Introduction
Worldwide, esophageal cancer is the eighth most
common cancer, but the sixth most common cause of cancer-associated
deaths (1). Esophageal cancer is
comprised of two pathologically and epidemiologically distinct
subtypes: Esophageal adenocarcinoma (EAC) and esophageal squamous
carcinoma (ESCC). EAC resembles a chromosomally unstable variant of
gastric adenocarcinoma, whereas ESCC shares many characteristics
with squamous cell carcinoma of the head and neck (2). Although ESCC is the most prevalent
subtype worldwide, EAC incidence rates have risen in developed
countries over the past four decades (3). The majority of patient admissions
exhibit locally advanced or metastatic disease, and as a result of
this late diagnosis, EAC is an aggressive and difficult disease to
treat, with an overall five-year survival rate of <20% (4). Early malignant tumor cell dissemination
contributes to the poor prognosis of EAC (5). Therefore, there is an urgent requirement
for research into the mechanisms of EAC tumor invasion and
metastasis. With the information gained from these studies, novel
treatment options can be explored.
Epithelial-mesenchymal transition (EMT) is a process
that induces the invasion and metastasis of epithelial cancer cells
(6–9).
During EMT, epithelial cells lose their apical-basal polarity and
intercellular adhesion, and gain mesenchymal cell characteristics,
including increased migratory activity and fibroblast-like
morphology. EMT is modulated by changes at the genomic, epigenomic,
transcriptomic and proteomic levels (6–9). In
previous studies investigating EMT in EAC, the downregulation of
epithelial markers, and the upregulation of mesenchymal markers
with concomitant transforming growth factor (TGF)-β1, were observed
in invasive margins compared with the central tumor (10). The signaling of bone morphogenetic
protein 4 (BMP4), a TGF-family protein, has been revealed to induce
the EMT-like phenotype in EAC cell lines (11). Additionally, a recent study has
demonstrated that AKT inhibition reduces EMT, whereas
glioma-associated oncogene (GLI) activation promotes EMT in EAC
(12). Furthermore, the
overexpression of EMT-promoting transcription factors, including
SLUG (13) and snail family
transcriptional repressor 2 (SNAL2) (14) have been associated with poor EAC
prognosis. Despite these recent research advances, the process of
EMT in EAC remains poorly understood.
Empty spiracles homeobox 2 (EMX2) is a member of the
Homeobox gene family, which is comprised of a large group of
developmental regulators that are vital for growth and
differentiation (15). EMX2 serves
important roles in cortical development (16), neurogenesis (17,18), early
hair cell development (19), the
stereociliary bundle orientation of sensory hair cells (20) and mammalian reproduction (21). Recent studies have indicated the
potential involvement of EMX2 in various different human cancer
types, including lung (22–25), gastric (26), endometrial (27) and colorectal cancer (28,29). It
has also been revealed that EMX2 inhibits the proliferation of lung
(23), gastric (26) and colorectal (29) cancer cells, and suppresses the
invasive phenotype of lung (23) and
colorectal (28,29) cancer cells. These results indicate a
possible role of EMX2 in the process of EMT. In the present study,
EMX2 expression was determined in EAC tissues and cell lines, and
the role of EMX2 in the EMT of EAC was investigated. The results
demonstrated that the downregulation of EMX2 was associated with
EMT in EAC, and that EMX2 may suppress EMT via the inhibition of
AKT signaling in the AKT/mTOR/S6K pathway.
Materials and methods
Tissue specimens
Tissue specimens were collected between March 2015
and February 2016 from 48 patients (age, 67.5±10.6; males 29;
females 19) who underwent surgical resection for EAC at the
University of California San Francisco Thoracic Oncology Program
(Table SI). Tissue samples were
snap-frozen in liquid nitrogen immediately after resection and
stored at −170°C prior to use. The present study was conducted in
accordance with the ethical standards of the Declaration of
Helsinki, as well as national and international guidelines, with
the approval by the institutional review board of the University of
California, San Francisco (UCSF). Studies involving patient tissues
were approved by the Committee on Human Research (CHR approval
number: H8714-11647-10) at UCSF, and written, informed consent was
obtained for each patient prior to tissue specimen collection.
Protein extraction and western blot
analysis
Total protein was extracted from OE19 and OE33 cells
using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher
Scientific, Inc.) and Complete Protease Inhibitor Cocktails (Roche
Diagnostics), according to the manufacturer's protocol. Protein
concentrations were determined using a Pierce BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.), and 10 µg protein/lane was
separated using SDS-PAGE on a 4–20% gel, prior to transfer to
Immobilon-P membranes (EMD Millipore). The membranes were then
blocked in 5% non-fat milk and incubated with primary antibodies
overnight at 4°C. The membranes were then incubated with the
appropriate secondary antibodies (goat anti-rabbit IgG HRP
conjugate 31460 at 1:20,000 or goat anti-mouse IgG HRP conjugate
31430 at 1:10,000; Thermo Fisher Scientific, Inc.), and signals
were detected using an ECL blotting analysis system. The primary
antibodies used were as follows: Cyclin D1 (cat. no. sc-8396 at
1:200 dilution), B-cell lymphoma 2 (BCL2; cat. no. sc-7382 at 1:200
dilution), and fas cell surface death receptor (FAS; cat. no.
sc-8009 at 1:500 dilution; all from Santa Cruz Biotechnology,
Inc.); phosphorylated (p) m-TOR (cat. no. ab1093; 1:200 dilution)
and p-S6K1 (cat. no. ab131436; 1:250 dilution) (both from Abcam);
N-cadherin (cat. no. 05-915; 1:200 dilution), E-cadherin (cat. no.
07-697; 1:100 dilution) and β-catenin (cat. no. 04-958; 1:200
dilution; all from Sigma-Aldrich, Merck KGaA); vimentin (cat. no.
3932; 1:200 dilution), and p-AKT (cat. no. 9271; 1:200 dilution;
both Cell Signaling Technology, Inc.); GAPDH (cat. no. sc32233;
1:500 dilution; Santa Cruz Biotechnology, Inc.) and EMX2 (cat. no.
PA5-84687; 1:400 dilution; Thermo Fisher Scientific, Inc.).
Cell culture and transfection
Human EAC cell lines OE19 and OE33 were purchased
from the American Type Culture Collection and cultured in RPMI-1640
medium (Thermo Fisher Scientific, Inc.). Cells were supplemented
with 10% FBS and penicillin/streptomycin (100 mg/ml) at 37°C in a
humidified 5% CO2 incubator. To establish cell lines
exhibiting EMX2 overexpression, cultured OE19 and OE33 cells were
transfected with empty pCDNA3.1 vector controls or with pCDNA3.1
vectors containing EMX2. Transfection was performed using
Lipofectamine® 2000 (Thermo Fisher Scientific,
Inc.).
Treatment with 5-aza-2′-deoxycytidine
(DAC)
To analyze the restoration of EMX2 gene expression,
cells were treated with 5 µM DAC (Sigma-Aldrich; Merck KGaA) for 4
days, as previously described (23).
Medium was replaced daily with fresh medium containing the same
dose of DAC. Control cells were cultured with fresh medium
containing DMSO. Cells were subsequently isolated for further
analysis using DNA and RNA extraction.
Quantitative methylation-specific PCR
(qMSP)
Formalin-fixed, paraffin-embedded tissues were
de-paraffinized in xylene (Sigma-Aldrich; Merck KGaA) and
rehydrated in graded ethanol prior to bisulfite treatment,
according to the manufacturer's protocol. Bisulfite conversion from
tissues and cells was performed, without the prerequisite DNA
purification step, using an EZ DNA Methylation-Direct kit (Zymo
Research Corp.). Genomic DNA was extracted using Qiagen DNeasy kits
(Qiagen, Inc.). qMSP was performed using the ABsolute Blue qPCR
SYBR-Green ROX Mix (Thermo Fisher Scientific, Inc.) with MSP
primers, which recognized either methylated or unmethylated EMX2
promoter sequences after bisulfite treatment. PCR conditions were
as follows: 10 min at 95°C; 40 cycles of 30 sec at 95°C, 30 sec at
50°C and 30 sec at 72°C; with a final 7 min extension at 72°C.
