Introduction
Renal cell carcinoma (RCC) accounts for 90% of all
cases of kidney cancer and kidney/renal pelvis cancer ranks seventh
for men [5% of all new cancer diagnoses in 2016] and tenth for
women [3% in 2016] (1,2). Epigenetic alterations, in particular
DNA-methylation, are vital for tissue- and cell-specific
differentiation of an organism and are stably passed on to daughter
cells. DNA methylation which is important for the normal
functioning of cells has also been demonstrated in many tumor
entities and often shows an association with the pathogenesis as
well as the progression of malignancies (3,4). In
regards to RCC, tumor-specific hypermethylation and hypomethylation
of numerous loci in a substantial number of genes have also been
described (5,6). The role of DNA methylation in RCC has
been a topic of past and present research aimed to improve our
understanding of the molecular basis of the pathological changes
concerning RCC (7,8). Due to the lack of valid biomarkers, DNA
methylation alterations have been discussed as early
diagnosticators, prognosticators, and predictors of RCC as well as
for molecular differentiation of histological subtypes (9–11).
Moreover, detection of increased promotor methylation for the
estimation of patient prognosis is suggested by a substantial
number of studies identifying candidate loci associated with worse
survival in all RCC subtypes (12).
Although the TCGA data set provides the basis for new developments
in the research of novel markers, significant clinical advances in
prognostic models have not been introduced in the past decade
(13). Therefore, there is still a
need for identification of new candidate markers with prognostic
relevance. In addition, a comparatively low number of studies have
been designed to analyze DNA methylation for prediction of targeted
therapeutic outcome in the management of advanced RCC (14–17).
Independent of the possible clinical application in terms of
prognosis or prediction, the functional relevance of many
alterations in regards to RCC development, progression as well as
response to therapeutic approaches remains to be clarified for a
large part of loci (18–21).
The present study aimed to ascertain whether DNA
methylation alterations of the sarcosine dehydrogenase
(SARDH) gene occur in RCC. The SARDH protein was recently
described to undergo changes in prostate cancer (PCa) showing
potential prognostic relevance. SARDH is a monomeric flavoprotein
of the mitochondrial matrix. The enzyme catalyzes the oxidative
demethylation of N-methylglycine (sarcosine) which is synthesized
by the glycine N-methyltransferase (GNMT) catalyzed by the transfer
of a methyl group from the donator S-adenosylmethionine to the
amino acid glycine. It has been suggested that sarcosine is a
possible marker for distinguishing healthy prostate tissue,
localized PCa, and already metastasized tumors (22). Significantly elevated sarcosine levels
have been found in 79% of the examined metastatic tumors, whereas
only 42% of localized tumors exhibit elevated sarcosine
concentrations. Benign prostatic tissues did not show any
detectable sarcosine levels. Notably, alterations in the SARDH/GNMT
system could be measured non-invasively in the urine by means of
sarcosine measurements potentially offering efficient access to
diagnostic relevant information. Moreover, the functional analysis
suggested an effect of SARDH as well as GNMT protein levels on
mortality and invasiveness of benign prostatic cells roughly
comparable to effects by known oncogenic factors of PCa such as E26
family of transcription factors and the androgen receptor (22).
The SARDH gene is encoded on chromosome
9q34.2. Decreased expression has been detected in hepatocellular
carcinoma and has been discussed as a potential prognostic marker
(23–25). We investigated whether DNA-methylation
alterations of SARDH as a potential surrogate of gene
expression alterations and corresponding gene
activation/inactivation can be found in RCC and cell line models of
other human tumors. Although in human tumor cell lines overall high
SARDH methylation values were detected, we found statistical
robust associations of DNA hypomethylation with adverse clinical
parameter and survival of patients.
Materials and methods
Primary cells and tumor cell
lines
Renal proximal tubular epithelial cells (RPTECs)
were obtained from Lonza (Basel, Switzerland) and renal, urothelial
and prostate cancer cell lines (ACHN, A498, 786-O, RCC-GS,
RCC-HS/RCC-EW, RCC-MF, RT112, CLS-439, EJ28, 5637, T24, and PC-3)
were purchased from Cell Line Services (CLS, Eppelheim, Germany).
Cells were immediately after receipt cultured solely for the
purpose of DNA extraction for a maximum number of 5 passages.
Therefore, further authentication of cell lines after storage of
DNA was not carried out. Note, that the renal cancer cell line
RCC-HS has been meanwhile identified by the supplier to be
identical to the renal cancer cell line RCC-EW.
Tissue samples
Fresh frozen tissue samples of 118 RCC tumor tissues
were subjected to methylation analyses (Table I). The tissue samples were obtained
between January 2001 and December 2005 at Eberhard Karls University
of Tübingen by kidney surgery. The degree of differentiation
(grading) and the histopathological subtype of each tumor sample
were determined by two pathologists. Pathological tissue
assessment, preparation, storage as well as oncological staging and
grading beside to the data management have been described before
(26). For analysis of tumor-specific
hypermethylation a subset of 82 tumors for which tumor-adjacent
normal tissues were available were subjected to statistical
analysis. Follow-up data were available for a subset of 57 tumors
and were used for survival analyses.
 | Table I.Clinicopathological parameters of the
RCC tumor samples using in univariate and bivariate logistic
regressions. |
Table I.
Clinicopathological parameters of the
RCC tumor samples using in univariate and bivariate logistic
regressions.
| Parameters | All RCCs n (%) |
|---|
| Total cases | 118 (100) |
| Histology |
|
|
ccRCC | 82 (69.5) |
|
papRCC | 23 (19.5) |
| Chrom.
RCC | 3 (2.5) |
| Mixed
histology | 6 (5.1) |
|
Other | 4 (3.4) |
| No
RCC | 0 (0) |
| Sex |
|
|
Female | 41 (34.7) |
|
Male | 77 (65.3) |
| Age |
|
|
Median | 64.5 |
| Range
(min-max) | (35–91) |
| Metastasis |
|
| M0 | 92 (78) |
| M+ | 26 (22) |
| Na | 0 (0) |
| Lymph node
metastasis |
|
| N0 | 103 (87.3) |
| N+ | 15 (12.7) |
| Na | 0 (0) |
|
T-classification |
|
|
pT1 | 11 (9.3) |
|
pT1a | 34 (28.8) |
|
pT1b | 22 (18.6) |
|
pT2 | 7 (5.9) |
|
pT3 | 5 (4.2) |
|
pT3a | 10 (8.5) |
|
pT3b | 24 (20.3) |
|
pT3c | 3 (2.5) |
|
pT4 | 1 (0.8) |
| Na | 1 (0.8) |
|
Differentiation |
|
| G1 | 23 (19.5%) |
|
G1-2 | 15 (12.7%) |
| G2 | 61 (51.7%) |
|
G2-3 | 9 (7.6%) |
| G3 | 10 (8.5%) |
| Na | Na |
| State of
diseasea |
|
|
Localized disease | 61 (51.7) |
|
Advanced disease | 56 (47.5) |
| Na | 1 (0.8) |
| State of
diseaseb |
|
|
Localized disease | 64 (54.2) |
|
Advanced disease | 53 (44.9) |
| Na | 1 (0.8) |
| Paired samples |
|
| No. of
patients | 82 (69.5) |
Isolation and conversion of DNA
Isolation of DNA and bisulfite conversion was
performed as described previously (27).
