Introduction
The incidence of thyroid cancer has increased
worldwide due to the increased use of diagnostic imaging and
surveillance (1), and creates a
great burden on the health care system. Papillary thyroid cancer
(PTC) is the most common endocrine malignancy and accounts for 80%
of cases of differentiated thyroid cancer worldwide (2). Consistent with the majority of
malignancies, thyroid carcinomas are usually associated with
oncogenes that lead to aberrant cell proliferation, migration and
invasion (3). However, a more
detailed account of how oncogenes overcome the natural balance of
tumor surveillance during the formation of PTC and contribute to
tumor progression is required, and revealing these oncogenes and
their mechanisms may provide approaches for cancer treatment and
further improvement of clinical care.
Protein phosphatase, Mg2+/Mn2+
dependent, 1D (PPM1D), also referred to as wild-type p53 inducible
protein 1 serine/threonine phosphatase, is a member of the protein
phosphatase 2C family, and is recognized as an oncogene due to its
roles in promoting tumorigenesis (4) and negative regulation of the DNA
damage response system (5–7). A number of studies have demonstrated
that PPM1D is involved in the development of a majority of human
cancer types, including hepatocellular carcinoma, breast cancer,
ovarian clear-cell carcinoma, bladder cancer and glioblastomas
(8–10). Furthermore, PPM1D gene amplification
and/or protein overexpression have been identified to contribute to
tumorigenesis in in vivo (11) and in vitro studies (12–14).
PPM1D protein overexpression was also identified to be
significantly associated with poor clinical outcome in
neuroblastoma and ovarian clear-cell carcinoma (15). Consecutive investigations have
revealed that the oncogenic properties of PPM1D are mediated by
inhibition of several tumor suppressor pathways, including p53, p38
mitogen-activated protein kinase (p38 MAPK), ataxia telangiectasia
mutated and checkpoint kinase 1, therefore contributing to
tumorigenesis, progression, invasion, distant metastasis and
evasion of apoptosis (10,16).
Cellular homeostasis highly relies on fine-tuning
signaling pathways that control the pace of cell proliferation and
apoptosis, thereby preventing oncogenic cellular transformation
through aberrant stress (17,18).
The tumor suppressor p53 has a vital role in these pathways by
transcriptionally upregulating target proteins, including WAF1, Bax
and MDM2, which act to initiate cell cycle arrest or cell death
under stresses. PPM1D was first identified as a target gene of p53
(19), but subsequent studies
revealed that p53 may also be inactivated by PPM1D-induced
dephosphorylation while cells switch from stress status to normal
homeostasis (10,20). Previous studies indicated that the
enhanced p53 pathway in PPM1D-knockout mice significantly impaired
tumorigenesis in several tumor models (20,21),
which draws attention to PPM1D as a potential anticancer
target.
Furthermore, PPM1D also indirectly inactivates p53
through p38 MAPK (16). p38 MAPK is
a component of the MAPK pathway, which is another protective
signaling pathway in response to cellular stress (22). It was reported that PPM1D directly
binds and inactivates p38 MAPK via dephosphorylation at Thr180
(23). In line with the
aforementioned, p38 inactivation paralleled with p53 deactivation
in vivo was also identified in a number of studies (24–26).
However, the current knowledge on PPM1D is mostly based on studies
on breast cancer or the subtypes of breast cancer, and whether
PPM1D has any oncogenic properties via deactivation of p38 and p53
signaling pathways in PTC has so far remained elusive.
In the present study, PPM1D expression was examined
in human PTC tissues as well as in paired adjacent non-cancerous
tissues and a significant association between PPM1D overexpression
and metastasis was revealed. The potential oncogenic properties of
PPM1D were also confirmed in thyroid cell lines. A further
mechanistic study indicated that the oncogenic activities of PPM1D
in thyroid cancer cells are mediated by negative regulation of the
p38 MAPK and p53 signaling pathways. These results contribute to
the understanding of the effect of PPM1D overexpression in
promoting PTC tumor progression, indicating that it may serve as a
potential target for clinical treatment.
Materials and methods
Tissue specimens
A total of 89 thyroid cancer samples were obtained
from patients who underwent surgery for thyroid cancer between
August 2012 and February 2015 at Shanghai Cancer Center of Fudan
University (Shanghai, China). Tissue specimens were frozen in
liquid nitrogen immediately after surgical resection and stored at
−80°C. All tissues were pathologically confirmed as PTC and final
histological classification was obtained from paraffin-embedded
sections. The study was performed in accordance with the
Declaration of Helsinki and approved by the Institutional Research
Ethics Committee of Shanghai Cancer Center, Fudan University
(Shanghai, China). Written informed consent was obtained from all
participants after reviewing the content and purpose of the
study.
Cell culture and treatments
The human PTC original cell lines TPC-1 and K-1 were
obtained from Dr Schweppe from the University of Colorado Cancer
Center. STR profiling was performed to confirm cell authentication.
All cells were grown in RPMI-1640 media (Sigma-Aldrich; Merck KGaA)
supplemented with 10% heat-inactivated fetal bovine serum (FBS;
Invitrogen; Thermo Fisher Scientific, Inc.), 100 IU/ml penicillin
and 10 µg/ml streptomycin. Cell culture was performed at 37°C in a
90% humidified atmosphere with 5% CO2. A MAPK inhibitor
(SB203580; Sigma-Aldrich; Merck KGaA) was dissolved in dimethyl
sulfoxide (DMSO), then added into cell culture medium at a
concentration of 100 nM for 24 h in order to inhibit p38 MAPK
activity.
Small interfering (si)RNA and
transfection
The specific siRNA targeting PPM1D and the scrambled
siRNA used as a negative control were designed and purchased from
GenePharma Co., Ltd. The sequences of the siRNA targeting PPM1D
(siPPM1D) and scrambled siRNA were as follows: siPPM1D-1:
5′-CCGCACTCGTGCTTGCTTGAA-3′; siPPM1D-2: 5′-GTCACGTAACATGTCACAT-3′;
negative control (NC): 5′-CCACCUCUGAUCGAUUUAUdTdT-3′. Cells were
collected for further analysis of protein depletion after 48 h of
transfection. All siRNAs were transfected into thyroid cancer cells
using Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) following the manufacturer's protocol.
Western blot analysis
Cell lysates were obtained from 1×106
cultured cells with a mixture of ProteoJET Mammalian
Cell Lysis Reagent (Fermentas; Thermo Fisher Scientific, Inc.),
phenylmethanesulfonyl fluoride and PhosSTOP (both from Roche
Applied Science). Protein estimation was performed according to the
Bradford method (Bio-Rad Laboratories, Inc.) with bovine serum
albumin (BSA; Sigma-Aldrich; Merck KGaA) as a standard. Total
protein (10 µg per lane) was resolved on 10–15% gradient pre-cast
gels (Sigma-Aldrich; Merck KGaA) and transferred to a
polyvinylidene difluoride membrane (EMD Millipore). After blocking
in 5% non-fat milk for 2 h at room temperature, the membrane was
probed with antibodies against human PPM1D (1:1,000 dilution;
product code ab31270; Abcam), p38 (1:1,000 dilution; product no.
9212), phosphorylated p-p38 (1:1,000 dilution; product no. 4511),
p53 (1:1,000 dilution; product no. 2527), Bax (1:1,000 dilution;
product no. 5023), Histone H3 (1:1,000 dilution; product no. 4499;
all from Cell Signaling Technology, Inc.) or GAPDH (1:5,000
dilution; product code ab9485; Abcam) overnight at 4°C.
Subsequently, the membrane was incubated with HRP-conjugated goat
anti-rabbit or HRP-conjugated goat anti-mouse IgG (1:10,000
dilution for both; product code ab205718 and product code ab19195,
respectively; Abcam) for 2 h at room temperature. The indicated
antibodies were detected with the SuperSignal West Pico ECL
Chemiluminescent kit (Thermo Fisher Scientific, Inc.) and protein
bands were quantified using ImageJ software (version 1.47v;
National Institutes of Health, Bethesda).
Isolation of nuclei
Approximately 1×107 cells were collected
and resuspended in hypotonic buffer (10 mM Tris, pH 7.9, 10 mM KCl,
1.5 mM MgCl2 and 0.05 mM DTT). Cell suspensions were
homogenized with a pre-chilled Dounce homogenizer with 20 strokes
and centrifuged at 100 × g for 15 min to retain the supernatant.
