Gastrointestinal stromal tumors (GISTs) are found in
the stomach (56%), small intestine (32%), colon and rectum (6%),
esophagus (0.7%), and other areas, such as the omentum, intestinal
membrane, pelvis and retroperitoneum (5.5%) (1). The symptoms of GISTs are often
non-specific, and dependent on the size and location of the mass.
Many small GISTs (<2 cm) are frequently found by endoscopy or
radiographic examination, and these patients typically have no
symptoms. The most common symptom observed is gastrointestinal
bleeding, which is observed in ~50% of patients, followed by
abdominal pain (20–50% of patients) and gastrointestinal
obstruction (10–30% of patients). Other symptoms include melena,
hematemesis, feeling satiated and a palpable abdominal mass.
Proximal gastric stromal tumors may cause difficulty in swallowing,
while tumors located in the pylorus may present as a gastric outlet
obstruction (2,3). At initial diagnosis, ~20% of patients
present with metastases (4), which
typically occur in the abdominal cavity or liver, whereas lung,
bone or brain metastases are rare. Lymph node metastasis occurs in
20–60% of children with GISTs and Carney's triad, whereas other
types are rare (<10%) (5,6). In
addition, GIST-induced expendable hypothyroidism and IGF-II
production-related non-islet cell tumor hypoglycemia have also been
reported. Therefore, patients with endocrine and metabolic symptoms
should also be included in differential diagnosis (7,8).
Studies have shown that non-coding RNAs [long
non-coding RNAs (lncRNA) and microRNAs (miRNAs)] are associated
with the invasion, proliferation and drug resistance of GIST cells
(11,12). Detection of circulating (ct)DNA may
also contribute to the early diagnosis of GISTs (13).
The global incidence of GISTs (1–1.5 cases/100,000
individuals) and the prevalence rate (13 cases/100,000 individuals)
are low (14). Incidence rates in
Shanghai, Hong Kong and Norway are relatively higher (1.9–2.1
cases/100,000 individuals), whereas the incidence in the Shanxi
Province is relatively lower (4.3 cases/1,000,000 individuals). The
median age of diagnosis is 60 years (range, 10–100), but may be
diagnosed at any age; ~50% of cases are identified in individuals
aged 30–59, there is no significant difference in occurrence
between males and females, and the incidence in individuals <20
years is rare (<0.5% of all cases) (15,16).
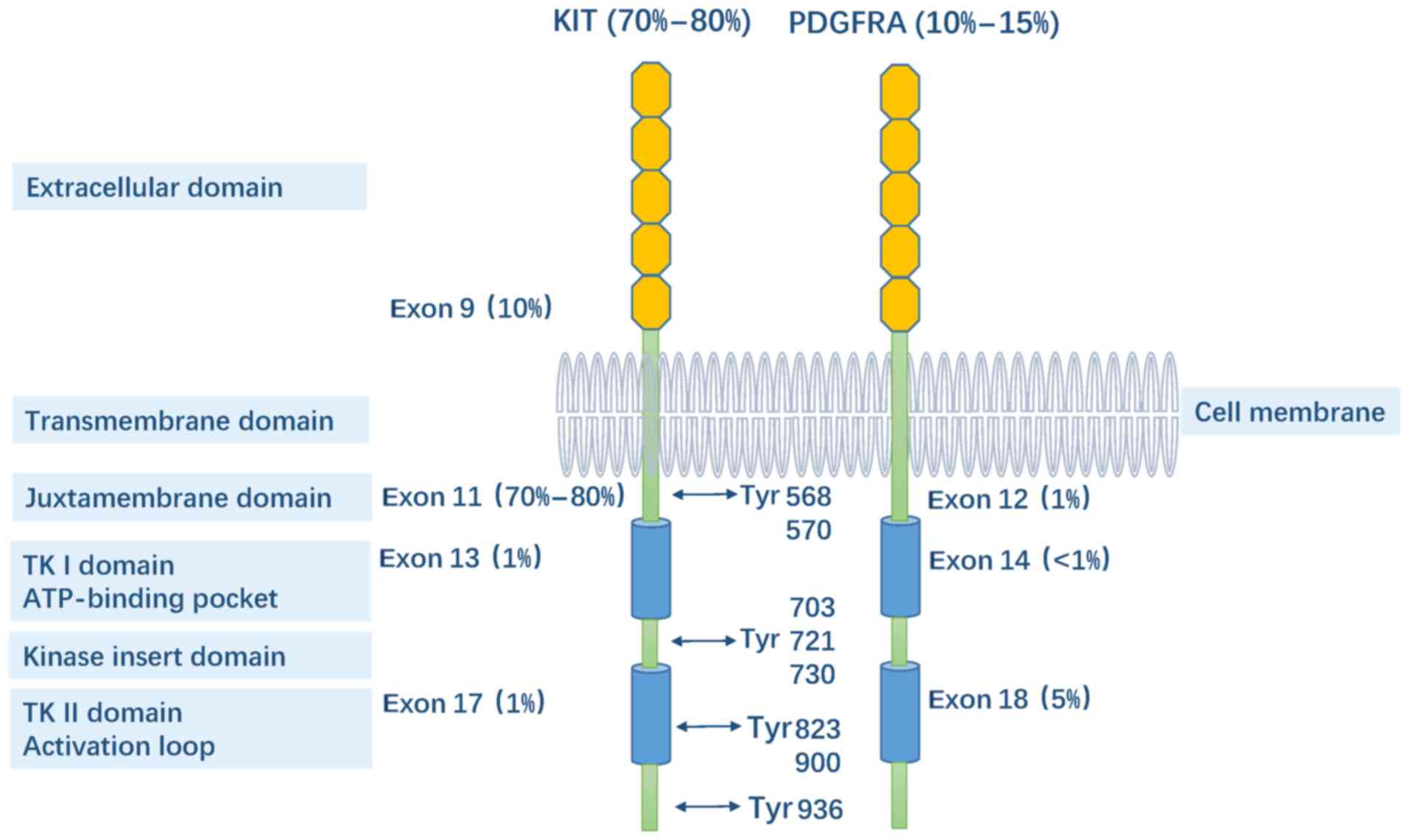
There are two types of KIT receptors; the wild-type
(145 kDa) and the mutant-type (125 kDa) receptor (17). The human KIT gene is a
proto-oncogene located on chromosome 4q12-13. Its products belong
to the receptor tyrosine kinase class III family, which also
includes PDGFRA, PDGFRB, CSF1R and FLT3 receptor (18). The receptor tyrosine kinase class
III family is characterized by an extracellular ligand binding
domain, which consists of five immunoglobulin-like regions. In
addition, the kinase structure includes a transmembrane domain, a
juxtamembrane membrane domain and an intracellular kinase domain
(Fig. 2). KIT is associated with
many human malignancies, including small cell lung cancer,
malignant melanoma, colorectal cancer and GISTs. More than 500
different KIT mutations have been discovered in human tumors but
only a few of these are considered driver mutations (19). As a KIT ligand, stem cell factor
(SCF) can induce receptor dimerization and activation of intrinsic
tyrosine kinase activity after binding to the receptor, thus
creating docking sites for signaling molecules containing SRC
homologous domain 2 (SH2). The SH2 domain is present in numerous
signal transduction molecules and consists of ~100 amino acids. It
can regulate cell growth by binding phosphorylated tyrosine
residues, including Tyr 568, Tyr 570, Tyr 703, Tyr 721, Tyr 730,
Tyr 823, Tyr 900 and Tyr 936. Phosphorylated tyrosine, together
with adjacent amino acid residues, forms a specific binding site
for downstream signaling molecules and activates specific
downstream signaling pathways, including the MAPK and PI3K/AKT
pathways. The former results in the upregulation of transcription
factors, such as MYC, ELK, CREB and FOS, whereas the latter results
in the downregulation of cell cycle inhibitors and the enhancement
of anti-apoptotic effects (20).
Phosphorylation of Tyr 568 serves an important role in the
activation of KIT and downstream signaling pathways. Phosphorylated
Tyr 568 activates the SRC family kinase and in turn promotes
activation of the KIT gene (21). SRC activity in tumors of patients
with GISTs is significantly higher compared with that noted in
normal tissues (22). Inhibition of
SRC family kinase activity weakens KIT activation, suggesting that
the activity of the SRC family kinase is necessary for the complete
activation of KIT. In addition, SRC family kinases activate SHC to
activate the downstream RAS/RAF/MEK/MAPK signaling pathway, which
is involved in KIT-mediated cell proliferation (21). Another important KIT phosphorylation
site is Tyr 721, which serves as a docking site for the regulatory
subunit p85 of PI3K (23). Tyr 703
and Tyr 936 serve as binding sites of Grb2 which are recruited to
activate the downstream PI3K/AKT and RAS/RAF/MEK/MAPK signaling
pathways (24).
The signal transducers and activators of
transcription (STAT) family of proteins is also associated with KIT
signaling (25). Binding of SCF to
the KIT receptor results in binding of JAK2 to c-kit, followed by
autophosphorylation leading to phosphorylation of STAT1, STAT3 and
STAT5. Phosphorylated STAT subsequently translocates to the nucleus
where it binds to the promoter region of target genes to regulate
transcription (26). At present, it
is hypothesized that cell cycle disorders caused by genomic
inactivation of cell cycle regulatory genes are a common mechanism
during the transition from low-risk to high-risk metastatic GISTs
(27).
Mutations in exon 11 are concentrated in the hot
spots of codon 550–560 at the 5′ end. The most frequently observed
type of mutation is deletion at codon 557–558 at the 5′ end,
followed by deletion mutations at codon 559 and point mutations
resulting in V575A (32). Imatinib
has desirable therapeutic effects in the majority of patients with
GISTs with mutations of the KIT gene. However, exon 11
mutations involving codon 557–558 deletion have also been
demonstrated to increase the malignant potential of a tumor and
reduce recurrence-free survival (RFS) (33–35). A
European multicenter analysis showed that deletion mutations in
exon 11 of the KIT gene at codon 557–558 may be used as a
reference factor predicting a less favorable prognosis (36). In another retrospective study,
moderate-risk patients with KIT exon 11 deletion mutations had
similar recurrence-free survival (RFS) to those with high-risk
diseases, whereas those with no KIT exon 11 deletion mutation
exhibited similar clinical manifestations to those of very low-risk
and low-risk patients. These findings also help to assess the
prognosis of patients at moderate risk with deletion mutations in
exon 11 deletion (37). Patients
with KIT exon 11 mutations exhibit improved drug responses and
higher overall survival compared with patients with KIT exon 9
mutations and those lacking KIT or PDGFRA mutations (31). This was also verified in phase I and
phase II trials in Europe (38).