Pyrosequencing
The SARDH gene is located on chromosome 9q34
between positions 136,528,684 and 136,605,077 according to the hg19
genomic assembly in the UCSC genome browser (28). For methylation analysis of
SARDH, eight CpG sites, located between positions
136,568,091 and 136,568,135, were identified to be suitable for
pyrosequencing. They were part of the CpG island designated as CpG
island 37 and located in the gene body of SARDH adjacent to
exon 12 or exon 1 of a putatively alternatively spliced transcript
displayed in the genome browser. Pyrosequencing was carried out for
relative quantitation of the CpG sites of interest applying the
universal reverse primer concept (29). Primer sequences are specified as
following: 5′-ATGGTTTATTTGAGGGATAGGTAGAA-3′ (forward primer),
5′-GGGACACCGCTGATCGTTTAACTAAAAACCACCTCTTTTCTTCCCAAATC-3′ (reverse
universal primer) and 5′-GGTGTATTAGTTTGTTAGTAGTTTG-3′ (sequencing
primer). PCR and pyrosequencing were carried out as previously
described (30).
Statistical methods
Clinicopathologic and experimental data were
collected in a relational database. All statistical analyses were
conducted by means of the statistical software packages R 3.03 and
R Studio 1.0.136 (https://www.R-project.org/). Distributions of
methylation values are presented by box plots using notches as an
estimate of the median confidence interval. Statistical
significance was assumed for P-values <0.05. For tumor-specific
hypermethylation analysis paired normal and tumor tissue samples
were analyzed using the two-sided paired t-test. Logistic
regression was applied for comparison of independent tumor samples
and analysis of possible associations with clinicopathological
parameters. P-values, odds ratios (ORs) and 95% confidence
intervals (Cis) were provided. Association of methylation and
clinicopathologic parameters with recurrence-free survival was
statistically evaluated using univariate Cox regression analysis
presenting P-values, hazard-ratios (HRs) and 95% CIs. For the
presentation of the univariate survival characteristic, a
Kaplan-Meier plot is presented. With respect to the low number of
patients in the survival subset of tumors, bivariate Cox regression
was carried out in pairwise combinations of dichotomized
methylation data and most relevant clinical covariates as a
surrogate for multivariate survival analysis of data.
Results
Analysis of SARDH methylation in
cancer cell lines models and primary normal cells
Twelve tumor cell lines and one primary cell as a
model for normal renal tissue were tested for DNA methylation. We
found increased methylation in large part in the tumor cell lines.
All cell lines representing tumors from the kidney, bladder, and
prostate demonstrated high levels of at least 70% up to about 95%
relative methylation (Fig. 1). Renal
proximal tubular epithelial cells (RPTECs) demonstrated a
substantial methylation level of 50%.
Analysis of paired tissue samples for
the detection of tumor-specific hypomethylation or
hypermethylation
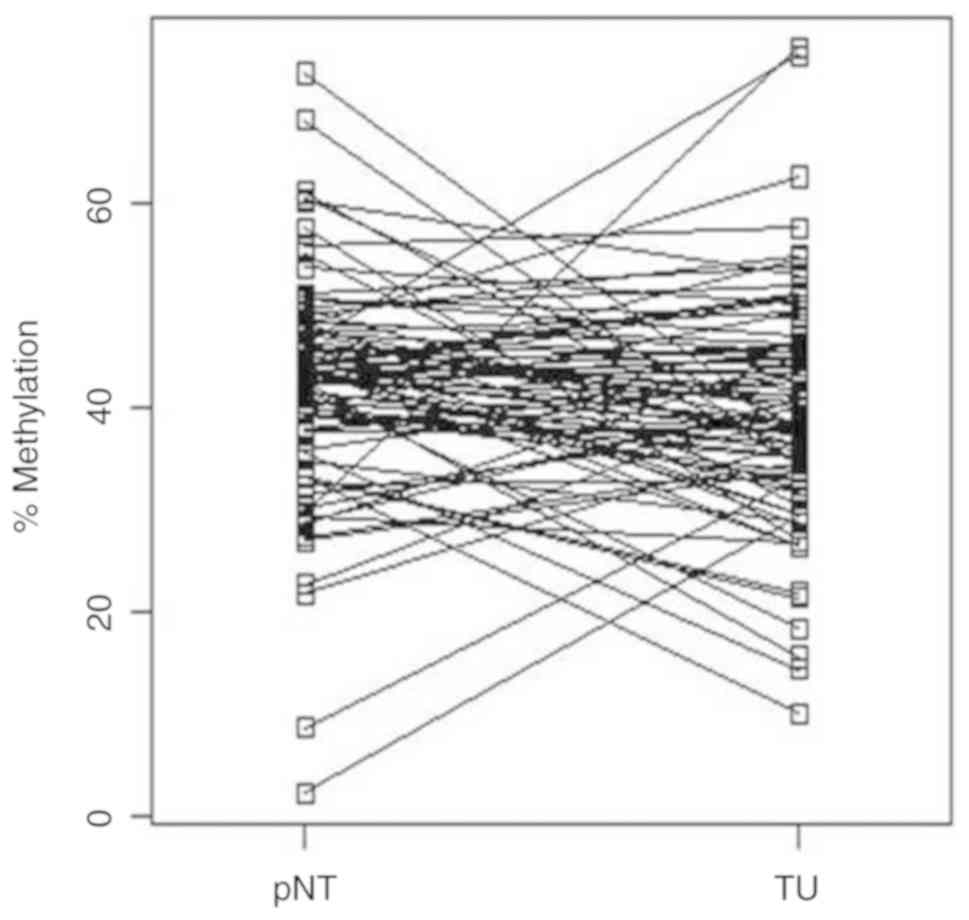
We investigated whether the comparison of
SARDH loci of histopathologic normal tissues with paired
tumoral tissue samples shows relevant alteration in DNA
methylation. We found both, tumor-specific hypermethylation as well
as hypomethylation in different subgroups of the tissue pairs,
showing overall a heterogeneous representation for methylation
alterations in the tumors (Fig. 2).
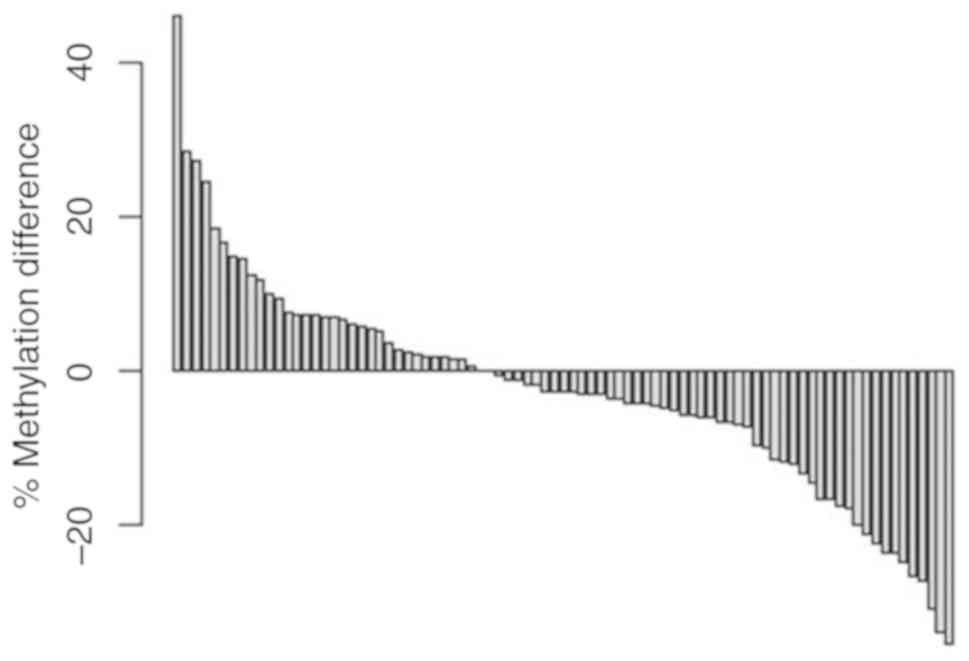
Thus, paired sorted difference analysis of methylation alterations
demonstrated a tumor-specific increase in methylation of more than
10% for ~15% of the tumors, while an estimated one-third of the
tumors showed hypomethylation of more than −20% relative
methylation (Fig. 3).