10X S100 buffer (0.3 M Tris, pH 7.9, 1.4 M KCl and 0.03 M
MgCl2) was added to the supernatant, followed by
centrifugation at 100,000 × g for 1 h and the supernatant was
obtained for analysis of cytoplasmic proteins. The nuclear pellet
was continuously lysed using high-salt extraction buffer (20 mM
Tris, pH 7.9, 25% glycerol, 1.2 M KCl, 1.5 mM MgCl2 and
0.2 mM EDTA, pH 8.0) with two gentle stokes in the Dounce
homogenizer. After centrifugation at 10,000 × g for 30 min, the
supernatants containing nuclear extract were collected for
analysis. All procedures were performed at 4°C.
Cell proliferation assay
Cell growth was determined using a Cell Counting
Kit-8 (CCK-8) assay (Sigma-Aldrich; Merck KGaA) according to the
manufacturer's protocol. In brief, cells were seeded in black
wall/clear-bottom 96-well plates at 6×103 cells in 100
µl/well and allowed to attach overnight. At the indicated
time-points, an aliquot of 10 µl CCK-8 solution was added to each
well and the plate was incubated for 4 h at 37°C. The absorbance
reading was performed at 450 nm using a spectrophotometer (Thermo
Scientific™ NanoDrop™ 8000; Thermo Fisher Scientific, Inc.). Five
replicates were used for each experimental condition. Cell-free
medium was used as the blank group and cells treated with the
solvent were used as a vehicle control. The viability of the cells
was calculated as follows: Cell viability (%) = (each
condition-blank group)/(vehicle control-blank group) ×100%.
Plate colony formation assay
Cells were trypsinized using 0.05% trypsin/EDTA
(Thermo Fisher Scientific, Inc.) and 2×103 cells were
seeded into 6-well plates and incubated at 37°C for 10 days.
Colonized cells were washed with PBS followed by fixation with 10%
methanol at room temperate (RT) for 5 min and staining with 5%
Giemsa (Sigma-Aldrich; Merck KGaA) at RT for 10 min. The number of
colonies >10 cells was counted under Leica DM4000B microscope
(Leica Microsystems) using magnification of ×1.25, images were
captured and scoring was performed.
Wound healing assay
Wound healing assays were performed to evaluate the
migration of the transfected TPC-1 and K-1 cells. Cells
(2×103) were seeded into 6-well plates. When cultured
cells reached 90% confluence in the 6-well plate, one scratch was
generated with a 200-µl pipette tip in each well to create a wound,
followed by two washes with PBS. Migration of cells into the
scraped area was recorded at 0 and 48 h after the scratches were
made under a Leica DM4000B microscope (Leica Microsystems) using
magnification of ×10.
Transwell invasion assays
Cell invasion was assessed using a modified
Transwell chamber system (BD Biosciences) according to the
manufacturer's protocols. In brief, cells were seeded onto
Matrigel®-coated membrane inserts with a pore size of 8
µm. Medium containing 10% FBS, which served as the chemoattractant,
was placed in the lower chamber. After 48 h of incubation, the
cells were fixed with 75% methanol for 10 min at RT and stained
with 0.1% crystal violet (Sigma-Aldrich; Merck KGaA) at RT for 10
min. The invaded cells on the lower surface of the filter that had
penetrated through the Matrigel®-coated membrane were
counted under an inverted microscope using magnification of
×10.
Apoptosis assay
Apoptotic cells were quantified by flow cytometry
using an Annexin V-FITC/propidium iodide (PI) double-staining assay
kit (BioVision, Inc.) according to the manufacturer's protocol. In
brief, the cells were transfected with indicated siRNAs for 24 h,
and were then harvested and washed 2 times with ice-cold PBS,
re-suspended in 100 µl binding buffer and stained in the dark with
50 µl Annexin V-FITC and 50 µl propidium iodide at room temperature
for 15 min. At least 10,000 events were recorded for each sample
and the percentages of cells (viable, apoptotic and necrotic) were
quantified by flow cytometry (Cytomics™ FC500 cytometer; Beckman
Coulter, Inc.).
Immunofluorescence
Cells were washed twice with ice-cold PBS and fixed
in 4% paraformaldehyde in PBS for 20 min at 4°C. After washing 3
times with PBS, cells were permeabilized with 0.1% Triton X-100 for
5 min at 4°C and incubated with 1% BSA in PBS for 30 min, followed
by incubation with primary antibody to p53 (1:1,000 dilution;
product no. 2527, Cell Signaling Technology, Inc.) at 4°C
overnight. The cells were then washed in PBS three times, followed
by incubation with the secondary antibodies, FITC-conjugated goat
anti-rabbit antibody (1:500 dilution; cat. no. 65-6111 Invitrogen;
Thermo Fisher Scientific, Inc.) for 1 h at room temperature in the
dark. Cells were washed with PBS and nuclei were stained with 0.5
µg/ml DAPI in PBS containing Tween-20 for 5 min. Samples were
mounted with immunofluorescence mounting medium (Dako Cytomation;
Agilent Technologies, Inc.) and images were captured under a
fluorescence microscope using magnification of ×20.
Immunohistochemical staining
Formalin-fixed and paraffin-embedded tissue sections
were deparaffinized in xylene and hydrated through descending
concentrations of ethanol prior to being placed in 3% hydrogen
peroxide for 10 min at room temperature to inhibit endogenous
peroxidase activity. The slides were incubated with blocking
solution (10% BSA in 1X phosphate-buffered saline) for 1 h at room
temperature, followed by incubation with primary antibody to PPM1D
(5 µg/ml; cat. no. PA5-72839, Invitrogen; Thermo Fisher Scientific,
Inc.) at 4°C overnight. A horseradish peroxidase-conjugated mouse
secondary antibody (1:2,000 dilution; cat no. 65-6120, Invitrogen;
Thermo Fisher Scientific, Inc.) was added, followed by incubation
for 60 min at room temperature, followed by development with
3,3′-diaminobenzidine (DAB Substrate Chromogen System; Dako;
Agilent Technologies, Inc.). Slides were fixed and images were
captured by using the Olympus IX71 inverted microscope using the
DP2-BSW Olympus image acquisition software system (Olympus, Corp.).
The staining results were determined on the basis of the percentage
of positive staining of tumor cell nuclei as follows: 0% (no
staining); 1 (≤10%); 2 (10–50%) and 3 (>50%). The staining
intensity was scored as follows: - (negative); + (moderately
positive); and ++ (strongly positive), as previously reported
(27). Two experienced pathologists
who were blinded to the clinicopathological data of the patients
confirmed the results.