Patients with exon 9 mutations, characterized by
codon 502–503 duplication had a higher recurrence and metastasis
rate, were significantly associated with larger tumor morphology,
tumor growth site (located in the small intestine) and spindle cell
type tumor cells (39). There is
also a relatively higher incidence of exon 9 mutations in men
(40). In a Japanese
population-based study, 4 patients with exon 9 mutations were also
found to be at high risk and all died of metastasis. These results
also suggest that mutation in exon 9 is associated with a less
favorable prognosis. However, 75% of Japanese patients with exon 9
mutations possessed a GIST located in the stomach, and the
conclusion that this mutation type is more common in the small
intestine suggested by Losata et al (41) was not observed, which may be related
to different ethnicities (42).
Controversially, no other studies have found any association
between exon 9 mutations and a poor prognosis (43). Mutation screening for exon 9 is
considered to have guiding significance for GIST treatment since
these claim a higher dose of imatinib to be effective, and thus
also is of great significance for the treatment. Besides after
progression to imatinib resistance, GISTs with KIT mutations at
exon 9 and wild-type GISTs seem to respond better to sunitinib
(44).
Typically, primary mutations occur in exons 9 and
11, whereas secondary mutations most frequently occur in exon 13
encoding the TK I domain and exon 17 encoding the TK II domain.
Most cases of metastatic secondary mutated GISTs have multiple
mutations at the same time, both between different metastases and
within the same metastatic deposit. Of the secondary mutations in
KIT, ~30–40% are secondary KIT exon 17 mutations, accounting for
resistance to imatinib or sunitinib in patients with GIST (45,46).
In these patients, regorafenib shows therapeutic efficacy (47). Conversely, patients with secondary
exon 13 mutations typically respond to sunitinib but not
regorafenib (48).
The majority of mutations in exon 13 of KIT in GISTs
are K642E mutations caused by base substitution (c.1945A>G;
c.1948G>A) (49). These base
substitution mutations result in constitutive activation of
tyrosine phosphorylation, independent of ligand binding, which
activates specific downstream signaling transduction pathways and
promotes cell proliferation (50).
The majority of mutations in exon 17 of KIT in GISTs are N822K
(70%), which also result in maintenance of tyrosine phosphorylation
activity (50). Less frequently
observed mutations (N822Y, N822K, N822H, D816F, D816Y, D820Y,
D820V, D816V and Y823D) have been identified in exon 17 (40,49).
D816V has also been found in several other human malignant tumors,
including acute myeloid leukemia (51), mastocytosis (52), germ cell tumors (53), sinus natural killer/T cell lymphoma
(54) and intracranial teratoma
(55), but is relatively rare in
GISTs. Mutations at codon 816 result in constitutive activation of
the receptor, which activates the downstream PI3K/AKT signaling
pathway. Most KIT exon 13 or exon 17 mutants have spindle-cell
morphology, and fewer tumors have epithelioid cell characteristics.
Gastric tumors with KIT exon 13 mutations are associated with
increased invasiveness, whereas small intestine tumors with KIT
exon 13 and exon 17 mutations did not result in any significant
differences in behaviors compared with other mutant small intestine
GISTs (49).
Furthermore, deletion mutations in chromosomes 14q
and 22q are considered early events in the progression of
KIT/PDGFRA mutant GISTs. Inactivation of dystrophin is hypothesized
to be a late event in the progression of GISTs. Dystrophin is a
tumor suppressor which is inactivated in 96% of metastatic GISTs
but not in low-risk GISTs (27).
KIT mutations and PDGFRA mutations are mutually
exclusive in GISTs. However, GISTs expressing KIT or PDGFRA
oncoprotein exhibit similar mechanisms of tumorigenesis and
progression (56). PDGFRA mutations
exist in 10–15% of GISTs and are found in exon 12 which encodes the
membrane proximal domain (1%), exon 14 which encodes the TK I
domain (N659Y, N659K; <1%) and exon 18 which encodes the TK II
domain (D824V, D842Y; 5%, I843_del, I843-H845_del, I843_del,
I843-H845_del, D842-H845_del, D842-M844_del, D842-H845_del,
D842-M844_del; 1%). Secondary mutations associated with secondary
resistance occur primarily in exons 14 and 18. The majority of the
PDGFRA mutations affect the TK II domain. These mutations alter the
activation loop, modulating the ATP binding pocket and causing
kinase activation leading to downstream signal transduction
pathways that promote cell survival and proliferation (56,57).
Protein kinase B (Akt), MAPK, and STAT1 and STAT3 in PDGFRA-mutant
GISTs are uniformly activated, resulting in the activation of the
same signaling pathways as KIT-mutant GISTs. PDGFRA-mutant GISTs
are primarily epithelioid and are predominantly present in the
stomach (58). Expression of CD117
in these tumors is weak or absent (56,59).
PDGFRA-mutant GISTs are less invasive, accounting for only 2.1% of
metastases in patients with GISTs (4,60).
The most common mutation type of PDGFRA in GISTs is
D842V mutation in exon 18 (accounting for 9.8% of all mutations and
65% of exon 18 mutations in PDGFRA). The substitution of a valine
at codon 842 by aspartic acid leads to resistance to first- and
second-line tyrosine kinase inhibitors (TKIs) such as imatinib and
sunitinib in patients, and therefore, these patients do not benefit
from treatment with TKIs (61). It
is also worth noting that in gastric tumors with a PDGFRA mutation,
the vast majority of the advanced cases carry the exon 18 PDGFRA
D842V mutation (36). Avapratinib
(BLU-285) is a novel inhibitor that specifically targets KIT exon
17 and PDGFRA D842 mutations. The results of the phase I NAVIGATOR
trial showed that the overall response rate (ORR) of patients with
PDGFRA D842V mutation was 84% and treatment caused tumor shrinkage
in 98% of cases. The NAVIGATOR phase I trial included four
different groups of patients with GISTs: i) GISTs treated with
second-line therapeutics; ii) GISTs being treated with third- or
fourth-line therapeutics that were regorafenib-naïve; iii) GISTs
being treated with fourth-line or more advanced line therapeutics;
and iv) PDGFRA D842V-mutated GISTs. In the second-line treatment
group, the reported avapritinib ORR was 25%. In patients being
treated with third- or fourth-line therapeutics that were
regorafenib-naïve, avapritinib was associated with an ORR of 26%.
In the patients being treated with fourth-line or more advanced
line therapeutics, ORR was 20%. In addition, in vitro
studies have shown that crenolanib exhibited improved
antiproliferative effects compared with imatinib for patients with
this type of mutation. Phase III clinical trials of crenolanib
(Randomized Trial of Crenolanib in Subjects with D842V Mutated
GIST; clinicaltrials.gov identifier,
NCT02847429), and phase III randomized trials of avapritinib and
regorafenib (VOYAGER, NCT03465722) are also on-going (62). The majority of GISTs with PDGFRA
mutations excluding D842V mutation still respond to imatinib,
therefore mutation screening is of great significance for the
treatment of GIST (63).
PDGFRA mutations are also found in exon 12
(juxtamembrane domain; JM) and exon 14 (TK I domain), but are
relatively rare (64). Among these,
exon 12 point mutation V561A is the second most common type of
PDGFRA mutation (65). The majority
of the exon 14 mutations of PDGFRA are N659K point mutations.
Clinical data showed that most tumors with this mutation were
located in the stomach, and most of these were epithelioid cells
with relatively good prognosis (66).
The majority of KIT and PDGFRA mutations are
sporadic and are only found in GIST tissues. However, there are
also patients with rare familial GISTs with autosomal dominant
inheritance, and affected family members have a 100% penetrance
rate (67). Clinical manifestations
include dysphagia, excessive skin pigmentation, urticaria
pigmentation and mastocytosis. These patients have the same type of
germline KIT and PDGFRA mutations as the somatic mutant GISTs. KIT
mutations in familial GIST patients have been found in exon 11
(V559A, W557R, D579 del) (68–70),
exon 17 (D820Y) (71) and exon 8
(D419 del) (72). The PDGFRA
mutation was also found in exon 18 (D846Y) (73). Histologically, familial GISTs are
basically the same as that of patients with sporadic GISTs, but
there are multiple lesions, primarily from the proliferation of
interstitial cells of Cajal. Identification of patients with
familial GISTs is important as the clinical management patterns are
different from those of GISTs with somatic mutations. Nevertheless,
currently there are no specific treatment options for such
patients. Considering the presence of multiple lesions,
conservative resection is often adopted in most medical
institutions.
