Introduction
Among women, cervical cancer is ranked 4th in global
cancer-associated deaths (1), with
over half a million deaths in 2012 (2). Cervical cancer can be broadly
categorized into squamous cell carcinoma, which constitutes the
majority of cases (70–80%) or adenocarcinoma, which comprises
10–15% of cases (3). Cervical
cancer is frequently caused by the oncovirus human papillomavirus
(HPV), mainly by types HPV-16 and −18 in ~70% of patients with
cervical cancer (3). The molecular
drivers of the formation and development of cervical cancer are
often due to recurrent mutations within the PI3K/AKT signaling
pathway (4–7) or from deactivating, loss-of-function
mutations in the tumor suppressor TP53 gene (p53 protein), which
produces a particularly aggressive phenotype (8). Additionally, altered expression levels
of microRNAs (miRNAs/miRs), which are short, regulatory, non-coding
RNAs, have been implicated in metastatic cervical cancer (9,10).
Among miRNAs associated with cervical cancer (11–13),
miR-3184 has been identified as a p53-responsive miRNA (14). However, its mechanistic role in
cervical oncogenesis remains unknown.
Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2), also
known as human epidermal growth factor receptor 2, is an
established oncogenic protein associated with tumorigenesis, cancer
progression and acquisition of therapy resistance in breast,
esophagogastric and colorectal cancer (15–17).
However, the involvement of ERBB2 in cervical cancer development is
less well known (18). Mithramycin
A is an antibacterial and anticancer agent that was first purified
from Streptomyces bacteria (19–21).
It binds to G/C-rich stretches of DNA within minor grooves
(19), which is one of its possible
mechanisms for its antitumor properties (22). Mithramycin A possesses a favorable
safety profile and inhibits tumor proliferation in vivo in
cervical cancer murine xenograft models (23). Another possible mechanism for the
antitumor effects of Mithramycin A is via p53 activation (24); however, in cervical cancer, the
influence of Mithramycin A on p53 and ERBB2 has not been
examined.
Phosphatidylinositol-4,5-bisphosphate 3-kinase
catalytic subunit α (PIK3CA) is the catalytic subunit of PI3K
(25). In humans, PIK3CA acts as a
proto-oncogene that supports proliferation and metastasis of tumor
cells (25), and PIK3CA copy number
gains are associated with higher-grade tumors and poor patient
prognosis in several malignancies, including gastric, thyroid and
prostate cancer (25). In patients
with cervical cancer, PIK3CA mutations are significantly
associated with shorter survival times (PIK3CA mutant median
survival of 67.1 months vs. non-mutant median survival of 90.3
months) (26). The aforementioned
evidence suggests that targeting PIK3CA activity may be a rational
approach in combating cervical cancer. Therefore, the present study
aimed to examine the association between the effects of the
p53-responsive miR-3184-5p, ERBB2 and PIK3CA in cervical
cancer.
Materials and methods
Patient samples
The present study was approved by the Ethics Review
Committee of the First Affiliated Hospital of Bengbu Medical
College (approval no. 201858; Bengbu, China). Patients who donated
cervical cancer tissues gave their informed consent in writing
prior to their involvement in the study. All specimens were
harvested following biopsy or surgery (cone biopsy, radical
trachelectomy or radical hysterectomy) at the aforementioned
hospital between March 2000 and March 2002 from female patients
(median age, 53 years; age range, 35–65 years) that had never
received chemo- or radiotherapy, yielding 65 pairs of primary
cervical cancer samples and matched healthy cervical tissue samples
(>2 cm from the tumor edge). Tumor specimens were staged by a
licensed pathologist according to the International Federation of
Gynecologists and Obstetricians (FIGO) staging system (27). The specimens were immediately kept
in liquid nitrogen and frozen at −80°C until further use.
Cell lines, culture conditions and
general materials
Human cervical cancer HeLa and SiHa cell lines were
obtained from the American Type Culture Collection. The
non-cancerous, HPV-immortalized H8 cell line of cervical epithelial
squamous cells was obtained from The Cell Bank of Type Culture
Collection of the Chinese Academy of Sciences. Cells were grown in
DMEM/F12 with 10% FBS (both from Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C in a 5% CO2 incubator. Reverse
transcription-quantitative PCR (RT-qPCR) was regularly employed to
check for contaminating mycoplasma (MycoITSF9 forward
primer, 5′-ACACCATGGGAGCTGGTAAT-3′ and reverse primer,
5′-CTTCATCGACTTTCAGACCCAAGGCA-3′), as previously described
(28). Mithramycin A and general
lab reagents were purchased from Sigma-Aldrich (Merck KGaA). For
in vitro experiments, Mithramycin A dissolved in DMSO at
concentrations of 0 (vehicle control), 50 and 100 nM was used to
treat cells for 48 h at 37°C in a 5% CO2 incubator.
Transfections
SiHa cells were transfected with plasmid vectors,
while HeLa cells were transfected with small-interfering RNAs
(siRNAs). pCMV6-XL4/5 plasmid vectors containing the
TrueClone® human cDNA sequences for human TP53
(NM_000546; cat. no. SC119832), PIK3CA (NM_006218; cat. no.
SC116227) or ERBB2 (NM_004448; cat. no. SC128161), and an
empty negative control (NC) plasmid vector (cat. no. PCMV6XL5) were
obtained from OriGene Technologies, Inc. For plasmid transfections,
cells were seeded in 6-well plates (1.5×105 cells/well)
and transiently transfected with 1 µg plasmid using
Lipofectamine® 3000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) for 20 min at room temperature and
then incubated in fresh medium at 37°C for an additional 48 h prior
to subsequent experimentation. siRNAs targeting human ERBB2
(ERBB2-siRNA; cat. no. sc-29405), human PIK3CA
(PIK3CA-siRNA; cat. no. sc-39127) and scrambled control (scr-siRNA;
cat. no. sc-37007) were obtained from Santa Cruz Biotechnology,
Inc. For siRNA transfections, cells were seeded in 6-well plates
(2×105 cells/well) and transiently transfected with 80
pmol siRNA using Lipofectamine 3000 transfection reagent for 7 h at
37°C and then incubated in fresh medium at 37°C for an additional
48 h prior to subsequent experimentation. miR-3184-5p (miRBase
accession no. MIMAT0015064) mirVana® miRNA mimic (cat.
no. 4464066) and mirVana® miRNA inhibitor (cat. no.
4464084), as well as the corresponding controls (cat. nos. 4464058
and 4464078, respectively), were obtained from AmbionÒ (Thermo
Fisher Scientific, Inc.). For miRNA mimic/inhibitor transfections,
HeLa and SiHa cells were seeded in 6-well plates (1×105
cells/well) and transiently transfected with 3 nM mimic or 10 nM
inhibitor using Lipofectamine 3000 transfection reagent for 48 h at
37°C prior to subsequent experimentation.