Statistical analysis
Values are expressed as the mean ± standard error of
the mean (SEM). Pearson's χ2 test was performed to
compare differences in clinicopathological parameters across groups
stratified by PPM1D protein expression. Univariate and multivariate
logistic regression analyses (Cox proportional hazards model) were
applied to assess the risk of lymph node metastasis and tumor size
of ≥1 cm, and the odds ratio (OR) and 95% CI were reported. A dot
plot displaying the distributions of the intensity of nuclear p53
across experimental groups was generated. Means between two groups
were compared using Student's t-test. For a comparison of more than
two group means, one-way analysis of variance (ANOVA) was applied
followed by Tukey's post hoc test. Statistical analysis was
performed using GraphPad Prism 5.1 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Correlation of clinicopathological
characteristics and expression of PPM1D in PTC tissue
specimens
To understand the clinicopathologic significance of
PPM1D expression in PTC, tissues from a total of 89 patients with
PTC who had undergone tumor resection were analyzed by
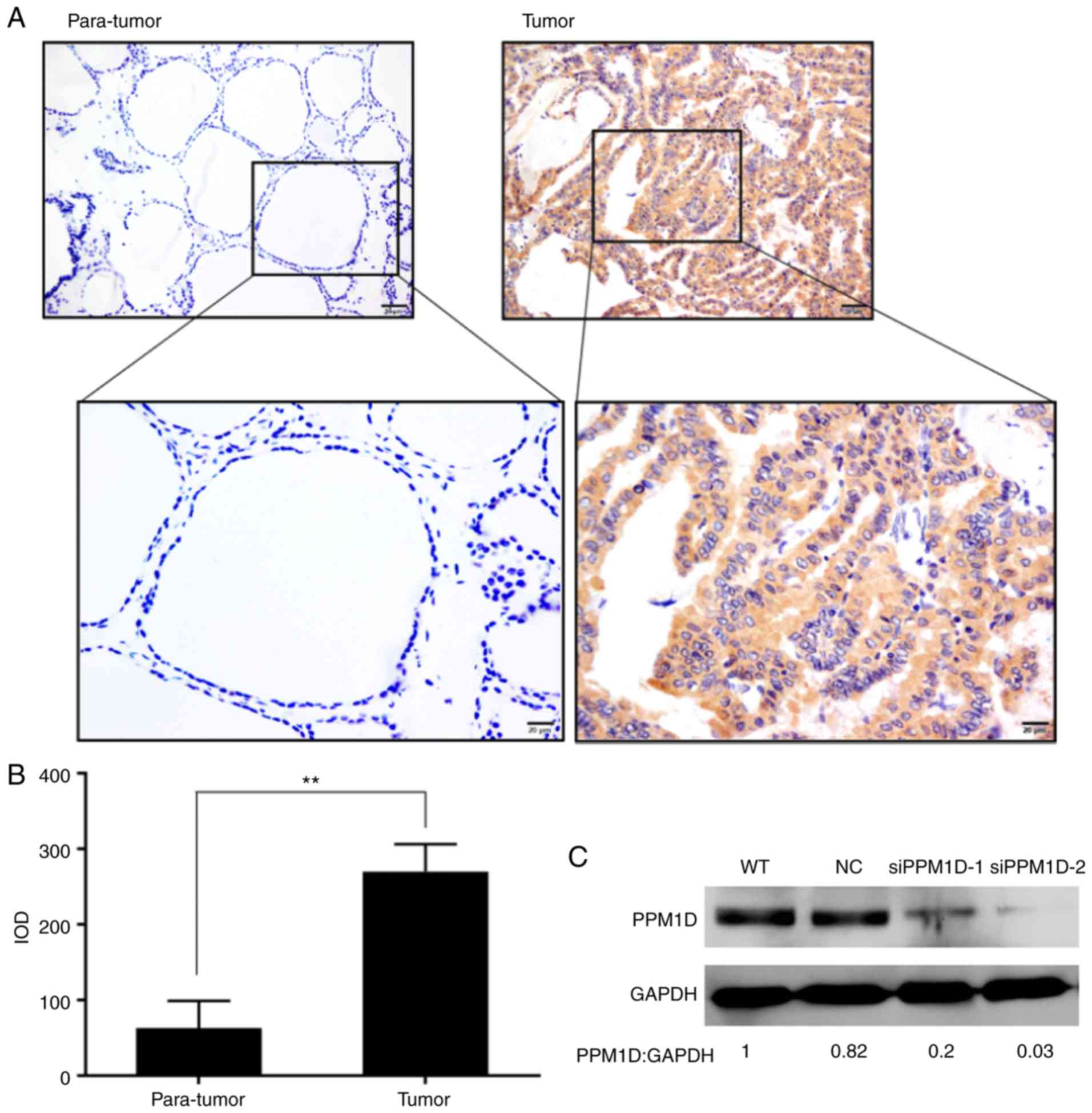
immunohistochemical staining. It was revealed that PPM1D expression
was located within the cytoplasm of the PTC cells, whereas only a
small number of scattered cells with positive staining were
observed in adjacent non-tumor tissues (Fig. 1A). In addition, the intensity of
PPM1D staining in cancer cells was significantly higher than that
in non-tumor cells (Fig. 1B). The
patients with PTC were classified into three groups including
negative (−), moderately positive (+), and strongly positive (++)
PPM1D expression; the associations between PPM1D expression and
clinicopathological characteristics were assessed (Table I). Pearson's χ2-test
indicated that PPM1D expression was significantly associated with
tumor size (P=0.016) and lymph node metastasis (P=0.039), whereas
no significant influence of PPM1D expression on other
clinicopathological features, including age, sex and TNM stage, was
observed (Table I). Next, in the
univariate analysis, clinical features including tumor size >1
cm, TNM stage III–IV and PPM1D protein expression (+) were
identified as risk factors of lymph node metastasis in PTC after
adjustment for age, sex and multifocal lesions (Table IIA). A subsequent multivariate
analysis revealed that a high level of PPM1D protein expression
(++) was a significant risk factor for large tumor size (Table IIB). Based on the aberrantly high
expression of PPM1D in PTC cells and its positive correlation with
tumor size and lymph node metastasis, further exploration of the
role of PPM1D in tumor formation, migration and invasion was
performed.
 | Table I.Clinicopathological parameters by
PPM1D protein expression in patients with PTC. |
Table I.
Clinicopathological parameters by
PPM1D protein expression in patients with PTC.
|
| PPM1D protein
expression |
|
|---|
|
|
|
|
|---|
| Clinicopathological
parameters | − | + | ++ | P-value |
|---|
| Age (years) |
|
|
| 0.256 |
|
<45 | 20 | 21 | 5 |
|
|
≥45 | 13 | 27 | 3 |
|
| Sex |
|
|
| 0.921 |
|
Male | 7 | 12 | 2 |
|
|
Female | 26 | 36 | 6 |
|
| Lymph node
metastasis |
|
|
| 0.039a |
|
Yes | 14 | 33 | 6 |
|
| No | 19 | 15 | 2 |
|
| Tumor size
(cm) |
|
|
| 0.016a |
| ≤1 | 14 | 30 | 1 |
|
|
>1 | 19 | 18 | 7 |
|
| Multifocal
lesions |
|
|
| 0.738 |
|
Yes | 5 | 10 | 1 |
|
| No | 28 | 38 | 7 |
|
| Bilateral |
|
|
| 0.635 |
|
Yes | 12 | 22 | 4 |
|
| No | 21 | 26 | 4 |
|
| Extrathyroidal
extension |
|
|
| 0.131 |
|
Yes | 1 | 6 | 2 |
|
| No | 32 | 42 | 6 |
|
| TNM stage |
|
|
| 0.310 |
|
I/II | 18 | 25 | 2 |
|
|
III/IV | 15 | 23 | 6 |
|
| Table II.Clinicopathological and molecular
factors in PTC. |
Table II.
Clinicopathological and molecular
factors in PTC.
| A,
Clinicopathological and molecular factors associated with lymph
node metastasis in PTC |
|---|
|
|---|
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variables | OR | 95% CI | P-value | AOR | 95% CI | P-value |
|---|
| Sex, female vs.
male | 1.469 | 0.548–3.937 | 0.445 |
|
|
|
| Age, <45 vs. ≥45
years | 2.214 | 0.933–5.257 | 0.072 |
|
|
|
| Multifocal lesions,
yes vs. no | 1.163 | 0.382–3.544 | 0.791 |
|
|
|
| Tumor size, >1
vs. ≤1 cm | 6.394 | 2.457–16.393 |
<0.001a | 6.394 | 2.208–18.182 | 0.001a |
| TNM stage, III–IV
vs. I–II | 3.048 | 1.258–7.384 | 0.014a | 2.659 | 0.950–7.444 | 0.063 |
| PPM1D protein
expression |
|
|
|
|
|
|
| − | Reference | Reference |
|
|
|
|
| + | 2.986 | 1.188–7.503 | 0.020a | 2.512 | 0.852–7.406 | 0.095 |
| ++ | 4.071 | 0.713–23.623 | 0.114 | 5.446 | 0.809–36.661 | 0.081 |
|
| B,
Clinicopathological and molecular factors associated with tumor
size >1 cm in PTC |
|
|
| Univariate
analysis | Multivariate
analysis |
|
|
|
|
|
Variables | OR | 95% CI | P-value | AOR | 95% CI | P-value |
|
| Sex, female vs.
male | 1.100 | 0.413–2.929 | 0.849 |
|
|
|
| Age, <45 vs. ≥45
years | 2.167 | 0.929–5.052 | 0.073 |
|
|
|
| Multifocal lesions,
yes vs. no | 1.808 | 0.596–5.495 | 0.296 |
|
|
|
| Extrathyroidal
extension, yes vs. no | 2.519 | 0.607–10.417 | 0.203 |
|
|
|
| Bilateral lesions,
yes vs. no | 11.765 | 4.202–32.258 |
<0.001a | 16.393 | 3.040–16.393 | 0.001a |
| PPM1D protein
expression |
|
|
|
|
|
|
| − | Reference | Reference |
|
|
|
|
| + | 0.442 | 0.179–1.092 | 0.077 | 0.455 | 0.137–1.512 | 0.199 |
| ++ | 5.158 | 0.568–46.834 | 0.145 | 17.218 | 1.393–212.831 | 0.027a |
PPM1D regulates thyroid cancer cell
growth
To investigate whether PPM1D is required for thyroid
cancer cell growth, two siRNAs targeting PPM1D, siPPM1D-1 and
siPPM1D-2, were used to knockdown PPM1D in TPC-1 and K-1 cells and
one scrambled siRNA was used for the NC groups. Western blot
analysis confirmed the efficient knockdown of PPM1D using the
siPPM1Ds (Fig. 1C). The cell
viability was then compared between the paired experimental groups.