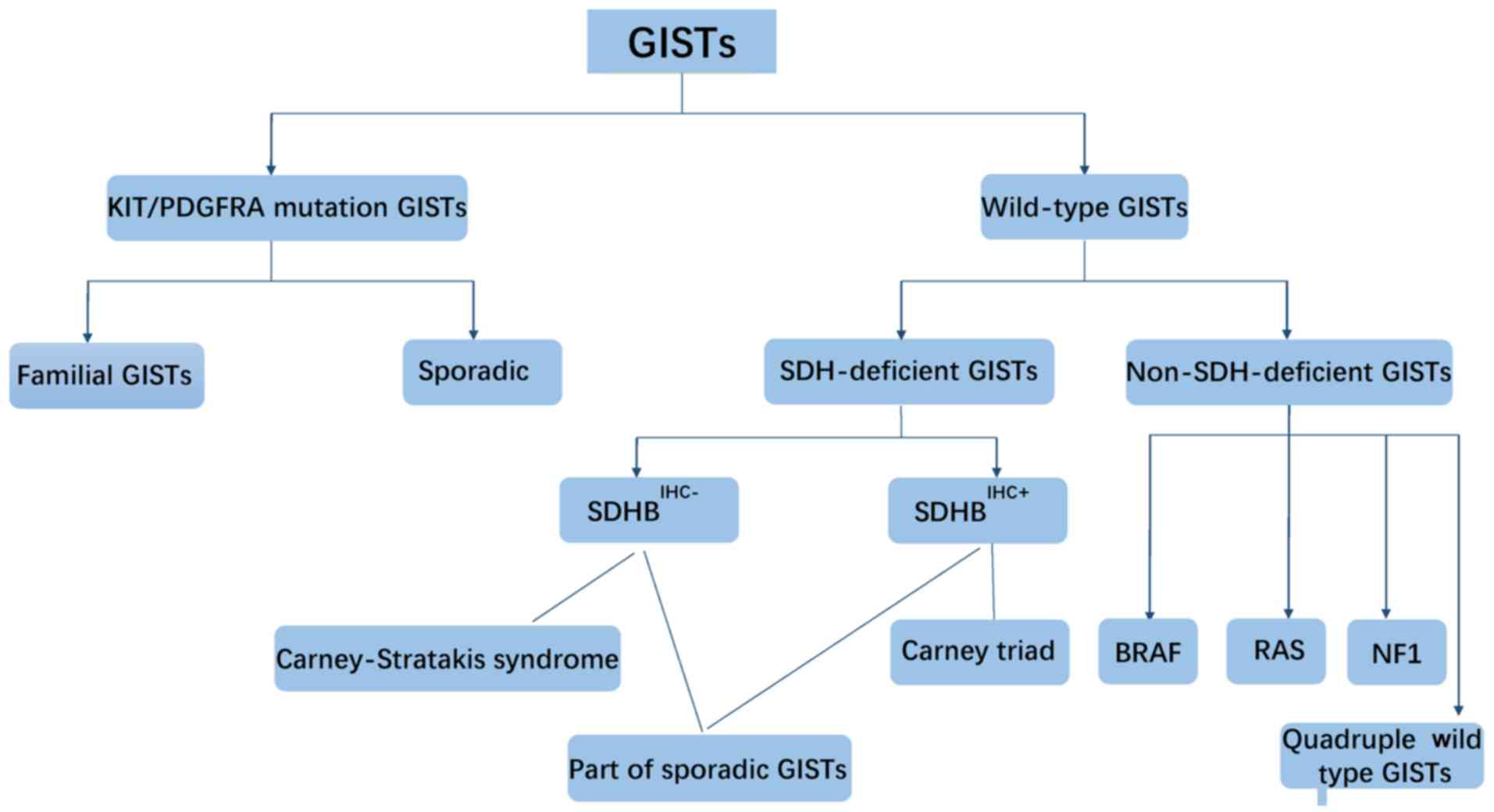
Most GISTs are driven by oncogenic KIT or PDGFRA
receptor tyrosine kinase activating mutations. However, ~10–15% of
GISTs lack KIT and PDGFRA mutations, and are referred to as
wild-type GISTs. Wild-type GISTs can be divided into succinate
dehydrogenase complex (SDH)-deficient and non-SDH-deficient GISTs
(63). Mutations of any of the four
SDH subunits or of members of the RAS pathway [(K/N/H-RAS, BRAF,
neurofibromatosis type 1 (NF1)] have been observed in wild-type
GISTs.
Whole-genome sequencing revealed that the genome of
wild-type GISTs exhibit a low mutation frequency, and only ~25% of
patients have chromosomal imbalances. Mutations in the 14q23.1
region of KIT/PDGFRA-mutant GISTs result in downregulation of tumor
suppressor RTN1, DAAM1 and DACT1, but mutations in the same region
in wild-type GISTs does not result in these effects (74). Wild-type GISTs tend to develop in
younger patients; ~85% of GISTs in patients <23 years of age are
wild-type. Lesions are primarily present in the stomach, and the
volume of the tumor is often small, but lymph node metastases are
more common. TKIs are often less effective in these cases (75–77).
Succinate dehydrogenase complex (SDH; also known as
mitochondrial complex II) deficiency is more common in wild-type
GISTs, accounting for ~5% of all GISTs. The lack of SDH is
considered to be an important feature of several human tumor
subtypes, including GISTs, paraganglioma, renal cell carcinoma and
pituitary adenomas (78). SDH is a
complex of enzymes located on the mitochondrial inner membrane, and
consists of four subunits (SDHA, SDHB, SDHC and SDHD) and two kinds
of succinate dehydrogenase assembly factors (SDHAF1 and SDHAF2),
which are components of the tricarboxylic acid cycle and the
respiratory electron transport chain.
Approximately 50% of subunit mutations are found in
SDH-deficient GISTs, and the most frequently observed being SDHA
which are primarily germline mutations, accounting for ~30% of all
mutations, with mutations in other subunits accounting for the
remaining ~20% (79). SDHA-mutant
GISTs can be individually identified by SDHA immunohistochemistry.
In the other 50% of cases, no subunit mutation is observed, but
immunohistochemistry showed a significant decrease or deletion of
SDHB protein expression, and the respiratory chain complex II
enzyme activity was lost as well. This may be due to epigenetic
modifications or related protein defects involved in maintaining
stability (80).
Defects in any of the subunits, particularly defects
in SDHB, will result in instability of the SDH complex, which in
turn affects the function of the complex. When the SDH complex is
dysfunctional, there is an accumulation of succinic acid in the
cytoplasm which results in the accumulation of hypoxia inducible
factor 1 subunit α (HIF1α). HIF1α transcriptionally upregulates
hypoxia-related tumorigenic processes and angiogenesis. Thus,
defects in energy metabolism are key carcinogenic mechanisms
(81). Therefore, if any of the SDH
subunits are mutated/inactivated, immunohistochemical staining of
SDHB is absent, and as such SDHB-negative staining is now
considered to be a highly sensitive marker for any SDH subunit
germline mutation (80,82).
SDH-deficient GISTs occur primarily in children and
patients with Carney-Stratakis syndrome and Carney triad which
affects children and younger adults. Patients are usually <40
years of age, while other GISTs are rarely seen in younger
individuals. SDH-deficient GISTs account for two-thirds of cases in
the 20–29 age group and are more common in females (16). Lesions are almost always located in
the stomach, and the antrum is the most common location, followed
by the posterior wall of the stomach and the fundus, and they have
a tendency for lymphatic invasion and lymph node metastasis
compared with other mutant GIST types (83). Immunohistochemically negative SDHA
GISTs are rare in children, are more common in males, and lack of
SDHA staining is associated with an increased rate of metastasis to
the liver; however, there is no difference in tumor size or mitosis
rate (79).
The Carney triad includes GISTs, pulmonary
chondromas and extra-adrenal paraganglioma. It was first described
as a triad of gastric leiomyosarcoma in 1977, and only few patients
exhibit a complete Carney triad. In the majority of cases, only two
of these tumors are observed (84).
Patients with Carney triad are typically younger women who are
susceptible to various tumors, including pheochromocytoma and other
non-functional bilateral or unilateral adrenal adenomas. Although
the Carney triad-associated GISTs lack mutations in any of the SDH
subunits, the function of the SDH complex is still impaired and may
be associated with hypermethylation of the SDHC promoter (85).
Carney-Stratakis syndrome, first described in 2002,
is a rare hereditary syndrome inherited by autosomal dominant
inheritance with incomplete penetrance. It is characterized by
multifocal gastric GISTs and multicentric paraganglioma (86). Germline mutations in SDHB, SDHC,
SDHD have been identified in patients with Carney-Stratakis
syndrome (87) and no mutations in
the coding sequence of the SDHA gene have been identified
(88).
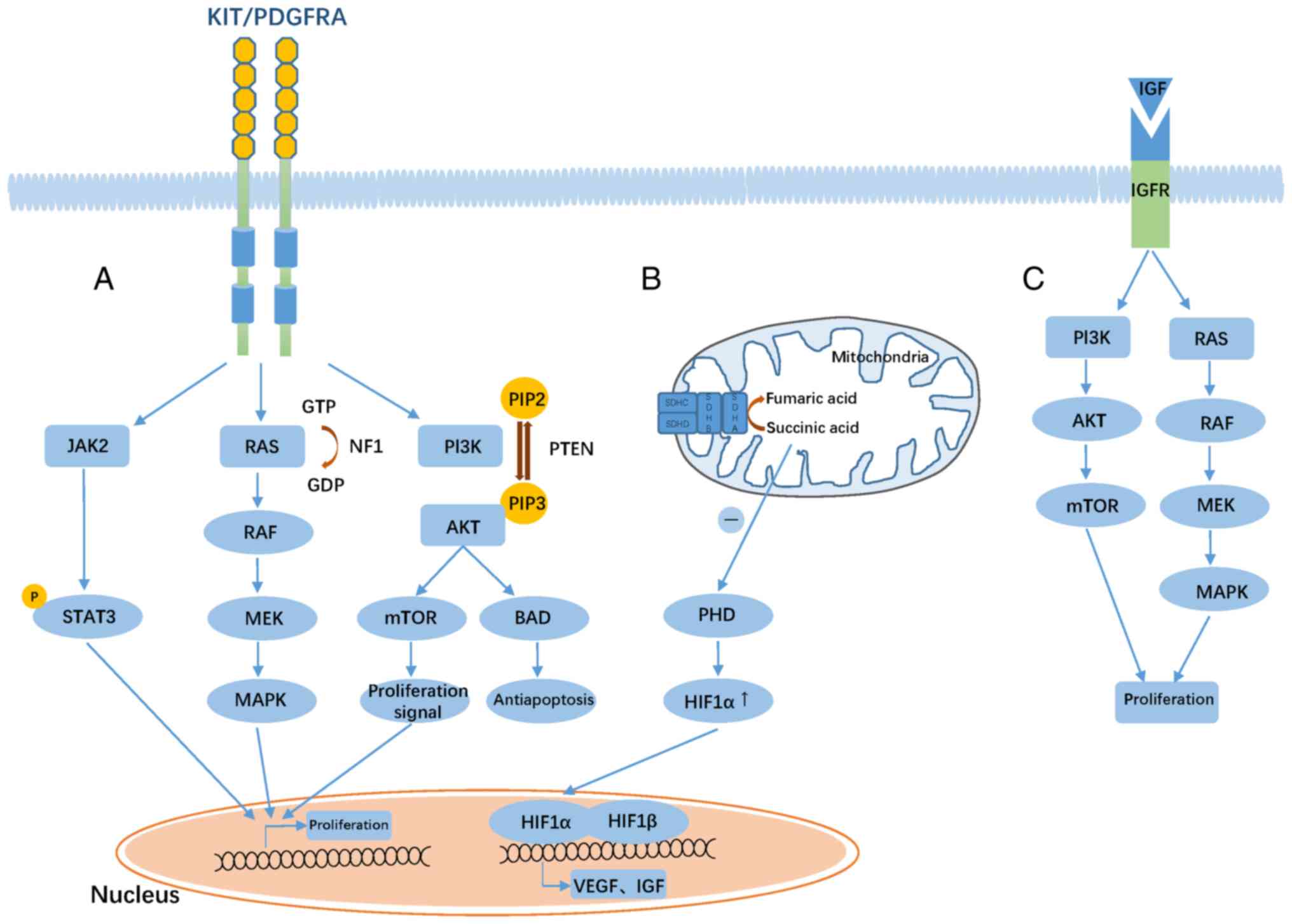
Upregulation of insulin like growth factor 1
receptor (IGF1R) expression in SDH-deficient tumors is considered
to be a feature of SDH-deficient GISTs (89). IGF1R is part of the insulin-like
growth factor family of proteins, which consists of two ligands
(IGF1 and IGF2), two receptors (IGF1R and IGF2R) and six IGF
binding proteins (IGFBPs). Binding of IGF and IGFR activates
downstream signaling, including the RAS/RAF/MAPK and PI3K/AKT
pathways (Fig. 3C) (90). Dysfunction of SDH leads to the
accumulation of succinic acid, which in turn inhibits the activity
of proline hydroxylase, resulting in the accumulation of HIF1α,
translocation of HIF1α to the nucleus and dimerization of HIF1β to
form an active transcription factor that induces expression of
glycolytic and angiogenic genes (including IGF and VEGF), which in
turn promotes cell proliferation via the RAS/RAF/MAPK and PI3K/AKT
signaling pathways (57,91–93).