RT-qPCR
TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to extract total RNA from cells or
tissue samples, of which a 100-ng aliquot from every sample was
used for cDNA preparation using the PrimeScript™ RT Reagent Kit
according to the manufacturer's protocol (Takara Bio, Inc.). A
SuperScript™ III Platinum™ SYBR™ Green One-Step qRT-PCR kit or
NCode™ SYBR™ Green miRNA qRT-PCR kit (both from Invitrogen; Thermo
Fisher Scientific, Inc.) was used to quantify mRNA or miR-3184-5p
expression, respectively, on an ABI PRISM™ 7000 Sequence Detection
System (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
mRNA qPCR thermocycling conditions were as follows: 95°C for 3 min,
followed by 45 cycles of 95°C for 15 sec and 60°C for 45 sec. The
miRNA qPCR thermocycling conditions were as follows: 50°C for 2 min
and 95°C for 2 min, followed by 40 cycles of 95°C for 15 sec and
60°C for 50 sec. The following primers were obtained from OriGene
Technologies, Inc., and Applied Biosystems (Thermo Fisher
Scientific, Inc.): ERBB2 forward, 5′-GGAAGTACACGATGCGGAGACT-3′ and
reverse, 5′-ACCTTCCTCAGCTCCGTCTCTT-3′; CD44 forward,
5′-CCAGAAGGAACAGTGGTTTGGC-3′ and reverse,
5′-ACTGTCCTCTGGGCTTGGTGTT-3′; CD133 forward,
5′-CACTACCAAGGACAAGGCGTTC-3′ and reverse,
5′-CAACGCCTCTTTGGTCTCCTTG-3′; Aldehyde Dehydrogenase 1 (ALDH1)
forward, 5′-CGGGAAAAGCAATCTGAAGAGGG-3′ and reverse,
5′-GATGCGGCTATACAACACTGGC-3′; SNAIL forward,
5′-TGCCCTCAAGATGCACATCCGA-3′ and reverse,
5′-GGGACAGGAGAAGGGCTTCTC−3′; vimentin forward,
5′-AGGCAAAGCAGGAGTCCACTGA-3′ and reverse,
5′-ATCTGGCGTTCCAGGGACTCAT-3′; E-cadherin forward,
5′-GCCTCCTGAAAAGAGAGTGGAAG-3′ and reverse,
5′-TGGCAGTGTCTCTCCAAATCCG-3′; PIK3CA forward,
5′-GAAGCACCTGAATAGGCAAGTCG-3′ and reverse,
5′-GAGCATCCATGAAATCTGGTCGC-3′; GAPDH forward,
5′-GCGAGATCCCTCCAAAATCAA−3′ and reverse,
5′-GTTCACACCCATGACGAACAT−3′; and small nuclear RNA (snRNA) U6
forward, 5′-CTCGCTTCGGCAGCACAT−3′ and reverse,
5′-TTTGCGTGTCATCCTTGCG-3′. The forward primer for miR-3185-5p
possessed the same sequence as the mature miR-3184-5p strand, while
the reverse primer for miR-3185-5p was the universal reverse primer
supplied with the NCode™ SYBR™ Green miRNA qRT-PCR Kit (Invitrogen;
Thermo Fisher Scientific, Inc.). GAPDH and snRNA U6 served as
internal standardization controls for mRNAs and miRNAs,
respectively. Relative expression levels were calculated using the
2−ΔΔCq method (29).
Western blotting (WB)
Cells or tissue samples were lysed using M-PER™
Mammalian Protein Extraction Reagent (Pierce; Thermo Fisher
Scientific, Inc.) and protein content was quantified using a
Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Inc.). Proteins
(30 µg/lane) were separated via 12% SDS-PAGE and transferred to
polyvinylidene difluoride (PVDF) membranes. The membranes were
rinsed, blocked with 5% skimmed milk in TBST (20 mM Tris, 150 mM
NaCl, 0.05% Tween-20) for 2 h at room temperature, and incubated at
4°C overnight with the following primary antibodies (all diluted
1:1,000 unless otherwise specified): ALDH1 (cat. no. ab9883;
Abcam), total AKT1 (1:2,000; cat. no. ab28422; Abcam),
phosphorylated (p-)AKT1 (S473; 1:5,000; cat. no. ab81283; Abcam),
CD44 (1:2,000; cat. no. ab157107; Abcam), CD133 (cat. no. ab19898;
Abcam), E-cadherin (1:50; cat. no. ab1416; Abcam), ERBB2 (cat. no.
ab16901; Abcam), GAPDH (cat. no. sc-25778; Santa Cruz
Biotechnology, Inc.), total mTOR (1:2,000; cat. no. ab2732; Abcam),
p-mTOR (S2448; cat. no. ab109268; Abcam), p21 (mouse monoclonal;
F-5; cat. no. sc-6246; Santa Cruz Biotechnology, Inc.), p53 (mouse
monoclonal; DO-1; cat. no. sc-126; Santa Cruz Biotechnology, Inc.),
PIK3CA (cat. no. ab40776; Abcam), SNAIL (cat. no. ab53519; Abcam)
and vimentin (cat. no. ab92547; Abcam). Membranes were then rinsed
and incubated for 1 h at room temperature with species-appropriate,
HRP-conjugated secondary antibodies (1:5,000; cat. nos. ab7090 and
ab97110; Abcam), rinsed and analyzed using an ECL Western Blotting
Detection kit (Amersham; Cytiva). ImageJ version 1.46 software
(National Institutes of Health) was used for blot densitometry
analysis.
Evaluation of invasion by Transwell
assay
BioCoat™ Matrigel® Invasion Chambers
(Corning, Inc.) were used to evaluate the invasive capacity of
cervical cancer cell lines according to previous studies (30,31).
Cervical cancer cells (5×104) that had undergone
transfection were plated into the top chamber in serum-free medium,
while complete medium (with 10% FBS) was added to the lower
chambers. The Invasion Chamber was incubated for 1 day at 37°C in a
5% CO2 incubator, after which a cotton swab was used to
remove cells from the top chamber. The remaining cells that had
invaded through the Transwell membrane were fixed with ice-cold
100% methanol for 5 min and stained with 0.05% Giemsa for 30 min at
room temperature. The cell numbers were counted and recorded using
a light microscope at ×400 magnification.
Quantification of cell viability
A Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc.) was employed to quantify cell viability. A
total of 1×104 cervical cancer cells/well seeded in
96-well culture plates were incubated for 72 h at 37°C, after which
CCK-8 reagent (10 µl/well) was added for a further 4 h at 37°C. A
microplate reader measured the colorimetric signal at 450 nm.
Assessment of sphere formation
Dissociated cells were plated in 6-well Ultra-Low
Attachment plates (Corning, Inc.) at a density of 5,000 cells/ml in
serum-free DMEM/F12 supplemented with N-2 supplement (Invitrogen;
Thermo Fisher Scientific, Inc.), epidermal growth factor (20
ng/ml), basic fibroblast growth factor (20 ng/ml) and heparin (4
mg/ml). A serum-free medium containing additional growth factors is
the standard medium used for sphere formation experiments (32–34).
Every well was replenished with new media every 3 days for 2 weeks
at room temperature, after which spheroids with diameters >50 µm
were counted manually using an inverted phase contrast light
microscopy at ×100 magnification.
Immunoprecipitation (IP)
For the initial IP step, 10 µg anti-ERBB3 antibody
or IgG control antibody (cat. nos. 4754 and 2729, respectively;
Cell Signaling Technology, Inc.) was incubated with protein G
magnetic beads according to the manufacturer's protocol (Pierce;
Thermo Fisher Scientific, Inc.) in 150 mM NaCl/100 mM HEPES (pH
8.2) buffer for 2 h at 4°C to form the anti-ERBB3-protein G beads
or anti-IgG-protein G beads, respectively. After washing, 300 µg
cell lysate protein (obtained using M-PER™ Mammalian Protein
Extraction Reagent; Pierce; Thermo Fisher Scientific, Inc.) was
immunoprecipitated on a rotator overnight at 4°C using 20 µl
antibody-protein G beads. The antibody-protein G beads were then
collected by pulse centrifugation (5 sec at 14,000 × g) at 4°C. The
antibody-protein G beads were washed with lysis buffer, SDS-sample
buffer with dithiothreitol (50 mM) was added, and the samples were
boiled for 4 min. The samples were electrophoresed on a 4–12%
bis-tris gel followed by transfer to a PVDF membrane. Co-IP with
ERBB2 or PI3K (p85) was assessed via WB, as aforementioned, using
an anti-ERBB2 antibody or anti-PI3K (p85) antibody (1:1,000; cat.
no. ab16901 and ab191606, respectively; Abcam).
TargetScan analysis and luciferase
reporter assay
The TargetScan database (www.targetscan.org) was used to search for candidate
miRNAs that may bind to the ERBB2 3′-untranslated region (UTR). A
pMirTarget firefly luciferase reporter plasmid (cat. no. PS100062)
containing the wild-type (WT) 3′-UTR of human ERBB2
(ERBB2-3′-UTRWT; cat. no. SC208188) was obtained from
OriGene Technologies, Inc. Mutations were introduced using a
QuikChange™ Site-Directed Mutagenesis kit (Agilent Technologies,
Inc.) into the putative miR-3184-5p binding site on
ERBB2-3′-UTRWT to create the mutant (MU)
ERBB2-3′-UTRMU. Subsequently, transfection of cervical
cancer cells using Lipofectamine 3000 transfection reagent was
performed using the following: One of the firefly luciferase
reporter plasmids (ERBB2-3′-UTRWT or
ERBB2-3′-UTRMU), a transfection standardization
pGL4.74[hRluc/TK] Renilla reporter plasmid (cat. no. E692;
Promega Corporation) and one of the miRNAs (miR-3184-5p mimic or
inhibitor, or their corresponding negative controls). After 1 day
of transfection, bioluminescence from luciferase activity was
analyzed using a Dual-Luciferase Reporter Assay System (cat. no.
