Glucose-6-phosphate dehydrogenase (G6PD) is
conventionally considered as the first and rate-limiting enzyme of
the pentose phosphate pathway (PPP), is present in the cytoplasm of
red blood cells and protects cells against oxidative damage
(1). PPP produces large quantities
of NADPH and ribose 5-phosphate for various cellular synthetic
functions, such as the synthesis of aliphatic acid and sterols. In
addition, this pathway ensures glutathione (GSH) reduction, which
enhances antioxidant defense and promotes cell proliferation
(2). Individuals with clinical G6PD
deficiency are prone to neonatal jaundice, infection or drug
hemolysis, chronic non-spherical red blood cell hemolytic anemia
and abnormal lipid metabolism (3).
Furthermore, mutations in G6PD have been previously demonstrated to
be the primary cause of certain diseases, such as chronic hemolytic
anemia (4).
It has been shown that the expression of G6PD in
tumor cells is higher compared with that in normal cells, and its
expression is associated with the overall survival of tumor
patients (2,5). Numerous studies have also demonstrated
increased G6PD activity in several types of cancer, including
bladder cancer, carcinoma of the endometrium, prostate cancer,
kidney cancer, stomach cancer, cholangiocarcinoma, colon
adenocarcinoma, lung cancer, cervical cancer, carcinoma of the
ovary, hepatocellular carcinoma (HCC), glioma, pancreatic cancer
and melanoma (6–17). Based on the findings of previous
studies, the aim of the present review was to focus on the
mechanism underlying the role of G6PD in tumorigenesis and tumor
development, as G6PD is hypothesized to become a topic of increased
interest with regards to cancer in the future.
G6PD is encoded by the G6PD gene, which is localized
on chromosome X in the high-density region xq28, and is 18 kb in
length, consisting of 13 exons and 12 introns. G6PD exhibits
significant genetic diversity and the full-length cDNA of the human
G6PD is 1,548 bp. The quaternary structure of G6PD is present as a
dimer of two identical monomers (18), and the active form is a dimer or
tetramer. Each monomer contains 514 amino acids in humans, and has
a catalytic coenzyme-binding site and a substrate-binding site that
binds to glucose-6-phosphate (G6P) (19,20).
G6PD is a housekeeping enzyme and is present in all tissues and
organs. To the best of our knowledge, the first description of the
physiological role and effects of G6PD were presented in 1931
(21). To date, >400 biochemical
variants and >200 genetic variants of G6PD have been described
(22). G6PD is an essential enzyme
of the PPP, which is a metabolic pathway parallel to glycolysis
that catalyzes the dehydrogenation of G6P to produce
6-phosphogluconate. 6-phosphogluconic acid undergoes a series of
chemical reactions to produce 6-phosphofructose, which in turn can
enter the glycolytic pathway or aerobic oxidation pathways. PPP has
three important functions: i) PPP is the only means of using
glucose to produce ribose 5-phosphate, and thus provides raw
materials for nucleic acid synthesis in vivo. PPP can
protect and stabilize DNA, which may make cancer cells more
resistant to chemoradiotherapy damage (23–26).
ii) PPP provides NADPH and stabilizes the antioxidant defense
NADP/NADPH balance. NAPDH acts as an antioxidant and is used to
detoxify high levels of reactive oxygen species (ROS) produced
during rapid cell multiplication, thereby promoting cell survival
(21,27). iii) Pentose enters glycolysis
through the PPP (Fig. 1). In
mammals, the PPP occurs solely in the cytoplasm, and is found to be
most active in the human liver, mammary gland and adrenal cortex.
NADPH and pentose supply are prerequisites for runaway growth and
proliferation of cells, particularly in tumor cells (28). In addition to serving a role in cell
proliferation and aging, G6PD may also be involved in the
transmission of apoptotic signals (29). Another study found that highly
glycosylated G6PD increases glucose uptake in the PPP and increases
its activity. Therefore, blocking the glycosylation of G6PD may
reduce the proliferation of cancer cells in vitro and impair
tumor growth in vivo (30).
Being a functional pathway independent of glycolysis
and oxidative phosphorylation, PPP also serves a crucial role in
the liver (31), adipose tissue
(32), gonads (33), bone marrow (34), red blood cells and other tissues
(1). Since red blood cells do not
contain mitochondria, the PPP is the only source of NADPH;
therefore, protection from oxidative damage largely relies on G6PD
(35). Tumor cell metabolism
involves a number of metabolic pathways, including glucose
transport, glycolysis, PPP, glutamine metabolism and the electron
transport chain (36). Glycolysis
refers to the process of transformation of glucose or glycogen into
lactic acid and releasing energy through several intermediate steps
in the absence of oxygen. Normally, cancer cells metabolize
glucose, lactate, glutamine, pyruvic acid, acetate and aliphatic
acids at markedly higher rates compared with normal cells (37). Cancer cells take advantage of
conventional oxidative metabolism and glycolytic metabolism at the
same time. However, even under conditions of sufficient oxygen,
proliferation of cancer cells is marked by increased glycolytic
metabolism (38). This phenomenon
is termed the Warburg effect. The Warburg effect, first described
by the German biochemist Otto Warburg, is a metabolic feature of
aerobic glycolysis under conditions of sufficient glucose in tumors
(5,39). Since the rate of ATP produced by the
subsequent steps of glycolysis is 100 times faster compared with
that produced by oxidative phosphorylation, as is seen in cancer,
cells that rely on oxidative phosphorylation have an advantage
since glucose concentrations are higher (40,41).
Although it is somewhat slower to convert glucose to ATP compared
with other routes, glucose is the most plentiful nutrient present
in the blood, and is a metabolic substrate commonly used by tumor
cells (39).
The metabolic biology of tumors is complex, and a
growing body of evidence suggests that cancer cells have a unique
metabolic program that allows for rapid cell proliferation
(42). Previous studies have
demonstrated that G6PD expression or enzymatic activity is
increased in several types of tumors, including colorectal
(43), myeloma (44), bladder (11), breast (45), gastrointestinal (46), esophageal (47) and prostate cancer (13) In addition, the instability of G6PD
expression is also associated with the degree of malignancy of the
tumor. There is increasing evidence that G6PD deficiency affects
nucleated cells and cellular pathophysiology, which includes cell
proliferation disorders, accelerating cell senescence, increasing
susceptibility to viral infections and impairing embryonic
development (48,49). The energetic and biosynthetic
demands of rapidly proliferating cells, and the relatively balanced
levels of ROS regulated by G6PD, may highlight a means of treatment
for patients with cancer.
The nutrient demands and energy flow rates of tumor
cells are often higher compared with those of normal cells. It was
previously confirmed that regulation of tumor cell metabolism may
affect the status of the cancer (50). Rapidly dividing cancer cells require
three basic metabolic events: The formation of ATP, macromolecular
synthesis and cell assembly, and an appropriate cellular redox
environment (51). G6PD serves a
key role in maintaining a normal redox potential in cells by
reducing NADP+ to NADPH in the PPP, the dysregulation of
which results in insufficient antioxidant defense (52). Changes in the G6PD state are
associated with numerous pathophysiological cellular alterations
and diseases, including oxygen deficits (53), inflammation (54), infection (55), septicemia (56), diabetes (7), high blood pressure (57) and kidney diseases (14), amongst others. G6PD affects tumor
development by regulating several metabolic pathways (14,54).