Primers were designed and purchased from Eurofins MWG Operon, Inc.
Sequences are as follows: ATCB forward,
5′-TGGTGATGGAGGAGGTTTAGTAAGT-3′ and reverse,
5′-AACCAATAAAACCTACTCCTCCCTTAA-3′; MSP-M forward,
5′-TAGTTTTTTGTTCGTTTCGCGTTTC-3′ and reverse,
5′-GAATTAAAATAAACGCCCCTACCGAC-3′; and MSP-U forward,
5′-GTTTTTTGTTTGTTTTGTGTTTTGA-3′ and reverse,
5′-CCAAATTAAAATAAACACCCCTACCAAC-3′.
All qMSP assays were performed in triplicate using
the ABI 7900HT Fast Real-Time PCR System (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Relative EMX2 methylation levels
were determined using the 2−∆∆Cq method (30) and normalized to the housekeeping gene,
ACTB, prior to calculating the ratio of tumor/paired normal tissue
per patient.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated using the Qiagen RNeasy kit,
and cDNA was transcribed from 500 ng RNA using the iScript cDNA
Synthesis kit (Bio-Rad Laboratories, Inc.) according to the
manufacturer's protocol. EMX2, Aurora Kinase (AURK)A, AURKB,
caspase (CASP)-3 and GAPDH mRNA expression were subsequently
examined using RT-qPCR with commercially available primers and
probes (Thermo Fisher Scientific, Inc.) and Relative Quantification
Software (Applied Biosystems; Thermo Fisher Scientific, Inc.). Gene
expression was normalized to GAPDH expression using the
2−∆∆Cq method (30).
Wound healing, Transwell invasion and
cell survival assays
In the wound healing migration assays, four scratch
wounds were made in each treatment condition and cell migration was
determined using the average differences in distance between 0–24
h. Transwell invasion assays (Thermo Fisher Scientific, Inc.) were
performed using six-well plates, according to the manufacturer's
protocol. EAC cells were placed on the upper layer of a cell
culture insert (Matrigel-coated membrane), and incubated for 24 h.
The migrated cells were then fixed with methanol and stained with
crystal violet. For each well, three representative fields of view
were selected to measure the invasion level.
To evaluate cell survival after chemotoxic
treatment, cells were plated in 96-well plates at a density of
500–1,000 cells/well. The medium was changed daily. Logarithmically
growing cells were treated with 5 µM cisplatin or DMSO (vehicle
control) for 24–72 h. Cell survival was assessed using
CellTiter-Glo Luminescent Cell Viability Assay reagent (Promega
Corporation), according to the manufacturer's protocol.
Luminescence was measured using a GloMax-96 Microplate Luminometer
(Promega Corporation). The percentage cell survival was calculated
based on the measurement of untreated cells as 100% using GraphPad
Prism 6.0 software (GraphPad Software, Inc.), which was also used
to generate dose-response curves and IC50 values. All
experiments were conducted in triplicate and data were standardized
to DMSO-treated cells.
Statistical analysis
Two-tailed Student's t-tests were performed for
wound healing (migration) and Transwell (invasion) assay analyses.
ANOVA and Scheffe tests were used to determine significance in
luciferase reporter and qPCR data. P<0.05 was considered to
indicate a statistically significant difference.
Results
EMX2 expression is downregulated by
methylation in the tissues of patients with EAC, and EAC cell
lines
To determine whether EMX2 served a role in the
progression of EAC, EMX2 protein expression was assessed in the EAC
tissues and paired adjacent normal tissues of 48 patients with EAC,
using western blot analysis. The results demonstrated that 79.2%
(38/48) of tumor samples exhibited lower EMX2 protein expression
compared with paired normal tissue expression (Fig. 1A and Fig.
S1).
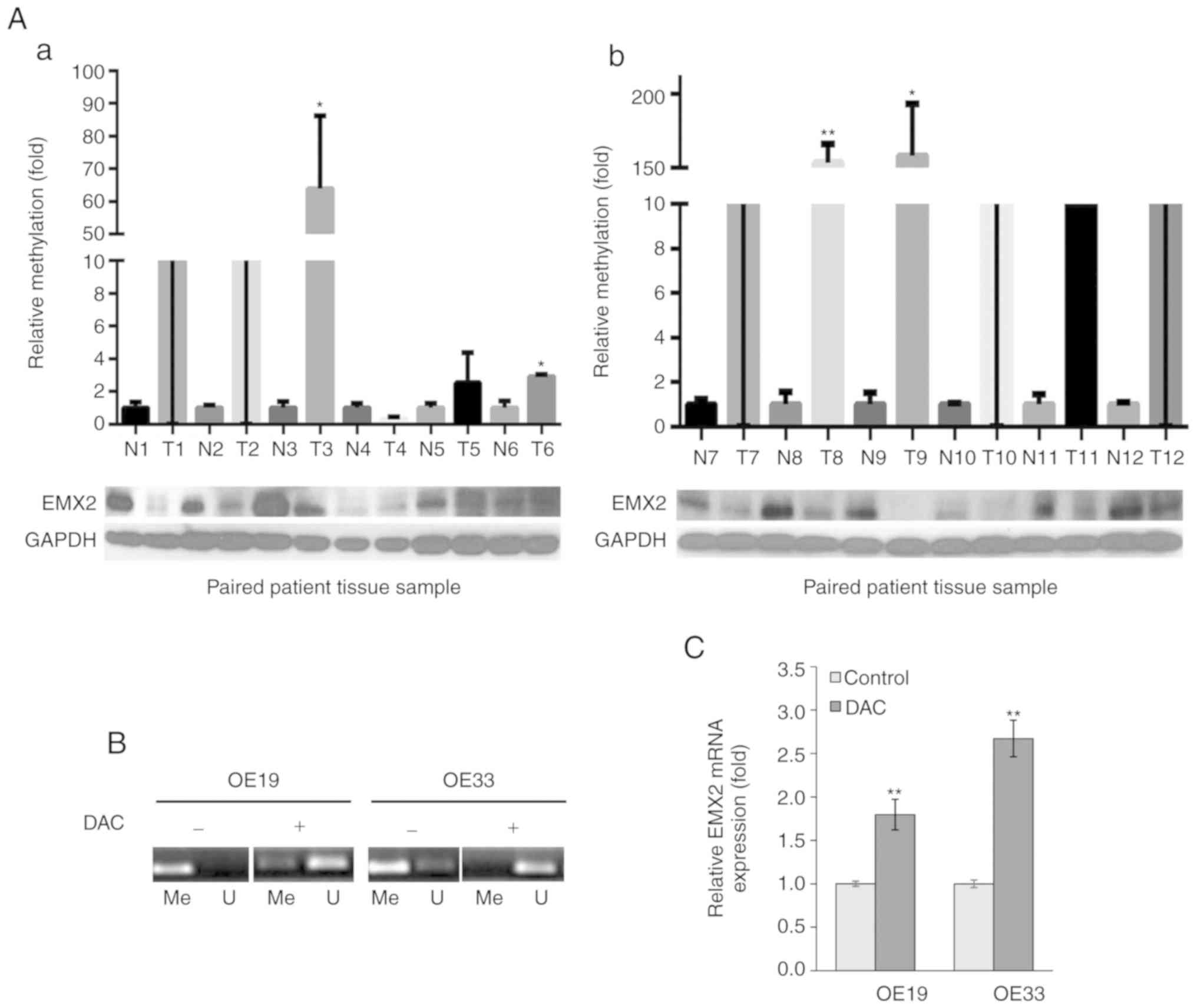 | Figure 1.EMX2 expression is downregulated by
methylation in EAC tissues and cell lines. (A) Western blot
analysis of EMX2 and qMSP in 12 matched pairs of N and T tissues.