E1910; Promega Corporation). Relative luciferase intensity was
obtained as the intensity of the firefly to Renilla
luciferase signal for each experimental condition, which was then
expressed relative to the respective control conditions, whose
values were taken as 1.0.
Statistical analysis
All data are expressed as the mean ± SEM, unless
otherwise specified. Data were analyzed using SPSS 24.0 (IBM
Corp.). Comparisons of expression levels in cervical cancer tissues
vs. matched healthy cervical tissues were assessed using a Wilcoxon
signed-rank test, while comparisons between independent tumor
tissue samples were assessed using a Mann-Whitney U test.
Kaplan-Meier survival analyses were stratified based on median
expression levels, with high levels being above the median and low
levels being below the median; comparisons between survival curves
were analyzed using a log-rank test. For in vitro
experiments, an unpaired two-tailed Student's t-test or one-way
ANOVA with Bonferroni post-hoc testing was employed for comparisons
between 2 or ≥3 groups, respectively. P<0.05 was considered to
indicate a statistically significant difference.
Results
ERBB2 expression is upregulated in
cervical cancer tissues and is associated with a poor
prognosis
The clinicopathological characteristics of the 65
recruited patients with cervical cancer are presented in Table SI. ERBB2 mRNA and protein
expression was significantly higher in cervical cancer tissues
compared with in matched healthy cervical tissues (Fig. 1A and B). Furthermore, tumor ERBB2
transcript and protein expression was significantly upregulated in
advanced FIGO stages compared with in early FIGO stages (Fig. 1C and D), as well as in lymph node
metastatic disease compared with in non-metastatic disease
(Fig. 1E and F). Subsequently, the
prognostic significance of ERBB2 expression on the survival of
patients with cervical cancer was evaluated. The median ERBB2
transcript expression was used to divide patients with cervical
cancer into the high (above the median) or low (below the median)
ERBB2 expression groups. High ERBB2 mRNA expression was associated
with a poorer survival outcome compared with low ERBB2 expression
(Fig. 1G). Overall, the data
demonstrated that ERBB2 expression was positively associated with
clinicopathological indicators of cervical cancer progression.
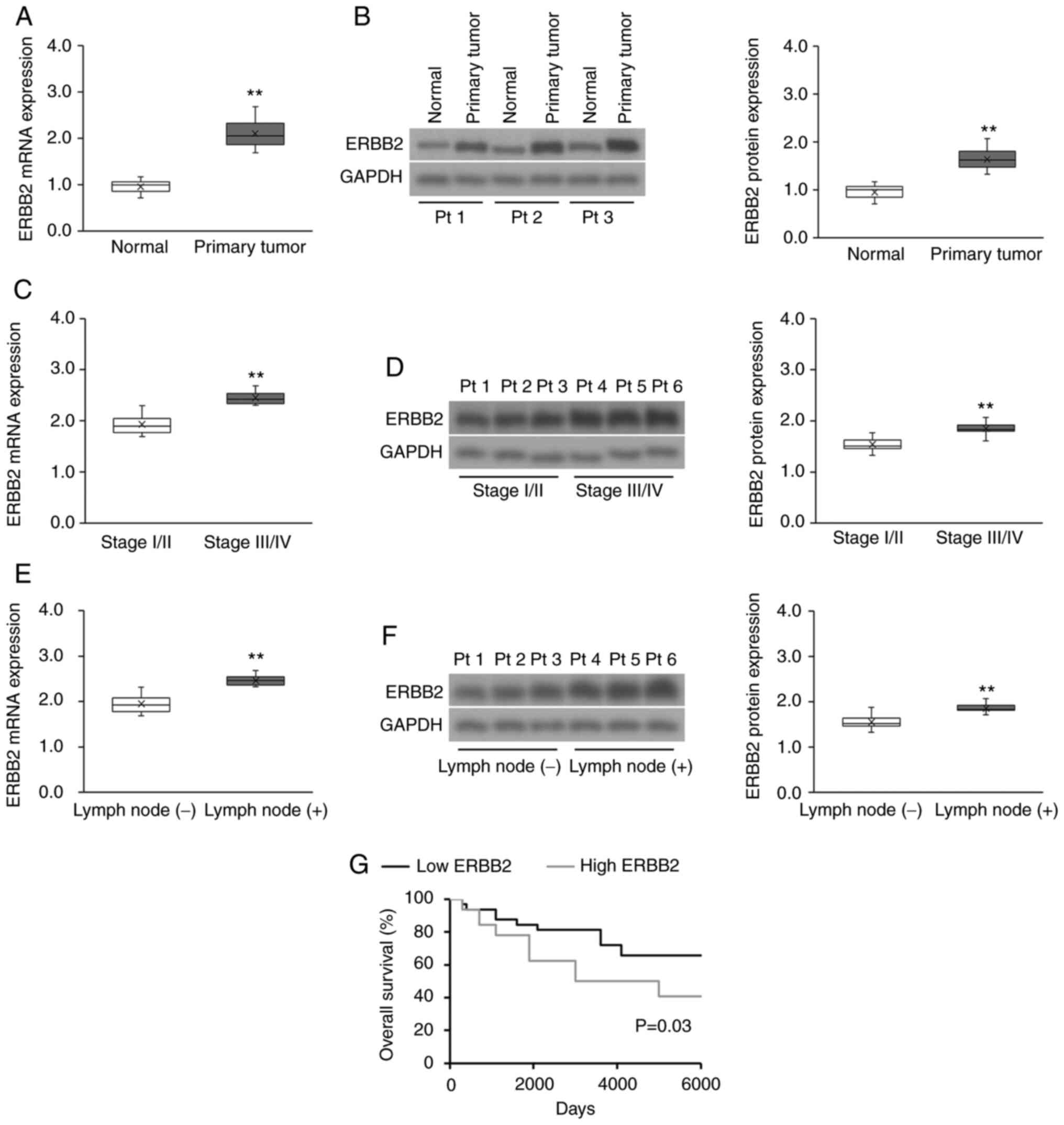 | Figure 1.ERBB2 expression is upregulated in
patient-derived cervical cancer tissues and is associated with a
poor prognosis. (A) RT-qPCR and (B) WB analysis of ERBB2 transcript
and protein expression, respectively, in patient-derived cervical
cancer tissues (n=65) vs. matched healthy cervical tissues (n=65).
Data were analyzed via Wilcoxon signed-rank test. (C) RT-qPCR and
(D) WB analysis of ERBB2 transcript and protein expression,
respectively, in stage I/II vs. stage III/IV patient-derived
cervical cancer tissues (n=43 stage I/II; n=22 Stage III/IV). Data
were analyzed via Mann-Whitney U test. (E) RT-qPCR and (F) WB
analysis of ERBB2 transcript and protein expression, respectively,
in lymph node metastatic and non-metastatic patient-derived
cervical cancer biopsies [n=46 lymph node (−); n=19 lymph node
(+)]. Data were analyzed via Mann-Whitney U test. (G) Survival
analysis using the Kaplan-Meier method according to high (above the
median) or low (below the median) ERBB2 mRNA expression (n=32 in
each cohort). The P-value was calculated using the log-rank test.
For purposes of comparison across cohorts, the median ERBB2 mRNA
and protein expression levels (normalized to the RT-qPCR
housekeeping control and WB loading control GAPDH) in the normal
cohort have been set to 1.0. Data in box plots are expressed as the
median ± IQRs (boxes) and absolute ranges (whiskers). n=3.
**P<0.01. RT-qPCR, reverse transcription-quantitative PCR; WB,
western blotting; ERBB2, Erb-B2 Receptor Tyrosine Kinase 2; Pt,
patient. |
Upregulated ERBB2 expression in cervical cancer cell
lines promotes viability, invasion and sphere formation. After
discovering the association between increased ERBB2 expression and
cervical cancer progression, the present study investigated the
effects of ERBB2 in cervical cancer cells. The SiHa and HeLa cell
lines were used as the cervical cancer cell lines, while the
non-cancerous cervical epithelial H8 cell line was used as the
negative control. In agreement with patient-derived samples, ERBB2
mRNA and protein expression was significantly higher in HeLa and
SiHa cells compared with in H8 cells (Fig. S1). The SiHa cell line, which
expressed comparatively less endogenous ERBB2 than the HeLa cell
line, underwent transient transfection with an ERBB2 overexpression
(OE) vector (ERBB2 vec) and an empty control plasmid (Ctrl vec). On
the other hand, the HeLa cell line, which expressed comparatively
high endogenous ERBB2 compared with the SiHa cell line, underwent
transient transfection with an ERBB2-targeting siRNA to knock down
(KD) its expression (siERBB2) or a control scrambled siRNA
(siCtrl). OE or KD of ERBB2 protein was validated via WB (Fig. 2A).