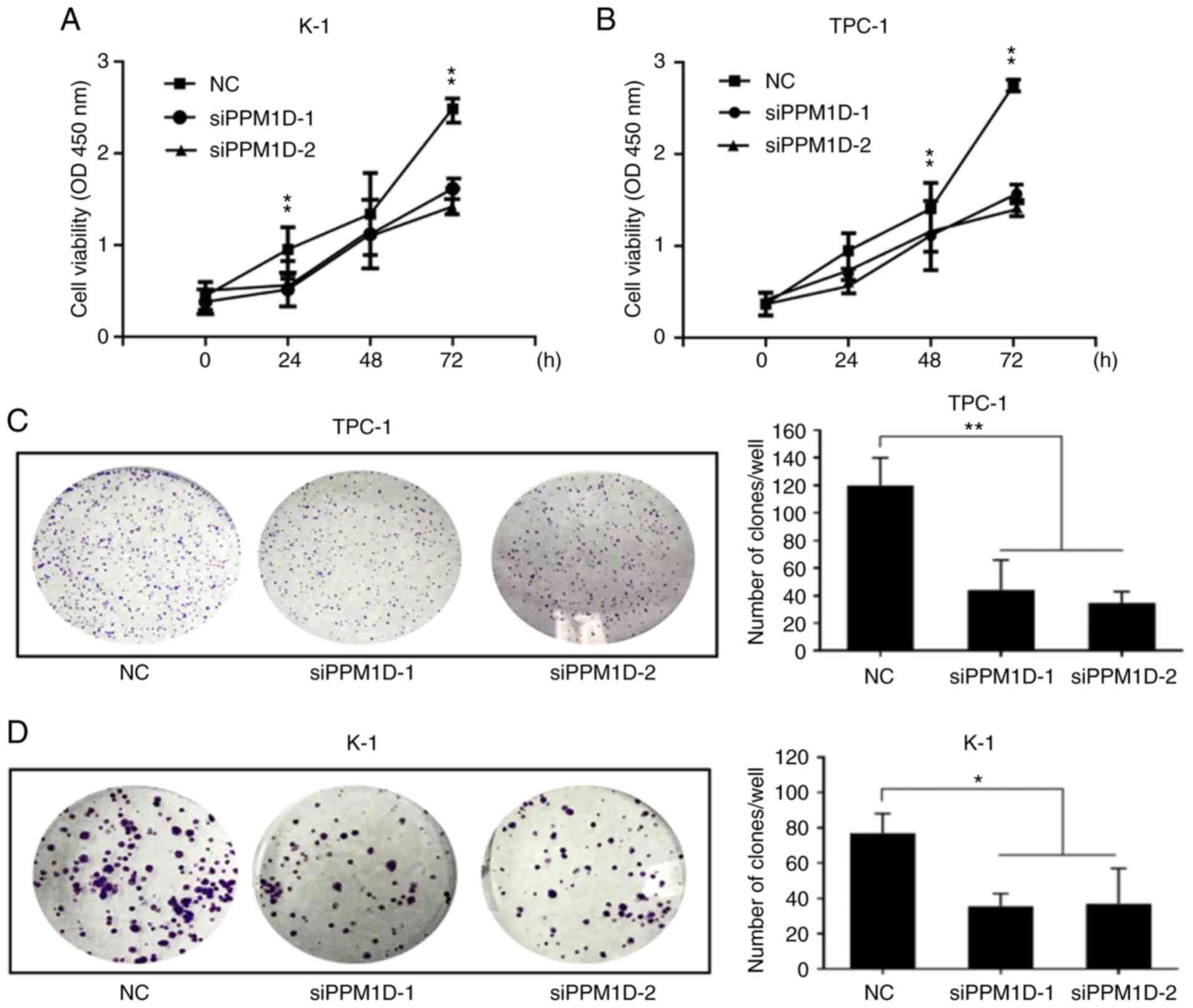
A CCK-8 assay revealed that PPM1D depletion led to a significant
decrease in cell viability compared with that in the NC groups at
72 h for each of the two cell lines (Fig. 2A and B). Colony formation assays
also indicated that PPM1D depletion reduced the colony number by
66.9 and 72.9% in TPC-1 cells and 56.6 and 52.8% in K-1 cells when
using siPPM1D-1 and siPPM1D-2, respectively (Fig. 2C and D). In addition, statistical
analysis revealed a significant decrease in the colony formation
ability of siPPM1D-treated cells compared with that of NC cells
(Fig. 2C and D). These results
indicated that PPM1D promoted cell proliferation in TPC-1 and K-1
cell lines.
Silencing of PPM1D inhibits the
invasive ability of thyroid cancer cells
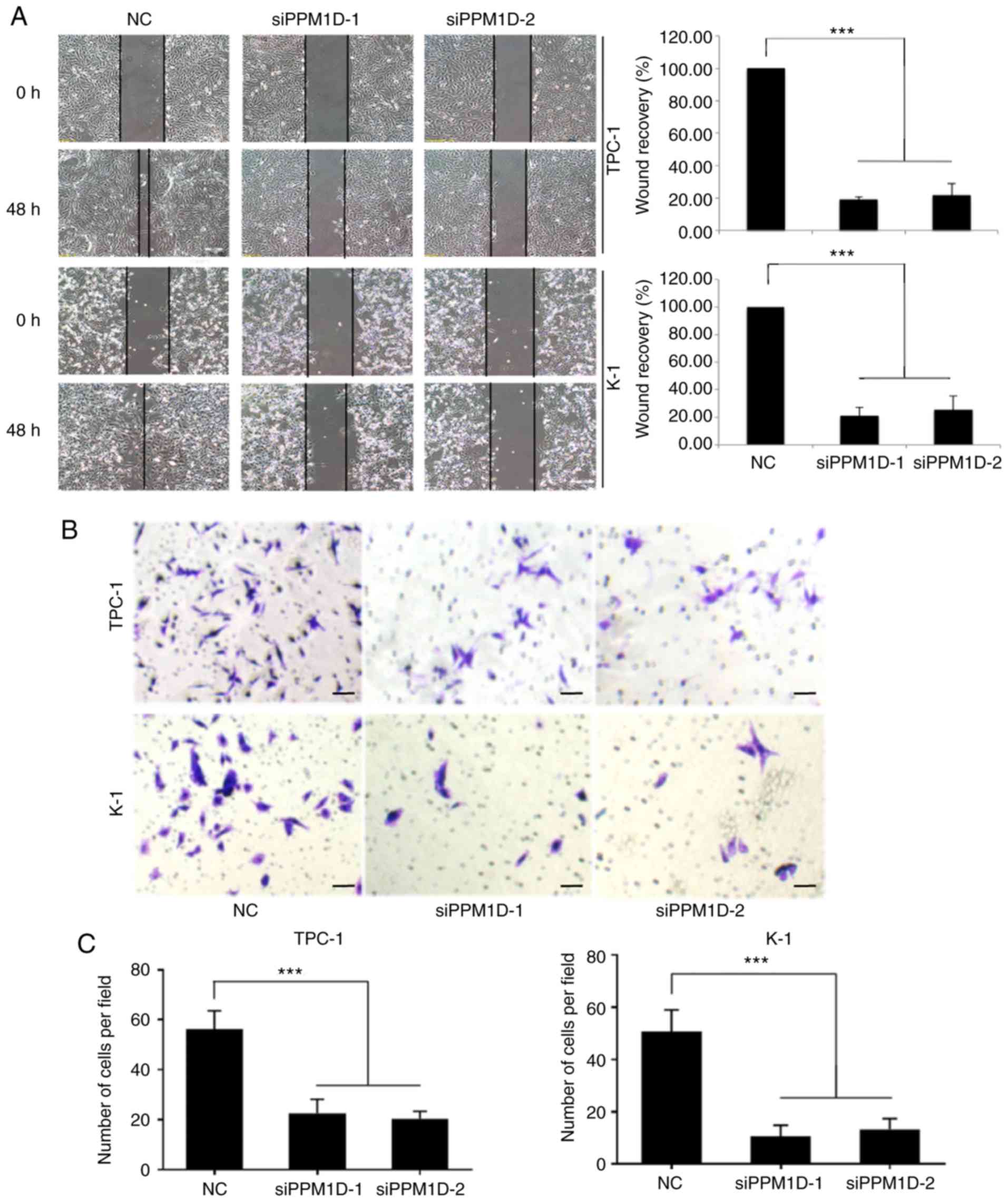
To investigate whether PPM1D is required for
migration and invasion of TPC-1 and K-1 cells, a wound-healing
assay and a Transwell invasion assay were performed. In the
wound-healing assay, cells were cultured in serum-free medium and
then allowed to migrate into a physical scratch. As presented in
Fig. 3A, in the NC group, the
cleared zone was almost filled with the cells at 48 h after the
scratch wound was made. However, the siPPM1D-treated cells
displayed a reduced ability to migrate and fill the cleared zone
compared with that in the NC group. The statistical analysis
(images on the right) indicated that the wound recovery rate was
significantly reduced in siPPM1D-transfected cells compared with
that in the NC group. The role of PPM1D in cell invasion was then
evaluated using a Matrigel invasion assay, a modification of the
Transwell assay. In brief, cells were placed onto Transwell with
Matrigel pre-coated polycarbonate filters. The results indicated
that cells in the control group and siRNA-treated cells were able
to transgress through the Matrigel-coated membrane to the lower
side of the filter, exhibiting invasive behavior. However, compared
to the control group, a reduced number of cells transfected with
siPPM1D passed though the Transwell insert (Fig. 3B). The quantitative data in Fig. 3C indicated a significant inhibition
of cell invasion in PPM1D-silenced cells. Collectively, these
results indicated that PPM1D is important for the migration and
invasion of thyroid cancer cells in vitro.