IGF1R expression has been observed in 88.75% of SDH-deficient
GISTs, whereas only in 1% of SDHB-positive patients. Thus detection
of IGF1R can help to identify SDH defective GISTs. IGF1 has been
shown to promote cell proliferation in vitro. Animal
experiments have shown that inhibiting IGF signaling reduced tumor
growth (94). Epidemiological
evidence suggests that IGF1 levels are associated with cancer risk
and prognosis (95). At present,
upregulation of IGF1R is observed in a number of different types of
cancer. Because of its role in the metabolism of cancer cells and
its potential correlation with the survival of malignant cells,
IGF1R has become a target of anticancer therapy (96–98).
In a phase I clinical trial, patients with solid tumors were
treated with IGF1R monoclonal antibody R1507 (also known as
RO4858696) and chemotherapeutics or targeted drugs. One of these
patients did not receive the full treatment, but the partial
response to the disease reached 3 years (99). However, the complexity of the IGF1R
pathway and its interaction with other signaling pathways has not
been fully elucidated. Further preclinical research is required to
understand the complex mechanisms to develop improved therapeutic
options.
Non-SDH-deficient GISTs include NF1 correlation,
BRAF mutation, RAS gene mutation and quadruple wild-type. GISTs
with BRAF/RAS or NF1 mutations can be collectively called RAS
pathway-mutant GISTs. The demographic characteristics of
non-SDH-deficient GISTs are similar to those of KIT/PDGFRA-mutant
GISTs, and are more frequently observed in adults and primary
exhibit a spindle-cell morphology (100). However, unlike KIT/PDGFRA-mutant
GISTs, non-SDH-deficient wild-type GIST are primarily located in
the small intestine.
NF1, also known as von Recklinghausen disease, is an
inherited cancer susceptibility syndrome, caused by a biallelic
deletion of the NF1 gene and is characterized by
neurological, cutaneous and skeletal lesions. It is one of the most
common genetic syndromes with an incidence of 1/2,500-3,000
individuals (101). Patients with
NF1 type have a higher risk of developing GISTs, and ~7% of NF1
patients have concurrent GISTs, which is considerably higher than
the normal population (102).
Patients with NF1-related GISTs are usually younger, and the
lesions are primarily located in the duodenum and small intestine
and exhibit a slower clinical progression. However, 15–20% of
patients with NF1-related GISTs exhibit malignant clinical
outcomes. The tumors are small in size, with a high positive rate
of CD117 and CD34 expression. Tumor tissues are composed of cells
with a spindle-cell like morphology, with a low mitotic rate
(100,103).
The RAS protein acts as a molecular switch that
switches between its active GTP binding state and inactive GDP
binding state. The majority of RAS mutations are observed in codons
12, 13 or 61. Mutations of the RAS gene or its regulatory factors
cause the RAS protein to remain active, leading to tumorigenesis.
Activated alleles of HRAS, NRAS and KRAS have been shown to possess
a similar phenotype to activated BRAF, CRAF, MEK1 or MEK2-driven
alleles in human tumors (112).
Miranda et al (113)
identified codon 12 (G12D) and codon 13 (G13D) mutations in the
KRAS gene in ~5% of patients with GISTs. G12D-mutant tumors
are accompanied by deletion mutations in KIT exon 11 (570–576 and
579), whereas patients with G13D mutations are accompanied by
PDGFRA gene D842V mutation. Additionally, KRAS and BRAF mutations
may affect the response of imatinib-sensitive KIT mutants to
imatinib therapy, thus it was suggested that KRAS and BRAF mutation
analysis should be introduced in clinical diagnosis of patients
with GISTs.
Quadruple wild-type GISTs are not associated with
SDH mutations, BRAF mutations or RAS mutations (114). Nannini et al (115) analyzed the genome of patients with
quadruple wild-type GISTs and found that they had significantly
different genome profiles from KIT/PDGFR mutation or SDH-deficient
GISTs. Molecular markers CALCRL and COL22A1, and specific
oncogenes, including NTRK2 and CDK6, and ETS were found in both
small intestine quadruple wild-type GISTs. Brenca et al
(116) found an ETV6-NTRK3 fusion
in a patient with rectal quadruple wild-type GISTs. Shi et
al (117) found a fusion of
the FGFR1 gene (FGFR1-HOOK3 and FGFR1-TACC1) in quadruple
wild-type GISTs. NF1 gene inactivation was also observed in
quadruple wild-type GISTs, but the patient did not show
neurofibromatosis type 1 symptoms (118). In addition, somatic gene mutations
such as TP53, MEN1, MAX, FGFR1, CTNND2 and CHD4 were found in
quadruple wild-type GISTs (119).
These findings may assist in the identification of new therapeutic
targets. In vitro experiments indicate that ETV6-NTRK3
fusion is associated with activation of downstream IGF1R signaling
and that tumor cells are susceptible to IGF1R inhibition. In
addition, ALK inhibitors, a target drug for ETV6-NTRK3 gene fusion,
have shown therapeutic potential (116). The Murine double-minute 2
inhibitor can inhibit the growth of TP53 wild-type GIST cells and
enhance the therapeutic response of GIST cells to TKI. This
suggests that the regulation of the p53 gene may be a potential
effective therapeutic strategy (120).
Extracellular DNA fragments in different body fluids
(plasma, serum, urine, and saliva) are referred to as cell free DNA
(cfDNA). The primary part of cfDNA in the blood is adsorbed to the
surface of white blood cells or red blood cells, and its half-life
is short because it is easily degraded by nucleases (121,122). Among these, cfDNA derived from
tumor cells is called ctDNA, which may be produced by apoptosis or
necrosis of cancer cells (123).
cfDNA is primarily composed of ctDNA, and normal extracellular DNA
accounts for only a small portion of cfDNA (124,125). The feasibility report of
plasma-based ctDNA analysis for tumors in GISTs was first published
at the ASCO annual meeting in 2013. The report showed that the
consistency of mutations in exon 9 and 11 of KIT between tumor
tissue and plasma was 84%. In particular, the consistency of KIT
mutations of exon 9 was 100%, whereas that of KIT exon 11 was only
79%. It is also noteworthy that a higher KIT mutation rate (47% vs.
12%) was detected in plasma by BEAMing compared with tumor tissue
(13). Maier et al (126) subsequently confirmed the presence
of KIT/PDGFRA mutant ctDNA in the plasma, and the quantity of
mutant ctDNA was associated with the clinical course of the
disease. Namløs et al (127) found that the detection rate of
tumor ctDNA mutations in the plasma of high-risk patients or
patients with metastatic diseases was higher compared with patients
with localized, or moderate- or low-risk GISTs. Jilg et al
(128) confirmed that
tumor-specific KIT and PDGFRA mutations could be detected in the
ctDNA of patients with active GISTs and were positively correlated
with disease activity. Other mutations indicating disease
progression, including BRAF, NRAS, PIK3CA, PTEN and CTNNB1
mutations, are found in ctDNA, which can influence treatment
regimens. All the above studies have demonstrated that ctDNA
detection in plasma has important guiding significance for the
diagnosis and prognosis of GISTs. However, liquid biopsy technology
as a complement to other commonly used clinical techniques still
has many limitations, and it is far from ready for use in clinical
practice; however, the prospects of this technology is
promising.
Non-coding RNA refers to RNA that does not encode
proteins. It has been found that various miRNAs and lncRNAs are
associated with the occurrence and development of GISTs.
MicroRNAs (miRNAs) are small non-coding RNAs which
participate in the regulation of post-transcriptional gene
expression (129). Dysregulation
of various miRNAs has been identified in many different types of
cancer, including gastric cancer (130), colorectal cancer (131), pancreatic cancer (132), and childhood glioma (133). miRNAs regulate target gene
expression by acting on key molecular pathways and mediate cell
invasion, migration, proliferation, apoptosis and drug resistance.
A German study found that miR-221 and miR-222 were downregulated in
wild-type and mutant GISTs, and in vitro experiments, they
confirmed that the expression of these miRNAs induced apoptosis
through a KIT/AKT/BCL2 signaling pathway (134). miR-17 (135), miR-20a (135), miR-21 (136), miR-133b (137), miR-137 (138), miR-152 (139), miR-218 (140), miR-494 (141), miR-518a (117), have all been confirmed to be
downregulated in GISTs. Overexpression of miR-218 also inhibits the
PI3K/AKT pathway and thereby increases the sensitivity of GIST
cells to imatinib (140).
Conversely, miR-125a and miR-196a were found to be upregulated in
GISTs. Overexpression of miR-125a is associated with imatinib
resistance (142). Overexpression
of miR-196a increases the invasiveness of cancer cells (143), and is associated with high risk
classification, a high rate of metastasis and reduced survival
rates (144). In addition, studies
have shown that circulating miRNAs can be used as biomarkers for
detection of various types of malignant tumors, including gastric
cancer (145), colorectal cancer
(146), and pancreatic cancer
(147). Serum circulating
miR-518e-5p has been found to be a potential non-invasive biomarker
for early detection and diagnosis of secondary imatinib-resistant
GISTs (148).
lncRNAs works by interacting with other cellular
molecules, including DNA, RNA binding protein and RNA. lncRNAs
serve an important role in the diagnosis, monitoring, prognosis and
evaluation of therapeutic reactivity of tumors (149). HOTAIR is one of the most widely
studied carcinogenic lncRNAs. Overexpression of HOTAIR is
associated with an increase in invasiveness of GIST cells and
knockdown of HOTAIR can inhibit the invasiveness of GIST cells
(143). HOTAIR also regulates the
progression of GISTs by inducing methylation of the promoter of the
pro-cadherin 10 gene in GIST cells (11). HOTAIR is upregulated in invasive
GISTs and mediates gene-specific DNA methylation. This further
confirms the role of HOTAIR in GISTs (150). Yan et al (151) confirmed that CCDC26 enhances the
sensitivity of imatinib by downregulating IGF-1R expression, and
showed that CCDC26 may be used as a therapeutic target to reverse
imatinib resistance in patients with GISTs.