Samples are presented with corresponding EMX2 protein levels. qMSP
results were normalized to paired normal tissues, and GAPDH was
used as an internal control for EMX2 protein expression. (a) N and
T pairs 1–6, with values of T1 and T2 in the broken region of the
y-axis. (b) N and T pairs 7–12, with values of T7, T10, T11 and T12
in the broken region of the y-axis. (B) DAC treatment reduced EMX2
promoter methylation in OE19 and OE33 cells. EAC cells were treated
with 5 µM DAC, followed by qMSP analysis of EMX2 promoter
methylation using reactions specific to methylated (Me) or
unmethylated (U) targets. (C) DAC treatment restored EMX2
expression in OE19 and OE33 cells. The EAC cells were treated with
5 µM DAC, followed by reverse transcription-quantitative PCR
examination of EMX2 mRNA expression. *P<0.05 and **P<0.01.
EMX2, empty spiracles homeobox 2; EAC, esophageal adenocarcinoma;
qMSP, quantitative methylation-specific polymerase chain reaction;
N, normal; T, tumor; DAC, 5-aza-2′-deoxycytidine. |
Homeobox (HOX) gene silencing by promoter
methylation has been previously reported for HOXA5 and HOXA9 in
breast cancer (31) and testicular
germ cell tumors (32), respectively.
Previous studies have revealed that the downregulation of EMX2 is
associated with the hypermethylation of its promoter in lung
(23) and gastric cancer (26). To determine whether promoter
hypermethylation was responsible for the downregulation of EMX2 in
EAC tissues, the EMX2 promoter methylation status was determined
using qMSP (33). Decreased EMX2
protein expression in EAC tissues was revealed to be associated
with hypermethylation of the EMX2 promoter in EAC when compared
with adjacent normal tissues (Fig.
1A).
EMX2 expression in two EAC cell lines, OE19 and
OE33, was also determined. When treated with the methyltransferase
inhibitor, DAC, the methylation levels of EMX2 promoter decreased
in OE19 and OE33 cells (Fig. 1B), and
the expression of EMX2 transcripts was activated (Fig. 1C). These data indicated that EMX2
expression is downregulated by promoter hypermethylation in EAC
tissues and cell lines.
EMX2 induces apoptotic marker
expression in EAC cells
Genes or proteins that are associated with apoptosis
or cell cycle regulation, in OE19 and OE33 cells, were assessed to
determine the biological function of EMX2 in EAC progression. EMX2
expression was induced via transient transfection with a EMX2
vector, and pCDNA3.1 was used as a control, as previously described
(23). mRNA expression, measured
using RT-qPCR, indicated that EMX2 expression increased by 4–5
orders of magnitude subsequent to transfection with an EMX2
expression vector (P<0.01; Fig.
2A).
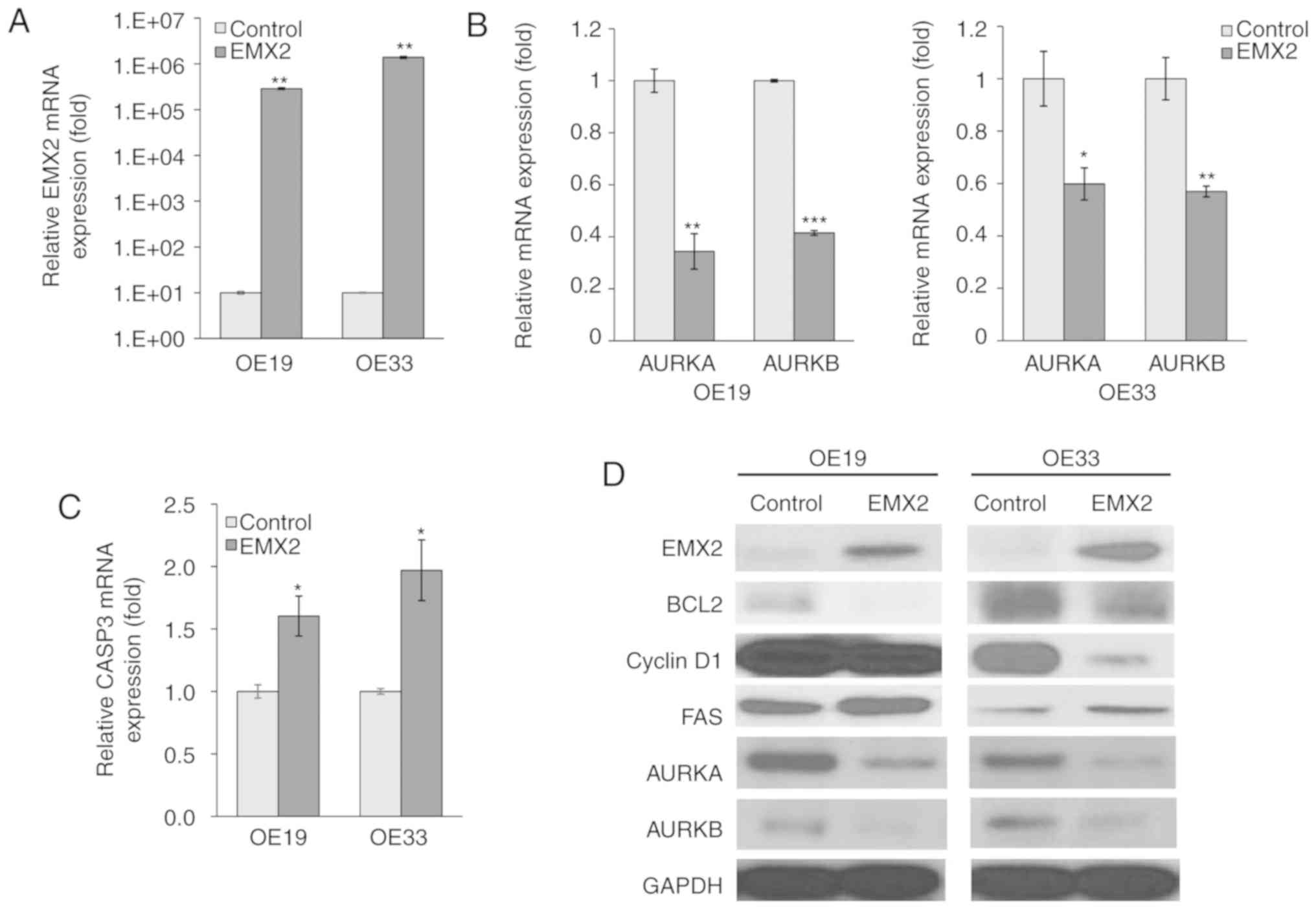 | Figure 2.EMX2 induces apoptotic marker
expression in EAC cells. (A) mRNA expression of EMX2 in two EAC
cell lines, OE19 and OE33, that were transfected with an empty
pCDNA3.1 vector control or EMX2, as determined using RT-qPCR. (B)
mRNA expression of AURKA and AURKB in OE19 and OE33 cells
transfected with an empty pCDNA3.1 vector control or EMX2, as
determined using RT-qPCR. (C) CASP3 mRNA expression in OE19 and
OE33 cells transfected with an empty pCDNA3.1 vector control or
EMX2, as determined using RT-qPCR. (D) Western blot analysis of key
cell cycle and apoptosis markers (BCL2, cyclin D1 and FAS), and
AURKA and AURKB protein expression in OE19 and OE33 cells
transfected with an empty pCDNA3.1 vector control or EMX2. GAPDH
was used as a loading control. *P<0.05, **P<0.01 and
***P<0.001. EMX2, empty spiracles homeobox 2; EAC, esophageal
adenocarcinoma; RT-q, reverse transcription-quantitative; AURK,
aurora kinase; CASP3, caspase-3; BCL2, B-cell lymphoma 2; FAS, fas
cell surface death receptor. |
AURKA and AURKB are essential regulators for
mitosis, and have been demonstrated to inhibit apoptosis (34–36). In
the present study, EMX2 expression significantly inhibited AURKA
and AURKB expression in OE19 (63% reduction; P<0.01; Fig. 2B) and OE33 (40% reduction; P<0.01;
Fig. 2B) cell lines. AURKA and AURKB
protein expression also decreased following EMX2 expression
(Fig. 2D). CASP3, an apoptosis
marker, was upregulated following EMX2 expression and exhibited a
1.5- and 2-fold upregulation in OE19 and OE33 cells, respectively
(P<0.01; Fig. 2C). Furthermore,
regulators of apoptosis and cell cycle progression, including BCL2
(37,38), cyclin D1 (39,40) and
FAS (41,42) were examined using western blot
analysis (Fig. 2D). The expression of
BLC2, a cell death suppressor, and cyclin D1, a protein required
for G1 phase progression in the cell cycle, were negatively
correlated with EMX2 expression. In contrast, the expression of
FAS, which can induce apoptosis, was positively associated with
EMX2. These results indicated that EMX2 promotes apoptosis and
inhibits cell cycle progression in EAC.