The expression and activity of G6PD has been shown to be associated
with the degree of malignancy of a variety of tumors. It has been
demonstrated that blocking 80% of the G6PD activity in tumor cells
significantly reduced cell proliferation, migration, invasion, and
colony-formation; in addition, tumor cell apoptosis was increased
(58). Using GEPIA (version 2019;
gepia.cancer-pku.cn) combined with customizable functional analysis
(such as tumor/normal tissue differences) to examine and mine data
on multiple types of tumors in Genotype Tissue Expression
(gtexportal.org/home/) and The Cancer
Genome Atlas (portal.gdc.cancer.gov/), the clinical role of G6PD was
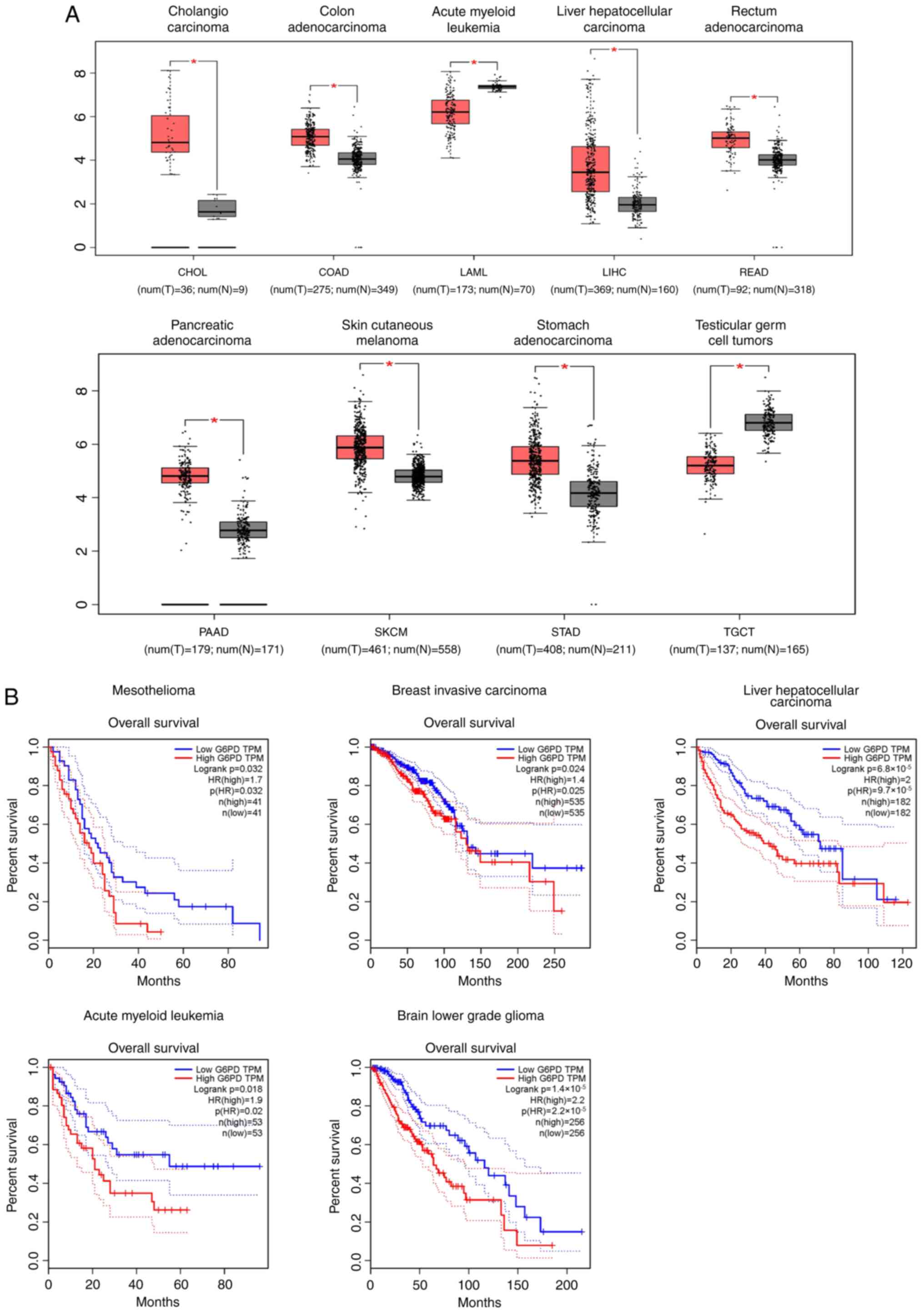
further explored (Fig. 2A and B).
Marked alterations of G6PD gene expression was observed in samples
from 9 types of tumors [cholangio carcinoma, colon adenocarcinoma,
acute myeloid leukemia (AML), HCC, rectal adenocarcinoma,
pancreatic adenocarcinoma, cutaneous melanoma, stomach
adenocarcinoma and testicular tumor] compared with normal tissues
(Fig. 2A). Furthermore, overall
survival analysis revealed that G6PD overexpression was associated
with a poor prognosis in certain types of cancer, including
mesothelioma, invasive breast carcinoma, HCC, AML and low-grade
brain glioma (Fig. 2B). Thus,
abnormal expression of GP6D is a common finding observed in
tumorigenesis, and G6PD is potentially carcinogenic.
Studies on several types of cancer have demonstrated
that overexpression of G6PD promotes tumorigenesis and development
(summarized in Table I).
In HCC, the phosphatase and tensin homologue (PTEN)
signaling pathway is downregulated compared with normal tissues,
and its ability to inhibit tumorigenesis is reduced (59). PTEN can inhibit the formation of
dimers of the G6PD holoenzyme (60). In advanced stage liver cancer, the
expression of G6PD is significantly increased, whereas expression
of PTEN is decreased (60).
Huh-sh-heterogeneous nuclear ribonucleoprotein (hnRNPK) is known to
regulate G6PD-mRNA splicing. It has been demonstrated that T-cell
leukemia 1 (Tcl1) affects cell resistance in HCC. Tcl1 directly
interacts with hnRNPK in vitro to competitively inhibit the
binding of hnRNPK and G6PD. Conversely, increasing evidence is
indicating that PTEN disrupts the interaction between Tcl1 and
hnRNPK by inactivating Tcl1 (59).
In summary, PTEN induces the expression of G6PD through the
Tcl1/hnRNPK/G6PD axis, and thus promotes hepatocarcinogenesis.
In HCC cell lines, it was found that the number of
β-galactosidase-positive cells and expression of p21 (a classical
aging marker) increased significantly following G6PD gene knockout,
which indicated that the decrease of G6PD delayed the growth of
tumor cells (60). The possible
implications of this in the treatment of liver cancer is that G6PD
inhibition may increase the sensitivity of liver cancer cells to
chemotherapeutic drugs (61).
Chronic hepatitis B virus (HBV) infection is considered to be
involved in the pathogenesis of HCC. Therefore, the role of G6PD in
HBV is also associated with its effect on liver cancer. It has been
reported that G6PD may be associated with HBV replication, due to
the fact that small interfering RNA-mediated silencing of G6PD
reduced HBV production. In addition, when G6PD was silenced, the
concentration of HBsAg and HBeAg secreted in the supernatant was
also significantly reduced (62).
Renal cell carcinoma (RCC) is the most common type
of renal malignancy, accounting for ~3% of all types of tumors
(63,64). RCC is the most aggressive type of
urinary tract tumor (63,65). Several studies have concluded that
RCC can be viewed as a metabolic disease, the occurrence of which
is affected by metabolic changes (66–68).
G6PD is not only upregulated in all types of RCC specimens, but
also exhibits increased activity in RCC cells (52). Elevated G6PD expression is
associated with shorter overall survival. It has been confirmed
that the median survival of the patients with high G6PD expression
was significantly shorter compared with that of patients with low
G6PD expression (69). G6PD
overexpression promotes RCC cell proliferation, whereas G6PD
silencing reduces the rate of RCC cell proliferation in
vitro and inhibits xenograft tumor development in vivo
(70). It was previously
demonstrated that G6PD may promote tumorigenesis by increasing the
rate of proliferation and enhancing antioxidant defense (71), whereas G6PD disorders render tumor
cells more adaptable to the immune response and more aggressive
(72). The pathways involving G6PD
in the proliferation of renal cancer cells are as follows: i) G6PD
stimulates ROS production through NADPH oxidase 4 (NOX4); ii) ROS
accumulation increases phospho-(p-)signal transducer and activator
of transcription 3 (STAT3); and iii) G6PD activates p-STAT3/STAT3
signaling and enhances cyclin D1 expression (73). Briefly, G6PD can stimulate RCC
tumorigenesis in vivo and in vitro by activating the
G6PD/ROS/p-STAT3/cyclin D1 signaling pathway in RCC (70).
A number of studies on hematological malignancies
have investigated the role of G6PD. It has been reported that NADPH
produced by G6PD may be used for lipogenic reactions in leukemia
cells (74). AML is a hematological
malignancy and it is the most common type of adult acute leukemia
(75), characterized by abnormal
growth of myeloid progenitor cells and inhibition of cell
differentiation (76). PPP, which
is closely associated with glycolysis, is also frequently altered
in AML. Glucose catabolism in leukemia cells may be mediated
through the PPP rather than the glycolysis pathway, indicating that
PPP may serve an important role in leukemia metabolism (74,77–80).
It has been observed that AML cells rely on glucose metabolism for
survival, and a high-flux of glucose is primarily maintained by PPP
(81). The suppression of G6PD
abolished glycolysis, weakened the effects of PPP and decreased the
glucose utilization rate. As a result, AML cells exhibited arrested
growth or death, and became more sensitive to chemotherapeutic
drugs (82–84). In vivo mouse models have
shown that enhanced glucose uptake accelerates the development of
leukemia in vivo (75,85,86).
Therefore, it may be concluded that G6PD is involved in tumor
invasion by affecting other enzymes.
In terms of protein interactions, the deacetylation
of G6PD by NAD-dependent deacetylase sirtuin 2 (SIRT2) can enhance
the production of NADPH and promote the proliferation of leukemia
cells (74). It is hypothesized
that the binding of G6PD and SIRT2 is disrupted in heat shock
protein 27 (HSPB1-L) knockout cells (87). When HSPB1 expression increases, it
can promote the binding of SIRT2 and G6PD, in-turn affecting tumor
cell proliferation. By activating the phosphoinositide
3-kinase/Akt/mammalian target of rapamycin signaling pathway, the
activity of G6PD is regulated by sterol-regulatory-element-binding
protein 1c, which ultimately promotes tumor cell transformation
(28).
The role of G6PD in several other types of cancer
are summarized below. In gastric cancer, the expression levels of
G6PD protein was found to be associated with the
Tumor-Node-Metastasis stage. The rate of G6PD protein expression in
gastric cancer tissues with positive lymph node metastasis was also
higher compared with that in lymph node-negative patients (46,88).