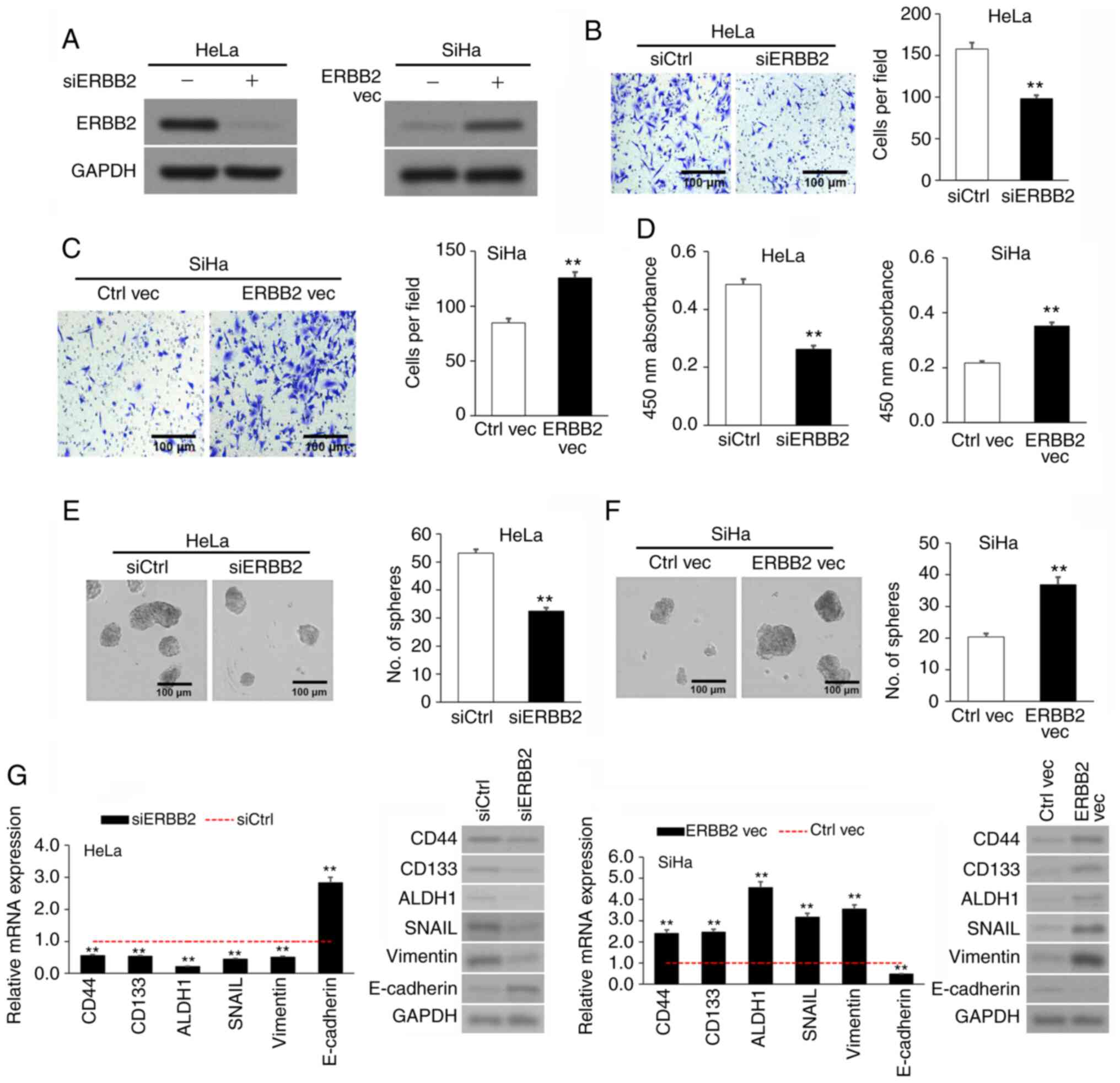 | Figure 2.ERBB2 overexpression in cervical
cancer cell lines stimulates viability, invasion and
sphere-formation. (A) Confirmation of ERBB2 KD in siERBB2 cells and
OE in ERBB2 vec cells via WB. GAPDH was used as the loading
control. (B) Invasion of siERBB2 vs. siCtrl cells via Transwell
assay. (C) Invasion of ERBB2 vec vs. Ctrl vec cells via Transwell
assay. (D) Cellular viability of siERBB2, siCtrl, ERBB2 vec and
Ctrl vec cells quantified using a Cell Counting Kit-8. (E)
Sphere-formation of siERBB2 vs. siCtrl cells. (F) Sphere-formation
of ERBB2 vec vs. Ctrl vec cells. (G) Analysis of
metastasis-associated and cancer stem cell biomarkers mRNA and
protein expression in siERBB2, siCtrl, ERBB2 vec and Ctrl vec cells
via RT-qPCR and WB, respectively. GAPDH was used as the RT-qPCR
housekeeping control and WB loading control. Data are expressed as
the mean ± SEM (n=3). **P<0.01 vs. siCtrl or Ctrl vec analyzed
via unpaired Student's t-test. KD, knockdown; OE, overexpression;
WB, western blotting; RT-qPCR, reverse transcription-quantitative
PCR; si, small interfering; Ctrl, control; vec, vector; ERBB2,
Erb-B2 Receptor Tyrosine Kinase 2. |
Invasion was assessed via Transwell assay. siERBB2
cells invaded significantly less than siCtrl cells (Fig. 2B), while ERBB2 vec cells invaded
significantly more than Ctrl vec cells (Fig. 2C), implying that upregulated ERBB2
expression stimulated the invasion of cervical cancer cells in
vitro. Cell viability was measured using the CCK-8 assay. Cell
viability in siERBB2 cells was significantly lower than that in
siCtrl cells, while cell viability in ERBB2 vec cells was
significantly higher than that in Ctrl vec cells (Fig. 2D). Cancer stem cell (CSC)-like
characterization was evaluated via sphere formation assays. siERBB2
cells had a significantly lower sphere-forming capacity than siCtrl
cells (Fig. 2E), while ERBB2 vec
cells had a significantly higher sphere-forming capacity than Ctrl
vec cells (Fig. 2F). The results
suggested that ERBB2 promoted the viability and stemness of
cervical cancer cell lines.
Since ERBB2 stimulated an invasive and CSC-like
phenotype in cervical cancer cells, the transcript and protein
expression levels of three metastasis-associated genes (SNAIL,
E-cadherin and vimentin) and three CSC biomarkers (CD44, CD133 and
ALDH1) (35,36) were measured following ERBB2 KD or
OE. The results revealed that siERBB2 cells expressed lower
expression levels of CD44, CD133, ALDH1, SNAIL and vimentin, but
higher expression levels of E-cadherin compared with siCtrl cells
(Fig. 2G). Conversely, ERBB2 vec
cells expressed higher expression levels of CD44, CD133, ALDH1,
SNAIL and vimentin, but lower expression levels of E-cadherin
compared with Ctrl vec cells (Fig.
2G). Overall, the current results supported a role for ERBB2 in
cervical cancer cell viability, invasion and stemness.
ERBB2 promotes cervical cancer cell
viability and invasion by stimulating PIK3CA activity
PI3K is an oncogenic protein complex that supports
the proliferation and metastasis of tumor cells (25). PI3K is formed by an active catalytic
subunit (p110, also known as PIK3CA) associating with a p85
regulatory subunit, which regulates the activity of PIK3CA
(37). The ERBB2-ERBB3 complex is
known to bind to p85, thereby promoting PIK3CA activity and AKT
phosphorylation (Fig. 3A) (38–40).