The progress made in the field of molecular biology
in the past 20 years has provided a deeper understanding of the
pathogenesis of GISTs. The characteristics of each molecular
subtype are summarized in Table I.
Receptor TKIs currently used to treat patients with GISTs are based
on these subtypes and the use of these drugs significantly prolong
life expectancy. Nevertheless, resistance to second-line
therapeutics is becoming increasingly common. Additionally, there
is no specific treatment for patients with wild-type GISTs. Thus an
improved understanding is required to further understand the
molecular mechanisms underlying the different subtypes of GISTs to
develop improved therapeutic options.
Not applicable.
The present study was supported by the Science and
Technology Projects of Zhejiang Province (LGF19H030007), the
Traditional Chinese Medicine Science and Technology Project of
Zhejiang Province (2018ZA109), the Natural Science Foundation of
Ningbo (2016A610157 and 2018A610371), the Medical Science and
Technology Project of Zhejiang Province (2020KY813) and the Medical
and Health Science and Technology Project of Zhejiang Province
(2018ZH026).
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
HD and ZY conceived and designed the study and
prepared the manuscript. XY, YY, XL and CH were responsible for the
literature search, data visualization and analysis. KG and YJ
retrieved the relevant literature and revised the manuscript. All
authors read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Søreide K, Sandvik OM, Søreide JA, Giljaca
V, Jureckova A and Bulusu VR: Global epidemiology of
gastrointestinal stromal tumours (GIST): A systematic review of
population-based cohort studies. Cancer Epidemiol. 40:39–46. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miettinen M, Sobin LH and Lasota J:
Gastrointestinal stromal tumors of the stomach: A
clinicopathologic, immunohistochemical, and molecular genetic study
of 1765 cases with long-term follow-up. Am J Surg Pathol. 29:52–68.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Briggler AM, Graham RP, Westin GF, Folpe
AL, Jaroszewski DE, Okuno SH and Halfdanarson TR: Clinicopathologic
features and outcomes of gastrointestinal stromal tumors arising
from the esophagus and gastroesophageal junction. J Gastrointest
Oncol. 9:718–727. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Emile JF, Brahimi S, Coindre JM, Bringuier
PP, Monges G, Samb P, Doucet L, Hostein I, Landi B, Buisine MP, et
al: Frequencies of KIT and PDGFRA mutations in the MolecGIST
prospective population-based study differ from those of advanced
GISTs. Med Oncol. 29:1765–1772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Agaimy A and Wünsch PH: Lymph node
metastasis in gastrointestinal stromal tumours (GIST) occurs
preferentially in young patients < or = 40 years: An overview
based on our case material and the literature. Langenbecks Arch
Surg. 394:375–381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang L, Smyrk TC, Young WF Jr, Stratakis
CA and Carney JA: Gastric stromal tumors in Carney triad are
different clinically, pathologically, and behaviorally from
sporadic gastric gastrointestinal stromal tumors: Findings in 104
cases. Am J Surg Pathol. 34:53–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pink D, Schoeler D, Lindner T,
Thuss-Patience PC, Kretzschmar A, Knipp H, Vanhoefer U and
Reichardt P: Severe hypoglycemia caused by paraneoplastic
production of IGF-II in patients with advanced gastrointestinal
stromal tumors: A report of two cases. J Clin Oncol. 23:6809–6811.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maynard MA and Huang SA: Thyroid hormone
inactivation in gastrointestinal stromal tumors. N Engl J Med.
371:86–87. 2014.PubMed/NCBI
|
|
9
|
Duensing A, Medeiros F, McConarty B,
Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD and
Fletcher JA: Mechanisms of oncogenic KIT signal transduction in
primary gastrointestinal stromal tumors (GISTs). Oncogene.
23:3999–4006. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ricci R: Syndromic gastrointestinal
stromal tumors. Hered Cancer Clin Pract. 14:152016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee NK, Lee JH, Kim WK, Yun S, Youn YH,
Park CH, Choi YY, Kim H and Lee SK: Promoter methylation of PCDH10
by HOTAIR regulates the progression of gastrointestinal stromal
tumors. Oncotarget. 7:75307–75318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kupcinskas J: Small molecules in rare
tumors: Emerging role of microRNAs in GIST. Int J Mol Sci.
19:192018. View Article : Google Scholar
|
|
13
|
Demetri GD, Jeffers M and Reichardt PG:
Mutational analysis of plasma DNA from patients (pts) in the phase
III GRID study of regorafenib (REG) versus placebo (PL) in tyrosine
kinase inhibitor (TKI)-refractory GIST: Correlating genotype with
clinical outcomes. Oncol Res Treat. 37:58. 2013.
|
|
14
|
Nilsson B, Bümming P, Meis-Kindblom JM,
Odén A, Dortok A, Gustavsson B, Sablinska K and Kindblom LG:
Gastrointestinal stromal tumors: The incidence, prevalence,
clinical course, and prognostication in the preimatinib mesylate
era-a population-based study in western Sweden. Cancer.
103:821–829. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Joensuu H, Vehtari A, Riihimäki J, Nishida
T, Steigen SE, Brabec P, Plank L, Nilsson B, Cirilli C, Braconi C,
et al: Risk of recurrence of gastrointestinal stromal tumour after
surgery: An analysis of pooled population-based cohorts. Lancet
Oncol. 13:265–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Janeway KA, Kim SY, Lodish M, Nosé V,
Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, et al: NIH
pediatric and wild-type GIST clinic: Defects in succinate
dehydrogenase in gastrointestinal stromal tumors lacking KIT and
PDGFRA mutations. Proc Natl Acad Sci USA. 108:314–318. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Broudy VC, Lin NL and Sabath DF: The fifth
immunoglobulin-like domain of the Kit receptor is required for
proteolytic cleavage from the cell surface. Cytokine. 15:188–195.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanks SK, Quinn AM and Hunter T: The
protein kinase family: Conserved features and deduced phylogeny of
the catalytic domains. Science. 241:42–52. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lennartsson J and Rönnstrand L: Stem cell
factor receptor/c-Kit: From basic science to clinical implications.
Physiol Rev. 92:1619–1649. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roskoski R Jr: Signaling by Kit
protein-tyrosine kinase-the stem cell factor receptor. Biochem
Biophys Res Commun. 337:1–13. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lennartsson J, Blume-Jensen P, Hermanson
M, Pontén E, Carlberg M and Rönnstrand L: Phosphorylation of Shc by
Src family kinases is necessary for stem cell factor receptor/c-kit
mediated activation of the Ras/MAP kinase pathway and c-fos
induction. Oncogene. 18:5546–5553. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rotert JV, Leupold J, Hohenberger P, Nowak
K and Allgayer H: Src activity is increased in gastrointestinal
stromal tumors-analysis of associations with clinical and other
molecular tumor characteristics. J Surg Oncol. 109:597–605. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Serve H, Hsu YC and Besmer P: Tyrosine
residue 719 of the c-kit receptor is essential for binding of the
P85 subunit of phosphatidylinositol (PI) 3-kinase and for
c-kit-associated PI 3-kinase activity in COS-1 cells. J Biol Chem.
269:6026–6030. 1994.PubMed/NCBI
|
|
24
|
Sun J, Pedersen M and Rönnstrand L: Gab2
is involved in differential phosphoinositide 3-kinase signaling by
two splice forms of c-Kit. J Biol Chem. 283:27444–27451. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deberry C, Mou S and Linnekin D: Stat1
associates with c-kit and is activated in response to stem cell
factor. Biochem J. 327:73–80. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brizzi MF, Blechman JM, Cavalloni G, Givol
D, Yarden Y and Pegoraro L: Protein kinase C-dependent release of a
functional whole extracellular domain of the mast cell growth
factor (MGF) receptor by MGF-dependent human myeloid cells.
Oncogene. 9:1583–1589. 1994.PubMed/NCBI
|
|
27
|
Heinrich MC, Patterson J, Beadling C, Wang
Y, Debiec-Rychter M, Dewaele B, Corless CL, Duensing A, Raut CP,
Rubin B, et al: Genomic aberrations in cell cycle genes predict
progression of KIT-mutant gastrointestinal stromal tumors (GISTs).
Clin Sarcoma Res. 9:32019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kinoshita K, Isozaki K, Hirota S, Nishida
T, Chen H, Nakahara M, Nagasawa Y, Ohashi A, Shinomura Y, Kitamura
Y and Matsuzawa Y: c-kit gene mutation at exon 17 or 13 is very
rare in sporadic gastrointestinal stromal tumors. J Gastroenterol
Hepatol. 18:147–151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rubin BP, Singer S, Tsao C, Duensing A,
Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, et al: KIT
activation is a ubiquitous feature of gastrointestinal stromal
tumors. Cancer Res. 61:8118–8121. 2001.PubMed/NCBI
|
|
31
|
Heinrich MC, Corless CL, Demetri GD,
Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den
Abbeele AD, Druker BJ, et al: Kinase mutations and imatinib
response in patients with metastatic gastrointestinal stromal
tumor. J Clin Oncol. 21:4342–4349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu CW, Lin S, Wang WL, Gao WB, Lv JY, Gao
JS, Zhang LY, Li Y, Wang L, Zhang YP and Tian YW: Analysis of
mutation of the c-Kit gene and PDGFRA in gastrointestinal stromal
tumors. Exp Ther Med. 10:1045–1051. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martín J, Poveda A, Llombart-Bosch A,
Ramos R, López-Guerrero JA, García del Muro J, Maurel J, Calabuig
S, Gutierrez A, González de Sande JL, et al Spanish Group for
Sarcoma Research, : Deletions affecting codons 557–558 of the c-KIT
gene indicate a poor prognosis in patients with completely resected
gastrointestinal stromal tumors: A study by the Spanish Group for
Sarcoma Research (GEIS). J Clin Oncol. 23:6190–6198. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wardelmann E, Losen I, Hans V, Neidt I,
Speidel N, Bierhoff E, Heinicke T, Pietsch T, Büttner R and
Merkelbach-Bruse S: Deletion of Trp-557 and Lys-558 in the
juxtamembrane domain of the c-kit protooncogene is associated with
metastatic behavior of gastrointestinal stromal tumors. Int J
Cancer. 106:887–895. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Singer S, Rubin BP, Lux ML, Chen CJ,
Demetri GD, Fletcher CD and Fletcher JA: Prognostic value of KIT
mutation type, mitotic activity, and histologic subtype in
gastrointestinal stromal tumors. J Clin Oncol. 20:3898–3905. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wozniak A, Rutkowski P, Schöffski P,
Ray-Coquard I, Hostein I, Schildhaus HU, Le Cesne A, Bylina E,
Limon J, Blay JY, et al: Tumor genotype is an independent
prognostic factor in primary gastrointestinal stromal tumors of
gastric origin: A European Multicenter Analysis based on
ConticaGIST. Clin Cancer Res. 20:6105–6116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Quek R, Farid M, Kanjanapan Y, Lim C, Tan
IB, Kesavan S, Lim TKH, Oon LL, Goh BK, Chan WH, et al: Prognostic
significance of KIT exon 11 deletion mutation in intermediate-risk
gastrointestinal stromal tumor. Asia Pac J Clin Oncol. 13:115–124.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Debiec-Rychter M, Dumez H, Judson I, Wasag
B, Verweij J, Brown M, Dimitrijevic S, Sciot R, Stul M, Vranck H,
et al: Use of c-KIT/PDGFRA mutational analysis to predict the
clinical response to imatinib in patients with advanced
gastrointestinal stromal tumours entered on phase I and II studies
of the EORTC soft tissue and bone sarcoma group. Eur J Cancer.