Colorectal cancer (CRC) is the third most commonly diagnosed type
of cancer in Western countries. Moreover, the morbidity rates of
CRC have rapidly increased in China over the past two decades
(61,89,90).
It has been reported that G6PD expression is upregulated in CRC
cells and patient specimens. Breast cancer has the highest
incidence amongst all types of cancer in women, and remains one of
the leading causes of cancer-related death (91). Upregulated expression of G6PD is
considered a predictor of a high risk of recurrence and metastasis
in patients with breast cancer (92). In prostate cancer, G6PD has been
used as a biomarker of diagnosis and prognosis for 30 years
(93). G6PD activity in patients
with prostate cancer is 4-fold higher compared with that in
patients with benign prostatic hyperplasia (93,94).
Previous studies have shown that the G6PD levels increase via the
PPP in prostate cancer (95–97).
In glioma, the activity and content of G6PD affects the efficacy of
chemotherapy and radiotherapy (52,98).
When human melanoma was investigated using a G6PD knockdown
xenograft mouse model, it was observed that G6PD expression was
closely associated with tumor formation, size and weight (99). In bladder cancer, knockdown of G6PD
inhibits tumor cell proliferation and colony formation (11).
Therefore, G6PD expression is frequently found to be
elevated in several different types of cancer, and its internal
mechanisms of action have become the focus of increasing
attention.
The mechanisms through which G6PD regulates tumor
development are yet to be fully elucidated. It is well established
that G6PD levels are aberrantly elevated in several different types
of cancer, and promotes the proliferation of cancer cells through
production of ribose-5-phosphate and NADPH as discussed above. The
most significant function of NADPH is to form two GSH molecules by
reducing one GSH disulfide molecule (100). In turn, GSH may be used to defend
against excessive accumulation of ROS (74). ROS, which are produced at low levels
by the electron transport chain as part of physiological cellular
metabolism, serve a physiologically important role in the
regulation of cell signaling, proliferation and differentiation.
When ROS production increases above a certain cell-dependent
threshold, it may damage cellular components, resulting in cell
death (101). Increased generation
of ROS in metabolically active cells requires an appropriate level
of antioxidants, such as the reduced form of GSH, produced by the
PPP (102). It has been shown that
ROS can contribute to tumorigenesis by activating various signaling
pathways that control cell growth and proliferation (103). However, excess ROS also induces
apoptosis (102). If ROS
production is higher than the antioxidant defense capacity of the
cell, it may lead to oxidative stress, thereby causing cell death.
Furthermore, as an organisms ages, the capacity to repair
ROS-induced injury becomes gradually weaker and the accumulation of
ROS in vivo results in terminal damage (100). That is, when the expression or
activity of G6PD gradually decreases, the antioxidant ability of
the PPP is weakened and the levels of ROS progressively increases.
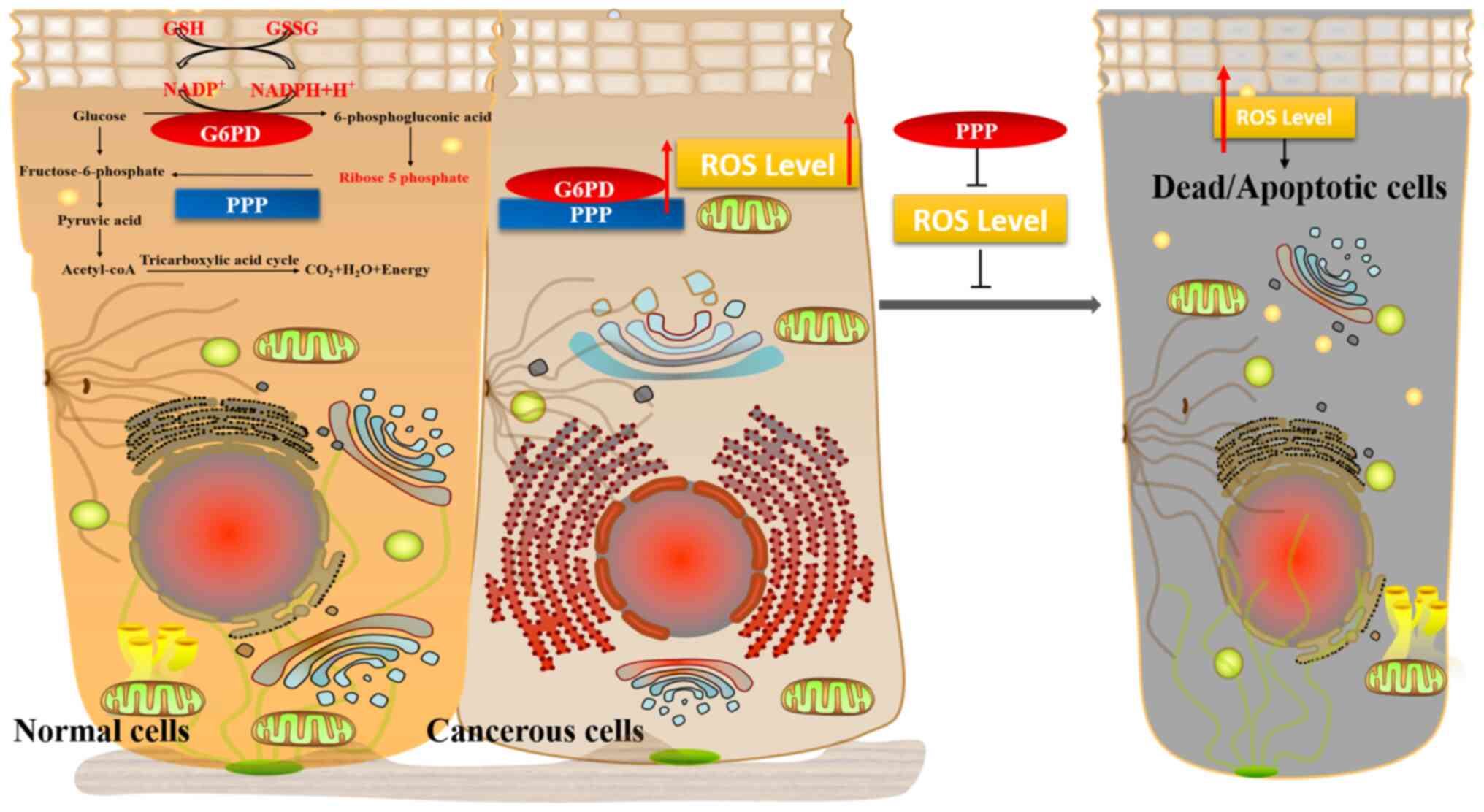
G6PD participates in the PPP to promote the production of GSH. As a
classic antioxidant, GSH can defend against excessive production of
ROS, thereby maintaining cells, particularly tumor cells, in a
proliferating state (Fig. 1). To
resist oxidative stress, cancer cells have enhanced antioxidant
programs (50). Thus, a key
molecule produced as a result of altered cancer metabolism is
NADPH, which is an antioxidant and forms part of the defense
against ROS (50). There are two
primary forms of NADPH production: i) NADH and NADP+
generate NADPH by catalysis of mitochondrial transhydrogenase and
ii) NADP+ produces NADPH by catalysis of a variety of
NADP+-dependent enzymes (104). Currently, increasing evidence is
suggesting that NAD (NAD+ or NADH) and NADP
(NADP+ or NADPH) can influence various biological
processes, including energy metabolism, mitochondrial functions,
calcium homeostasis, antioxidation/generation of oxidative stress,
gene expression, immunological functions, aging and cell death
(104–107). In fact, the sources of NADPH
generation may determine the different biological effects of NADPH:
The NADPH catalyzed by the mitochondrial enzymes primarily
contribute to antioxidation and biosynthesis, whereas the NADPH
catalyzed by the cytosolic enzymes contributes to NADPH
oxidase-dependent ROS generation when NADPH oxidase (NOX) is
activated (104). The NOX family
consists of seven enzymatic isoforms, and is an enzyme complex with
the unique function of producing superoxide anions and ROS through
consumption of NADPH (108). The
process of NOX-mediated production of ROS is primarily through
one-electron reduction of oxygen to superoxide depending on NADPH
(109). NADPH is produced by
glucose, once inside the cells as an intermediate of glycolysis, to
generate glucose 6-phosphate (G6P). G6P continues to pass through
the glycolytic pathway or is split with a hexose-phosphate shunt to
produce NADPH (108). In tumor
cells, ROS promotes cell proliferation via redox reactions. ROS
modulates several signaling pathways associated with cellular
transformation, inflammation, tumor survival, proliferation,
invasion, angiogenesis and metastasis of cancer (110). For maintaining cell proliferation,
the levels of ROS in cancer cells is elevated compared with normal
cells (105,106,111,112). Excessive accumulation of ROS may
lead to cell apoptosis or even death; however, tumor cells may
maintain proliferation without being affected by ROS, primarily due
to the ability of anti-oxidative stress with the participation of
NADPH (50,74,101,102). In the PPP pathway, NADPH maintains
the reduced state of GSH, thereby inducing resistance to ROS
(102). This process is involved
in the anti-ROS effects of NADPH (102). Therefore, it may be concluded that
a moderate increase in G6PD activity is beneficial for avoiding the
harmful effects of ROS. Of note, G6PD activity and its expression
are increased by oncogenes, such as KRAS (113), and suppressed by tumor
suppressors, such as P53 or PTEN (60,114).