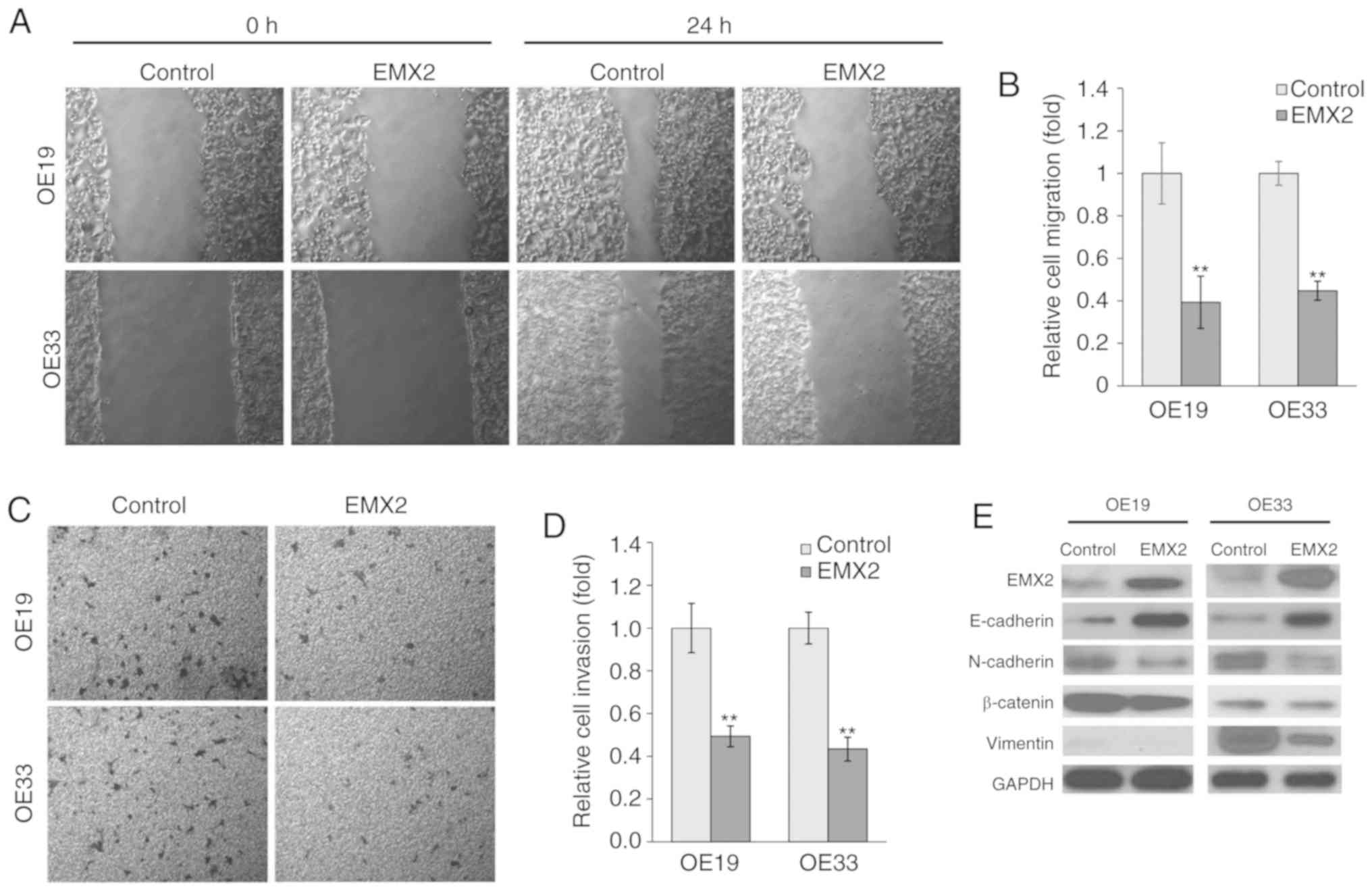
EMX2 attenuates EAC cell migration and
invasion
To determine the role of EMX2 in EAC cell migration
and invasion, wound healing and Transwell invasion assays were
performed in OE19 and OE33 cells. Over a period of 24 h, OE19 and
OE33 cells exhibited significantly slower migration in cells
transfected with EMX2 (Fig. 3A and
B). Statistical analysis indicated that the migration of
OE19-EMX2 and OE33-EMX2 cells was ~40% of the migration exhibited
by the vector control cells (P<0.01). Furthermore, in the
Transwell invasion assays, OE19 and OE33 cells transfected with
EMX2 demonstrated decreased invasion of >50% compared with cells
transfected with the vector control (P<0.01; Fig. 3C and D).
EMX2 inhibits EMT marker expression in
EAC
An important hallmark of EMT is the loss of
E-cadherin expression, a cell-to-cell adhesion molecule that is
encoded by CDH, a tumor suppressor gene. It has been previously
revealed that E-cadherin expression is reduced in EAC, and this
decreased expression is associated with an increased frequency of
lymph node metastasis and decreased patient survival (43). A previous study indicated that
E-cadherin protein expression was reduced in the tumor tissues of
patients with EAC. In contrast, two mesenchymal markers, N-cadherin
and vimentin, and cell adhesion and signaling molecule, β-catenin,
were overexpressed (12). Due to the
fact that cell migration and invasion have been associated with
altered levels of EMT biomarkers, the effect of EMX2 expression on
epithelial features of EAC cells was assessed in the present study.
The expression of epithelial markers, including E-cadherin, and of
mesenchymal markers, including N-cadherin and vimentin, were
examined. As presented in Fig. 3E,
EMX2 expression in OE19 and OE33 cells increased E-cadherin
expression and decreased N-cadherin, vimentin and β-catenin
expression. These results indicated that EMX2 suppresses EMT in
EAC.
EMX2 inhibits EMT by suppressing
AKT/mTOR/S6K1 signaling in EAC
The present study also investigated the mechanism by
which EMX2 inhibits EMT in EAC. AKT genes are overexpressed in EAC,
and their expression has been associated with the pathologic
response of EAC treated with neoadjuvant therapy (44). A previous study revealed the
upregulation of the AKT pathway proteins p-AKT, p-mTOR and p-S6K1
in the tumor tissues of patients with EAC (12). The results of the present study
demonstrated that the upregulation of AKT pathway markers was
correlated with EMX2 downregulation in EAC tumor tissues (Fig. 4A). These results prompted the
hypothesis that EMX2 inhibits EMT by suppressing the AKT signaling
cascade. In OE19 and OE33 cells, EMX2 overexpression decreased
p-MTOR, p-AKT and p-S6K1 levels (Fig.
