkovic DJ, Harhaji LJ, Vuckovic O, Stosic-Grujicic S,
Mostarica Stojkovic M and Trajkovic V: Anti-glioma action of aloe
emodin: The role of ERK inhibition. Cell Mol Life Sci. 62:589–598.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan Y, Su X, Liang Y, Zhang J, Shi C, Lu
Y, Gu L and Fu L: Emodin azide methyl anthraquinone derivative
triggers mitochondrial-dependent cell apoptosis involving in
caspase-8- mediated bid cleavage. Mol Cancer Ther. 7:1688–1697.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yan YY, Zheng LS, Zhang X, Chen LK, Singh
S, Wang F, Zhang JY, Liang YJ, Dai CL, Gu LQ, et al: Blockade of
Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone
derivative induces proteasomal degradation of Her2/neu. Mol Pharm.
8:1687–1697. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yan YY, Fu LW, Zhang W, Ma HS, Ma CG,
Liang YJ, Liu BY, Yu JZ, Wu QZ and Dong YM: Emodin azide methyl
anthraquinone derivative induced G0/G1 arrest in
HER2/neu-overexpressing MDA-MB-453 breast cancer cells. J BUON.
19:650–655. 2014.PubMed/NCBI
|
|
50
|
Wen-Feng W, Feng-Sen Z, Wen-Na Z, Ze-Dong
B, Hui-Jun Y, Jing-Wei S and Yao-Feng Y: The synthesis, structural
study and anticancer activity evaluation of emodin derivatives
containing conjugative groups. Med Chem. 9:545–552. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xing JY, Song GP, Deng JP, Jiang LZ, Xiong
P, Yang BJ and Liu SS: Antitumor effects and mechanism of novel
emodin rhamnoside derivatives against human cancer cells in vitro.
PLoS One. 10:e01447812015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Thimmegowda NR, Park C, Shwetha B,
Sakchaisri K, Liu K, Hwang J, Lee S, Jeong SJ, Soung NK, Jang JH,
et al: Synthesis and antitumor activity of natural compound aloe
emodin derivatives. Chem Biol Drug Des. 85:638–644. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gebhardt R, Lerche KS, Götschel F, Günther
R, Kolander J, Teich L, Zellmer S, Hofmann HJ, Eger K, Hecht A and
Gaunitz F: 4-Aminoethylamino-emodin-a novel potent inhibitor of
GSK-3beta-acts as an insulin-sensitizer avoiding downstream effects
of activated beta-catenin. J Cell Mol Med. 14:1276–1293. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang X, Zhao W, Hu X, Hao X, Hong F, Wang
J, Xiang L, Zhu Y, Yuan Y, Ho RJ, et al: Synthesis,
characterization, and anticancer activity of novel lipophilic
emodin cationic derivatives. Chem Biol Drug Des. 86:1451–1457.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee YR, Chen TC, Lee CC, Chen CL, Ahmed
Ali AA, Tikhomirov A, Guh JH, Yu DS and Huang HS: Ring fusion
strategy for synthesis and lead optimization of sulfur-substituted
anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivatives as promising
scaffold of antitumor agents. Eur J Med Chem. 102:661–676. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Silva JAF, Bruni-Cardoso A, Augusto TM,
Damas-Souza DM, Barbosa GO, Felisbino SL, Stach-Machado DR and
Carvalho HF: Macrophage roles in the clearance of apoptotic cells
and control of inflammation in the prostate gland after castration.
Prostate. 78:95–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dart DA, Uysal-Onganer P and Jiang WG:
Prostate-specific PTen deletion in mice activates inflammatory
microRNA expression pathways in the epithelium early in hyperplasia
development. Oncogenesis. 6:4002017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mantovani A: Cancer: An infernal triangle.
Nature. 448:547–548. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Baek HS, Park N, Kwon YJ, Ye DJ, Shin S
and Chun YJ: Annexin A5 suppresses cyclooxygenase-2 expression by
downregulating the protein kinase C-ζ-nuclear factor-κB signaling
pathway in prostate cancer cells. Oncotarget. 8:74263–74275. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhu P, Baek SH, Bourk EM, Ohgi KA,
Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld
MG and Rose DW: Macrophage/cancer cell interactions mediate hormone
resistance by a nuclear receptor derepression pathway. Cell.
124:615–629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chen G, Huang AC, Zhang W, Zhang G, Wu M,
Xu W, Yu Z, Yang J, Wang B, Sun H, et al: Exosomal PD-L1
contributes to immunosuppression and is associated with anti-PD-1
response. Nature. 560:382–386. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Winograd-Katz SE, Fässler R, Geiger B and
Legate KR: The integrin adhesome: From genes and proteins to human
disease. Nat Rev Mol Cell Biol. 15:273–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Eke I, Dickreuter E and Cordes N: Enhanced
radiosensitivity of head and neck squamous cell carcinoma cells by
β1 integrin inhibition. Radiother Oncol. 104:235–242. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wu XQ, Dai Y, Yang Y, Huang C, Meng XM, Wu
BM and Li J: Emerging role of microRNAs in regulating macrophage
activation and polarization in immune response and inflammation.
Immunology. 148:237–248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ouimet M, Ediriweera HN, Gundra UM, Sheedy
FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C,
Fullerton MD, Cecchini K, et al: MicroRNA-33-dependent regulation
of macrophage metabolism directs immune cell polarization in
atherosclerosis. J Clin Invest. 125:4334–4348. 2015. View Article : Google Scholar : PubMed/NCBI
|