PPM1D negatively regulates the p38 and
p53 signaling pathways
Next, it was investigated how PPM1D affects tumor
cell behavior. Since PPM1D was first indicated to interact with p53
(19) and also reported to be
involved in p38 MAPK activation, changes in the levels of key
proteins in the p38 and p53 signaling pathways in thyroid cancer
cells transfected with siPPM1D or scrambled siRNA were analyzed.
The western blot results indicated that the protein levels of p-p38
MAPK, p53 and Bax were increased after treatment with siPPM1D
(Fig. 4A and B, upper panel).
Compared with the negative control group, knockdown of PPM1D
increased the protein levels of p-p38 MAPK by >3-fold in TPC-1
cells and >1.8-fold in K-1 cells with concomitant upregulation
of p53 by >1.7-fold in TPC-1 cells and >14-folds in K-1 cells
and Bax by >3-fold in TPC-1 cells and >2-fold in K-1 cells
(Fig. 4A and B, lower panel). These
data indicated that PPM1D may exert its functions, at least
partially, by inhibiting the p38 and p53 pathways.
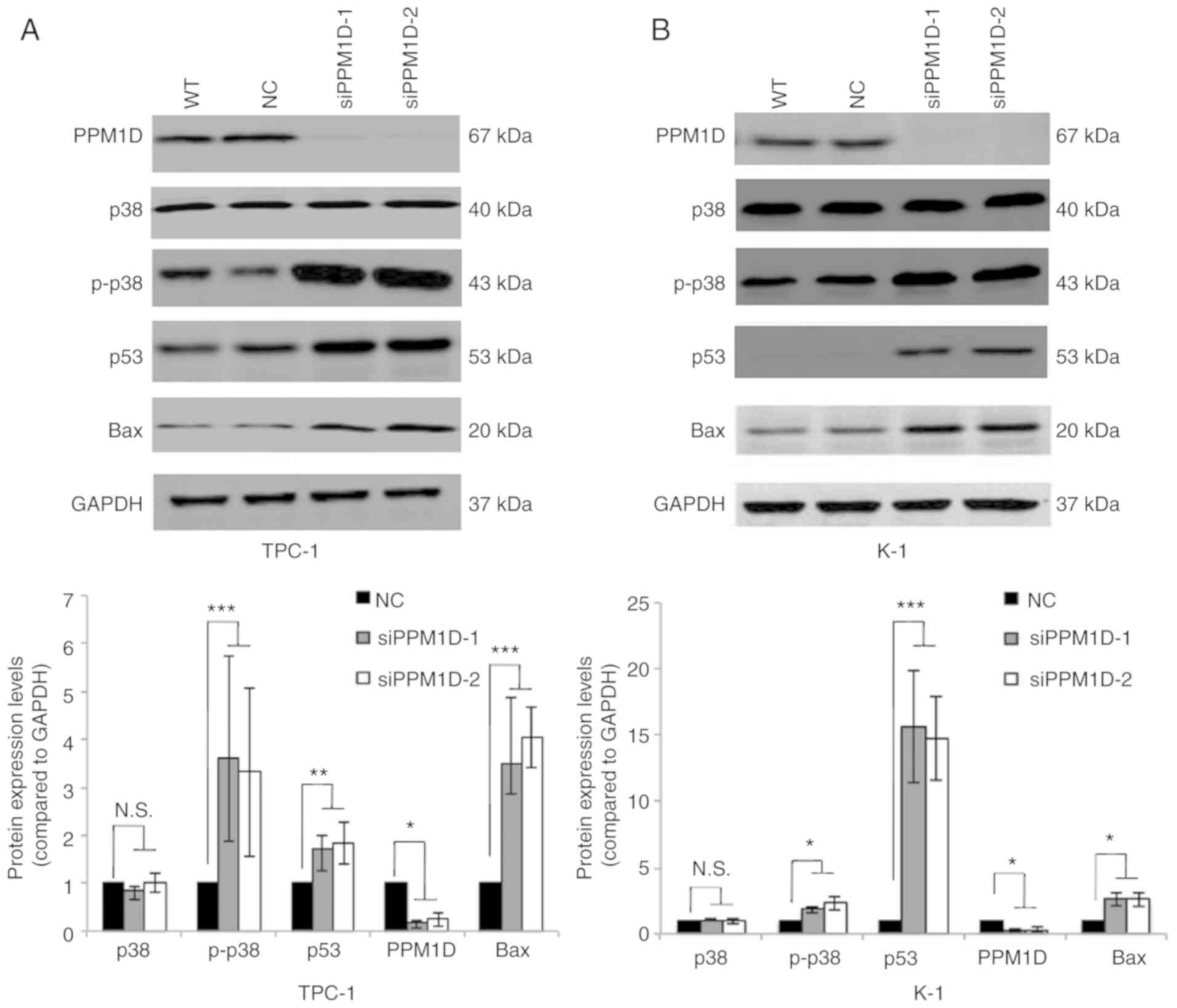 | Figure 4.PPM1D suppresses p38 MAPK and p53
signaling pathways. p-p38 MAPK, p53 and Bax protein levels were
upregulated in siPPM1D-treated thyroid cancer cells. (A and B)
Western blot analysis of protein levels in TPC-1 and K-1 cells
transfected with siPPM1Ds or scrambled RNA for 24 h (upper panel)
and relative quantitative analysis of the protein levels in each
group (lower panel). Values are expressed as the mean ± SEM of
three independent experiments. P-values were determined using ANOVA
test followed by Tukey's post hoc test. *P<0.05, **P<0.01,
***P<0.001. N.S., no significance; PPM1D, protein phosphatase,
Mg2+/Mn2+ dependent, 1D; p-p38,
phosphorylated p38; MAPK mitogen-activated protein kinase; WT,
wild-type; NC, negative control; siPPM1D-1/siPPM1D-2: siRNAs
against PPM1D; siRNA, small interfering RNA. |
Inhibition of p38 activity reverses
the inhibitory effect of PPM1D depletion on cell
proliferation/invasion
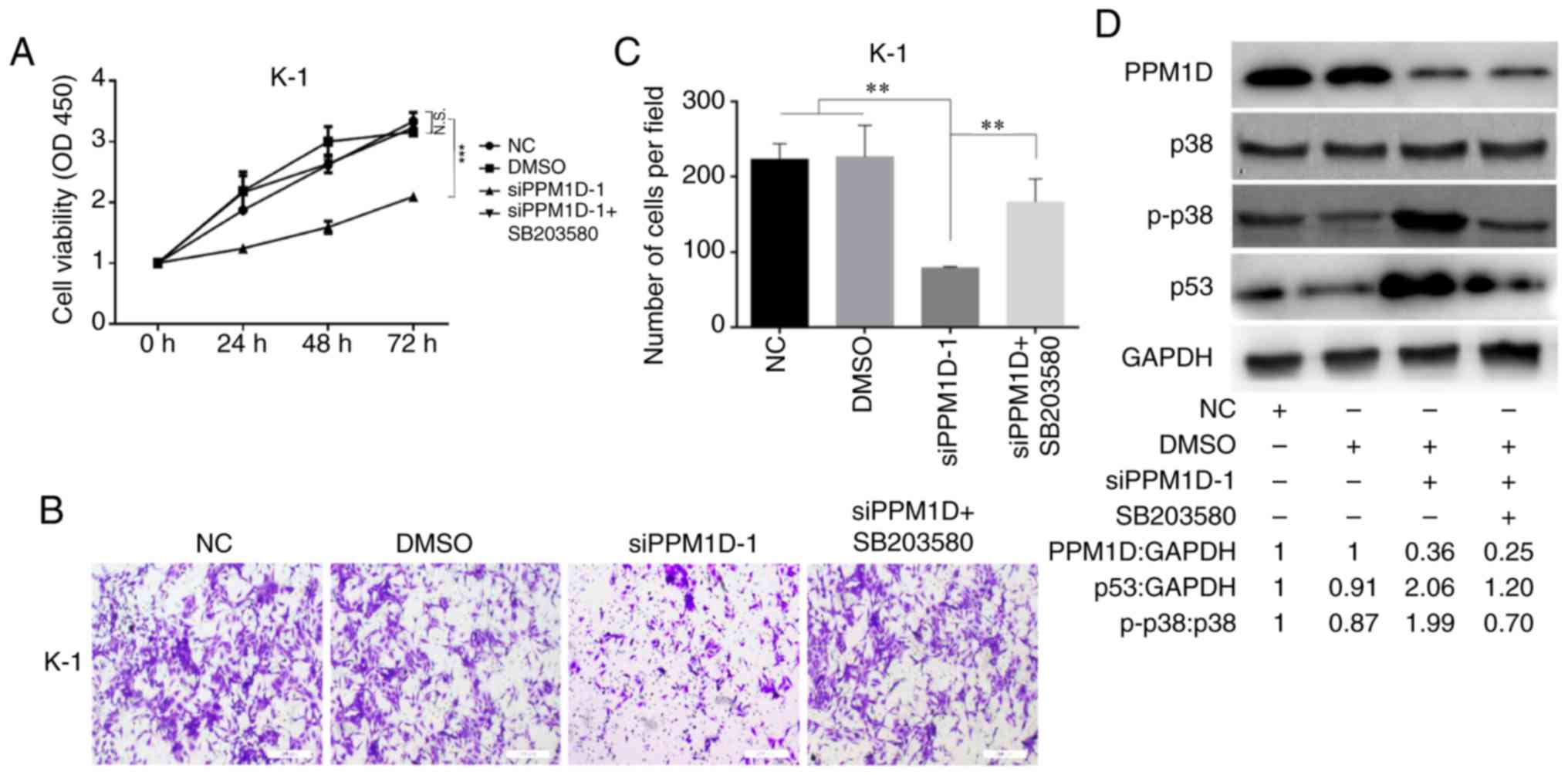
Since overexpression of PPM1D was revealed to
inactivate p38 (10), which may
suppress the activity of p53, it was next examined whether PPM1D
exerts its oncogenic role through regulation of p38 MAPK activity.
After transfection with siPPM1D, K-1 cells were treated with or
without SB203580, a chemical inhibitor of p38, and cell
proliferation and invasion were compared among different groups at
the indicated time-points. The results indicated decreased cell
growth in response to PPM1D knockdown compared with that in the
vehicle treatment DMSO or control group. However, the growth of
siPPM1D-transfected cells was enhanced to a level similar to that
of the control group when co-treated with the inhibitor across all