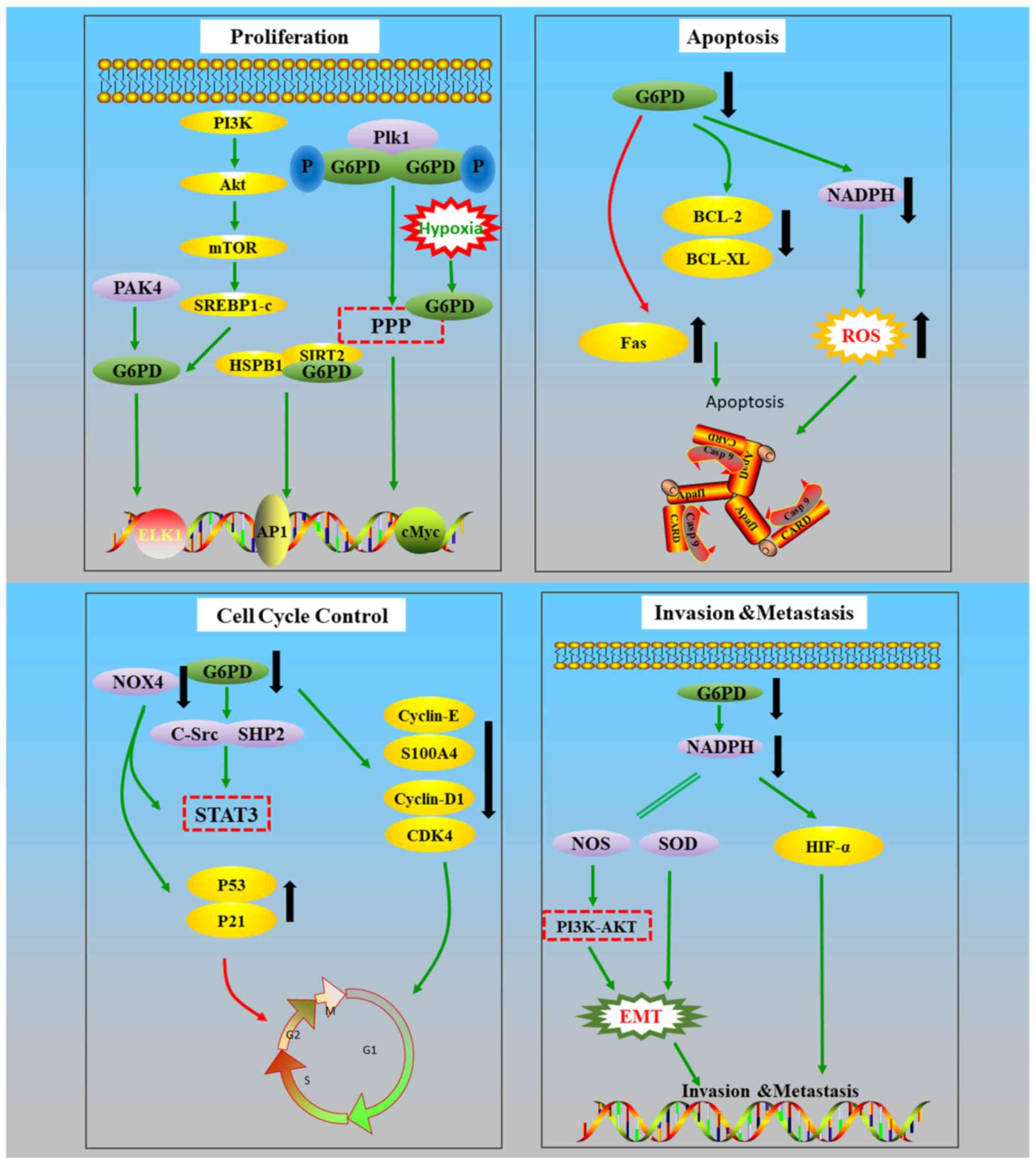
Below, a summary of the studies investigating the underlying
mechanisms, with a focus on proliferation, cell cycle progression,
apoptosis, invasion and migration is provided (Fig. 3).
G6PD promotes epithelial-mesenchymal transition
(EMT) by activating the STAT3 pathway. and EMT serves a key role in
enhancing tumor cell invasion and metastasis (61). G6PD is important for several
cellular processes and enzymes using NADPH, including enzymes in
the antioxidant system (21),
including nitric oxide synthase and superoxide dismutase, both of
which are NADPH-dependent enzymes (115) and can promote EMT and invasion of
pancreatic cancer cells (116).
Regarding cell apoptosis, the expression levels of
Bcl-2 and Bcl-xL, which are both inhibitors of apoptosis, were
found to be reduced in cells with low G6PD expression levels.
However, the expression of Fas was increased, and Fas can bind to
the cytokines of the death receptor TNFRSF6/Fas and induce
apoptosis caused by T-cell-mediated cytotoxicity. Athanogene 3
(BAG3) protein directly binds to G6PD to exert a tumor
suppressor-like function in HCCs (117). The Bcl-2 associated BAG3 protein
is involved in several cellular functions, including cell cycle,
autophagy, cell growth and pathogen replication (118,119). Through Bcl-2, BAG3 suppresses
dimerization and activity of G6PD. It is hypothesized that G6PD may
affect apoptosis by interacting with apoptosis-related factors
(71,99).
Although there are no definitive conclusions on the
role of G6PD in regulating the cell cycle, the results of other
studies in this area are summarized in the present review. It has
been established that in cells lacking G6PD, downregulation of the
cell cycle proteins cyclin D1 and E, and S100a4 is observed
(99). The expression of cyclin D1
is decreased following G6PD knockout (120), and this affected cell growth and
division via regulation of the G1/S transition (63,121).
Additionally, G6PD overexpression can activate the STAT3 pathway,
primarily through increasing the levels of p-STAT3. When the
expression of G6PD is low, protein tyrosine kinase (c-SRC) and
protein tyrosine phosphatase (SHP2) are suppressed. As c-SRC and
SHP2 can regulate the DNA-binding activity of STAT3, G6PD serves an
important indirect role in DNA-binding via the above mentioned
pathways, thus affecting cell cycle progression and suppressing
cell proliferation (120). After
silencing G6PD and NOX4, the expression levels of p53 and p21 were
upregulated and the cell cycle was arrested in the G1/S phase. At
the same time, the protein levels of cyclin D1 and cyclin-dependent
kinase 4 were decreased. Upregulation of p53 may be achieved by
downregulating PAK4, resulting in S phase arrest of the cell cycle
(122). PAK4 can also directly
interact with G6PD, and then enhance the activity of G6PD to
promote glucose intake and the production of NADPH, thereby
promoting tumor growth (123).
These results indicate that the cell cycle is also affected by this
pathway. Polo-like kinase 1 (Plk1), a serine/threonine kinase,
regulates cell mitosis (124).
G6PD is activated when Plk1 binds to G6PD. Moreover, G6PD-mediated
PPP may be involved in the regulation of the cell cycle by Plk1
(124). By contrast, p53 inhibits
the proliferation of tumor cells by repressing the formation of
G6PD dimers (70). The mechanism by
which G6PD affects the cell cycle is debated. In G6PD-deficient
cells, there was a noticeable delay in cell proliferation, which
was found to resemble cell senescence (29).
In an anoxic environment, G6PD also serves a
regulatory role in the survival of tumor cells. Abnormal
acetylation was found to be associated with the growth and invasion
of cancer cells in vitro and in vivo (125,126). Under hypoxic conditions, the
glycosylation of G6PD is activated, thus activating the PPP
(30). Hypoxia-inducible factor-1α
(HIF-1α) is present in abundant quantities in a variety of human
tumor cells, and serves an important role in promoting tumor
angiogenesis, accelerating tumor cell proliferation, and promoting
invasion and metastasis (127). It
has been suggested that the G6PD gene may impair the stability and
expression of HIF-1α by downregulating the levels of NADPH, which
may in-turn affect the hypoxia response of tumor cells (127). However, the detailed mechanisms by
which G6PD affects tumor cells remain unknown.
Investigation of the mechanism by which G6PD
promotes tumorigenesis and tumor development may improve our
understanding of the association between G6PD and cancer, and may
also provide theoretical guidance for targeted therapy.
Successful treatment, recurrence and resistance of
cancer are commonly encountered problems. The results of
traditional methods of cancer treatment (including surgical
chemotherapy, radiotherapy, use of traditional Chinese medicines
and biological therapy) are not optimal. Although first-line
treatment with cytotoxic chemotherapy has achieved a high rate of
remission, the majority of the patients eventually succumb to the
disease. In order to increase the cure rates, improve the
prognosis, and to minimize the suffering of the patients, more
effective and less toxic cancer treatments are urgently required.