Thus, IP was used to validate if the ERBB2-ERBB3 complex associated
with p85 in cervical cancer cells. ERBB2 and p85 were enriched in
the anti-ERBB3 IP fraction compared with in the anti-IgG control IP
fraction; these effects were diminished by ERBB2-KD and potentiated
by ERBB2-OE (Fig. 3B). PIK3CA
transcript and protein expression was analyzed via RT-qPCR and WB,
respectively. siERBB2 cells expressed similar levels of PIK3CA
transcript and protein to siCtrl cells, as well as ERBB2 vec cells
to Ctrl vec cells (Fig. 3C-F). To
validate these findings, an OE vector was used to overexpress
PIK3CA (PIK3CA vec) in HeLa cells and a PIK3CA-targeting siRNA
(siPIK3CA) was used to silence PIK3CA in SiHa cells (Fig. S2A and B). Additional
co-transfection of siERBB2 cells with PIK3CA vec significantly
upregulated PIK3CA transcript and protein expression in HeLa cells,
while additional co-transfection of ERBB2 vec cells with siPIK3CA
significantly downregulated PIK3CA transcript and protein
expression in SiHa cells (Fig.
3C-F).
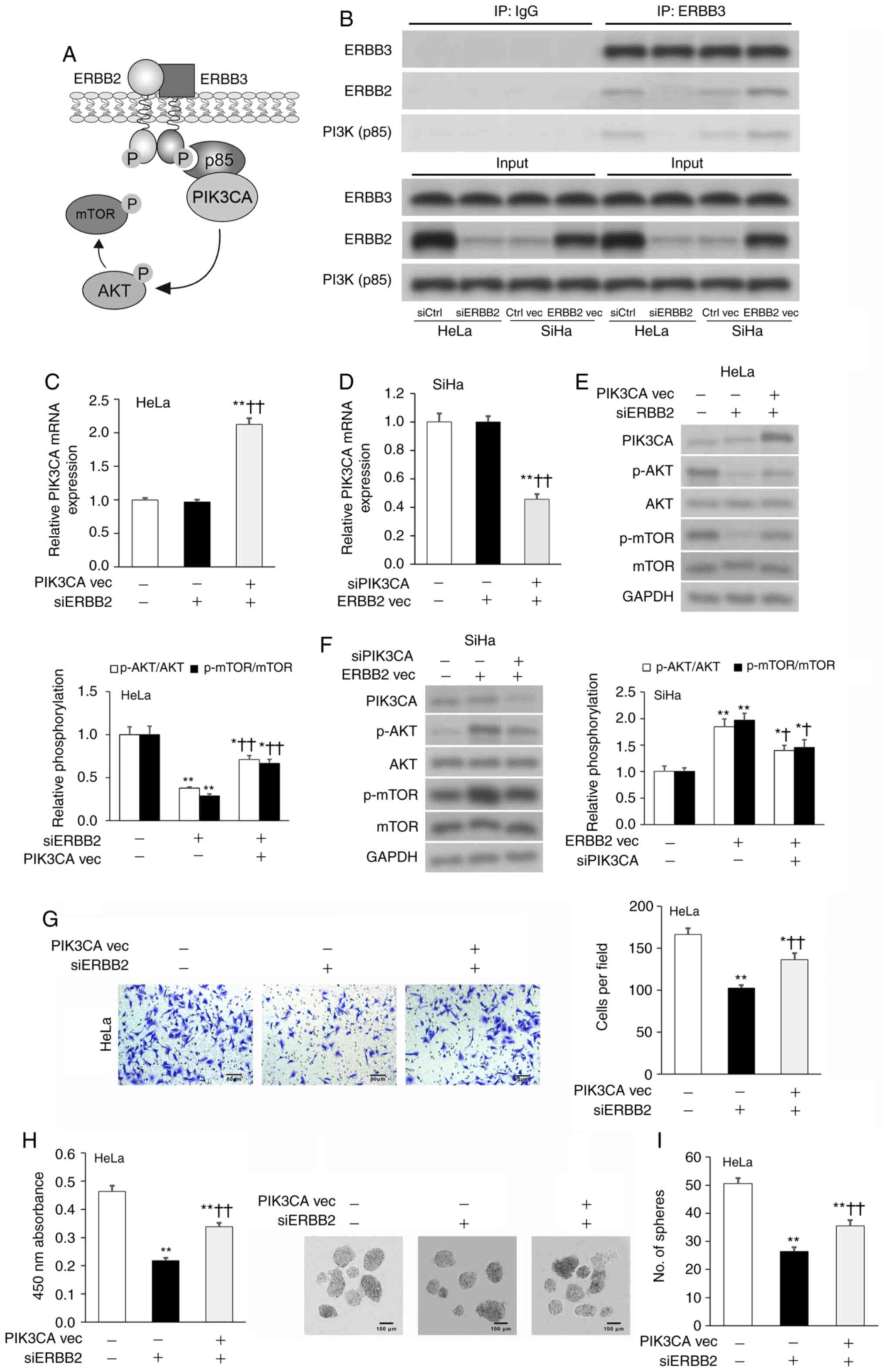 | Figure 3.ERBB2 controls cervical cancer cell
viability and invasion by regulating PIK3CA protein expression. (A)
Schematic diagram of the ERBB2-ERRB3 complex interacting with
PI3K(p85), thereby promoting the downstream phosphorylation of AKT
and mTOR. (B) IP in cervical cancer cell lysates with antibodies
against ERBB3 or IgG control. Expression levels of ERBB3, ERBB2 and
PI3K(p85) in the IP fraction were assessed via WB. PIK3CA mRNA
expression in transfected (C) HeLa and (D) SiHa cells assessed via
RT-qPCR. GAPDH was used as the housekeeping control. PIK3CA,
p-AKT/AKT and p-mTOR/mTOR protein expression in transfected (E)
HeLa and (F) SiHa cells assessed via WB. GAPDH was used as the
loading control. (G) Invasion of transfected HeLa cells assessed
via Transwell assay. (H) Cellular viability of transfected HeLa
cells quantified using Cell Counting Kit-8. (I) Sphere-formation of
transfected HeLa cells. Data are expressed as the mean ± SEM (n=3).
*P<0.05 and **P<0.01 vs. siCtrl or Ctrl vec;
†P<0.05 and ††P<0.01 vs. siERBB2 or
ERBB2 vec. Data were analyzed via one-way ANOVA. IP,
immunoprecipitation; WB, western blotting; RT-qPCR, reverse
transcription-quantitative PCR; si, small interfering; vec, vector;
ERBB2, Erb-B2 Receptor Tyrosine Kinase 2; p-, phosphorylated;
PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic
subunit α. |
As AKT is directly phosphorylated by PIK3CA
(41) and mTOR is a known
downstream target of the ERBB2-PIK3CA-AKT axis (42), the expression levels of AKT
phosphorylation (p-AKT/AKT) and mTOR phosphorylation (p-mTOR/mTOR)
were assessed as indicators of PIK3CA activity. siERBB2 cells
expressed significantly lower AKT and mTOR phosphorylation levels
compared with siCtrl cells, while ERBB2 vec cells expressed
significantly higher AKT and mTOR phosphorylation levels compared
with Ctrl vec cells (Fig. 3E and
F). The addition of PIK3CA vec partially rescued
siERBB2-induced downregulation of AKT and mTOR phosphorylation
levels, while the addition of siPIK3CA partially abrogated ERBB2
vec-induced upregulation of AKT and mTOR phosphorylation levels
(Fig. 3E and F). Since modulating
ERBB2 expression significantly affected ERBB3-PI3K(p85) binding and
downstream AKT/mTOR phosphorylation, but did not change PIK3CA
expression, the present results indicated that ERBB2 promoted
PIK3CA activity via regulating the ERBB3-PI3K(p85) interaction.
Subsequently, the current study examined if PIK3CA
participated in ERBB2-facilitated stimulation of HeLa cell
viability, invasion and sphere-formation. PIK3CA vec resulted in a
partial rescuing of ERBB2-siRNA-mediated attenuation of cellular
viability, invasion and sphere-formation (Fig. 3G-I). Overall, the present findings
suggested that ERBB2 promoted an aggressive phenotype in cervical
cancer cells via stimulating PIK3CA activity.
miR-3184-5p attenuates cervical cancer
cell viability and invasion by targeting ERBB2
The TargetScan database was used to search for
candidate miRNAs that may regulate ERBB2. miR-3184-5p was
identified as a candidate miRNA, with a putative binding site on
the ERBB2 3′-UTR (Fig. 4A).