4B). These findings indicated that EMX2 inhibits EMT in EAC
cells, at least in part, by suppressing the AKT/mTOR/S6K1 signaling
pathway.
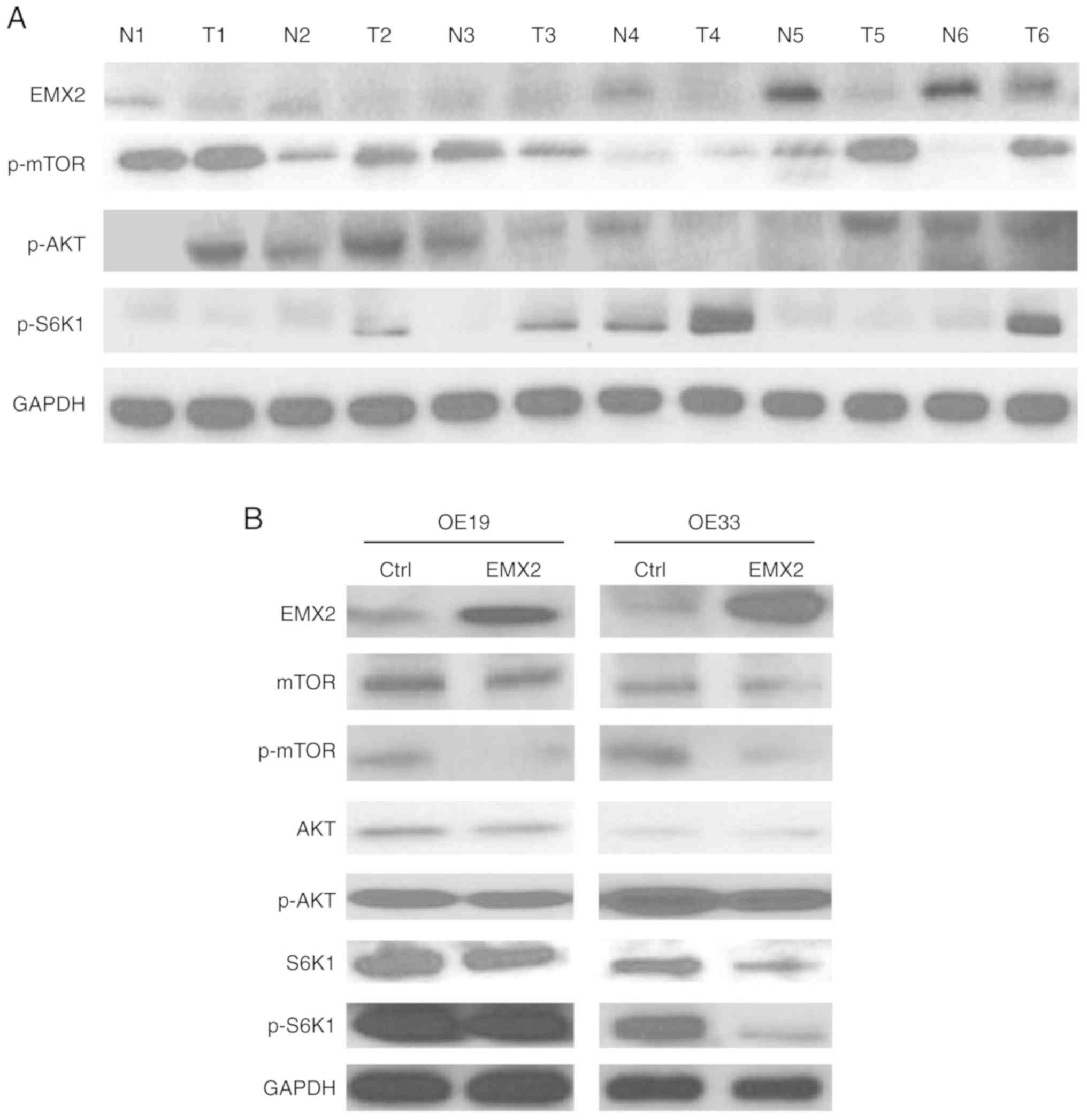 | Figure 4.EMX2 inhibits EMT by suppressing
AKT/mTOR/S6K1 signaling in EAC. (A) Western blot analysis of EMX2
and AKT signaling pathway proteins (p-mTOR, p-AKT and p-S6K1) in
matched pairs of N and T tissues. GAPDH was used as a loading
control. (B) Western blot analysis of EMX2 and AKT signaling
pathway proteins (mTOR, AKT, S6K1, and p-mTOR, p-AKT and p-S6K1) in
OE19 and O33 cells transfected with a vector control or EMX2. EMX2,
empty spiracles homeobox 2; EMT, epithelial-mesenchymal transition;
EAC, esophageal adenocarcinoma; p, phosphorylated; N, normal; T,
tumor. |
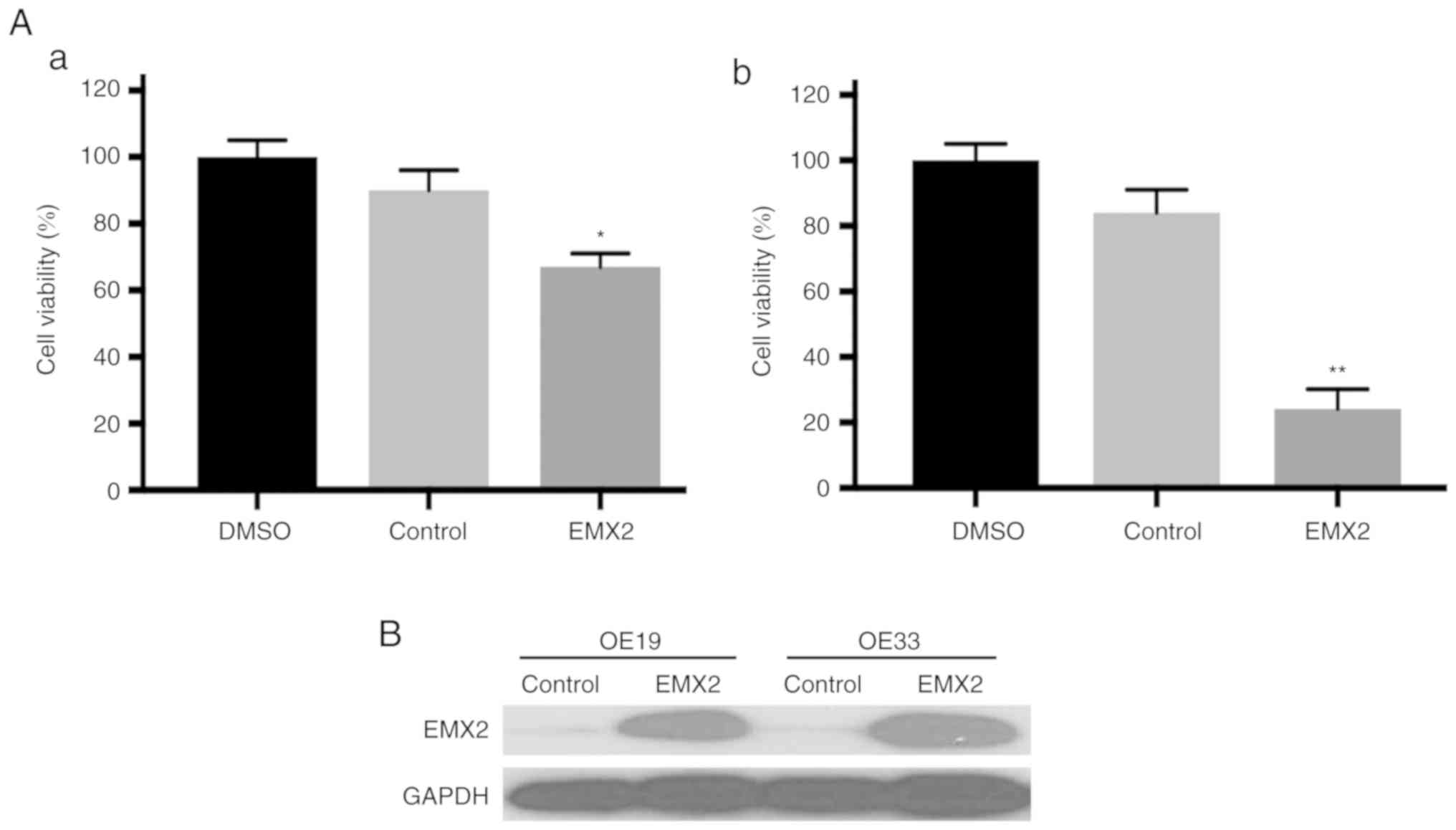
EMX2 sensitizes EAC cells to
cisplatin
It has been revealed that EMT serves a critical role
in drug resistance. Therefore, in the present study, whether EMX2
overexpression could sensitize EAC cells to cisplatin, a widely
used chemotherapy agent for EAC, was assessed (45). A cell viability assay revealed that
OE19 and OE33 cells transfected with a control vector were
resistant to cisplatin treatment. In contrast, when treated with
cisplatin, the viability of EMX2-overexpressed OE19 and OE33 cells
was reduced by at least 30% compared with cells transfected with a
vector control (Fig. 5A). EMX2
protein expression was further confirmed using western blot
analysis (Fig. 5B). These results
indicated that EMX2 overexpression can sensitize EAC cells to
cisplatin.