40:689–695. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Antonescu CR, Sommer G, Sarran L,
Tschernyavsky SJ, Riedel E, Woodruff JM, Robson M, Maki R, Brennan
MF, Ladanyi M, et al: Association of KIT exon 9 mutations with
nongastric primary site and aggressive behavior: KIT mutation
analysis and clinical correlates of 120 gastrointestinal stromal
tumors. Clin Cancer Res. 9:3329–3337. 2003.PubMed/NCBI
|
|
40
|
Wozniak A, Rutkowski P, Piskorz A,
Ciwoniuk M, Osuch C, Bylina E, Sygut J, Chosia M, Rys J, Urbanczyk
K, et al: Prognostic value of KIT/PDGFRA mutations in
gastrointestinal stromal tumours (GIST): Polish clinical GIST
registry experience. Ann Oncol. 23:353–360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lasota J, Wozniak A, Sarlomo-Rikala M, Rys
J, Kordek R, Nassar A, Sobin LH and Miettinen M: Mutations in exons
9 and 13 of KIT gene are rare events in gastrointestinal stromal
tumors. A study of 200 cases. Am J Pathol. 157:1091–1095. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sakurai S, Oguni S, Hironaka M, Fukayama
M, Morinaga S and Saito K: Mutations in c-kit gene exons 9 and 13
in gastrointestinal stromal tumors among Japanese. Jpn J Cancer
Res. 92:494–498. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Künstlinger H, Huss S, Merkelbach-Bruse S,
Binot E, Kleine MA, Loeser H, Mittler J, Hartmann W, Hohenberger P,
Reichardt P, et al: Gastrointestinal stromal tumors with KIT exon 9
mutations: Update on genotype-phenotype correlation and validation
of a high-resolution melting assay for mutational testing. Am J
Surg Pathol. 37:1648–1659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mulet-Margalef N and Garcia-Del-Muro X:
Sunitinib in the treatment of gastrointestinal stromal tumor:
Patient selection and perspectives. Onco Targets Ther. 9:7573–7582.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yeh CN, Chen TW, Tseng JH, Liu YY, Wang
SY, Tsai CY, Chiang KC, Hwang TL, Jan YY and Chen MF: Surgical
management in metastatic gastrointestinal stromal tumor (GIST)
patients after imatinib mesylate treatment. J Surg Oncol.
102:599–603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gao J, Tian Y, Li J, Sun N, Yuan J and
Shen L: Secondary mutations of c-KIT contribute to acquired
resistance to imatinib and decrease efficacy of sunitinib in
Chinese patients with gastrointestinal stromal tumors. Med Oncol.
30:5222013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yeh CN, Chen MH, Chen YY, Yang CY, Yen CC,
Tzen CY, Chen LT and Chen JS: A phase II trial of regorafenib in
patients with metastatic and/or a unresectable gastrointestinal
stromal tumor harboring secondary mutations of exon 17. Oncotarget.
8:44121–44130. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mazzocca A, Napolitano A, Silletta M,
Spalato Ceruso M, Santini D, Tonini G and Vincenzi B: New frontiers
in the medical management of gastrointestinal stromal tumours. Ther
Adv Med Oncol. 11:17588359198419462019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lasota J, Corless CL, Heinrich MC,
Debiec-Rychter M, Sciot R, Wardelmann E, Merkelbach-Bruse S,
Schildhaus HU, Steigen SE, Stachura J, et al: Clinicopathologic
profile of gastrointestinal stromal tumors (GISTs) with primary KIT
exon 13 or exon 17 mutations: A multicenter study on 54 cases. Mod
Pathol. 21:476–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lux ML, Rubin BP, Biase TL, Chen CJ,
Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD and Fletcher
JA: KIT extracellular and kinase domain mutations in
gastrointestinal stromal tumors. Am J Pathol. 156:791–795. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ning ZQ, Li J and Arceci RJ: Activating
mutations of c-kit at codon 816 confer drug resistance in human
leukemia cells. Leuk Lymphoma. 41:513–522. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nagata H, Worobec AS, Oh CK, Chowdhury BA,
Tannenbaum S, Suzuki Y and Metcalfe DD: Identification of a point
mutation in the catalytic domain of the protooncogene c-kit in
peripheral blood mononuclear cells of patients who have
mastocytosis with an associated hematologic disorder. Proc Natl
Acad Sci USA. 92:10560–10564. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tian Q, Frierson HF Jr, Krystal GW and
Moskaluk CA: Activating c-kit gene mutations in human germ cell
tumors. Am J Pathol. 154:1643–1647. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hongyo T, Li T, Syaifudin M, Baskar R,
Ikeda H, Kanakura Y, Aozasa K and Nomura T: Specific c-kit
mutations in sinonasal natural killer/T-cell lymphoma in China and
Japan. Cancer Res. 60:2345–2347. 2000.PubMed/NCBI
|
|
55
|
Sakuma Y, Sakurai S, Oguni S, Satoh M,
Hironaka M and Saito K: c-kit gene mutations in intracranial
germinomas. Cancer Sci. 95:716–720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Joensuu H, Hohenberger P and Corless CL:
Gastrointestinal stromal tumour. Lancet. 382:973–983. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wardelmann E, Hrychyk A, Merkelbach-Bruse
S, Pauls K, Goldstein J, Hohenberger P, Losen I, Manegold C,
Büttner R and Pietsch T: Association of platelet-derived growth
factor receptor alpha mutations with gastric primary site and
epithelioid or mixed cell morphology in gastrointestinal stromal
tumors. J Mol Diagn. 6:197–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Debiec-Rychter M, Wasag B, Stul M, De
Wever I, Van Oosterom A, Hagemeijer A and Sciot R: Gastrointestinal
stromal tumours (GISTs) negative for KIT (CD117 antigen)
immunoreactivity. J Pathol. 202:430–438. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lasota J, Dansonka-Mieszkowska A, Sobin LH
and Miettinen M: A great majority of GISTs with PDGFRA mutations
represent gastric tumors of low or no malignant potential. Lab
Invest. 84:874–883. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Indio V, Astolfi A, Tarantino G, Urbini M,
Patterson J, Nannini M, Saponara M, Gatto L, Santini D, do Valle
IF, et al: Integrated molecular characterization of
gastrointestinal stromal tumors (GIST) harboring the rare D842V
mutation in PDGFRA gene. Int J Mol Sci. 19:E7322018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Heinrich M, von Mehren M, Jones RL, Bauer
S, Kang YK, Schöffski P, Eskens F, Serrano C, Cassier PA, Mir O, et
al: Avapritinib is highly active and well-tolerated in patients
(pts) with advanced GIST driven by diverse variety of oncogenic
mutations in KIT and PDGFRA. Presented at: 2018 CTOS Annual
Meeting; November 15, 2018 Rome, Italy (abstract 3027631).
https://www.blueprintmedicines.com/wp-content/uploads/2019/01/CTOS-Avapritinib-Update-Nov-2018.pdf
|
|
63
|
Medeiros F, Corless CL, Duensing A,
Hornick JL, Oliveira AM, Heinrich MC, Fletcher JA and Fletcher CD:
KIT-negative gastrointestinal stromal tumors: Proof of concept and
therapeutic implications. Am J Surg Pathol. 28:889–894. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lasota J and Miettinen M: KIT and PDGFRA
mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn
Pathol. 23:91–102. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Corless CL, Schroeder A, Griffith D, Town
A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J and
Heinrich MC: PDGFRA mutations in gastrointestinal stromal tumors:
Frequency, spectrum and in vitro sensitivity to imatinib. J Clin
Oncol. 23:5357–5364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lasota J, Stachura J and Miettinen M:
GISTs with PDGFRA exon 14 mutations represent subset of clinically
favorable gastric tumors with epithelioid morphology. Lab Invest.
86:94–100. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nishida T, Hirota S, Taniguchi M,
Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T,
Takabayashi A, Matsuda H and Kitamura Y: Familial gastrointestinal
stromal tumours with germline mutation of the KIT gene. Nat Genet.
19:323–324. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
68
|
Beghini A, Tibiletti MG, Roversi G,
Chiaravalli AM, Serio G, Capella C and Larizza L: Germline mutation
in the juxtamembrane domain of the kit gene in a family with
gastrointestinal stromal tumors and urticaria pigmentosa. Cancer.
92:657–662. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lasota J and Miettinen M: A new familial
GIST identified. Am J Surg Pathol. 30:13422006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Robson ME, Glogowski E, Sommer G,
Antonescu CR, Nafa K, Maki RG, Ellis N, Besmer P, Brennan M and
Offit K: Pleomorphic characteristics of a germ-line KIT mutation in
a large kindred with gastrointestinal stromal tumors,
hyperpigmentation, and dysphagia. Clin Cancer Res. 10:1250–1254.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hirota S, Nishida T, Isozaki K, Taniguchi
M, Nishikawa K, Ohashi A, Takabayashi A, Obayashi T, Okuno T,
Kinoshita K, et al: Familial gastrointestinal stromal tumors
associated with dysphagia and novel type germline mutation of KIT
gene. Gastroenterology. 122:1493–1499. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hartmann K, Wardelmann E, Ma Y,
Merkelbach-Bruse S, Preussner LM, Woolery C, Baldus SE, Heinicke T,
Thiele J, Buettner R and Longley BJ: Novel germline mutation of KIT
associated with familial gastrointestinal stromal tumors and
mastocytosis. Gastroenterology. 129:1042–1046. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Chompret A, Kannengiesser C, Barrois M,
Terrier P, Dahan P, Tursz T, Lenoir GM and Bressac-De Paillerets B:
PDGFRA germline mutation in a family with multiple cases of
gastrointestinal stromal tumor. Gastroenterology. 126:318–321.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Astolfi A, Nannini M, Pantaleo MA, Di
Battista M, Heinrich MC, Santini D, Catena F, Corless CL, Maleddu
A, Saponara M, et al: A molecular portrait of gastrointestinal
stromal tumors: An integrative analysis of gene expression
profiling and high-resolution genomic copy number. Lab Invest.