Molecular targeted cancer treatment may hold promise as a
therapeutic approach. The expression of G6PD may represent a
potential treatment option. As the effect of G6PD activity on the
cell cycle has been demonstrated, several pathways that involve
G6PD may be targeted to improve the efficacy of treatment. The
interactions between G6PD and multiple signaling pathways directly
or indirectly, affects cell cycle progression of tumor cells,
eventually altering cell proliferation or apoptosis. Ongoing
studies in different types of cancer, and investigation of G6PD
specificity may highlight potential avenues to overcome these
problems. The PPP may also affect the adaptation of tumor cells to
the changes in the microenvironment, and enable them to survive
longer compared with normal cells (128,129). Although the detailed mechanisms of
the functional mechanisms of G6PD are not fully understood, further
investigations should address the following possibilities: i) The
defense of tumor cells against oxidative stress may be weakened by
inhibiting G6PD to interrupt the energy supply, leading to
increased sensitivity of tumor cells to oxidants. ii) Based on the
principle of metabolic reprogramming, by adjusting the activity of
G6PD to affect the role of PPP in biosynthesis, the resistance of
tumor cells to drugs may be overcome. iii) Alternatively, certain
types of cancer cells may adapt to high levels of carcinogenic
signals by destroying their aging or apoptotic induction circuits.
G6PD may be involved in this process, or it may enable tumor cells
to escape certain events that have a negative effect on cancer cell
proliferation.
The metabolic reprogramming alters the tumor
microenvironment affecting cancer cells and other immune cells,
such as macrophages, T lymphocytes and myeloid-derived suppressor
cells (5,130). Targeting metabolic transformation
or metabolism-regulated signaling pathways in tumor development may
be a potential anticancer treatment strategy, alone or in
combination with immunotherapy. At present, as one of the basic
characteristics of tumor cells, metabolic programming has been
recognized as a new cancer hallmark (130). In order to meet the needs of rapid
proliferation for energy and to counteract the increased oxidative
stress, tumor cells are reprogrammed, and this includes changes to
their glucose, fat and other metabolic pathways. Ribose-5-phosphate
and NADPH are important products catalyzed by G6PD. The former is
the raw material of nucleic acid synthesis (108), which is the basis of cell
division, proliferation and maturation, whereas the latter is
involved in fatty acid, nucleic acid and ROS metabolism, amongst
other functions, acting as a hydrogen donor in all these processes
(104). In this context, G6PD
serves an important role in promoting biosynthesis, and an increase
of G6PD activity further promotes the activity of PPP. As a result,
a large quantity of glucose is consumed, which is involved in
numerous biosynthetic processes and this produces a large set of
reductants (23–26). Thus, it is hypothesized that
metabolic reprogramming by PPP may be of value as a biomarker for
detection of cancer and as a target for the development of novel
anticancer treatments (36).
G6PD inhibitors have been developed, and
1-bromoacety l-3,3-dinitroazetidine (RRx-001) is one of these.
RRx-001 is both a G6PD inhibitor and a multifunctional anticancer
drug that has been tested in phase III clinical trials (131). RRx-001 has been evaluated in ~300
patients in 9 clinical trials (131), and the results were promising,
with no associated hematological toxicities reported. Based on the
proven antitumor activity and favorable toxicity profile in the
phase II clinical trial, RRx-001 has been approved by the FDA and
EMA, and may be used in phase III multi-center studies in subjects
with relapsed/refractory solid tumors (131,132). However, in vitro
experiments have shown that high concentrations (7–10 µM) of
RRx-001 reduce the viability of peripheral mononuclear blood cells
by 30–70%, indicating that normal cells are not entirely refractory
to RRx-001 (133). Polydatin is
another known G6PD inhibitor, which may cause ROS accumulation and
enhanced endoplasmic reticulum stress by inhibiting G6PD (134). Experiments have shown that
polydatin is non-toxic to animals at a dose of 200 mg/kg (134). Phase II clinical trials
demonstrated that it is also well tolerated in humans at a dose of
40 mg twice a day for 90 days. Therefore, polydatin may be used
with confidence in clinical treatment (134). Thus, it appears that the toxicity
of G6PD inhibitors to normal cells is limited, but not completely
absent. However, G6PD is still hypothesized to serve as a promising
target for anti-cancer treatments.
In summary, G6PD may be characterized as a
participant rather than a bystander in the process of
tumorigenesis, and downregulation of G6PD may enhance the
sensitivity of certain types of tumors to chemotherapeutic drugs.
Therefore, G6PD may serve as an important target for cancer therapy
and for overcoming resistance to chemotherapy.
We would like to thank Ms Mengjie Guo (School of
Medicine and Holistic Integrative Medicine, Nanjing University of
Chinese Medicine, Nanjing, China) for her outstanding efforts in
the revision of this manuscript.
This work was supported by grants from the National
Natural Science Foundation of China (grant nos. 81670200 and
81970196); Innovation Team of Six Talent Peaks Project in Jiangsu
Province (grant no. TD-SWYY-015); and A Project Funded by the
Priority Academic Program Development of Jiangsu Higher Education
Institutions (Integration of Chinese and Western Medicine).
The data used for the analyses described in this
manuscript were obtained from the GTEx (version 2019;
gepia.cancer-pku.cn) and The Cancer Genome Atlas (portal.gdc.cancer.gov/).
YY and CG conceived the subject of the review. RL
and WW wrote the manuscript, analyzed the data, and plotted the
graphs. CG and YY edited the manuscript. All authors have read and
approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Luzzatto L and Arese P: Favism and
Glucose-6-phosphate dehydrogenase deficiency. N Engl J Med.
378:60–71. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang HC, Wu YH, Liu HY, Stern A and Chiu
DT: What has passed is prolog: New cellular and physiological roles
of G6PD. Free Radic Res. 50:1047–1064. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luzzatto L and Seneca E: G6PD deficiency:
A classic example of pharmacogenetics with on-going clinical
implications. Br J Haematol. 164:469–480. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Longo L, Vanegas OC, Patel M, Rosti V, Li
H, Waka J, Merghoub T, Pandolfi PP, Notaro R, Manova K and Luzzatto
L: Maternally transmitted severe glucose 6-phosphate dehydrogenase
deficiency is an embryonic lethal. EMBO J. 21:4229–4239. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun L, Suo C, Li ST, Zhang H and Gao P:
Metabolic reprogramming for cancer cells and their
microenvironment: Beyond the Warburg Effect. Biochim Biophys Acta
Rev Cancer. 1870:51–66. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ho HY, Cheng ML and Chiu DT:
Glucose-6-phosphate dehydrogenase-beyond the realm of red cell
biology. Free Radic Res. 48:1028–1048. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heymann AD, Cohen Y and Chodick G:
Glucose-6-phosphate dehydrogenase deficiency and type 2 diabetes.
Diabetes Care. 35:e582012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carette C, Dubois-Laforgue D, Gautier JF
and Timsit J: Diabetes mellitus and glucose-6-phosphate
dehydrogenase deficiency: from one crisis to another. Diabetes
Metab. 37:79–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wan GH, Tsai SC and Chiu DT: Decreased
blood activity of glucose-6-phosphate dehydrogenase associates with
increased risk for diabetes mellitus. Endocrine. 19:191–195. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang Z, Jiang C, Feng Y, Chen R, Lin X,
Zhang Z, Han L, Chen X, Li H, Guo Y and Jiang W: Effects of G6PD
activity inhibition on the viability, ROS generation and mechanical
properties of cervical cancer cells. Biochim Biophys Acta.
1863:2245–2254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen X, Xu Z, Zhu Z, Chen A, Fu G, Wang Y,
Pan H and Jin B: Modulation of G6PD affects bladder cancer via ROS
accumulation and the AKT pathway in vitro. Int J Oncol.
53:1703–1712. 2018.PubMed/NCBI
|
|
12
|
Ai G, Dachineni R, Kumar DR, Alfonso LF,
Marimuthu S and Bhat GJ: Aspirin inhibits glucose-6-phosphate
dehydrogenase activity in HCT 116 cells through acetylation:
Identification of aspirin-acetylated sites. Mol Med Rep.
14:1726–1732. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen J, Cao S, Situ B, Zhong J, Hu Y, Li
S, Huang J, Xu J, Wu S, Lin J, et al: Metabolic reprogramming-based
characterization of circulating tumor cells in prostate cancer. J
Exp Clin Cancer Res. 37:1272018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spencer NY and Stanton RC: Glucose
6-phosphate dehydrogenase and the kidney. Curr Opin Nephrol
Hypertens. 26:43–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shan CL, Lu Z, Li Z, Sheng H, Fan J, Qi Q,
Liu S and Zhang S: 4-hydroxyphenylpyruvate dioxygenase promotes
lung cancer growth via pentose phosphate pathway (PPP) flux
mediated by LKB1-AMPK/HDAC10/G6PD axis. Cell Death Dis. 10:5252019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu Y, Gao W, Zhang Y, Wu S, Liu Y, Deng X,
Xie L, Yang J, Yu H, Su J and Sun L: ABT737 reverses cisplatin
resistance by targeting glucose metabolism of human ovarian cancer
cells. Int J Oncol. 53:1055–1068. 2018.PubMed/NCBI
|
|
17
|
Minchenko OH, Garmash IA, Minchenko DO,
Kuznetsova AY and Ratushna OO: Inhibition of IRE1 modifie s hypoxic
regulation of G6PD, GPI, TKT, TALDO1, PGLS and RPIA genes
expression in U87 glioma cells. Ukr Biochem J. 89:38–49. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kiani F, Schwarzl S, Fischer S and Efferth
T: Three-dimensional modeling of Glucose-6-phosphate
dehydrogenase-deficient variants from German ancestry. PLoS One.