Correspondingly, paired t-test analysis for comparison of mean
relative methylation values for the tumor (39.2%) and corresponding
normal tissue group (40.9%) did not demonstrate a significant
difference (two-sided paired t-test, P=0.084).
Statistical association of SARDH
methylation and histological and clinicopathological parameters of
the tumors
To identify possible statistical associations of
SARDH methylation with clinicopathological parameters we
carried out bivariate logistic regression analyses following
dichotomization of tumors if necessary. The mean relative
methylation values, P-values, odds ratios (ORs) and 95% confidence
intervals (CIs) are summarized in Table
II.
| Table II.Overview of the comparison of
SARDH methylation levels in regards to the different tumor
characteristics. |
Table II.
Overview of the comparison of
SARDH methylation levels in regards to the different tumor
characteristics.
| SARDH
methylation | Relative
methylation (%) (mean values of the categories) | P-value | ORa | 95% CI |
|---|
| Grade of tumor |
| 0.260 | 0.976 | 0.936–1.017 |
|
G1-2 | 41.0 |
|
|
|
| G3 | 36.9 |
|
|
|
| Stage of tumor |
| 0.002 | 0.944 | 0.908–0.977 |
|
T1-T2 | 43.1 |
|
|
|
|
T3-T4 | 35.5 |
|
|
|
| Lymph node
status |
| 0.020 | 0.946 | 0.902–0.990 |
| N0 | 41.5 |
|
|
|
| N1 | 32.9 |
|
|
|
| Distant
metastases |
| 0.003 | 0.938 | 0.897–0.975 |
| M0 | 42.4 |
|
|
|
| M1 | 33.2 |
|
|
|
| State of
disease |
| <0.001 | 0.919 | 0.879–0.956 |
|
Localized | 44.8 |
|
|
|
|
Advancedb | 34.9 |
|
|
|
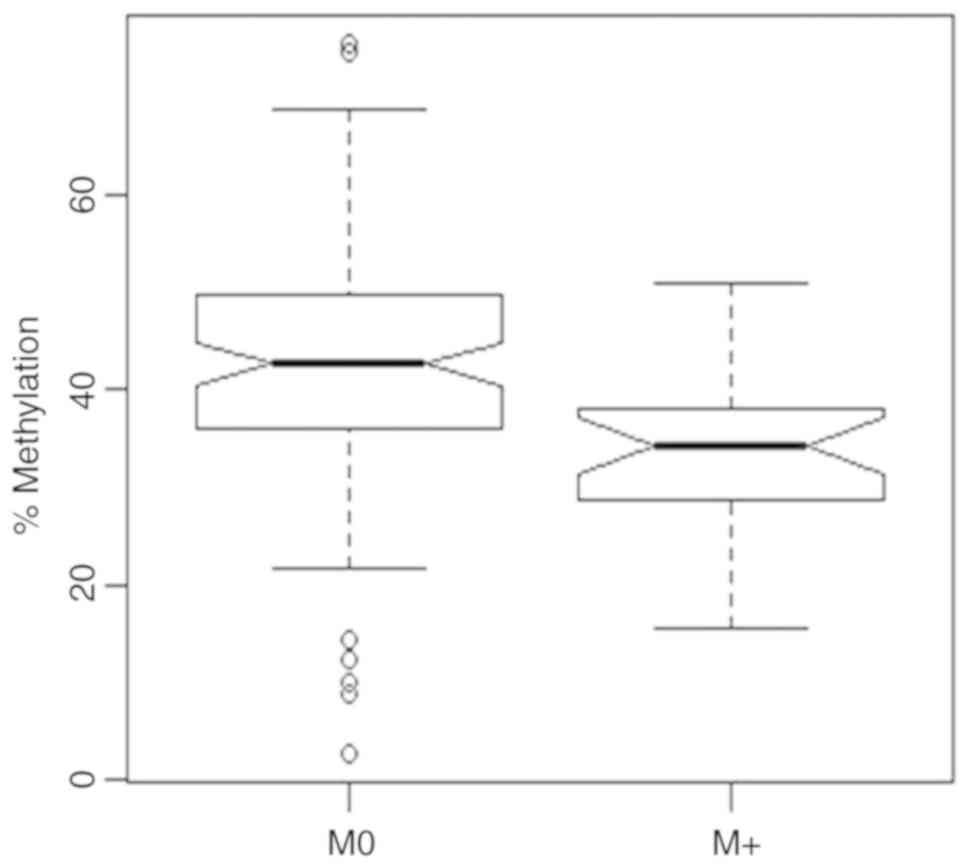
Distant metastasis
Comparison of relative methylation values
corresponding to the tumors showing distant metastasis (M+) with
primary non-metastatic tumors (M0) demonstrated a substantial
hypomethylation for the M+ group (Fig.
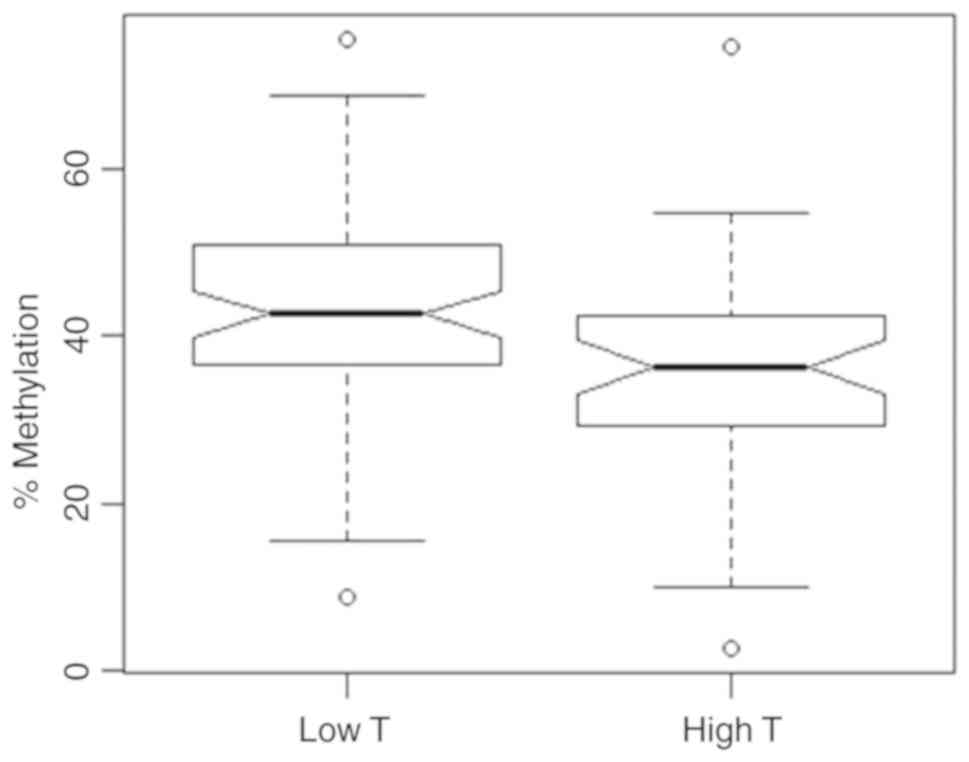
4). Relative mean methylation values of 33.2% (M+) and 42.4%
(M0) were observed showing a significant substantial difference
between both groups (bivariate logistic regression, P=0.003,
OR=0.938, 95% CI 0.897–0.975) while age was not detected as a
significant parameter in the bivariate model.