time-points (Fig. 5A). Furthermore,
the invasive capability was reduced in PPM1D-knockdown cells;
however, concurrent inhibition of p38 restored the invasiveness of
PPM1D-silenced K-1 cells (Fig. 5B).
The number of invaded cells was significantly increased in the
PPM1D-silencing group with p38 inhibition as compared with that in
the group with PPM1D silencing only (Fig. 5C). Furthermore, the inhibitory
effect of pharmacological inhibition of p38 on PPM1D
silencing-induced expression of p53 was determined by western blot
analysis. The results indicated that p-p38 MAPK and p53 were
increased in PPM1D-depleted cells but remained suppressed in the
presence of p38 inhibitor (Fig.
5D). Considering the crosstalk between the p53 and p38
signaling pathways during cell oncogenic transformation, these
results indicated that the oncogenic properties of PPM1D were
exerted via the p38 signaling pathway and that the p53 pathway has
a direct and/or indirect contribution.
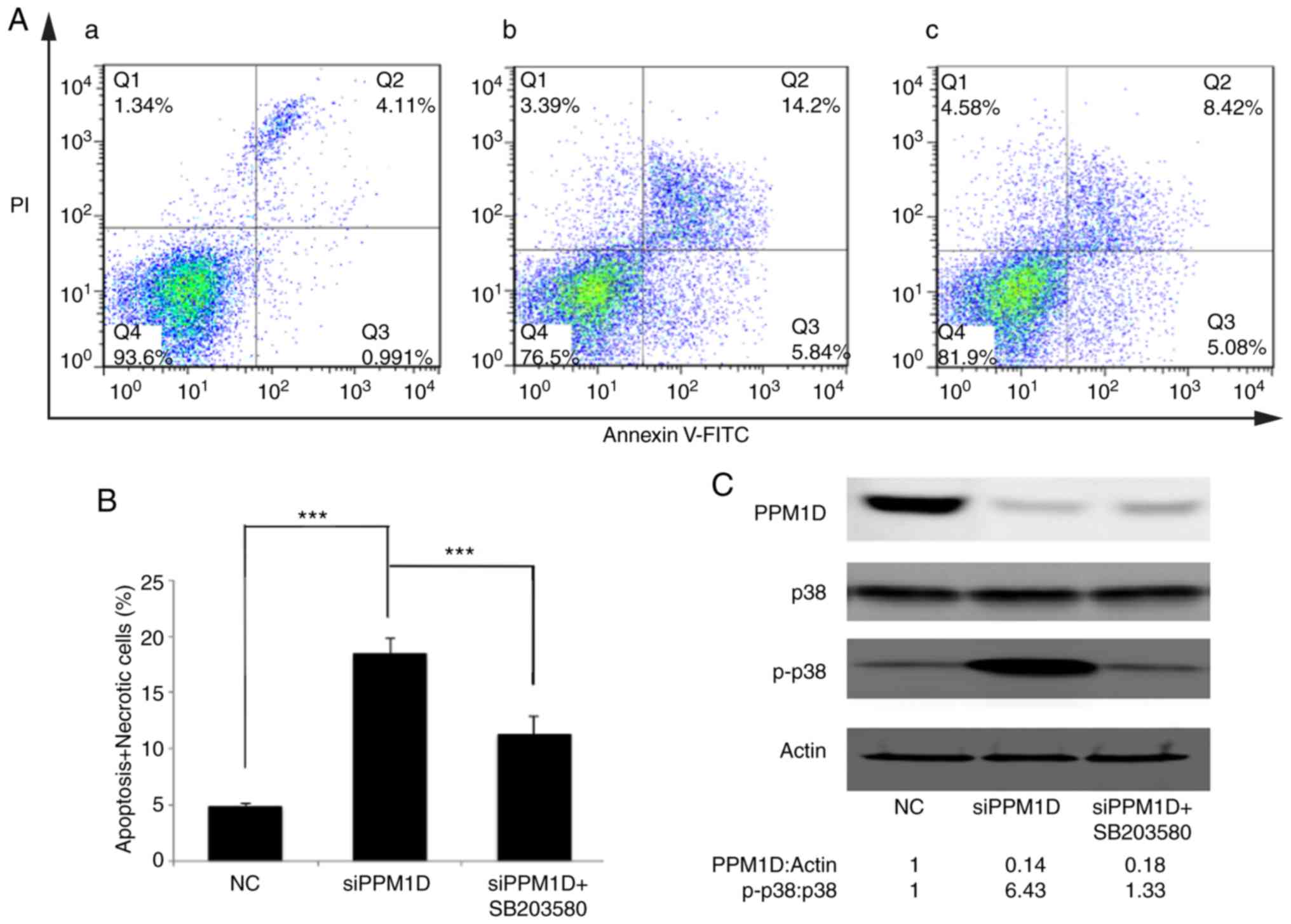
Inhibition of p38 activity counteracts
PPM1D depletion-induced thyroid cancer cell apoptosis
The present study attempted to assess the effect of
PPM1D in deregulating cell apoptosis and whether this was dependent
on the p38 signaling pathway. PPM1D expression was silenced in K-1
cells. Concurrently, p38 activity was inhibited by SB203580. Cell
apoptosis was detected by flow cytometry. As presented in Fig. 6A, viable cells were Annexin V-FITC-
and PI-negative (Fig. 6A-Q4), dead
cells were Annexin V-FITC-negative and PI-positive (Fig. 6A-Q1), cells in early apoptosis were
Annexin V-FITC-positive and PI-negative (Fig. 6A-Q3), while cells in late apoptosis
or already dead were Annexin V-FITC- and PI double-positive
(Fig. 6A-Q2). The basal level of
cell apoptosis was ~5%, as presented in Fig. 6A-a. Cell apoptosis was further
evaluated in PPM1D-depleted K-1 cells with or without
pharmacological inhibition of p38 MAPK activity and it was revealed
that in K-1 cells, transfection with siPPM1D alone increased the
number of apoptotic cells to 20.04% (Fig. 6A-b), but concurrent inhibition of
p38 MAPK activity in PPM1D-depleted cells reduced cell apoptosis to
13.5% (Fig. 6A-c). These results
indicated that PPM1D suppressed cell apoptosis by inhibiting the
activity of p38 MAPK. All Annexin V-FITC-positive cells were
combined to calculate the proportion of apoptotic cells. The
statistical analysis based on the flow cytometric data indicated
that cell apoptosis was significantly enhanced in
siPPM1D-transfected K-1 cells compared with the NC group. By
contrast, cell apoptosis was decreased when p38 activity was
blocked in PPM1D-knockdown cells compared with the group with
siPPM1D transfection alone (Fig.