90:1285–1294. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wada R, Arai H, Kure S, Peng WX and Naito
Z: ‘Wild type’ GIST: Clinicopathological features and clinical
practice. Pathol Int. 66:431–437. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Prakash S, Sarran L, Socci N, DeMatteo RP,
Eisenstat J, Greco AM, Maki RG, Wexler LH, LaQuaglia MP, Besmer P
and Antonescu CR: Gastrointestinal stromal tumors in children and
young adults: A clinicopathologic, molecular, and genomic study of
15 cases and review of the literature. J Pediatr Hematol Oncol.
27:179–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Janeway KA, Liegl B, Harlow A, Le C,
Perez-Atayde A, Kozakewich H, Corless CL, Heinrich MC and Fletcher
JA: Pediatric KIT wild-type and platelet-derived growth factor
receptor alpha-wild-type gastrointestinal stromal tumors share KIT
activation but not mechanisms of genetic progression with adult
gastrointestinal stromal tumors. Cancer Res. 67:9084–9088. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gill AJ, Lipton L, Taylor J, Benn DE,
Richardson AL, Frydenberg M, Shapiro J, Clifton-Bligh RJ, Chow CW
and Bogwitz M: Germline SDHC mutation presenting as recurrent SDH
deficient GIST and renal carcinoma. Pathology. 45:689–691. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Miettinen M, Killian JK, Wang ZF, Lasota
J, Lau C, Jones L, Walker R, Pineda M, Zhu YJ, Kim SY, et al:
Immunohistochemical loss of succinate dehydrogenase subunit A
(SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA
germline mutation. Am J Surg Pathol. 37:234–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Barletta JA and Hornick JL: Succinate
dehydrogenase-deficient tumors: Diagnostic advances and clinical
implications. Adv Anat Pathol. 19:193–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gill AJ: Succinate dehydrogenase (SDH) and
mitochondrial driven neoplasia. Pathology. 44:285–292. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gaal J, Stratakis CA, Carney JA, Ball ER,
Korpershoek E, Lodish MB, Levy I, Xekouki P, van Nederveen FH, den
Bakker MA, et al: SDHB immunohistochemistry: A useful tool in the
diagnosis of Carney-Stratakis and Carney triad gastrointestinal
stromal tumors. Mod Pathol. 24:147–151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Miettinen M, Wang ZF, Sarlomo-Rikala M,
Osuch C, Rutkowski P and Lasota J: Succinate
dehydrogenase-deficient GISTs: A clinicopathologic,
immunohistochemical, and molecular genetic study of 66 gastric
GISTs with predilection to young age. Am J Surg Pathol.
35:1712–1721. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Carney JA, Sheps SG, Go VL and Gordon H:
The triad of gastric leiomyosarcoma, functioning extra-adrenal
paraganglioma and pulmonary chondroma. N Engl J Med. 296:1517–1518.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Boikos SA, Pappo AS, Killian JK, LaQuaglia
MP, Weldon CB, George S, Trent JC, von Mehren M, Wright JA,
Schiffman JD, et al: Molecular subtypes of KIT/PDGFRA wild-type
gastrointestinal stromal tumors: A report from the national
institutes of health gastrointestinal stromal tumor clinic. JAMA
Oncol. 2:922–928. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Carney JA and Stratakis CA: Familial
paraganglioma and gastric stromal sarcoma: A new syndrome distinct
from the Carney triad. Am J Med Genet. 108:132–139. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
McWhinney SR, Pasini B and Stratakis CA;
International Carney Triad and Carney-Stratakis Syndrome
Consortium, : Familial gastrointestinal stromal tumors and
germ-line mutations. N Engl J Med. 357:1054–1056. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Pasini B, McWhinney SR, Bei T, Matyakhina
L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie
G, et al: Clinical and molecular genetics of patients with the
Carney-Stratakis syndrome and germline mutations of the genes
coding for the succinate dehydrogenase subunits SDHB, SDHC, and
SDHD. Eur J Hum Genet. 16:79–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chou A, Chen J, Clarkson A, Samra JS,
Clifton-Bligh RJ, Hugh TJ and Gill AJ: Succinate
dehydrogenase-deficient GISTs are characterized by IGF1R
overexpression. Mod Pathol. 25:1307–1313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
LeRoith D and Roberts CT Jr: The
insulin-like growth factor system and cancer. Cancer Lett.
195:127–137. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Covello KL and Simon MC: HIFs, hypoxia,
and vascular development. Curr Top Dev Biol. 62:37–54. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pugh CW and Ratcliffe PJ: Regulation of
angiogenesis by hypoxia: Role of the HIF system. Nat Med.
9:677–684. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lasota J, Wang Z, Kim SY, Helman L and
Miettinen M: Expression of the receptor for type I insulin-like
growth factor (IGF1R) in gastrointestinal stromal tumors: An
immunohistochemical study of 1078 cases with diagnostic and
therapeutic implications. Am J Surg Pathol. 37:114–119. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wu Y, Cui K, Miyoshi K, Hennighausen L,
Green JE, Setser J, LeRoith D and Yakar S: Reduced circulating
insulin-like growth factor I levels delay the onset of chemically
and genetically induced mammary tumors. Cancer Res. 63:4384–4388.
2003.PubMed/NCBI
|
|
95
|
Gallagher EJ and LeRoith D: Minireview:
IGF, insulin, and cancer. Endocrinology. 152:2546–2551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Gotlieb WH, Bruchim I, Gu J, Shi Y,
Camirand A, Blouin MJ, Zhao Y and Pollak MN: Insulin-like growth
factor receptor I targeting in epithelial ovarian cancer. Gynecol
Oncol. 100:389–396. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sarfstein R, Maor S, Reizner N,
Abramovitch S and Werner H: Transcriptional regulation of the
insulin-like growth factor-I receptor gene in breast cancer. Mol
Cell Endocrinol. 252:241–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wu JD, Haugk K, Woodke L, Nelson P,
Coleman I and Plymate SR: Interaction of IGF signaling and the
androgen receptor in prostate cancer progression. J Cell Biochem.
99:392–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Mahadevan D, Sutton GR, Arteta-Bulos R,
Bowden CJ, Miller PJ, Swart RE, Walker MS, Haluska P, Munster PN,
Marshall J, et al: Phase 1b study of safety, tolerability and
efficacy of R1507, a monoclonal antibody to IGF-1R in combination
with multiple standard oncology regimens in patients with advanced
solid malignancies. Cancer Chemother Pharmacol. 73:467–473. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Miettinen M, Fetsch JF, Sobin LH and
Lasota J: Gastrointestinal stromal tumors in patients with
neurofibromatosis 1: A clinicopathologic and molecular genetic
study of 45 cases. Am J Surg Pathol. 30:90–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ferner RE: Neurofibromatosis 1 and
neurofibromatosis 2: A twenty first century perspective. Lancet
Neurol. 6:340–351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Maertens O, Prenen H, Debiec-Rychter M,
Wozniak A, Sciot R, Pauwels P, De Wever I, Vermeesch JR, de Raedt
T, De Paepe A, et al: Molecular pathogenesis of multiple
gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet.
15:1015–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Andersson J, Sihto H, Meis-Kindblom JM,
Joensuu H, Nupponen N and Kindblom LG: NF1-associated
gastrointestinal stromal tumors have unique clinical, phenotypic,
and genotypic characteristics. Am J Surg Pathol. 29:1170–1176.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Mussi C, Schildhaus HU, Gronchi A,
Wardelmann E and Hohenberger P: Therapeutic consequences from
molecular biology for gastrointestinal stromal tumor patients
affected by neurofibromatosis type 1. Clin Cancer Res.
14:4550–4555. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Kalender M, Sevinc A, Tutar E, Sirikci A
and Camci C: Effect of sunitinib on metastatic gastrointestinal
stromal tumor in patients with neurofibromatosis type 1: A case
report. World J Gastroenterol. 13:2629–2632. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Cui Y and Guadagno TM: B-Raf (V600E)
signaling deregulates the mitotic spindle checkpoint through
stabilizing Mps1 levels in melanoma cells. Oncogene. 27:3122–3133.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Matos P, Oliveira C, Velho S, Gonçalves V,
da Costa LT, Moyer MP, Seruca R and Jordan P: B-Raf(V600E)
cooperates with alternative spliced Rac1b to sustain colorectal
cancer cell survival. Gastroenterology. 135:899–906. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Huss S, Pasternack H, Ihle MA,
Merkelbach-Bruse S, Heitkötter B, Hartmann W, Trautmann M,
Gevensleben H, Büttner R, Schildhaus HU and Wardelmann E:
Clinicopathological and molecular features of a large cohort of
gastrointestinal stromal tumors (GISTs) and review of the
literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare
events. Hum Pathol. 62:206–214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Hostein I, Faur N, Primois C, Boury F,
Denard J, Emile JF, Bringuier PP, Scoazec JY and Coindre JM: BRAF
mutation status in gastrointestinal stromal tumors. Am J Clin
Pathol. 133:141–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Rossi S, Gasparotto D, Miceli R,
Toffolatti L, Gallina G, Scaramel E, Marzotto A, Boscato E,
Messerini L, Bearzi I, et al: KIT, PDGFRA, and BRAF mutational
spectrum impacts on the natural history of imatinib-naive localized
GIST: A population-based study. Am J Surg Pathol. 39:922–930. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Simanshu DK, Nissley DV and McCormick F:
RAS Proteins and their regulators in human disease. Cell.