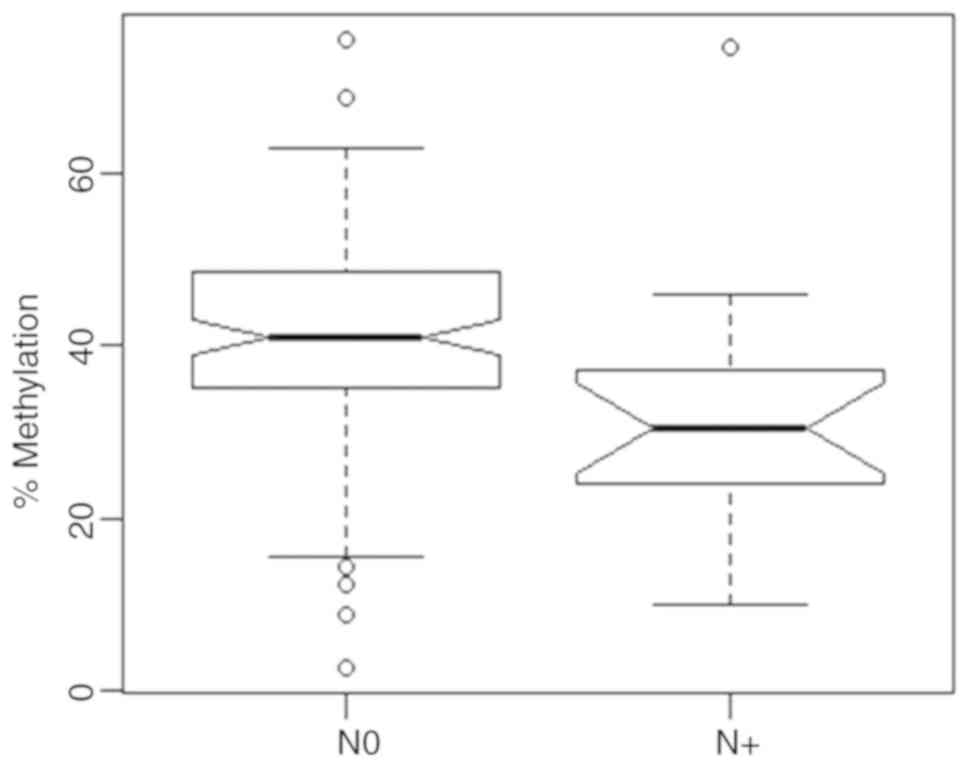
Lymph node status
We analyzed relative methylation of SARDH in
relation to lymph node status (Fig.
5) and found that patients with lymph node metastases (N1) had
a statistically significantly lower methylation with a mean value
of 32.9% in comparison with tumors from patients without lymph node
metastasis (N0), who exhibited a mean value of 41.5% (bivariate
logistic regression, P=0.020, OR=0.946, 95% CI 0.902–0.990). Age
was no significant parameter in the bivariate statistical
model.
Tumor stage
Patients showing high-tumor stages (T3-T4) showed a
significantly reduced SARDH methylation of 35.5% (Fig. 6) when compared with low-stage tumors
(T1-T2) demonstrating mean relative methylation of 43.1% (bivariate
logistic regression, P=0.002, OR=0.944, 95% CI 0.908–0.977). The
covariate age was not detected as a significant parameter.
Comparison of localized and advanced
tumors
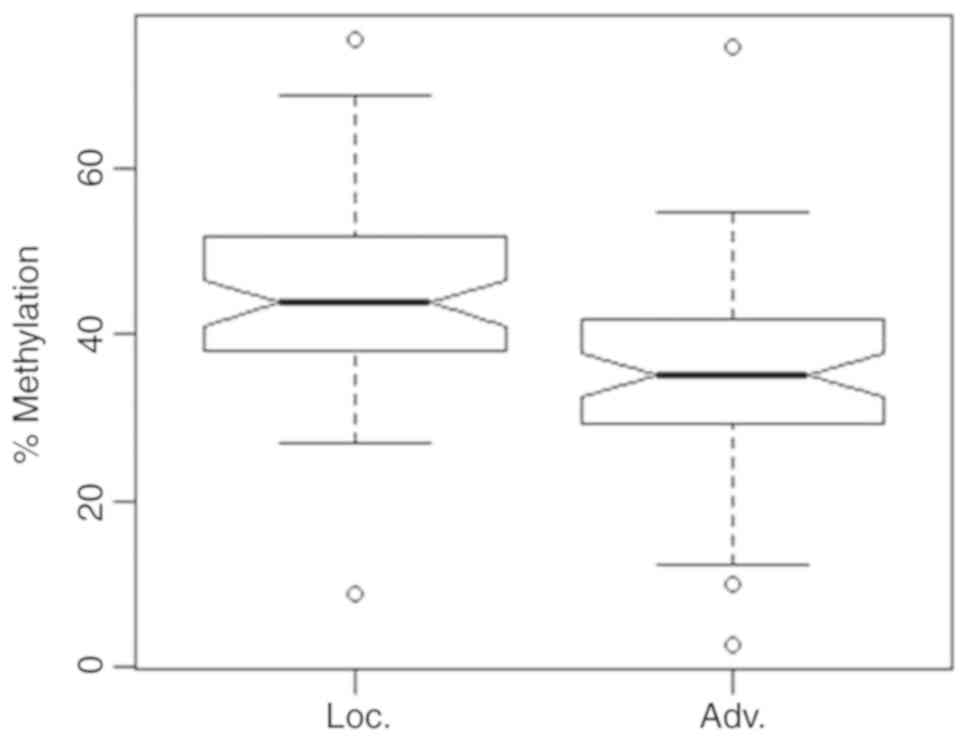
Dichotomization of tumors into a localized tumor
group (pT2, N0, M0, G1-2) and an advanced tumor group (pT ≥3 and/or
N1, M1 or G 2–3) for bivariate statistical comparison revealed
significantly lower methylation values of 34.9% for advanced tumors
in comparison to localized tumors showing a mean value of 44.8%
(Fig. 7, bivariate logistic
regression, P<0.001, OR=0.919, 95% CI 0.879–0.956). No
significance was observed for the covariate age in bivariate
analysis.