2:e6252007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kotaka M, Gover S, Vandeputte-Rutten L, Au
SW, Lam VM and Adams MJ: Structural studies of glucose-6-phosphate
and NADP+ binding to human glucose-6-phosphate
dehydrogenase. Acta Crystallogr D Biol Crystallogr. 61:495–504.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Au SW, Gover S, Lam VM and Adams MJ: Human
glucose-6-phosphate dehydrogenase: The crystal structure reveals a
structural NADP(+) molecule and provides insights into enzyme
deficiency. Structure. 8:293–303. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stanton RC: Glucose-6-phosphate
dehydrogenase, NADPH, and cell survival. IUBMB life. 64:362–369.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Minucci A, Moradkhani K, Hwang MJ, Zuppi
C, Giardina B and Capoluongo E: Glucose-6-phosphate dehydrogenase
(G6PD) mutations database: Review of the ‘old’ and update of the
new mutations. Blood Cells Mol Dis. 48:154–165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramos-Montoya A, Lee WN, Bassilian S, Lim
S, Trebukhina RV, Kazhyna MV, Ciudad CJ, Noé V, Centelles JJ and
Cascante M: Pentose phosphate cycle oxidative and nonoxidative
balance: A new vulnerable target for overcoming drug resistance in
cancer. Int J Cancer. 119:2733–2741. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brizel DM, Schroeder T, Scher RL, Walenta
S, Clough RW, Dewhirst MW and Mueller-Klieser W: Elevated tumor
lactate concentrations predict for an increased risk of metastases
in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 51:349–353.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Seyfried TN, Sanderson TM, El-Abbadi MM,
McGowan R and Mukherjee P: Role of glucose and ketone bodies in the
metabolic control of experimental brain cancer. Br J Cancer.
89:1375–1382. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu RH, Pelicano H, Zhou Y, Carew JS, Feng
L, Bhalla KN, Keating MJ and Huang P: Inhibition of glycolysis in
cancer cells: A novel strategy to overcome drug resistance
associated with mitochondrial respiratory defect and hypoxia.
Cancer Res. 65:613–621. 2005.PubMed/NCBI
|
|
27
|
Patra KC and Hay N: The pentose phosphate
pathway and cancer. Trends Biochem Sci. 39:347–354. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cernaj IE: Simultaneous dual targeting of
Par-4 and G6PD: A promising new approach in cancer therapy?
Quintessence of a literature review on survival requirements of
tumor cells. Cancer Cell Int. 16:872016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ho HY, Cheng ML and Chiu DT:
Glucose-6-phosphate dehydrogenase-from oxidative stress to cellular
functions and degenerative diseases. Redox Rep. 12:109–118. 2013.
View Article : Google Scholar
|
|
30
|
Rao X, Duan X, Mao W, Li X, Li Z, Li Q,
Zheng Z, Xu H, Chen M, Wang PG, et al: O-GlcNAcylation of G6PD
promotes the pentose phosphate pathway and tumor growth. Nat
Commun. 6:84682015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jin ES, Sherry AD and Malloy CR:
Interaction between the pentose phosphate pathway and
gluconeogenesis from glycerol in the liver. J Biol Chem.
289:32593–32603. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park YJ, Choe SS, Sohn JH and Kim JB: The
role of glucose-6-phosphate dehydrogenase in adipose tissue
inflammation in obesity. Adipocyte. 6:147–153. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Keran EE and Barker KL: Regulation of
glucose-6-phosphate dehydrogenase activity in uterine tissue in
organ culture. Endocrinology. 99:1386–1397. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chandra R, Villanueva E, Feketova E,
Machiedo GW, Haskó G, Deitch EA and Spolarics Z: Endotoxemia
down-regulates bone marrow lymphopoiesis but stimulates
myelopoiesis: The effect of G6PD deficiency. J Leukoc Biol.
83:1541–1550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van Zwieten R, Verhoeven AJ and Roos D:
Inborn defects in the antioxidant systems of human red blood cells.
Free Radic Biol Med. 67:377–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schulze A and Harris AL: How cancer
metabolism is tuned for proliferation and vulnerable to disruption.
Nature. 491:364–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Martinez-Outschoorn UE, Peiris-Pages M,
Pestell RG, Sotgia F and Lisanti MP: Cancer metabolism: A
therapeutic perspective. Nat Rev Clin Oncol. 14:11–31. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Icard P, Shulman S, Farhat D, Steyaert JM,
Alifano M and Lincet H: How the Warburg effect supports
aggressiveness and drug resistance of cancer cells? Drug Resist
Updat. 38:1–11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boroughs LK and DeBerardinis RJ: Metabolic
pathways promoting cancer cell survival and growth. Nat Cell Biol.
17:351–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pfeiffer T, Schuster S and Bonhoeffer S:
Cooperation and competition in the evolution of ATP-producing
pathways. Science. 292:504–507. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cox E and Bonner J: Ecology. The
advantages of togetherness. Science. 292:448–449. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Israelsen WJ and Vander Heiden MG: ATP
consumption promotes cancer metabolism. Cell. 143:669–671. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ju HQ, Lu YX, Wu QN, Liu J, Zeng ZL, Mo
HY, Chen Y, Tian T, Wang Y, Kang TB, et al: Disrupting
G6PD-mediated Redox homeostasis enhances chemosensitivity in
colorectal cancer. Oncogene. 36:6282–6292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
McBrayer SK, Yarrington M, Qian J, Feng G,
Shanmugam M, Gandhi V, Krett NL and Rosen ST: Integrative gene
expression profiling reveals G6PD-mediated resistance to
RNA-directed nucleoside analogues in B-cell neoplasms. PLoS One.
7:e414552012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang HS, Zhang ZG, Du GY, Sun HL, Liu HY,
Zhou Z, Gou XM, Wu XH, Yu XY and Huang YH: Nrf2 promotes breast
cancer cell migration via up-regulation of G6PD/HIF-1α/Notch1 axis.
J Cell Mol Med. 23:3451–3463. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang J, Yuan W and Chen Z, Wu S, Chen J,
Ge J, Hou F and Chen Z: Overexpression of G6PD is associated with
poor clinical outcome in gastric cancer. Tumour Biol. 33:95–101.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang X, Li X, Zhang X, Fan R, Gu H, Shi Y
and Liu H: Glucose-6-phosphate dehydrogenase expression is
correlated with poor clinical prognosis in esophageal squamous cell
carcinoma. Eur J Surg Oncol. 41:1293–1299. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang HC, Chen TL, Wu YH, Cheng KP, Lin YH,
Cheng ML, Ho HY, Lo SJ and Chiu DT: Glucose 6-phosphate
dehydrogenase deficiency enhances germ cell apoptosis and causes
defective embryogenesis in Caenorhabditis elegans. Cell Death Dis.
4:e6162013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu YH, Lee YH, Shih HY, Chen SH, Cheng YC
and Tsun-Yee Chiu D: Glucose-6-phosphate dehydrogenase is
indispensable in embryonic development by modulation of
epithelial-mesenchymal transition via the NOX/Smad3/miR-200b axis.
Cell Death Dis. 9:102018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tong X, Zhao F and Thompson CB: The
molecular determinants of de novo nucleotide biosynthesis in cancer
cells. Curr Opin Genet Dev. 19:32–37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Frederiks WM, Bosch KS, Hoeben KA, van
Marle J and Langbein S: Renal cell carcinoma and oxidative stress:
The lack of peroxisomes. Acta Histochem. 112:364–371. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chettimada S, Joshi SR, Alzoubi A, Gebb
SA, McMurtry IF, Gupte R and Gupte SA: Glucose-6-phosphate
dehydrogenase plays a critical role in hypoxia-induced
CD133+ progenitor cells self-renewal and stimulates
their accumulation in the lungs of pulmonary hypertensive rats. Am
J Physiol Lung Cell Mol Physiol. 307:L545–L556. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Peiró C, Romacho T, Azcutia V, Villalobos
L, Fernández E, Bolaños JP, Moncada S and Sánchez-Ferrer CF:
Inflammation, glucose, and vascular cell damage: The role of the
pentose phosphate pathway. Cardiovasc Diabetol. 15:822016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wu YH, Tseng CP, Cheng ML, Ho HY, Shih SR
and Chiu DT: Glucose-6-phosphate dehydrogenase deficiency enhances
human coronavirus 229E infection. J Infect Dis. 197:812–816. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rostami-Far Z, Ghadiri K, Rostami-Far M,
Shaveisi-Zadeh F, Amiri A and Rahimian Zarif B: Glucose-6-phosphate
dehydrogenase deficiency (G6PD) as a risk factor of male neonatal
sepsis. J Med Life. 9:34–38. 2016.PubMed/NCBI
|
|
57
|
Zhao J, Zhang X, Guan T, Wang X, Zhang H,
Zeng X, Dai Q, Wang Y, Zhou L and Ma X: The association between
glucose-6-phosphate dehydrogenase deficiency and abnormal blood
pressure among prepregnant reproductive-age Chinese females.