Therefore, miR-3184-5p was selected for further investigation. The
results revealed that miR-3184-5p expression in HeLa and SiHa cell
lines was significantly lower compared with in the non-cancerous H8
cell line (Fig. 4B). Direct binding
between miR-3184-5p and the ERBB2-3′-UTR was measured using the
luciferase reporter plasmids ERBB2-3′-UTRWT or
ERBB2-3′-UTRMU, which possessed WT or MU miR-3184-5p
binding locations, respectively. In these experiments, HeLa and
SiHa cells underwent co-transfection with the aforementioned WT or
MU reporter plasmids in addition to either miR-3184-5p mimics or
inhibitors (Fig. S3). The
luciferase signal emitted by the WT reporter plasmid was
significantly lower when transfected with miR-3184-5p mimics and
significantly higher when transfected with miR-3184-5p inhibitors
(Fig. 4C and D). On the other hand,
the signal emitted by the MU reporter plasmid was not affected by
miR-3184-5p mimics or inhibitors (Fig.
4C and D). These findings suggested that miR-3184-5p may
directly bind to ERBB2-3′-UTRWT to regulate its
expression. Additionally, ERBB2 and p-AKT protein expression was
decreased in HeLa cells transfected with miR-3184-5p mimics
(Fig. 4E), but was increased in
SiHa cells transfected with miR-3184-5p inhibitors (Fig. 4F). Furthermore, ERBB2-OE in HeLa
cultures rescued miR-3184-5p-induced attenuation of cellular
viability, invasion and sphere-formation (Fig. 4G-I). These experiments demonstrated
that miR-3184-5p directly interacted with the ERBB2 3′-UTR and
inhibited ERBB2 expression, thereby promoting a less aggressive
cervical cancer phenotype.
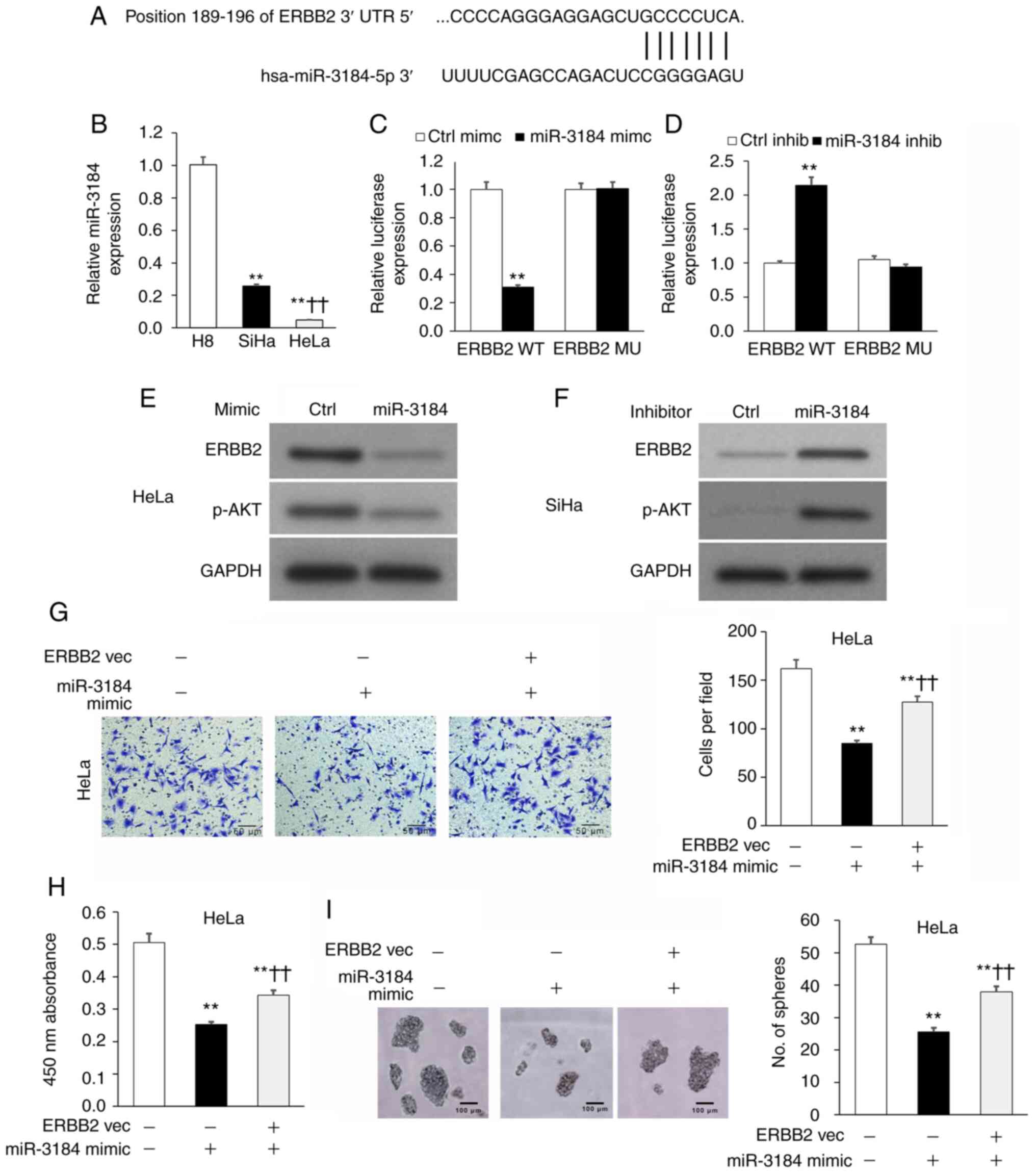 | Figure 4.miR-3184-5p attenuates cervical
cancer cell viability and invasion by targeting ERBB2. (A) Putative
binding location for miR-3184-5p on ERBB2 3′-UTR via TargetScan
analysis. (B) miR-3184-5p expression in HeLa and SiHa cervical
cancer cell lines compared with in the non-cancerous human H8
cervical epithelial cell line assessed via RT-qPCR. U6 was used as
the housekeeping control. **P<0.01 vs. H8;
††P<0.01 vs. SiHa. Luciferase reporter assay of
ERBB2-3′-UTRWT or ERBB2-3′-UTRMU in (C) HeLa
or (D) SiHa cells transfected with miR-3184-5p mimic or inhibitor,
respectively. **P<0.01 vs. Ctrl mimic or Ctrl inhib. WB of (E)
HeLa and (F) SiHa cells transfected with miR-3184-5p mimic or
inhibitor, respectively. (G) Invasion of transfected HeLa cells
assessed via Transwell assay. (H) Cellular viability of transfected
HeLa cells quantified using Cell Counting Kit-8. (I)
Sphere-formation of transfected HeLa cells. Data are expressed as
the mean ± SEM (n=3). **P<0.01 vs. Ctrl vec;
††P<0.01 vs. miR-3184-5p mimic. Data were analyzed
via one-way ANOVA. UTR, untranslated region; WT, wild-type; MU,
mutant; Ctrl, control; inhib, inhibitor; WB, western blotting;
RT-qPCR, reverse transcription-quantitative PCR; miR, microRNA;
vec, vector; ERBB2, Erb-B2 Receptor Tyrosine Kinase 2; PIK3CA,
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
α. |
miR-3184-5p expression is lower in
patient-derived cervical cancer biopsy tissues and low miR-3184-5p
expression is associated with a poor prognosis
The association of miR-3184-5p with
clinicopathological cervical cancer characteristics was assessed in
patient-derived biopsies. miR-3184-5p expression was significantly
lower in cervical cancer biopsy specimens compared with in
neighboring matched healthy tissues (Fig. S4A) and significantly lower in
patients with late-stage (Fig.
S4B) and lymph node metastatic disease (Fig. S4C) compared with in patients with
early stage and no lymph node metastasis. Kaplan-Meier analysis
revealed that low miR-3184-5p expression was associated with poorer
patient survival outcomes compared with high miR-3184-5p expression
(Fig. S4D). Overall, the data
demonstrated that miR-3184-5p expression was negatively associated
with clinicopathological indicators of cervical cancer
progression.