6B). PPM1D knockdown and the reduced protein levels of p-p38
were confirmed by western blot analysis (Fig. 6C). Collectively, these results
indicated that PPM1D was involved in the downregulation of p38
MAPK-mediated cell apoptosis.
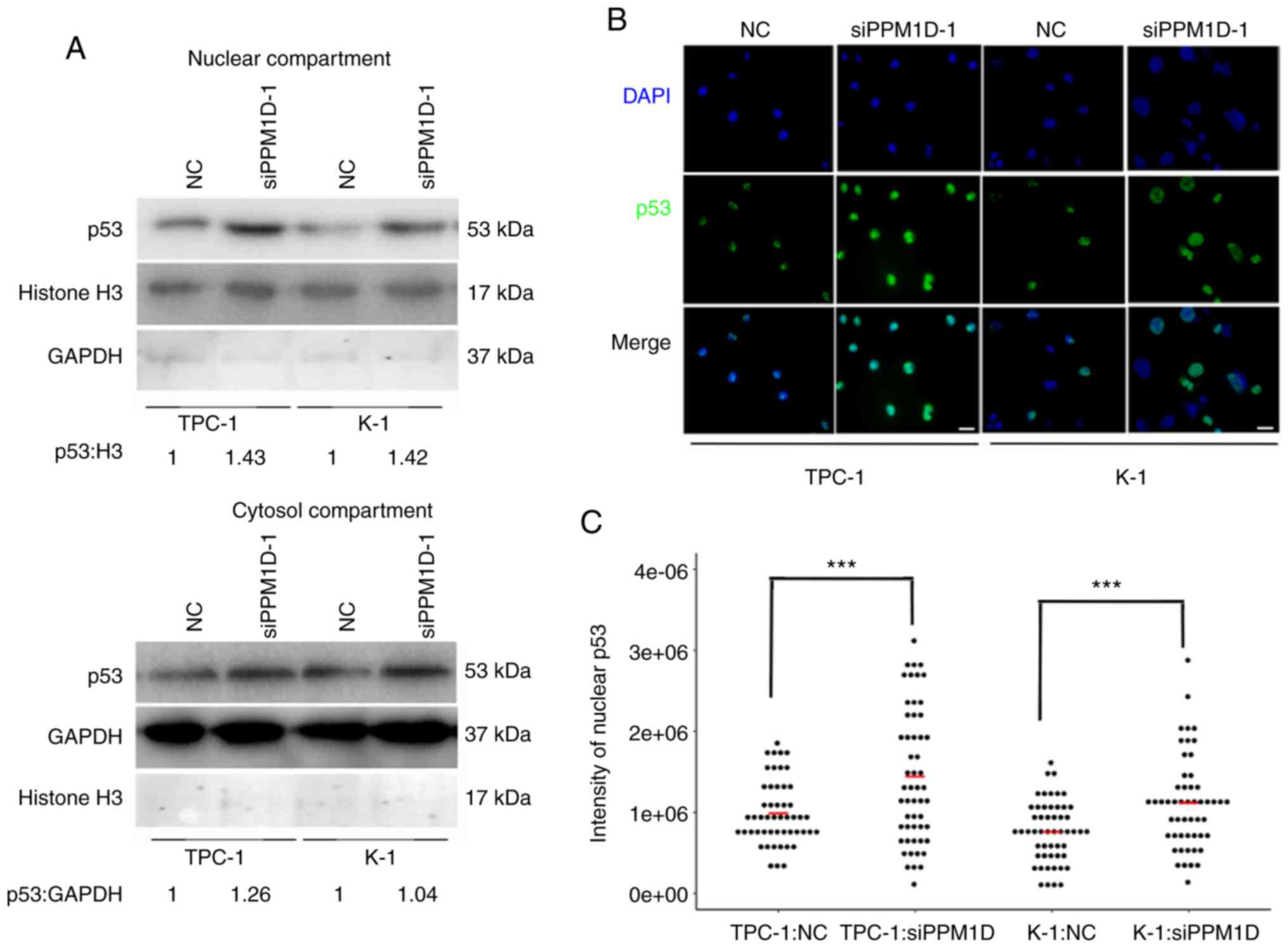
PPM1D regulates p53 nuclear
translocation in thyroid cancer cells
Since p53 is able to transiently accumulate in the
nucleus and act as a transcription factor of Bax in response to
cell stress (19), it was further
investigated whether downregulation of PPM1D increases p53 in the
nucleus. The protein levels of p53 in the cytosolic and nuclear
extract of TPC-1 and K-1 cells after PPM1D siRNA transfection were
assessed by western blot analysis. In line with aforementioned
findings, the two cell lines treated with siPPM1D exhibited
increased p53 protein levels compared with those in the control
groups (Fig. 7A). In addition, the
increased accumulation of p53 in siPPM1D-transfected cells compared
with that in the control groups was more evident in the nucleus
compared with that in the cytosol, indicating that p53 translocates
to the nucleus in response to PPM1D depletion (Fig. 7A, upper vs. lower panel). Next, to
examine the nuclear distribution of p53, an indirect
immunofluorescence assay was performed. In the imagines, p53 and
nuclei were indicated by green and blue color, respectively
(Fig. 7B). As indicated in a dot
plot, the intensity of p53-positive nuclei was significantly
increased after transfection with siPPM1D compared with that in the
control groups (Fig. 7C),
indicating more nuclear retention of p53 in PPM1D-knockdown cells.
Collectively, the negative regulation of the p53 signaling pathway
by PPM1D may lead to less nuclear accumulation of p53.
Discussion
In the present study, PPM1D was identified as a
molecular marker of metastasis and poor prognosis in patients with
PTC and the oncogenic properties of PPM1D were also confirmed in
vitro. PPM1D was indicated to promote cancer progression in
PTC, as the analysis of 89 PTC patient samples indicated that high
PPM1D protein expression was significantly associated with tumor
size and lymph node metastasis. It was also demonstrated that PPM1D
was overexpressed in PTC patient tissues compared with that in
paired adjacent non-cancerous tissues. These results were in line
with numerous previous studies reporting on the oncogenic
properties of PPM1D in various types of cancer (5,28–30).
These previous studies indicated that these features were largely
associated with the ability of PPM1D to modulate the p38 MAPK and
p53 signaling network.
In addition, the present study demonstrated that
PPM1D exerted an essential influence on thyroid cancer cell
proliferation and invasion in vitro. The growth rates of
TPC-1 and K-1 cells transfected with siPPM1D were significantly
decreased compared with those of control cells. Furthermore,
significant decreases in colony formation and migration were
observed in PPM1D-silenced cells. Of note, the cell proliferation
of PPM1D-silenced K-1 cells was successfully restored by inhibiting
the activity of p38 MAPK, indicating that PPM1D promotes cancer
cell progression at least partially via regulation of p38 MAPK
activity.