Comparison of low- and high-grade
tumor groups
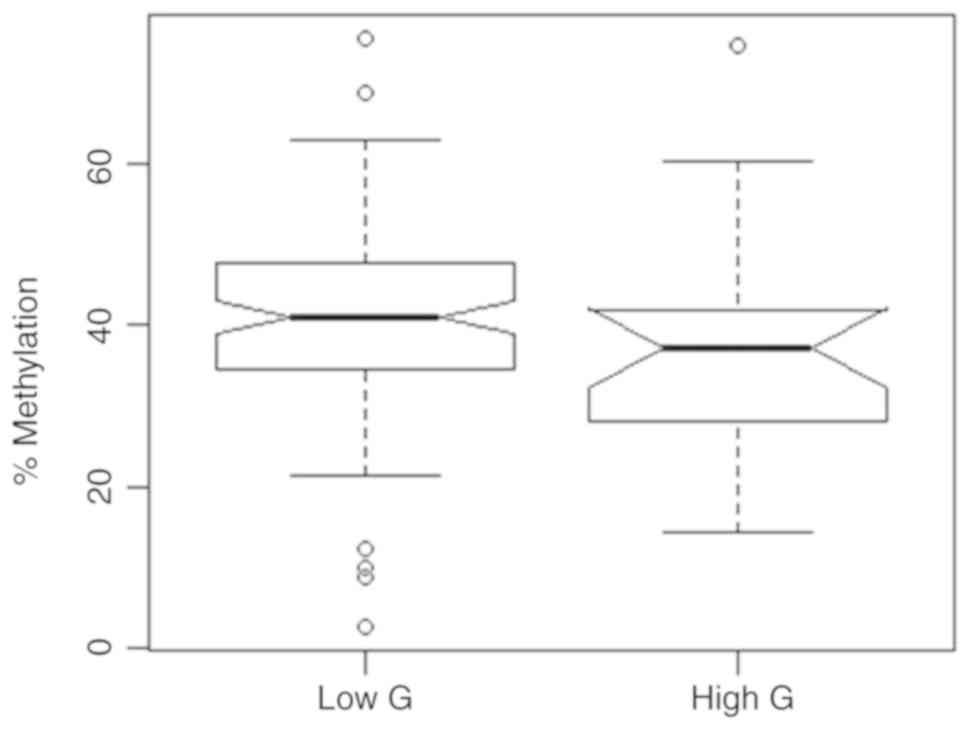
Comparing tumor groups including G1-G2
classifications for the low-grade and G2-3 or higher
classifications for the high-grade tumor group exhibited no
statistically significant difference in mean relative methylation
(Fig. 8). The corresponding values
were 41.0 and 36.9% for the low- and high-grade groups (bivariate
logistic regression, P=0.260, OR=0.976, 95% CI 0.936–1.017). In
addition, age demonstrated no significant effect on methylation in
the bivariate model.
Comparison of normal and tumor
samples
The analysis of the paired tissue samples showed
that on average both the paired normal tissue samples (pNT) and the
corresponding tumor samples (TU) exhibited high methylation
demonstrating no statistically significant difference between pNT
and TU samples (Fig. 2).
Statistical association of SARDH
methylation with recurrence-free survival
We evaluated the known prognostic factors of distant
metastasis (M), lymph node status (N), the degree of tumor
differentiation (G), tumor stage (T) as well as SARDH
methylation for their association with the recurrence-free survival
of patients. Univariate Cox regression analysis demonstrated that
SARDH methylation, distant metastases (M1), positive lymph
nodes (N1), poor tumor differentiation (G3) and high tumor stage
(T3) were significant parameters for poorer outcome as indicated by
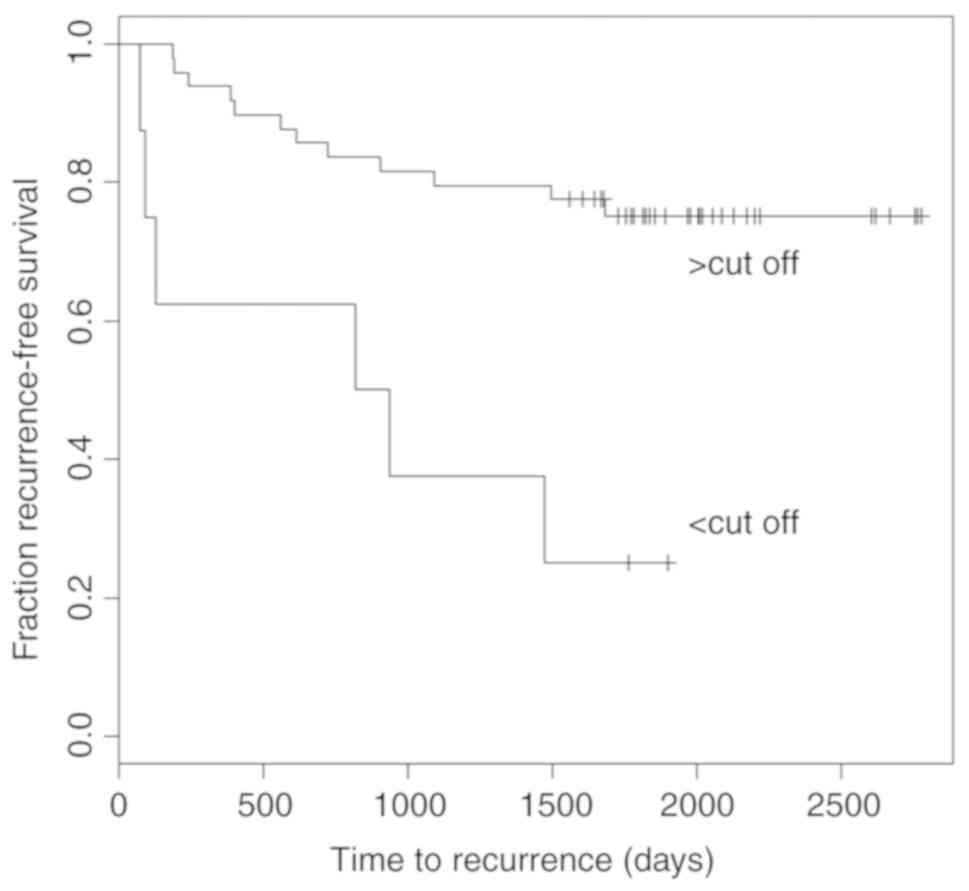
high hazard ratios (HRs) of ~3.2 to 14 (Table III). Kaplan Meier analysis revealed
that patients with SARDH methylation below the statistical
determined optimum threshold value of 31.7% were at higher risk for
early recurrence (P<0.005, Fig. 9)
showing a median time to recurrence of 878 days (28.8 months). In
contrast, patients exhibiting higher SARDH methylation
demonstrated in less than half of cases a recurrence event within
the maximum observation period.
| Table III.Evaluation of patient recurrence-free
survival in the univariate model. |
Table III.
Evaluation of patient recurrence-free
survival in the univariate model.
| Recurrence-free
survivala | P-value | HR | 95% CI |
|---|
| Optimal
thresholdb | 0.000095 | 0.09 |
0.031–0.315 |
| Distant metastasis
(M0 vs. M1) | 0.001241 | 5.55 | 1.962–15.720 |
| Lymph node status
(N0 vs. N1) | 0.203067 | 2.29 | 0.640–8.193 |
| Grade of the tumor
(G1-2 vs. G3) | 0.000123 | 9.32 | 2.982–29.140 |
| Stage of the tumor
(T1 vs. T3) | 0.004072 | 6.43 | 1.806–22.880 |
Whether SARDH methylation provides prognostic
information independent of the analyzed clinical parameters was
investigated using pairwise bivariate Cox regression analyses to
circumvent statistical limitations due to insufficient patient
numbers in subsets needed for multivariate Cox regression
analysis.