Discussion
Despite recent advances in cancer research, the
survival rates of patients with EAC remain low, and EAC continues
to be a poorly understood disease. EMT is an important mechanism in
epithelial cancer cell invasion and metastasis, with local invasion
and metastasis occurring early in the pathogenesis of EAC. Although
EMT has been indicated to serve a role in the early metastasis of
EAC, the regulatory mechanisms of EMT in EAC remain
undetermined.
The results of the present study demonstrated, to
the best of our knowledge, for the first time, that EMX2 suppresses
EMT in EAC. EMX2 is downregulated in a variety of cancer types and
has been revealed to be associated with the EMT process. In a
previous study, it was revealed that the overexpression of EMX2 in
lung squamous cell carcinoma cells inhibited cell migration and
downregulated EMT markers, whereas the knockdown of endogenous EMX2
promoted cell migration and upregulated EMT markers (25). Zhang et al (29) demonstrated that the overexpression of
EMX2 significantly inhibited cell migration and invasion, and
upregulated EMT marker expression, in a human colorectal cancer
cell line. The present study revealed that EMX2 is downregulated in
EAC tissues and cell lines, and the restoration of EMX2 in EAC cell
lines induced the expression of apoptotic markers, inhibited cell
migration and invasion and induced E-cadherin expression while
inhibiting mesenchymal marker expression. These data indicated that
EMX2 suppresses EMT in EAC.
In a previous study with lung squamous cell
carcinoma (25), 12 cell lines were
screened and the majority exhibited low EMX2 expression. Only two
cell lines had high EMX2 expression and were used for silencing
experiments. For EAC, the available cell lines were limited and
none of the tested cell lines exhibited a high level of EMX2
expression. As revealed in Fig. S1,
the esophageal adenocarcinomas had much lower EMX2 expression
compared with the adjacent normal tissues, and some were below the
detection limit. The two cell lines used in this study, OE19 and
OE33, had barely detectable EMX2 (Fig.
2A and 2D). Due to the intrinsic
low level of EMX2 expression, the silencing experiment was
infeasible.
To illustrate the relationship between EMX2 promoter
methylation and gene expression, the methylation levels of EMX2 in
OE19 and OE33 cells with or without 5-aza-2′-deoxycytidine (DAC)
treatment were measured. The promoter regions of EMX2 in OE19 and
OE33 cells were highly methylated, which revealed decreased
methylation upon DAC treatment (Fig.
1B). Consequently, EMX2 expression was increased (Fig. 1C). These results demonstrated that the
downregulation of EMX2 in EAC tissues is correlated with
hypermethylation of its promoter, and suggested that epigenetic
silencing is an important mechanism for the downregulation of EMX2
in EAC. It corresponds with previous findings regarding epigenetic
silencing of EMX2 in lung (23) and
gastric cancer (26). Previous
studies have revealed that in a variety of cancer types, the
inhibition of Homeobox genes by hypermethylation of their promoters
contributes to tumorigenesis (46).
Furthermore, a recent study by Kikuchi et al (47) demonstrated that HOP homeobox promoter
methylation independently predicts poor prognosis in human
epidermal growth factor receptor 2-negative breast cancer. These
results highlighted the clinical value of Homeobox gene epigenetic
silencing, and the requirement for future investigations into the
clinical value of EMX2 epigenetic silencing.
The PI3K/AKT and mTOR signaling pathways are crucial
to numerous aspects of cell growth, survival and EMT, and have been
implicated in tumor cell motility, invasion and metastasis
(48–51). For instance, AKT activation was
revealed to induce EMT in human oral squamous cell carcinomas
(52), and subsequently it was
revealed that AKT induced EMT by activating NF-κB (53). Conversely, the inhibition of AKT
activity reversed EMT in these cells (54). AKT genes are overexpressed in EAC, and
their expression has been associated with the degree of pathologic
response during neoadjuvant therapy for EAC (44). S6K1 is positively regulated by mTOR,
and the activation of mTOR/S6K1 signaling has been suggested to be
involved in EAC progression (55). A
previous study revealed that AKT signaling proteins were
upregulated in the tumor tissues of patients with EAC, and AKT
inhibition reduced EMT and cell cycle activity in EAC cell lines
(12). In the present study, the
observation that the loss of EMX2 is correlated with increased
levels of p-AKT, p-mTOR and p-S6K1 in the tumor tissues is
consistent with the hypothesis that the loss of EMX2 may be
associated with the upregulation of AKT signaling pathways
(Fig. 4A). A limitation of the study
is that due to limited amount of patient samples, the total protein
levels of AKT/mTOR/S6K1 were not assessed. It is unknown whether
the elevated levels of p-AKT, p-mTOR and p-S6K1 were due to
increased total protein levels or increased phosphorylation per
se. In OE19 and OE33 cancer cell lines, the phosphorylation
levels of these proteins upon EMX2 expression were decreased more
prominently than the changes in their total protein levels
(Fig. 4B), suggesting that the
suppression of AKT/mTOR/S6K1 pathway by EMX2 was not simply due to
direct inhibition of AKT/mTOR/S6K1 gene expression in these cells.
These data support the notion that EMX2 inhibits EMT via inhibition
of AKT/mTOR/S6K1 signaling in EAC cells.
AKT/mTOR/S6K1 is a major hub of signal transduction
pathways, and is interconnected with multiple regulatory factors.
Since EMX2 is a homeobox protein, it is speculated that it may
function as a specific transcription factor to regulate the
expression of genes upstream of the AKT/mTOR/S6K1 pathway.
Recently, the induction of EMT by GLI activation was reported in
colon (56) and breast cancer
(57). A previous study revealed that
GLI is upregulated and is correlated with EMT and the activation of
AKT pathway markers both in EAC cell lines and tissue samples
(12). These studies indicated that
AKT and GLI activation may be a general mechanism in EMT.
Currently, it is unknown whether EMX2 functions upstream or
downstream of GLI, which is the focus of the ongoing research.
Previous studies investigating EMT in EAC have
revealed that the reduced expression of the E-cadherin-catenin
complex, and upregulation of AKT genes in EAC, is correlated with
poor prognosis (43,44). The results of the present study
demonstrated that EMX2 induced the upregulation of E-cadherin and
inhibited AKT signaling in OE19 and OE33 cells. Furthermore, the
downregulation of EMX2 in lung cancer and colorectal cancer has
been associated with decreased overall survival (22,24,28).