Mithramycin A-induced p53 activation
boosts miR-3184-5p expression, which lowers ERBB2 expression and
attenuates the viability and invasion of cervical cancer cell
lines
Since the tumor suppressor p53 stimulates miR-3184
expression (43), it was
hypothesized that incubation of cervical cancer cells with the p53
activator Mithramycin A (44) would
enhance miR-3184-5p expression and would consequently suppress
ERBB2 expression, cellular viability and invasion. Indeed,
incubation of HeLa and SiHa cultures with Mithramycin A increased
the expression levels of p53 and its downstream mediator p21 in a
dose-dependent manner, while decreasing ERBB2 expression, compared
with the vehicle control cultures (Fig.
5A). As predicted, HeLa and SiHa cells expressed a concurrent
increase in miR-3184-5p (Fig. 5B).
Subsequently, HeLa and SiHa cultures underwent transfection with a
TP53 OE plasmid to test whether p53 controlled miR-3184-5p and
ERBB2 expression. As expected, TP53 OE in HeLa and SiHa cultures
decreased ERBB2 expression (Fig.
5C) and increased miR-3184-5p expression (Fig. 5D). Lastly, incubation of HeLa and
SiHa cultures with Mithramycin A significantly decreased cell
viability, invasion and sphere-formation compared with vehicle
control cultures (Fig. 5E and F).
Overall, these experiments indicated that Mithramycin A-induced p53
activation promoted miR-3184-5p expression, which in turn inhibited
ERBB2 expression, as well as viability and invasion of cervical
cancer cells (Fig. 5G).
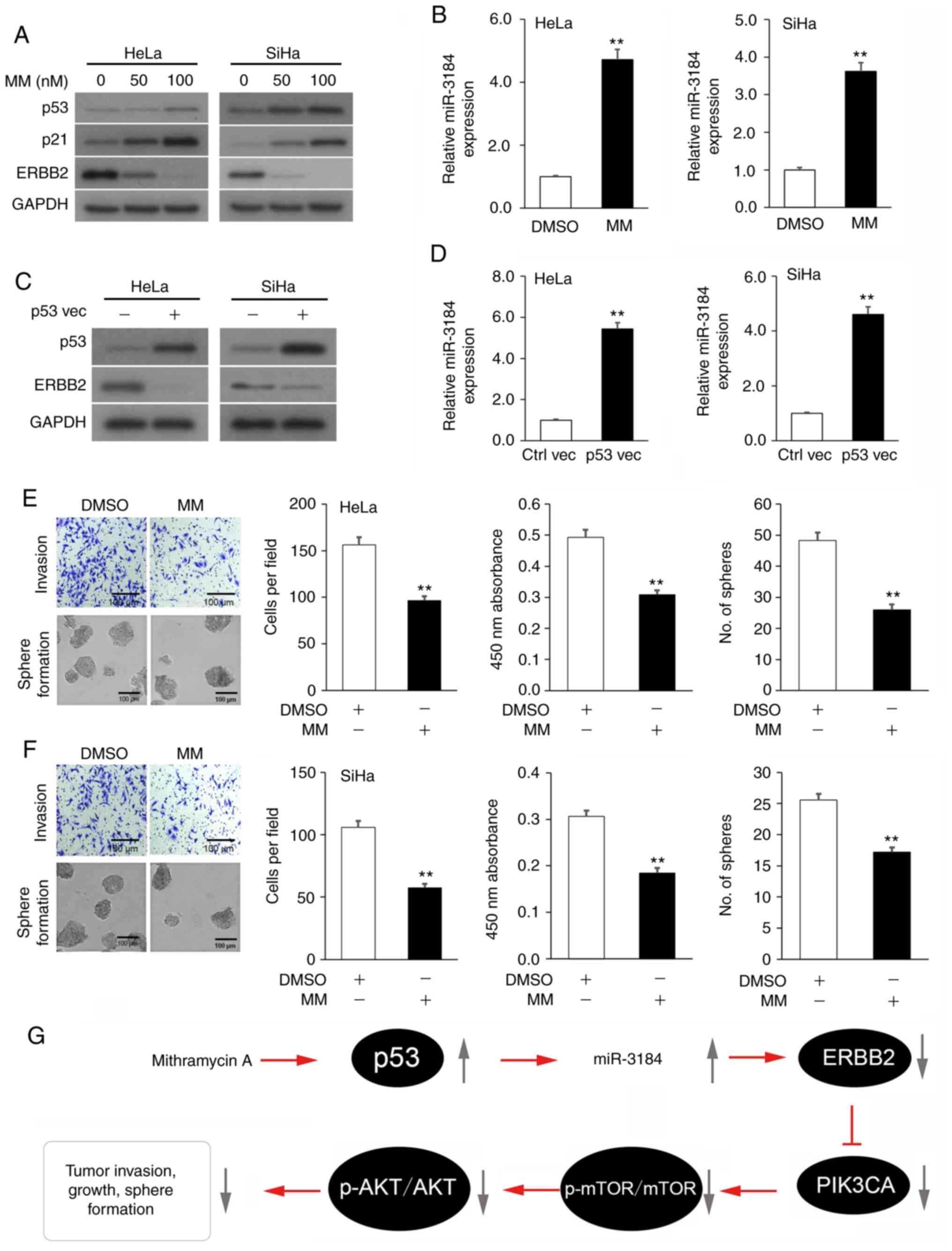 | Figure 5.p53-activating Mithramycin A boosts
miR-3184-5p expression, which lowers ERBB2 expression and
attenuates viability and invasion of cervical cancer cell lines.
(A) p53, p21 and ERBB2 protein expression in cervical cancer
cultures incubated with MM or vehicle (DMSO) assessed via WB. GAPDH
was used as the loading control. (B) miR-3184-5p expression in
cervical cancer cultures incubated with MM or vehicle assessed via
RT-qPCR. U6 was used as the housekeeping control. (C) p53 and ERBB2
protein expression in cervical cancer cultures transfected with a
p53 overexpression plasmid or empty plasmid control assessed via
WB. GAPDH was used as the loading control. (D) miR-3184-5p
expression in cervical cancer cultures transfected with a p53
overexpression plasmid or empty plasmid control assessed via
RT-qPCR. U6 was used as the housekeeping control. (E)
Representative images of Transwell and sphere-formation assays in
(E) HeLa and (F) SiHa cells, and quantitative analysis of
viability, invasion and sphere-formation of cells treated with MM
or vehicle. (G) Schematic diagram of the p53 activator MM rescuing
miR-3184-5p expression, thereby suppressing ERBB2 transcription.
This attenuates PIK3CA activity, which stimulates cervical cancer
cell viability, invasion and sphere-formation. Data are expressed
as the mean ± SEM (n=3). **P<0.01 analyzed via unpaired
Student's t-test. MM, Mithramycin A; Ctrl, control; WB, western
blotting; RT-qPCR, reverse transcription-quantitative PCR; miR,
microRNA; vec, vector; ERBB2, Erb-B2 Receptor Tyrosine Kinase 2;
p-, phosphorylated; PIK3CA, phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit α. |
Discussion
Cervical cancer continues to be among the more
prevalent types of cancer in women globally and was responsible for
over a quarter million mortalities in 2012 (2). To develop improved cervical cancer
treatment options, a deeper knowledge of its molecular determinants
is required. The ERBB2 gene is frequently mutated in several
solid tumors, with the following types of tumor exhibiting the
highest incidence of ERBB2 mutations: Bladder cancer
(16.6%), small intestine cancer (8.6%), ampullar cancer (6.5%),
non-melanoma skin cancer (6.1%) and cervical cancer (5.5%)
(45). ERBB2 upregulation has been
associated with higher tumor grades and more advanced staging in
patients with bladder cancer (46).
On a cellular level, ERBB2 is known to increase the aggressiveness
of breast tumor cells and help them acquire resistance to
conventional chemotherapies (47–49).
Molecularly, ERBB2 is known to heterodimerize with other HER/EGFR
family members to affect downstream cascades associated with
anti-apoptotic mechanisms, cell proliferation and cell invasiveness
(50). For example, ERBB2
stimulates the downstream oncogenic,
multidrug-resistance-conferring PI3K/AKT axis in breast
adenocarcinoma cells through its heterodimerization with ERBB3
(38,39,51).