To confirm that PPM1D accelerates thyroid cancer
cell progression through inhibition of the p38 MAPK and/or p53
signaling pathways in thyroid cancer cells, the association between
PPM1D levels and the activities of p38 MAPK and p53 were
delineated. The present study revealed that PPM1D knockdown in K-1
and TPC-1 cells led to increased protein levels of p53 and Bax, as
well as enhanced p38 MAPK phosphorylation. As aforementioned, it is
essential to consider the crosstalk between the p38 MAPK and p53
signaling pathways, and in this light, it was further demonstrated
that knockdown of PPM1D induced apoptosis, which was partially
reversed by additional inhibition of p38 MAPK activities. The
present results demonstrated that silencing of PPM1D caused a
significant upregulation of the p38 MAPK signaling pathway, which
promoted the apoptosis of thyroid cancer cells. The mechanism
underlying how PPM1D mediates p53 activity was further
investigated, revealing that PPM1D may regulate nuclear
translocation of p53. Since Bax is transcriptionally regulated by
p53, it is possible that PPM1D deregulates Bax protein expression
through the p53 signaling pathway. Since activation of p38 and
expression of p53 are events that are commonly associated with
apoptosis and that are situated downstream of the actual trigger of
apoptosis, the direct target of PPM1D should be explored in the
future.
In conclusion, the present results provide novel
insight into the cancer biology of PTC; PPM1D was indicated to be a
risk factor of metastasis and may serve as a potential therapeutic
target. The downstream targets of PPM1D and the mechanisms
underlying how these targets mediate PPM1D oncogenic activities
remains to be further investigated.
Acknowledgements
We sincerely thank the University of Colorado Cancer
Center Cell Bank for providing two thyroid cancer cell lines
including K1 and TPC-1.
Funding
The present study was supported by funds from the
National Natural Science Foundation of China (grant nos. 81272934,
81572622, and 81772854 to QHJ; grant nos. 81472498 and 81772851 to
YLW; grant no. 81502317 to WJW; grant no. 81702753 to TL); the
Shanghai Municipal Planning Commission of Science and Research Fund
for Young Scholars (grant no. 20154Y0050 to JX).
Availability of data and materials
Please contact the author for data requests.
Authors' contributions
QHJ conceived and designed the experiments. LTH
selected the patients and collected the clinical samples. ZWL, DW
and LTH performed the experiments. WJW analyzed the data and
interpreted the results data along with TL and JX. TL, LTH and JX
contributed the reagents/materials/analysis tools. ZWL and QHJ
wrote the manuscript. YLW and YW critically revised manuscript for
important intellectual content. All authors read and approved the
final version of manuscript to be published and agree to be
accountable for all aspects of the research in ensuring that
questions in terms of the accuracy or integrity of any part of the
research are appropriately investigated and resolved.
Ethics approval and consent to
participate
For the use of the clinical materials for research
purposes, the study was conducted in accordance with the
Declaration of Helsinki. The Institutional Research Ethics
Committee of Shanghai Cancer Center, Fudan University, approved the
study, and informed consent was obtained from all participants
and/or their legal guardians.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Glossary
Abbreviations
Abbreviations:
|
PTC
|
papillary thyroid carcinoma
|
|
PPM1D
|
protein phosphatase,
Mg2+/Mn2+ dependent, 1D
|
References
|
1
|
Cabanillas ME, McFadden DG and Durante C:
Thyroid cancer. Lancet. 388:2783–2795. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lubitz CC and Sosa JA: The changing
landscape of papillary thyroid cancer: Epidemiology, management,
and the implications for patients. Cancer. 122:3754–3759. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Landa I, Ibrahimpasic T, Boucai L, Sinha
R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho C, Krishnamoorthy GP,
Xu B, et al: Genomic and transcriptomic hallmarks of poorly
differentiated and anaplastic thyroid cancers. J Clin Invest.
126:1052–1066. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Le Guezennec X and Bulavin DV: WIP1
phosphatase at the crossroads of cancer and aging. Trends Biochem
Sci. 35:109–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li J, Yang Y, Peng Y, Austin RJ, van
Eyndhoven WG, Nguyen KCQ, Gabriele T, McCurrachM E, Marks JR, Hoey
T, et al: Oncogenic properties of PPM1D located within a breast
cancer amplification epicenter at 17q23. Nat Genet. 31:133–134.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tamura S, Toriumi S, Saito JI, Awano K,
Kudo TA and Kobayashi T: PP2C family members play key roles in
regulation of cell survival and apoptosis. Cancer Sci. 97:563–567.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pullen KE, Ng HL, Sung PY, Good MC, Smith
SM and Alber T: An alternate conformation and a third metal in
PstP/Ppp, the M. tuberculosis PP2C-Family Ser/Thr protein
phosphatase. Structure. 12:1947–1954. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang ZP, Tian Y and Lin J: Role of
wild-type p53-induced phosphatase 1 in cancer. Oncol Lett.
14:3893–3898. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harrison M, Li J, Degenhardt Y, Hoey T and
Powers S: Wip1-deficient mice are resistant to common cancer genes.
Trends Mol Med. 10:359–361. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bulavin DV, Demidov ON, Saito S,
Kauraniemi P, Phillips C, Amundson SA, Ambrosino C, Sauter G,
Nebreda AR, Anderson CW, et al: Amplification of PPM1D in human
tumors abrogates p53 tumor-suppressor activity. Nat Genet.
31:210–215. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cancer Genome Atlas Research N, .
Integrated genomic characterization of papillary thyroid carcinoma.
Cell. 159:676–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chew J, Biswas S, Shreeram S, Humaidi M,
Wong HET, Dhillion MK, Teo H, Hazra A, Fang CC, López-Collazo D, et
al: WIP1 phosphatase is a negative regulator of NF-kappaB
signalling. Nat Cell Biol. 11:659–666. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan DS, Maryou B, Lambros K, Rayter S,
Natrajan R, Vatcheva R, Gao Q, Marchiò C, Geyer FC, Savage K, et
al: PPM1D is a potential therapeutic target in ovarian clear cell
carcinomas. Clin Cancer Res. 15:2269–2280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goloudina AR, Kochetkova EY, Pospelova TV
and Demidov ON: Wip1 phosphatase: Between p53 and MAPK kinases
pathways. Oncotarget. 7:31563–31571. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gorrini C, Harris IS and Mak TW:
Modulation of oxidative stress as an anticancer strategy. Nat Rev
Drug Discov. 12:931–947. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fiscella M, Zhang H, Fan S, Sakaguchi K,
Shen S, Mercer WE, Vande Woude GF, O'Connor PM and Appella E: Wip1,
a novel human protein phosphatase that is induced in response to
ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci
USA. 94:6048–6053. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu X, Nannenga B and Donehower LA: PPM1D
dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints.
Genes Dev. 19:1162–1174. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Demidov ON, Timofeev O, Lwin HNY, Kek C,
Appella E and Bulavin DV: Wip1 phosphatase regulates p53-dependent
apoptosis of stem cells and tumorigenesis in the mouse intestine.
Cell Stem Cell. 1:180–190. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dudgeon C, Shreeram S, Tanoue K, Mazur SJ,
Sayadi A, Robinson RC, Appella E and Bulavin DV: Genetic variants
and mutations of PPM1D control the response to DNA damage. Cell
Cycle. 12:2656–2664. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu Y, Li N, Xiang R and Sun P: Emerging
roles of the p38 MAPK and PI3K/AKT/mTOR pathways in
oncogene-induced senescence. Trends Biochem Sci. 39:268–276. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takekawa M, Adachi M, Nakahata A, Nakayama
I, Itoh F, Tsukuda H, Taya Y and Imai K: p53-inducible wip1
phosphatase mediates a negative feedback regulation of p38 MAPK-p53
signaling in response to UV radiation. EMBO J. 19:6517–6526. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bulavin DV, Phillips C, Nannenga B,
Timofeev O, Donehower LA, Anderson CW, Appella E and Fornace AJ Jr:
Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis
through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf)
pathway. Nat Genet. 36:343–350. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wen D, Liao T, Ma B, Qu N, Shi RL, Lu ZW,
Wang YL, Wei WJ and Ji QH: Downregulation of CSN6 attenuates
papillary thyroid carcinoma progression by reducing
Wnt/beta-catenin signaling and sensitizes cancer cells to FH535
therapy. Cancer Med. 7:285–296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fallahi P, Mazzi V, Vita R, Ferrari SM,
Materazzi G, Galleri D, Benvenga S, Miccoli P and Antonelli A: New
therapies for dedifferentiated papillary thyroid cancer. Int J Mol
Sci. 16:6153–6182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang YL, Liu X, Gao SY, Feng H, Jiang YP,
Wang SS, Yang J, Jiang J, Ma XR, Tang YJ, et al: WIP1 stimulates
migration and invasion of salivary adenoid cystic carcinoma by
inducing MMP-9 and VEGF-C. Oncotarget. 6:9031–9044. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun GG, Wang YD, Liu Q and Hu WN:
Expression of Wip1 in kidney carcinoma and its correlation with
tumor metastasis and clinical significance. Pathol Oncol Res.
21:219–224. 2015. View Article : Google Scholar : PubMed/NCBI
|