170:17–33. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Miranda C, Nucifora M, Molinari F, Conca
E, Anania MC, Bordoni A, Saletti P, Mazzucchelli L, Pilotti S,
Pierotti MA, et al: KRAS and BRAF mutations predict primary
resistance to imatinib in gastrointestinal stromal tumors. Clin
Cancer Res. 18:1769–1776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Pantaleo MA, Nannini M, Corless CL and
Heinrich MC: Quadruple wild-type (WT) GIST: Defining the subset of
GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling
pathways. Cancer Med. 4:101–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Nannini M, Astolfi A, Urbini M, Indio V,
Santini D, Heinrich MC, Corless CL, Ceccarelli C, Saponara M,
Mandrioli A, et al: Integrated genomic study of quadruple-WT GIST
(KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer.
14:6852014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Brenca M, Rossi S, Polano M, Gasparotto D,
Zanatta L, Racanelli D, Valori L, Lamon S, Dei Tos AP and Maestro
R: Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion
involved in GIST. J Pathol. 238:543–549. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Shi Y, Gao X, Hu Q, Li X, Xu J, Lu S, Liu
Y, Xu C, Jiang D, Lin J, et al: PIK3C2A is a gene-specific target
of microRNA-518a-5p in imatinib mesylate-resistant gastrointestinal
stromal tumor. Lab Invest. 96:652–660. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Belinsky MG, Rink L, Cai KQ, Capuzzi SJ,
Hoang Y, Chien J, Godwin AK and von Mehren M: Somatic loss of
function mutations in neurofibromin 1 and MYC associated factor X
genes identified by exome-wide sequencing in a wild-type GIST case.
BMC Cancer. 15:8872015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Pantaleo MA, Urbini M, Indio V, Ravegnini
G, Nannini M, De Luca M, Tarantino G, Angelini S, Gronchi A,
Vincenzi B, et al: Genome-wide analysis identifies MEN1 and MAX
mutations and a neuroendocrine-like molecular heterogeneity in
quadruple WT GIST. Mol Cancer Res. 15:553–562. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Henze J, Mühlenberg T, Simon S, Grabellus
F, Rubin B, Taeger G, Schuler M, Treckmann J, Debiec-Rychter M,
Taguchi T, et al: p53 modulation as a therapeutic strategy in
gastrointestinal stromal tumors. PLoS One. 7:e377762012. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Stewart CM and Tsui DWY: Circulating
cell-free DNA for non-invasive cancer management. Cancer Genet.
228-229:169–179. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wan JCM, Massie C, Garcia-Corbacho J,
Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R and Rosenfeld N:
Liquid biopsies come of age: Towards implementation of circulating
tumour DNA. Nat Rev Cancer. 17:223–238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kidess E and Jeffrey SS: Circulating tumor
cells versus tumor-derived cell-free DNA: Rivals or partners in
cancer care in the era of single-cell analysis? Genome Med.
5:702013. View
Article : Google Scholar : PubMed/NCBI
|
|
124
|
Fiala C and Diamandis EP: Utility of
circulating tumor DNA in cancer diagnostics with emphasis on early
detection. BMC Med. 16:1662018. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Elazezy M and Joosse SA: Techniques of
using circulating tumor DNA as a liquid biopsy component in cancer
management. Comput Struct Biotechnol J. 16:370–378. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Maier J, Lange T, Kerle I, Specht K,
Bruegel M, Wickenhauser C, Jost P, Niederwieser D, Peschel C,
Duyster J and von Bubnoff N: Detection of mutant free circulating
tumor DNA in the plasma of patients with gastrointestinal stromal
tumor harboring activating mutations of CKIT or PDGFRA. Clin Cancer
Res. 19:4854–4867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Namløs HM, Boye K, Mishkin SJ, Barøy T,
Lorenz S, Bjerkehagen B, Stratford EW, Munthe E, Kudlow BA,
Myklebost O and Meza-Zepeda LA: Noninvasive detection of ctDNA
reveals intratumor heterogeneity and is associated with tumor
burden in gastrointestinal stromal tumor. Mol Cancer Ther.
17:2473–2480. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Jilg S, Rassner M, Maier J, Waldeck S,
Kehl V, Follo M, Philipp U, Sauter A, Specht K, Mitschke J, et al:
Circulating cKIT and PDGFRA DNA indicates disease activity in
gastrointestinal stromal tumor (GIST). Int J Cancer. 145:2292–2303.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Steponaitiene R, Kupcinskas J, Langner C,
Balaguer F, Venclauskas L, Pauzas H, Tamelis A, Skieceviciene J,
Kupcinskas L, Malfertheiner P and Link A: Epigenetic silencing of
miR-137 is a frequent event in gastric carcinogenesis. Mol
Carcinog. 55:376–386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Balaguer F, Link A, Lozano JJ, Cuatrecasas
M, Nagasaka T, Boland CR and Goel A: Epigenetic silencing of
miR-137 is an early event in colorectal carcinogenesis. Cancer Res.
70:6609–6618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Rizzato C, Campa D, Talar-Wojnarowska R,
Halloran C, Kupcinskas J, Butturini G, Mohelníková-Duchoňová B,
Sperti C, Tjaden C, Ghaneh P, et al: Association of genetic
polymorphisms with survival of pancreatic ductal adenocarcinoma
patients. Carcinogenesis. 37:957–964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Catanzaro G, Sabato C, Russo M, Rosa A,
Abballe L, Besharat ZM, Po A, Miele E, Bellavia D, Chiacchiarini M,
et al: Loss of miR-107, miR-181c and miR-29a-3p promote activation
of Notch2 signaling in pediatric high-grade gliomas (pHGGs). Int J
Mol Sci. 18:E27422017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ihle MA, Trautmann M, Kuenstlinger H, Huss
S, Heydt C, Fassunke J, Wardelmann E, Bauer S, Schildhaus HU,
Buettner R and Merkelbach-Bruse S: miRNA-221 and miRNA-222 induce
apoptosis via the KIT/AKT signalling pathway in gastrointestinal
stromal tumours. Mol Oncol. 9:1421–1433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Gits CM, van Kuijk PF, Jonkers MB, Boersma
AW, van Ijcken WF, Wozniak A, Sciot R, Rutkowski P, Schöffski P,
Taguchi T, et al: MiR-17-92 and miR-221/222 cluster members target
KIT and ETV1 in human gastrointestinal stromal tumours. Br J
Cancer. 109:1625–1635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Cao CL, Niu HJ, Kang SP, Cong CL and Kang
SR: miRNA-21 sensitizes gastrointestinal stromal tumors (GISTs)
cells to imatinib via targeting B-cell lymphoma 2 (Bcl-2). Eur Rev
Med Pharmacol Sci. 20:3574–3581. 2016.PubMed/NCBI
|
|
137
|
Yamamoto H, Kohashi K, Fujita A and Oda Y:
Fascin-1 overexpression and miR-133b downregulation in the
progression of gastrointestinal stromal tumor. Mod Pathol.
26:563–571. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liu S, Cui J, Liao G, Zhang Y, Ye K, Lu T,
Qi J and Wan G: MiR-137 regulates epithelial-mesenchymal transition
in gastrointestinal stromal tumor. Tumour Biol. 35:9131–9138. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Lu HJ, Yan J, Jin PY, Zheng GH, Qin SM, Wu
DM, Lu J and Zheng YL: MicroRNA-152 inhibits tumor cell growth
while inducing apoptosis via the transcriptional repression of
cathepsin L in gastrointestinal stromal tumor. Cancer Biomark.
21:711–722. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Fan R, Zhong J, Zheng S, Wang Z, Xu Y, Li
S, Zhou J and Yuan F: MicroRNA-218 inhibits gastrointestinal
stromal tumor cell and invasion by targeting KIT. Tumour Biol.
35:4209–4217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Yun S, Kim WK, Kwon Y, Jang M, Bauer S and
Kim H: Survivin is a novel transcription regulator of KIT and is
downregulated by miRNA-494 in gastrointestinal stromal tumors. Int
J Cancer. 142:2080–2093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Huang WK, Akçakaya P, Gangaev A, Lee L,
Zeljic K, Hajeri P, Berglund E, Ghaderi M, Åhlén J, Bränström R, et
al: miR-125a-5p regulation increases phosphorylation of FAK that
contributes to imatinib resistance in gastrointestinal stromal
tumors. Exp Cell Res. 371:287–296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Niinuma T, Suzuki H, Nojima M, Nosho K,
Yamamoto H, Takamaru H, Yamamoto E, Maruyama R, Nobuoka T, Miyazaki
Y, et al: Upregulation of miR-196a and HOTAIR drive malignant
character in gastrointestinal stromal tumors. Cancer Res.
72:1126–1136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Akçakaya P, Caramuta S, Åhlen J, Ghaderi
M, Berglund E, Östman A, Bränström R, Larsson C and Lui WO:
microRNA expression signatures of gastrointestinal stromal tumours:
Associations with imatinib resistance and patient outcome. Br J
Cancer. 111:2091–2102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Juzėnas S, Saltenienė V, Kupcinskas J,
Link A, Kiudelis G, Jonaitis L, Jarmalaite S, Kupcinskas L,
Malfertheiner P and Skieceviciene J: Analysis of deregulated
microRNAs and their target genes in gastric cancer. PLoS One.
10:e01323272015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
ElSharawy A, Röder C, Becker T, Habermann
JK, Schreiber S, Rosenstiel P and Kalthoff H: Concentration of
circulating miRNA-containing particles in serum enhances miRNA
detection and reflects CRC tissue-related deregulations.
Oncotarget. 7:75353–75365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Alemar B, Izetti P, Gregório C, Macedo GS,
Castro MA, Osvaldt AB, Matte U and Ashton-Prolla P: miRNA-21 and
miRNA-34a Are potential minimally invasive biomarkers for the
diagnosis of pancreatic ductal adenocarcinoma. Pancreas. 45:84–92.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Kou Y, Yang R and Wang Q: Serum
miR-518e-5p is a potential biomarker for secondary
imatinib-resistant gastrointestinal stromal tumor. J Biosci.
43:1015–1023. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Schmitt AM and Chang HY: Long noncoding
RNAs in cancer pathways. Cancer Cell. 29:452–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Bure I, Geer S, Knopf J, Roas M, Henze S,
Ströbel P, Agaimy A, Wiemann S, Hoheisel JD, Hartmann A, et al:
Long noncoding RNA HOTAIR is upregulated in an aggressive subgroup
of gastrointestinal stromal tumors (GIST) and mediates the
establishment of gene-specific DNA methylation patterns. Genes
Chromosomes Cancer. 57:584–597. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Yan J, Chen D, Chen X, Sun X, Dong Q, Hu
C, Zhou F and Chen W: Downregulation of lncRNA CCDC26 contributes
to imatinib resistance in human gastrointestinal stromal tumors
through IGF-1R upregulation. Braz J Med Biol Res. 52:e83992019.
View Article : Google Scholar : PubMed/NCBI
|