Hypertens Res. 42:75–84. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li D, Zhu Y, Tang Q, Lu H, Li H, Yang Y,
Li Z and Tong S: A new G6PD knockdown tumor-cell line with reduced
proliferation and increased susceptibility to oxidative stress.
Cancer Biother Radiopharm. 24:81–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang XQ, Ongkeko WM, Chen L, Yang ZF, Lu
P, Chen KK, Lopez JP, Poon RT and Fan ST: Octamer 4 (Oct4) mediates
chemotherapeutic drug resistance in liver cancer cells through a
potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology.
52:528–539. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hong XH, Song RP, Song HW, Zheng T, Wang
J, Liang Y, Qi S, Lu Z, Song X, Jiang H, et al: PTEN antagonises
Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to
hepatocarcinogenesis. Gut. 63:1635–1647. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lu M, Lu L, Dong Q, Yu G, Chen J, Qin L,
Wang L, Zhu W and Jia H: Elevated G6PD expression contributes to
migration and invasion of hepatocellular carcinoma cells by
inducing epithelial-mesenchymal transition. Acta Biochim Biophys
Sin (Shanghai). 50:370–380. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hu H, Ding X, Yang Y, Zhang H, Li H, Tong
S, An X, Zhong Q, Liu X, Ma L, et al: Changes in
glucose-6-phosphate dehydrogenase expression results in altered
behavior of HBV-associated liver cancer cells. Am J Physiol
Gastrointest Liver Physiol. 307:G611–G622. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lima MS, Pereira RA, Costa RS, Tucci S,
Dantas M, Muglia VF, Ravinal RC and Barros-Silva GE: The prognostic
value of cyclin D1 in renal cell carcinoma. Int Urol Nephrol.
46:905–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lindblad P: Epidemiology of renal cell
carcinoma. Scand J Surg. 93:88–96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Linehan WM, Srinivasan R and Schmidt LS:
The genetic basis of kidney cancer: A metabolic disease. Nat Rev
Urol. 7:277–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Vavallo A, Simone S, Lucarelli G,
Rutigliano M, Galleggiante V, Grandaliano G, Gesualdo L, Campagna
M, Cariello M, Ranieri E, et al: Pre-existing type 2 diabetes
mellitus is an independent risk factor for mortality and
progression in patients with renal cell carcinoma. Medicine
(Baltimore). 93:e1832014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Cancer Genome Atlas Research Network, .
Comprehensive molecular characterization of clear cell renal cell
carcinoma. Nature. 499:43–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang Q, Yi X, Yang Z, Han Q, Di X, Chen
F, Wang Y, Yi Z, Kuang Y and Zhu Y: Overexpression of G6PD
represents a potential prognostic factor in clear cell renal cell
carcinoma. J Cancer. 8:665–673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang Q, Yang Z, Han Q, Bai H, Wang Y, Yi
X, Yi Z, Yang L, Jiang L, Song X, et al: G6PD promotes renal cell
carcinoma proliferation through positive feedback regulation of
p-STAT3. Oncotarget. 8:109043–109060. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yang HC, Wu YH, Yen WC, Liu HY, Hwang TL,
Stern A and Chiu DT: The redox role of G6PD in cell growth, cell
death, and cancer. Cells. 8:10552019. View Article : Google Scholar
|
|
72
|
Langbein S, Frederiks WM, zur Hausen A,
Popa J, Lehmann J, Weiss C, Alken P and Coy JF: Metastasis is
promoted by a bioenergetic switch: New targets for progressive
renal cell cancer. Int J Cancer. 122:2422–2428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Shasha L, Priceman SJ, Xin H, Zhang W,
Deng J, Liu Y, Huang J, Zhu W, Chen M, Hu W, et al: Icaritin
inhibits JAK/STAT3 signaling and growth of renal cell carcinoma.
PLoS One. 8:e816572013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xu SN, Wang TS, Li X and Wang YP: SIRT2
activates G6PD to enhance NADPH production and promote leukaemia
cell proliferation. Sci Rep. 6:327342016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Saito Y, Chapple RH, Lin A, Kitano A and
Nakada D: AMPK protects Leukemia-initiating cells in myeloid
leukemias from metabolic stress in the bone marrow. Cell Stem Cell.
17:585–596. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Xu Q, Simpson SE, Scialla TJ, Bagg A and
Carroll M: Survival of acute myeloid leukemia cells requires PI3
kinase activation. Blood. 102:972–980. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chen Y, Xu Q, Ji D, Wei Y, Chen H, Li T,
Wan B, Yuan L, Huang R and Chen G: Inhibition of pentose phosphate
pathway suppresses acute myelogenous leukemia. Tumour Biol.
37:6027–6034. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lowman XH, McDonnell MA, Kosloske A,
Odumade OA, Jenness C, Karim CB, Jemmerson R and Kelekar A: The
proapoptotic function of Noxa in human leukemia cells is regulated
by the kinase Cdk5 and by glucose. Mol Cell. 40:823–833. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yamamoto T, Takano N, Ishiwata K, Ohmura
M, Nagahata Y, Matsuura T, Kamata A, Sakamoto K, Nakanishi T, Kubo
A, et al: Reduced methylation of PFKFB3 in cancer cells shunts
glucose towards the pentose phosphate pathway. Nat Commun.
5:34802014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shan C, Elf S, Ji Q, Kang HB, Zhou L,
Hitosugi T, Jin L, Lin R, Zhang L, Seo JH, et al: Lysine
acetylation activates 6-phosphogluconate dehydrogenase to promote
tumor growth. Mol Cell. 55:552–565. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Poulain L, Sujobert P, Zylbersztejn F,
Barreau S, Stuani L, Lambert M, Palama TL, Chesnais V, Birsen R,
Vergez F, et al: High mTORC1 activity drives glycolysis addiction
and sensitivity to G6PD inhibition in acute myeloid leukemia cells.
Leukemia. 31:2326–2335. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hulleman E, Kazemier KM, Holleman A,
VanderWeele DJ, Rudin CM, Broekhuis MJ, Evans WE, Pieters R and Den
Boer ML: Inhibition of glycolysis modulates prednisolone resistance
in acute lymphoblastic leukemia cells. Blood. 113:2014–2021. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Meynet O, Bénetéau M, Jacquin MA, Pradelli
LA, Cornille A, Carles M and Ricci JE: Glycolysis inhibition
targets Mcl-1 to restore sensitivity of lymphoma cells to
ABT-737-induced apoptosis. Leukemia. 26:1145–1147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gregory MA, D'Alessandro A,
Alvarez-Calderon F, Kim J, Nemkov T, Adane B, Rozhok AI, Kumar A,
Kumar V, Pollyea DA, et al: ATM/G6PD-driven redox metabolism
promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc
Natl Acad Sci USA. 113:E6669–E6678. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Gottschalk S, Anderson N, Hainz C,
Eckhardt SG and Serkova NJ: Imatinib (STI571)-mediated changes in
glucose metabolism in human leukemia BCR-ABL-positive cells. Clin
Cancer Res. 10:6661–6668. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Karnauskas R, Niu Q, Talapatra S, Plas DR,
Greene ME, Crispino JD and Rudin CM: Bcl-x(L) and Akt cooperate to
promote leukemogenesis in vivo. Oncogene. 22:688–698. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ye H, Huang H, Cao F, Chen M, Zheng X and
Zhan R: HSPB1 enhances SIRT2-mediated G6PD activation and promotes
glioma cell proliferation. PLoS One. 11:e01642852016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tao L, Yu H, Liang R, Jia R, Wang J, Jiang
K and Wang Z: Rev-erbα inhibits proliferation by reducing
glycolytic flux and pentose phosphate pathway in human gastric
cancer cells. Oncogenesis. 8:572019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Gu MJ, Huang QC, Bao CZ, Li YJ, Li XQ, Ye
D, Ye ZH, Chen K and Wang JB: Attributable causes of colorectal
cancer in China. BMC Cancer. 18:382018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhang Y, Chen Z and Li J: The current
status of treatment for colorectal cancer in China: A systematic
review. Medicine (Baltimore). 96:e82422017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ishikawa M, Inoue T, Shirai T, Takamatsu
K, Kunihiro S, Ishii H and Nishikata T: Simultaneous expression of
cancer stem Cell-like properties and Cancer-associated
Fibroblast-like properties in a primary culture of breast cancer
cells. Cancers (Basel). 6:1570–1578. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tsouko E, Khan AS, White MA, Han JJ, Shi
Y, Merchant FA, Sharpe MA, Xin L and Frigo DE: Regulation of the
pentose phosphate pathway by an androgen Receptor-mTOR-mediated
mechanism and its role in prostate cancer cell growth. Oncogenesis.