Pairwise comparison with states of distant
metastasis, lymph node metastasis, grade, stage, diameter, age and
sex of the patients revealed that methylation remained a
significant parameter in all bivariate regression models showing
remarkably constant low HR values between 0.04 and 0.2 (Table IV).
| Table IV.Results of the pairwise bivariate
survival analyses. |
Table IV.
Results of the pairwise bivariate
survival analyses.
| Recurrence-free
survivala | P-value | HR | 95% CI |
|---|
| Methylation vs.
distant metastasis (M0/M1) |
|
|
|
|
Methylation | 0.0001506 | 0.09 | 0.024–0.305 |
| Distant
metastasis | 0.0015724 | 6.24 | 2.005–19.410 |
| Methylation vs.
lymph node status (N0 vs. N1) |
|
|
|
|
Methylation | 0.0004066 | 0.10 | 0.027–0.356 |
| Lymph
node status | 0.9928786 | 0.99 | 0.238–4.153 |
| Methylation vs.
grading (G1-2/G3) |
|
|
|
|
Methylation | 0.0134751 | 0.20 | 0.056–0.717 |
|
Grading | 0.0059307 | 5.83 | 1.660–20.450 |
| Methylation vs.
tumor stage (T1/T3) |
|
|
|
|
Methylation | 0.0088421 | 0.19 | 0.057–0.663 |
| Stage
of tumor | 0.0319277 | 4.41 | 1.137–17.080 |
| Methylation vs.
dichotomized tumor diameter |
|
|
|
|
Methylation | 0.0003234 | 0.04 | 0.007–0.230 |
| Tumor
diameter | 0.1358545 | 3.03 | 0.706–13.000 |
| Methylation vs.
dichotomized patient age |
|
|
|
|
Methylation | 0.0001626 | 0.05 | 0.010–0.235 |
|
Age | 0.1281683 | 0.34 | 0.083–1.368 |
In silico analysis of SARDH
methylation using TCGA KIRC data
To validate our results obtained by pyrosequencing
analysis we used in silico analysis of the KIRC dataset from
TCGA. We found seven evaluable loci exhibiting in two cases
statistically significant tumor-specific hypermethylation
(cg27114512, cg14163119) and in one case hypomethylation
(cg13824009) using paired t-test and Bonferroni-Hochberg correction
in the genome-wide analysis (Table
V). However, a detailed view using assorted paired difference
plots showed that all of the identified loci include
hypermethylated and hypomethylated subsets in varying
proportions.
| Table V.In silico validation of
SARDH methylation results using TCGA KIRC data. |
Table V.
In silico validation of
SARDH methylation results using TCGA KIRC data.
|
|
|
Clinicopathologya |
|---|
|
|
|
|
|---|
| Genomic
position | T | N | M | G |
|---|
|
|
|
|
|
|---|
| Label | Start position | P-value | OR | P-value | OR | P-value | OR | P-value | OR |
|---|
| cg13824009 | 135,537,683 | <0.001 | 0.007 | 0.795 | 1.399 | 0.0562 | 0.052 | 4.291 | 0.001 |
| cg14524643 | 135,538,907 | 0.095 | 0.077 | 0.338 | 0.236 | 0.928 | 1.190 | 0.059 | 0.053 |
| cg13709982 | 135,557,560 | 0.002 | 0.010 | 0.902 | 1.181 | 0.451 | 0.269 | 0.002 | 0.011 |
| cg27114512 | 135,557,966 | 0.047 | 0.052 | 0.687 | 1.788 | 0.267 | 0.126 | 0.022 | 0.032 |
| cg14163119 | 135,571,032 | 0.016 | 36.467 | 0.534 | 0.405 | 0.008 | 111.286 | 0.063 | 16.419 |
| cg14360014 | 135,593,537 | 0.226 | 0.282 | 0.767 | 0.743 | 0.518 | 2.234 | 0.812 | 1.272 |
| cg21122774 | 135,594,817 | 0.003 | 17.526 | 0.264 | 0.345 | <0.001 | 79.832 | 0.003 | 18.937 |
Statistical analysis for a possible association of
methylation of loci and clinical parameters of patients using
univariate logistic regression analysis revealed that two loci
(cg21122774, cg14163119), annotated to exon 1 and intron 8
respectively, showed high ORs for distant metastasis as well as
high stage and high-grade tumors, indicating that higher
methylation demonstrated an association with unfavorable
clinicopathology (Table V). In
contrast, cg13709982 and cg27114512 (adjacent to exon 13), as well
as cg13824009 (adjacent to exon 17), exhibited low ORs indicating a
possible association of hypomethylated tumors with adverse clinical
parameters (Table V). Univariate
survival analysis for cg21122774 showed that hypermethylated tumors
were associated with a worse survival of patients. Similar to the
analysis of clinicopathological parameters, cg13709982 and
cg13824009 showed a significant association of hypomethylated loci
with worse clinical outcome of patients (Table V).
Discussion
Metabolic as well as expression studies have
postulated sarcosine dehydrogenase (SARDH) as an effector of
tumor progression both in prostate and hepatocellular carcinoma
(22–25). Here we investigated whether DNA
methylation alterations of SARDH occur in renal cell
carcinoma (RCC) and can be utilized as a potential prognosticator
for the clinical course of patients.
Comparing RCC with paired normal tissue samples
demonstrated a heterogeneous pattern of methylation alterations.
While overall high methylation values of approximately 30 to 60%
were observed, subsets of tissue pairs showed hypermethylation as
well as hypomethylation and a large part of tissues demonstrated
low or no alteration in the loci investigated. Thus neither
hypomethylation nor hypermethylation in these loci seemed to be
associated with the development of RCC.
Notably, the TCGA KIRC dataset included seven
evaluable CpG sites from which cg27114512 and cg14163119 showed
clear hypermethylation while cg13824009 demonstrated
hypomethylation. The hypermethylated sites are located to exon 1
and 2 of the long transcript while the hypomethylated CpG site is
annotated to the region of exon 12 in case of the long transcript
as well as to exon 1 of an alternative shorter transcript. Taking
into account that this region is also adjacent to the CpG island
analyzed in part by our pyrosequencing analysis, the in
silico analysis of KIRC data obviously support our results.
We also statistically investigated whether tumor
subgroups stratified for the most important histological and
clinical parameters exhibit methylation alterations of
SARDH. First, we found no significant difference in mean
methylation comparing the clear cell and papillary histological
entities (data not shown). In contrast, methylation in primary
tumors without distant metastasis (M0) and metastasized tumors (M+)
showed a clear SARDH hypomethylation of the M+ tumors which
was independent of the covariate age of the patients, giving rise
to the assumption that hypomethylation of this locus may be
indicative of an adverse clinical outcome of patients. In line,
hypomethylation, in addition, turned out to be significantly
associated with lymphogenic metastasis, high stage of tumors as
well as the state of progressive tumors. In silico
validation analyzing the KIRC data also demonstrated that
hypomethylation at exon 12 was associated with the positive status
of distant metastasis as well as high-stage and high-grade tumors,
thus supporting our findings for the adjacent loci measured in our
study. Interestingly, hypermethylated loci identified in the KIRC
in silico analysis neighboring exon 1 and 2 were also found
to be associated with the positive status of distant
metastasis.