Therefore, it can be suggested that the downregulation of EMX2 may
be associated with a worse EAC prognosis. The manipulation of the
EMX2 pathway could be used to prevent EAC progression. It may take
years for this goal to be reached, however, two recent studies have
made this more feasible. A previous study demonstrated that
adenoviral EMX2 delivery in gastric cancer suppressed cancer cell
proliferation and improved overall survival in vivo
(26). Falcone et al reported
that EMX2 overexpression exhibited antioncogenic activity and
induced the collapse of glioblastoma cells in vitro and
in vivo (58). Recent studies
have also indicated an unexpected role of EMT in cancer
chemoresistance (59–61). A previous study demonstrating that the
knockdown of EMX2 expression in lung squamous cell carcinomas cells
promoted chemoresistance (25), and
the results of the present study revealing that EMX2 overexpression
sensitized EAC cell lines to the chemotoxic reagent cisplatin, are
consistent with these data. These observations indicate that the
role of EMX2 in chemotherapy sensitivity may be general in
different types of cancer. At present, the study on the role of
EMX2 in chemosensitivity is limited. With further investigation,
EMX2 may become a novel antitumor target, synergistic with
chemotherapy options, for patients with EAC.
In summary, the present study revealed that EMX2 is
epigenetically silenced in EAC, and a strong association exists
between EMX2 and EMT, apoptotic and cell cycle markers and the
AKT/mTOR/S6K1 pathway in EAC. These findings suggest that silencing
EMX2 may prove critical in promoting cell survival, metastasis and
resistance to chemotherapy. Restoration of EMX2 expression via the
alleviation of epigenetic silencing may serve as a potential
therapeutic strategy for the treatment of human EAC. With the
advances in pharmaceutical development of safe and potent
epigenetic regulatory reagents, this potential should be
investigated in the future.
Supplementary Material
Supporting Data
Acknowledgements
The authors acknowledge the important contributions
of the other members of the University of California, San Francisco
Thoracic Oncology Program and the research team of Dr Biao He,
including Fleur Leguay, B.S.; Ngoc Hoang, B.S.; and Luis Acevedo,
B.S.
Funding
The present study was supported by the Eileen D.
Ludwig Endowed Fund for Thoracic Oncology Research (to BH); the
Science and Technology Support Project of Hebei Province
(132077127D to LW), the Fund for International Scientific and
Technological Cooperation in Hebei Province (18397771D to LW), the
Fund for Scientific and Technological Activities of Overseas
Scholars by Hebei Provincial Office of Human and Social Affairs
(CY201613 to LW); the Science and Technology Planning Project of
Zhejiang Province Science and Technology Department (2018C37091 to
YH), and the Zhejiang Provincial Natural Science Foundation of
China (LY15H160048 to FD), the Jiaxing Science and Technology
Project (2015BZ12001 to FD) and the Jiaxing Nanhu District Science
and Technology Project (2018QC03 to FD).
Availability of data and materials
The materials and datasets used in this study are
available upon request.
Authors' contributions
JJ performed cell culture, western blot and data
analysis, and drafted the manuscript. LW, YZ, ZT and YH carried out
western blot, qRT-PCR, cell viability, wound healing, and Transwell
invasion assays. LW and YH also conducted data and statistical
analyses and helped to draft the manuscript. BH, YH and FD
conceived of the study, participated in its design and
coordination, and drafted the manuscript. All authors read and
approved the final manuscript and agree to be to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethical approval and consent to
participate
The present study was conducted in accordance with
the ethical standards of the Declaration of Helsinki, as well as
national and international guidelines, with the approval by the
institutional review board of the University of California, San
Francisco (UCSF). Studies involving patient tissues were approved
by the Committee on Human Research (CHR approval number:
H8714-11647-10) at UCSF, and written, informed consent was obtained
for each patient prior to tissue specimen collection.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DAC
|
5-aza-2′-deoxycytidine
|
|
EAC
|
esophageal adenocarcinoma
|
|
EMT
|
epithelial-mesenchymal transition
|
|
N
|
normal
|
|
p
|
phosphorylated
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
qMSP
|
quantitative methylation-specific
polymerase chain reaction
|
|
T
|
tumor
|
|
UCSF
|
University of California San
Francisco
|
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cancer Genome Atlas Research Network;
Analysis Working Group: Asan University; BC Cancer Agency; Brigham
and Women's Hospital; Broad Institute; Brown University; Case
Western Reserve University; Dana-Farber Cancer Institute; Duke
University; Greater Poland Cancer Centre, et al, . Integrated
genomic characterization of oesophageal carcinoma. Nature.
541:169–175. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arnold M, Soerjomataram I, Ferlay J and
Forman D: Global incidence of oesophageal cancer by histological
subtype in 2012. Gut. 64:381–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lagarde SM, ten Kate FJ, Richel DJ,
Offerhaus GJ and van Lanschot JJ: Molecular prognostic factors in
adenocarcinoma of the esophagus and gastroesophageal junction. Ann
Surg Oncol. 14:977–991. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: Emt: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: Acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rees JR, Onwuegbusi BA, Save VE, Alderson
D and Fitzgerald RC: In vivo and in vitro evidence for transforming
growth factor-beta1-mediated epithelial to mesenchymal transition
in esophageal adenocarcinoma. Cancer Res. 66:9583–9590. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kestens C, Siersema PD, Offerhaus GJ and
van Baal JW: BMP4 signaling is able to induce an
epithelial-mesenchymal transition-like phenotype in Barrett's
esophagus and esophageal adenocarcinoma through induction of
SNAIL2. PLoS One. 11:e01557542016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang L, Jin JQ, Zhou Y, Tian Z, Jablons DM
and He B: Gli is activated and promotes epithelial-mesenchymal
transition in human esophageal adenocarcinoma. Oncotarget.
9:853–865. 2017.PubMed/NCBI
|
|
13
|
Jethwa P, Naqvi M, Hardy RG, Hotchin NA,
Roberts S, Spychal R and Tselepis C: Overexpression of slug is
associated with malignant progression of esophageal adenocarcinoma.
World J Gastroenterol. 14:1044–1052. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Allott EH, Morine MJ, Lysaght J,
McGarrigle SA, Donohoe CL, Reynolds JV, Roche HM and Pidgeon GP:
Elevated tumor expression of PAI-1 and SNAI2 in obese esophageal
adenocarcinoma patients and impact on prognosis. Clin Transl
Gastroenterol. 3:e122012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abate-Shen C: Deregulated homeobox gene
expression in cancer: Cause or consequence? Nat Rev Cancer.
2:777–785. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cecchi C: Emx2: A gene responsible for
cortical development, regionalization and area specification. Gene.
291:1–9. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brancaccio M, Pivetta C, Granzotto M,
Filippis C and Mallamaci A: Emx2 and Foxg1 inhibit gliogenesis and
promote neuronogenesis. Stem Cells. 28:1206–1218. 2010.PubMed/NCBI
|
|
18
|
Falcone C, Filippis C, Granzotto M and
Mallamaci A: Emx2 expression levels in NSCs modulate astrogenesis
rates by regulating EgfR and Fgf9. Glia. 63:412–422. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Holley M, Rhodes C, Kneebone A, Herde MK,
Fleming M and Steel KP: Emx2 and early hair cell development in the
mouse inner ear. Dev Biol. 340:547–556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang T, Kindt K and Wu DK: Transcription
factor Emx2 controls stereociliary bundle orientation of sensory
hair cells. Elife. 6:e236612017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taylor HS and Fei X: Emx2 regulates
mammalian reproduction by altering endometrial cell proliferation.