3:e1032014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zampella EJ, Bradley EL Jr and Pretlow TG
II: Glucose-6-phosphate dehydrogenase: A possible clinical
indicator for prostatic carcinoma. Cancer. 49:384–387. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Pretlow TG II, Harris BE, Bradley EL Jr,
Bueschen AJ, Lloyd KL and Pretlow TP: Enzyme activities in
prostatic carcinoma related to Gleason grades. Cancer Res.
45:442–446. 1985.PubMed/NCBI
|
|
95
|
Ros S, Santos CR, Moco S, Baenke F, Kelly
G, Howell M, Zamboni N and Schulze A: Functional metabolic screen
identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as
an important regulator of prostate cancer cell survival. Cancer
Discov. 2:328–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kaushik AK, Vareed SK, Basu S, Putluri V,
Putluri N, Panzitt K, Brennan CA, Chinnaiyan AM, Vergara IA, Erho
N, et al: Metabolomic profiling identifies biochemical pathways
associated with Castration-resistant prostate cancer. J Proteome
Res. 13:1088–1100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nna E, Tothill IE, Ludeman L and Bailey T:
Endogenous control genes in prostate cells: Evaluation of gene
expression using ‘Real-Time’ quantitative polymerase chain
reaction. Med Princ Pract. 19:433–439. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Frederiks WM, Vizan P, Bosch KS,
Vreeling-Sindelarova H, Boren J and Cascante M: Elevated activity
of the oxidative and non-oxidative pentose phosphate pathway in
(pre)neoplastic lesions in rat liver. Int J Exp Pathol. 89:232–240.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hu T, Zhang C, Tang Q, Su Y, Li B, Chen L,
Zhang Z, Cai T and Zhu Y: Variant G6PD levels promote tumor cell
proliferation or apoptosis via the STAT3/5 pathway in the human
melanoma xenograft mouse model. BMC Cancer. 13:2512013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Nobrega-Pereira S, Fernandez-Marcos PJ,
Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Viña J
and Serrano M: G6PD protects from oxidative damage and improves
healthspan in mice. Nat Commun. 7:108942016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hamanaka RB and Chandel NS: Mitochondrial
reactive oxygen species regulate cellular signaling and dictate
biological outcomes. Trends Biochem Sci. 35:505–513. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Maryanovich M and Gross A: A ROS rheostat
for cell fate regulation. Trends Cell Biol. 23:129–134. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Weinberg F, Hamanaka R, Wheaton WW,
Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger
GR and Chandel NS: Mitochondrial metabolism and ROS generation are
essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA.
107:8788–8793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ying W: NAD+/NADH and
NADP+/NADPH in cellular functions and cell death:
Regulation and biological consequences. Antioxid Redox Signal.
10:179–206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Peter B, Bogan KL and Charles B:
NAD+ metabolism in health and disease. Trends Biochem
Sci. 32:12–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Berger F, Ramírez-Hernández MH and Ziegler
M: The new life of a centenarian: Signalling functions of NAD(P).
Trends Biochem Sci. 29:111–118. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Pollak N, Dölle C and Ziegler M: The power
to reduce: Pyridine nucleotides-small molecules with a multitude of
functions. Biochem J. 402:205–218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Rastogi R, Geng X, Li F and Ding Y: NOX
Activation by subunit interaction and underlying mechanisms in
disease. Front Cell Neurosci. 10:3012017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Meitzler JL, Antony S, Wu Y, Juhasz A, Liu
H, Jiang G, Lu J, Roy K and Doroshow JH: NADPH Oxidases: A
perspective on reactive oxygen species production in tumor biology.
Antioxid Redox Signal. 20:2873–2889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Prasad S, Gupta SC and Tyagi AK: Reactive
oxygen species (ROS) and cancer: Role of antioxidative
nutraceuticals. Cancer Lett. 387:95–105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: Physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Mittler R: ROS are good. Trends Plant Sci.
22:11–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
de Atauri P, Benito A, Vizán P, Zanuy M,
Mangues R, Marín S and Cascante M: Carbon metabolism and the sign
of control coefficients in metabolic adaptations underlying K-ras
transformation. Biochim Biophys Acta. 1807:746–754. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Jiang P, Du W, Wang X, Mancuso A, Gao X,
Wu M and Yang X: p53 regulates biosynthesis through direct
inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol.
13:310–316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Fujita M, Imadome K, Endo S, Shoji Y,
Yamada S and Imai T: Nitric oxide increases the invasion of
pancreatic cancer cells via activation of the PI3K-AKT and RhoA
pathways after carbon ion irradiation. FEBS Lett. 588:3240–3250.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Li W, Cao L, Han L, Xu Q and Ma Q:
Superoxide dismutase promotes the epithelial-mesenchymal transition
of pancreatic cancer cells via activation of the H2O2/ERK/NF-NF-κB
axis. Int J Oncol. 46:2613–2620. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Kong DH, Li S, Du ZX, Liu C, Liu BQ, Li C,
Zong ZH and Wang HQ: BAG3 elevation inhibits cell proliferation via
direct interaction with G6PD in hepatocellular carcinomas.
Oncotarget. 7:700–711. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Rosati A, Graziano V, De Laurenzi V,
Pascale M and Turco MC: BAG3: A multifaceted protein that regulates
major cell pathways. Cell Death Dis. 2:e1412011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Behl C: BAG3 and friends: Co-chaperones in
selective autophagy during aging and disease. Autophagy. 7:795–798.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Cai T, Kuang Y, Zhang C, Zhang Z, Chen L,
Li B, Li Y, Wang Y, Yang H, Han Q and Zhu Y: Glucose-6-phosphate
dehydrogenase and NADPH oxidase 4 control STAT3 activity in
melanoma cells through a pathway involving reactive oxygen species,
c-SRC and SHP2. Am J Cancer Res. 5:1610–1620. 2015.PubMed/NCBI
|
|
121
|
Hedberg Y, Ljungberg B, Roos G and
Landberg G: Expression of cyclin D1, D3, E, and p27 in human renal
cell carcinoma analysed by tissue microarray. Br J Cancer.
88:1417–1423. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Sun X, Liu B, Wang J, Li J and Ji WY:
Inhibition of p21-activated kinase 4 expression suppresses the
proliferation of Hep-2 laryngeal carcinoma cells via activation of
the ATM/Chk1/2/p53 pathway. Int J Oncol. 42:683–689. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhang X, Zhang X, Li Y, Shao Y, Xiao J,
Zhu G and Li F: PAK4 regulates G6PD activity by p53 degradation
involving colon cancer cell growth. Cell Death Dis. 8:e28202017.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ma X, Wang L, Huang D, Li Y, Yang D, Li T,
Li F, Sun L, Wei H, He K, et al: Polo-like kinase 1 coordinates
biosynthesis during cell cycle progression by directly activating
pentose phosphate pathway. Nat Commun. 8:15062017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Gu Y, Mi W, Ge Y, Liu H, Fan Q, Han C,
Yang J, Han F, Lu X and Yu W: GlcNAcylation plays an essential role
in breast cancer metastasis. Cancer Res. 70:6344–6351. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Mi W, Gu Y, Han C, Liu H, Fan Q, Zhang X,
Cong Q and Yu W: O-GlcNAcylation is a novel regulator of lung and
colon cancer malignancy. Biochim Biophys Acta. 1812:514–519. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Tokuda K, Baron B, Yamashiro C, Kuramitsu
Y, Kitagawa T, Kobayashi M, Sonoda KH and Kimura K: Up-regulation
of the pentose phosphate pathway and HIF-1α expression during
neural progenitor cell induction following glutamate treatment in
rat ex vivo retina. Cell Biol Int. 44:137–144. 2020. View Article : Google Scholar
|
|
128
|
Reina-Campos M, Moscat J and Diaz-Meco M:
Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol.
48:47–53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Singh L, Aldosary S, Saeedan AS, Ansari MN
and Kaithwas G: Prolyl hydroxylase 2: A promising target to inhibit
hypoxia-induced cellular metabolism in cancer cells. Drug Discov
Today. 23:1873–1882. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Cabrales P: RRx-001 acts as a dual small
molecule checkpoint inhibitor by downregulating CD47 on cancer
cells and SIRP-α on Monocytes/Macrophages. Transl Oncol.
12:626–632. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Yalcin O, Oronsky B, Carvalho LJ, Kuypers
FA, Scicinski J and Cabrales P: From METS to malaria: RRx-001, a
multi-faceted anticancer agent with activity in cerebral malaria.
Malar J. 14:2182015. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Das DS, Ray A, Das A, Song Y, Tian Z,
Oronsky B, Richardson P, Scicinski J, Chauhan D and Anderson KC: A
novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis
and overcomes drug resistance in multiple myeloma cells. Leukemia.
30:2187–2197. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Mele L, Paino F, Papaccio F, Regad T,
Boocock D, Stiuso P, Lombardi A, Liccardo D, Aquino G, Barbieri A,
et al: A new inhibitor of glucose-6-phosphate dehydrogenase blocks
pentose phosphate pathway and suppresses malignant proliferation
and metastasis in vivo. Cell Death Dis. 9:5722018. View Article : Google Scholar : PubMed/NCBI
|