In line, our survival analyses indicated that
SARDH methylation may serve as an independent predictor of
recurrence-free survival. Although our limited patient cohort did
not permit multivariate Cox regression analysis, our surrogate
multiple pairwise bivariate Cox regression clearly revealed
methylation to be independent of all of the clinical covariates
remaining a highly significant parameter in the statistical models
and showing remarkable alteration of patient hazards. In
silico univariate survival analysis of KIRC data also
demonstrated association both hypermethylated and hypomethylated
loci with the recurrence-free survival of patients. Thus, similar
to the characteristics of associations observed for
clinicopathological parameters, loci in the region of exon 1–7
preferably demonstrated association of hypermethylation with worse
clinical prognosis while loci in the region of exon 7–17 adjacent
to the two annotated CpG islands showed an association of
hypomethylation with adverse outcome.
Thus, overall, the comparison of our
pyrosequencing-based results with TCGA KIRC data for seven loci
distributed over the genomic region of SARDH revealed that
statistical associations with clinicopathology as well as the
survival of patients do agree well for the loci showing closest
proximity to the location of our analysis. On the other hand, the
possible relevance of SARDH methylation for clinical outcome
of patients together with the heterogeneity of methylation as
indicated by the TCGA KIRC data point to the necessity of gene-wide
methylation analyses in future studies.
Moreover, our primary analysis of various human
tumor cell lines representing renal, bladder and prostatic cancers
evenly demonstrated high relative methylation values for
SARDH. Considering that we observed high methylation and
corresponding low mRNA expression for all renal tumor cell lines
but only a part of bladder cancer cell lines (data not shown),
detailed methylation, expression, and re-expression analysis will
be required for future functional analyses.
Interestingly, the mechanism of sarcosine-induced
tumor progression to date has neither been elucidated for prostate
cancer (31) nor other human
malignancies have been functionally analyzed in detail. Thus,
making molecular assumptions of the consequences of SARDH
DNA-methylation alterations in RCC appear to be difficult at the
current time point.
On the other hand, our statistical analyses of
clinical data point to a contribution of SARDH to the
progression of RCC and support previous studies underlining the
relevance of the gene for human cancers.
In conclusion, our analyses in human cancer cell
lines of prostate, bladder and renal tumors showed uniformly high
methylation values of 70–95% for cancer cells and 50% for normal
renal epithelial tubular cells, which supports the role of DNA
methylation as an important epigenetic alteration in these human
malignancies. We carried out DNA methylation analyses of CpG
islands (CGIs) of the sarcosine dehydrogenase (SARDH) gene,
which is involved in the metabolism of the amino acid derivative
sarcosine. Interestingly, we were able to discover that the
SARDH-CGIs analyzed by us showed strong methylation in human
tumor and peritumoral tissues of the kidney. On the other hand DNA
hypomethylation in tumors was statistically associated with more
aggressive tumor behaviors. Aggressive carcinomas are thus
characterized by a hypomethylation of the SARDH-CGIs
characterized here. The hypomethylation of the SARDH-CGIs is
a strong influencing factor which increases the risk of an
unfavorable course of disease about four to five times. The
comparison of papillary and clear-cell tumors showed no methylation
differences. However, both patients with high tumor stages (T3) and
patients with positive lymph nodes (N1) had significantly lower
methylation values than patients with low tumor stages (T1) and
without lymph node involvement (N0). Hypomethylation of the
SARDH-CGIs was also associated with the patient
clinicopathological status with regard to remote metastasis (M) and
tumor status. The patients presenting with metastasis (M1) had a
lower SARDH methylation than those without distant
metastases (M0). Patients with a significant hypomethylation of
SARDH had an approximately 2-fold increased risk of distant
metastasis (Table II). Thus, the
methylation status of the SARDH-CGIs analyzed means a
statistically detectable risk-change in its clinicopathological
outcome.
Whether the methylation status of SARDH also
affects the survival of a patient or not was another question
addressed in our study. First, in the univariate analysis of
recurrence-free survival using the Cox proportional hazard model,
patients with low SARDH methylation showed a significantly
shortened recurrence-free survival as opposed to those with high
methylation. The low-methylated patients had a 5-fold increase in
their disease compared to highly methylated patients. In the
survival time analysis, patients who had already metastasized (M1)
when diagnosed were included. These patients have a higher risk of
a re-emergence of their disease, which makes the statement
concerning the independence of the marker to be tested more
difficult. On the other hand, metastatic patients are treated
surgically, thus the effect of the marker on survival is also of
great importance for this collective. For our investigation, this
meant that the independence of our marker had to be checked by
additional multivariate survival analysis. Due to the low cohort
size, this was carried out by means of paired combinations of
bivariate survival time analyzes. On the basis of the largely
constant HRs and P-values (Table
IV), it can be concluded that the methylation status of the
SARDH-CGIs has a knock-on effect on a patient survival.
Remarkably, the pairwise bivariate survival analyses revealed that
SARDH is an independent factor for RCC, which correlates
significantly with the course of the disease, independently of the
classical clinical prognostic factors (distance metastasis, lymph
node status, degree of differentiation, tumor stage, tumor diameter
and age of the patient).
It is to be assumed that SARDH plays an
important role in the clinical behavior of RCC and the
recurrence-free survival of patients. The exact functions and
connections would have to be evaluated in the future using
appropriate functional models. If SARDH can also prove to be
an important factor of influence for RCC in follow-up studies, it
would certainly be possible to routinely test the patients for
their SARDH-gene methylation. Methylations are easy and fast
to detect, which makes the introduction of a clinical test
procedure feasible.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HT and JS played the main role in the conception and
design of this research. JCC, ND, MM, AS and JH carried out the
acquisition of the data as the main participant colleagues. JS
served as the main researcher in the analysis and interpretation of
the data. The main responsibility for drafting and contribution in
writing of the manuscript were conducted by JCC, MM and IP. IP was
also involved in the conception of the study. Critical revision of
the manuscript for important intellectual content was accomplished
by CB, JS and IP. HT, MAK, CB, AS and JH were the most prominent
authors in the acquisition of funding, administrative, technical
and material support, as well as supervision and major
classification and interpretation of the patients. In order to
emphasize the participation of all of the mentioned authors in each
part of this manuscript, we would like to define it as the clear
active participation of all of them in accuracy and integrity of
any part of it.
Ethics approval and consent to
participate
The Ethics Committee ‘Ethik-Komission an der
Medizinischen Fakultät der Eberhard-Karls-Universität am
Universitätsklinikum Tübingen (Head Professor Lucht)’ and
‘Ethikkommission der Medizinischen Hochschule (Head Professor
Tröger)’ approved the study (ethics votes No. 128/2003V and
1213-2011). Written informed consent was obtained from all
participating patients. Informed consent was obtained for
publication of patient data without compromising anonymity or
confidentiality.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pal SK, Ghate SR, Li N, Swallow E, Peeples
M, Zichlin ML, Perez JR, Agarwal N and Vogelzang NJ: Real-world
survival outcomes and prognostic factors among patients receiving
first targeted therapy for advanced renal cell carcinoma: A
SEER-medicare database analysis. Clin Genitourin Cancer.
15:e573–e582. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Issa JP: CpG island methylator phenotype
in cancer. Nat Rev Cancer. 4:988–993. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lasseigne BN and Brooks JD: The role of
DNA methylation in renal cell carcinoma. Mol Diagn Ther.
22:431–442. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mendoza-Pérez J, Gu J, Herrera LA, Tannir
NM, Matin SF, Karam JA, Huang M, Chang DW, Wood CG and Wu X:
Genomic DNA hypomethylation and risk of renal cell carcinoma: A
case-control study. Clin Cancer Res. 22:2074–2082. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network, .