Mol Endocrinol. 19:2839–2846. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giroux Leprieur E, Hirata T, Mo M, Chen Z,
Okamoto J, Clement G, Li H, Wislez M, Jablons DM and He B: The
homeobox gene EMX2 is a prognostic and predictive marker in
malignant pleural mesothelioma. Lung Cancer. 85:465–471. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Okamoto J, Hirata T, Chen Z, Zhou HM,
Mikami I, Li H, Yagui-Beltran A, Johansson M, Coussens LM, Clement
G, et al: EMX2 is epigenetically silenced and suppresses growth in
human lung cancer. Oncogene. 29:5969–5975. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Okamoto J, Kratz JR, Hirata T, Mikami I,
Raz D, Segal M, Chen Z, Zhou HM, Pham P, Li H, et al:
Downregulation of EMX2 is associated with clinical outcomes in lung
adenocarcinoma patients. Clin Lung Cancer. 12:237–244. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yue D, Li H, Che J, Zhang Y, Tolani B, Mo
M, Zhang H, Zheng Q, Yang Y, Cheng R, et al: EMX2 is a predictive
marker for adjuvant chemotherapy in lung squamous cell carcinomas.
PLoS One. 10:e01321342015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Mo M, Chen Z, Chen Z, Sheng Q, Mu H,
Zhang F, Zhang Y, Zhi XY, Li H, et al: Adenoviral delivery of the
EMX2 gene suppresses growth in human gastric cancer. PLoS One.
7:e459702012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qiu H, Yan Q, Luo X, Zhang H, Bao W and
Wan X: EMX2 is downregulated in endometrial cancer and correlated
with tumor progression. Int J Gynecol Pathol. 32:193–198. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aykut B, Ochs M, Radhakrishnan P, Brill A,
Höcker H, Schwarz S, Weissinger D, Kehm R, Kulu Y, Ulrich A and
Schneider M: EMX2 gene expression predicts liver metastasis and
survival in colorectal cancer. BMC Cancer. 17:5552017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Cao G, Yuan QG, Li JH and Yang
WB: Empty spiracles homeobox 2 (EMX2) inhibits the invasion and
tumorigenesis in colorectal cancer cells. Oncol Res. 25:537–544.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Raman V, Tamori A, Vali M, Zeller K, Korz
D and Sukumar S: HOXA5 regulates expression of the progesterone
receptor. J Biol Chem. 275:26551–26555. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lind GE, Skotheim RI, Fraga MF, Abeler VM,
Esteller M and Lothe RA: Novel epigenetically deregulated genes in
testicular cancer include homeobox genes and SCGB3A1 (HIN-1). J
Pathol. 210:441–449. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Galm O and Herman JG: Methylation-specific
polymerase chain reaction. Methods Mol Med. 113:279–291.
2005.PubMed/NCBI
|
|
34
|
Andrews PD, Knatko E, Moore WJ and Swedlow
JR: Mitotic mechanics: The auroras come into view. Curr Opin Cell
Biol. 15:672–683. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gavriilidis P, Giakoustidis A and
Giakoustidis D: Aurora kinases and potential medical applications
of aurora kinase inhibitors: A review. J Clin Med Res. 7:742–751.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goldenson B and Crispino JD: The aurora
kinases in cell cycle and leukemia. Oncogene. 34:537–545. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kirkin V, Joos S and Zornig M: The role of
Bcl-2 family members in tumorigenesis. Biochim Biophys Acta.
1644:229–249. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zinkel S, Gross A and Yang E: BCL2 family
in DNA damage and cell cycle control. Cell Death Differ.
13:1351–1359. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Alao JP: The regulation of cyclin D1
degradation: Roles in cancer development and the potential for
therapeutic invention. Mol Cancer. 6:242007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Musgrove EA, Caldon CE, Barraclough J,
Stone A and Sutherland RL: Cyclin D as a therapeutic target in
cancer. Nat Rev Cancer. 11:558–572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
O'Brien DI, Nally K, Kelly RG, O'Connor
TM, Shanahan F and O'Connell J: Targeting the Fas/Fas ligand
pathway in cancer. Expert Opin Ther Targets. 9:1031–1044. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Peter ME, Hadji A, Murmann AE, Brockway S,
Putzbach W, Pattanayak A and Ceppi P: The role of CD95 and CD95
ligand in cancer. Cell Death Differ. 22:885–886. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Krishnadath KK, Tilanus HW, van
Blankenstein M, Hop WC, Kremers ED, Dinjens WN and Bosman FT:
Reduced expression of the cadherin-catenin complex in oesophageal
adenocarcinoma correlates with poor prognosis. J Pathol.
182:331–338. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saeed N, Shridhar R, Hoffe S, Almhanna K
and Meredith KL: AKT expression is associated with degree of
pathologic response in adenocarcinoma of the esophagus treated with
neoadjuvant therapy. J Gastrointest Oncol. 7:158–165.
2016.PubMed/NCBI
|
|
45
|
Rubenstein JH and Shaheen NJ:
Epidemiology, diagnosis, and management of esophageal
adenocarcinoma. Gastroenterology. 149:302–317 e301. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rodrigues MF, Esteves CM, Xavier FC and
Nunes FD: Methylation status of homeobox genes in common human
cancers. Genomics. 108:185–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kikuchi M, Katoh H, Waraya M, Tanaka Y,
Ishii S, Tanaka T, Nishizawa N, Yokoi K, Minatani N, Ema A, et al:
Epigenetic silencing of HOPX contributes to cancer aggressiveness
in breast cancer. Cancer Lett. 384:70–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 169:361–371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zarogoulidis P, Lampaki S, Turner JF,
Huang H, Kakolyris S, Syrigos K and Zarogoulidis K: mTOR pathway: A
current, up-to-date mini-review (Review). Oncol Lett. 8:2367–2370.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou H and Huang S: Role of mTOR signaling
in tumor cell motility, invasion and metastasis. Curr Protein Pept
Sci. 12:30–42. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Grille SJ, Bellacosa A, Upson J,
Klein-Szanto AJ, van Roy F, Lee-Kwon W, Donowitz M, Tsichlis PN and
Larue L: The protein kinase Akt induces epithelial mesenchymal
transition and promotes enhanced motility and invasiveness of
squamous cell carcinoma lines. Cancer Res. 63:2172–2178.
2003.PubMed/NCBI
|
|
53
|
Julien S, Puig I, Caretti E, Bonaventure
J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A
and Larue L: Activation of NF-kappaB by Akt upregulates Snail
expression and induces epithelium mesenchyme transition. Oncogene.
26:7445–7456. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hong KO, Kim JH, Hong JS, Yoon HJ, Lee JI,
Hong SP and Hong SD: Inhibition of Akt activity induces the
mesenchymal-to-epithelial reverting transition with restoring
E-cadherin expression in KB and KOSCC-25B oral squamous cell
carcinoma cells. J Exp Clin Cancer Res. 28:282009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG,
Lang JY, Li CW, Hsu JL, Miller SA, Wang X, et al: The crosstalk of
mTOR/S6K1 and hedgehog pathways. Cancer Cell. 21:374–387. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Varnat F, Duquet A, Malerba M, Zbinden M,
Mas C, Gervaz P, Ruiz I and Altaba A: Human colon cancer epithelial
cells harbour active HEDGEHOG-GLI signalling that is essential for
tumour growth, recurrence, metastasis and stem cell survival and
expansion. EMBO Mol Med. 1:338–351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Neelakantan D, Zhou H, Oliphant MUJ, Zhang
X, Simon LM, Henke DM, Shaw CA, Wu MF, Hilsenbeck SG, White LD, et
al: EMT cells increase breast cancer metastasis via paracrine GLI
activation in neighbouring tumour cells. Nat Commun. 8:157732017.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Falcone C, Daga A, Leanza G and Mallamaci
A: Emx2 as a novel tool to suppress glioblastoma. Oncotarget.
7:41005–41016. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang J, Wei Q, Wang X, Tang S, Liu H,
Zhang F, Mohammed MK, Huang J, Guo D, Lu M, et al: Transition to
resistance: An unexpected role of the EMT in cancer
chemoresistance. Genes Dis. 3:3–6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|