Comprehensive molecular characterization of clear cell renal cell
carcinoma. Nature. 499:43–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herman JG, Latif F, Weng Y, Lerman MI,
Zbar B, Liu S, Samid D, Duan DS, Gnarra JR and Linehan WM:
Silencing of the VHL tumor-suppressor gene by DNA methylation in
renal carcinoma. Proc Natl Acad Sci USA. 91:9700–9704. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ricketts CJ, De Cubas AA, Fan H, Smith CC,
Lang M, Reznik E, Bowlby R, Gibb EA, Akbani R, Beroukhim R, et al:
The Cancer genome atlas comprehensive molecular characterization of
renal cell carcinoma. Cell Rep. 23:313–326 e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Battagli C, Uzzo RG, Dulaimi E, Ibanez de
Caceres I, Krassenstein R, Al-Saleem T, Greenberg RE and Cairns P:
Promoter hypermethylation of tumor suppressor genes in urine from
kidney cancer patients. Cancer Res. 63:8695–8699. 2003.PubMed/NCBI
|
|
10
|
Slater AA, Alokail M, Gentle D, Yao M,
Kovacs G, Maher ER and Latif F: DNA methylation profiling
distinguishes histological subtypes of renal cell carcinoma.
Epigenetics. 8:252–267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malouf GG, Su X, Zhang J, Creighton CJ, Ho
TH, Lu Y, Raynal NJ, Karam JA, Tamboli P, Allanick F, et al: DNA
methylation signature reveals cell ontogeny of renal cell
carcinomas. Clin Cancer Res. 22:6236–6246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morris MR, Ricketts CJ, Gentle D, McRonald
F, Carli N, Khalili H, Brown M, Kishida T, Yao M, Banks RE, et al:
Genome-wide methylation analysis identifies epigenetically
inactivated candidate tumour suppressor genes in renal cell
carcinoma. Oncogene. 30:1390–1401. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klatte T, Rossi SH and Stewart GD:
Prognostic factors and prognostic models for renal cell carcinoma:
A literature review. World J Urol. 36:1943–1952. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choueiri TK, Fay AP, Gagnon R, Lin Y,
Bahamon B, Brown V, Rosenberg JE, Hutson TE, Baker-Neblett KL,
Carpenter C, et al: The role of aberrant VHL/HIF pathway elements
in predicting clinical outcome to pazopanib therapy in patients
with metastatic clear-cell renal cell carcinoma. Clin Cancer Res.
19:5218–5226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stewart GD, O'Mahony FC, Laird A, Eory L,
Lubbock AL, Mackay A, Nanda J, O'Donnell M, Mullen P, McNeill SA,
et al: Sunitinib treatment exacerbates intratumoral heterogeneity
in metastatic renal cancer. Clin Cancer Res. 21:4212–4223. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peters I, Dubrowinskaja N, Abbas M, Seidel
C, Kogosov M, Scherer R, Gebauer K, Merseburger AS, Kuczyk MA,
Grünwald V and Serth J: DNA methylation biomarkers predict
progression-free and overall survival of metastatic renal cell
cancer (mRCC) treated with antiangiogenic therapies. PLoS One.
9:e914402014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dubrowinskaja N, Gebauer K, Peters I,
Hennenlotter J, Abbas M, Scherer R, Tezval H, Merseburger AS,
Stenzl A, Grünwald V, et al: Neurofilament Heavy polypeptide CpG
island methylation associates with prognosis of renal cell
carcinoma and prediction of antivascular endothelial growth factor
therapy response. Cancer Med. 3:300–309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu J, Jin L, Li Y, He T, Liu J, Shi B,
Yang S, Gui Y, Mao X, Lai Y and Ni L: Multilocular cystic renal
cell carcinoma: A case report and review of the literature. Mol
Clin Oncol. 8:326–329. 2018.PubMed/NCBI
|
|
19
|
Kubiak-Wlekly A and Niemir ZI:
Neprilysin-structure of the gene and protein product and the
localization of expression. Pol Merkur Lekarski. 27:48–50. 2009.(In
Polish). PubMed/NCBI
|
|
20
|
van Vlodrop IJ, Niessen HE, Derks S,
Baldewijns MM, van Criekinge W, Herman JG and van Engeland M:
Analysis of promoter CpG island hypermethylation in cancer:
Location, location, location! Clin Cancer Res. 17:4225–4231. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morris MR, Ricketts C, Gentle D,
Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F and
Maher ER: Identification of candidate tumour suppressor genes
frequently methylated in renal cell carcinoma. Oncogene.
29:2104–2117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sreekumar A, Poisson LM, Rajendiran TM,
Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, et al:
Metabolomic profiles delineate potential role for sarcosine in
prostate cancer progression. Nature. 457:910–914. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martínez-Chantar ML, Vázquez-Chantada M,
Ariz U, Martínez N, Varela M, Luka Z, Capdevila A, Rodríguez J,
Aransay AM, Matthiesen R, et al: Loss of the glycine
N-methyltransferase gene leads to steatosis and hepatocellular
carcinoma in mice. Hepatology. 47:1191–1199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen YM, Shiu JY, Tzeng SJ, Shih LS, Chen
YJ, Lui WY and Chen PH: Characterization of
glycine-N-methyltransferase-gene expression in human hepatocellular
carcinoma. Int J Cancer. 75:787–793. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lim SO, Park SJ, Kim W, Park SG, Kim HJ,
Kim YI, Sohn TS, Noh JH and Jung G: Proteome analysis of
hepatocellular carcinoma. Biochem Biophys Res Commun.
291:1031–1037. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Waalkes S, Atschekzei F, Kramer MW,
Hennenlotter J, Vetter G, Becker JU, Stenzl A, Merseburger AS,
Schrader AJ, Kuczyk MA and Serth J: Fibronectin 1 mRNA expression
correlates with advanced disease in renal cancer. BMC Cancer.
10:5032010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gebauer K, Peters I, Dubrowinskaja N,
Hennenlotter J, Abbas M, Scherer R, Tezval H, Merseburger AS,
Stenzl A, Kuczyk MA and Serth J: Hsa-mir-124-3 CpG island
methylation is associated with advanced tumours and disease
recurrence of patients with clear cell renal cell carcinoma. Br J
Cancer. 108:131–138. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Eschenbrenner M and Jorns MS: Cloning and
mapping of the cDNA for human sarcosine dehydrogenase, a
flavoenzyme defective in patients with sarcosinemia. Genomics.
59:300–308. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Colella S, Shen L, Baggerly KA, Issa JP
and Krahe R: Sensitive and quantitative universal Pyrosequencing
methylation analysis of CpG sites. Biotechniques. 35:146–150. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tezval H, Dubrowinskaja N, Peters I, Reese
C, Serth K, Atschekzei F, Hennenlotter J, Stenzl A, Kuczyk MA and
Serth J: Tumor specific epigenetic silencing of corticotropin
releasing hormone -binding protein in renal cell carcinoma:
Association of hypermethylation and metastasis. PLoS One.
11:e01638732016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Heger Z, Merlos Rodrigo MA, Michalek P,
Polanska H, Masarik M, Vit V, Plevova M, Pacik D, Eckschlager T,
Stiborova M and Adam V: Sarcosine up-regulates expression of genes
involved in cell cycle progression of metastatic models of prostate
cancer. PLoS One. 11:e01658302016. View Article : Google Scholar : PubMed/NCBI
|