In contrast to other solid tumors, the relevance of
ERBB2 expression in cervical cancer has not been fully
investigated. Therefore, the present study examined the role of
ERBB2 in cervical cancer. First, ERBB2 expression was analyzed in
patient-derived cervical cancer and matched healthy cervical
tissues, which revealed that elevated ERBB2 expression in tumors
was associated with poorer patient survival outcomes compared with
low ERBB2 expression. In HeLa and SiHa cell lines, OE of ERBB2
increased cell viability, invasion and sphere-formation, while KD
of ERBB2 produced the opposite effect. The present results revealed
that OE of ERBB2 drove activation of PIK3CA and downstream AKT
phosphorylation, which are both crucial oncoproteins, via promoting
ERBB3-PI3K(p85) binding. Notably, ERBB2 mutations frequently
co-occur with PIK3CA mutations in malignant tumors (21.4%)
(45). The current data highlighted
the relevance of upregulated ERBB2 expression in cervical cancer,
which may foster an aggressive cervical cancer phenotype by driving
PIK3CA/p-AKT axis activity.
ERBB2 expression in cancer cells is regulated via
numerous cellular pathways, such as the estrogen receptor-PAX2
pathway, the MKK6/p38 MAPK pathway and the Neu differentiation
factor pathway (52–54). miRNA-mediated regulation of ERBB2
expression is of increasing interest to cancer research; for
instance, it has been demonstrated that miR-34a and miR-155 can
suppress ERBB2-induced pro-malignant effects in breast cancer cells
(55,56). The possibility of miRNA-dependent
aberrant ERBB2 expression in cervical cancer has not been
investigated. The present study revealed that miR-3184-5p
expression was lower in cervical cancer biopsies compared with in
matched control cervical tissues. Furthermore, a direct interaction
between miR-3184-5p and ERBB2 3′-UTR was demonstrated, suggesting
that miR-3184-5p may block ERBB2 to promote a less aggressive
cervical cancer phenotype. The current study revealed a
miR-3184-5p-dependent pathway for the negative regulation of ERBB2
in cervical cancer. This novel discovery suggested that
normalization of miR-3184-5p expression may ameliorate the cervical
cancer phenotype and that this miRNA may be used as a possible
therapeutic avenue.
Mithramycin A attenuates the proliferation of
diverse types of cancer, such as cervical cancer, via lowering the
levels of the transcription factor SP1 (23,57,58),
and stimulates p53 activation in vitro and in vivo
(24). Since the tumor suppressor
p53 induces miR-3184-5p expression (43), the present study hypothesized that
Mithramycin A may restore miR-3184-5p expression in a p53-dependent
manner. Indeed, treatment of HeLa and SiHa cultures with
Mithramycin A increased p53 and miR-3184-5p expression, while
lowering ERBB2 expression, which led to decreased cell viability,
invasion and sphere-formation. Notably, ERBB2 mutations
frequently co-occur with TP53 mutations in malignant tumors
(59.5%) (45). Mithramycin A
possesses a favorable safety profile following long-term treatment
in mouse cervical cancer xenograft models (23), advocating Mithramycin A as a
potential cervical cancer treatment option.
There are a few limitations to the present study.
First, the study was limited to an in vitro investigation of
the p53/miR-3184-5p/ERBB2 axis in patient-derived cervical cancer
samples and established cell lines. Future work should examine the
role of this axis in animal models of cervical cancer. Second, p53,
as a key tumor suppressor, has a multitude of downstream targets
that regulate cell proliferation, migration, metastasis and cell
cycle progression (59); however,
it is infeasible to study all p53 downstream signaling pathways in
a single study. Therefore, the scope of the current study was
solely focused on identifying and establishing the effects of the
p53/miR-3184-5p/ERBB2 axis in cervical cancer cells.
In conclusion, the present study reported the
involvement of the p53/miR-3184-5p/ERBB2 axis in promoting cervical
cancer development via PIK3CA. Treatment of cervical cancer cells
with the p53 activator Mithramycin A restored the levels of the
endogenous ERBB2 inhibitor miR-3184-5p and may constitute a novel
treatment strategy for cervical cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Bengbu Medical College (grant no. BYKF1726),
the Anhui Provincial Natural Science Foundation (grant no.
1908085MH252) and the Key Program of Natural Science Research of
Higher Education of Anhui Province (grant no. KJ2019A0346).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL conceived and designed the study. YL, JZ, NW, FL
and LW performed the experiments. YZ, JL, XZ, SG and HW analyzed
the data. HL drafted the manuscript. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics Review
Committee of the First Affiliated Hospital of Bengbu Medical
College (approval no. 201858; Bengbu, China). Patients who donated
cervical cancer tissues gave their informed consent in writing
prior to their involvement in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA A Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA A
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar
|
|
3
|
Colombo N, Carinelli S, Colombo A, Marini
C, Rollo D and Sessa C; ESMO Guidelines Working Group, : Cervical
cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment
and follow-up. Ann Oncol. 23 (Suppl 7):vii27–vii32. 2012.
View Article : Google Scholar
|
|
4
|
Manzo-Merino J, Contreras-Paredes A,
Vázquez-Ulloa E, Rocha-Zavaleta L, Fuentes-Gonzalez AM and Lizano
M: The role of signaling pathways in cervical cancer and molecular
therapeutic targets. Arch Med Res. 45:525–539. 2014. View Article : Google Scholar
|
|
5
|
Konno Y, Dong P, Xiong Y, Suzuki F, Lu J,
Cai M, Watari H, Mitamura T, Hosaka M, Hanley SJ, et al:
MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation,
invasion and stem cell-like phenotype of aggressive endometrial
cancer cells. Oncotarget. 5:6049–6062. 2014. View Article : Google Scholar
|
|
6
|
Liang X, Liu Y, Zeng L, Yu C, Hu Z, Zhou Q
and Yang Z: miR-101 inhibits the G1-to-S phase transition of
cervical cancer cells by targeting Fos. Int J Gynecol Cancer.
24:1165–1172. 2014. View Article : Google Scholar
|
|
7
|
Cheung T, Leung J, Chung TK, Lam S, To K
and Wong Y: C-fos overexpression is associated with the
pathoneogenesis of invasive cervical cancer. Gynecol Obstet Invest.
43:200–203. 1997. View Article : Google Scholar
|
|
8
|
Dong P, Ihira K, Hamada J, Watari H,
Yamada T, Hosaka M, Hanley SJ, Kudo M and Sakuragi N: Reactivating
p53 functions by suppressing its novel inhibitor iASPP: A potential
therapeutic opportunity in p53 wild-type tumors. Oncotarget.
6:19968–19975. 2015. View Article : Google Scholar
|
|
9
|
Dong P, Xiong Y, Watari H, Hanley SJ,
Konno Y, Ihira K, Suzuki F, Yamada T, Kudo M, Yue J and Sakuragi N:
Suppression of iASPP-dependent aggressiveness in cervical cancer
through reversal of methylation silencing of microRNA-124. Sci Rep.
6:354802016. View Article : Google Scholar
|
|
10
|
Xiong Y, Sun F, Dong P, Watari H, Yue J,
Yu MF, Lan CY, Wang Y and Ma ZB: iASPP induces EMT and cisplatin
resistance in human cervical cancer through miR-20a-FBXL5/BTG3
signaling. J Exp Clin Cancer Res. 36:482017. View Article : Google Scholar
|
|
11
|
Romaine SP, Tomaszewski M, Condorelli G
and Samani NJ: MicroRNAs in cardiovascular disease: An introduction
for clinicians. Heart. 101:921–928. 2015. View Article : Google Scholar
|
|
12
|
Sun NX, Ye C, Zhao Q, Zhang Q, Xu C, Wang
SB, Jin ZJ, Sun SH, Wang F and Li W: Long noncoding RNA-EBIC
promotes tumor cell invasion by binding to EZH2 and repressing
E-cadherin in cervical cancer. PLoS One. 9:e1003402014. View Article : Google Scholar
|
|
13
|
Hu X, Schwarz JK, Lewis JS Jr, Huettner
PC, Rader JS, Deasy JO, Grigsby PW and Wang X: A microRNA
expression signature for cervical cancer prognosis. Cancer Res.
70:1441–1448. 2010. View Article : Google Scholar
|
|
14
|
Hattori H, Janky RS, Nietfeld W, Aerts S,
Madan Babu M and Venkitaraman AR: p53 shapes genome-wide and cell
type-specific changes in microRNA expression during the human DNA
damage response. Cell Cycle. 13:2572–2586. 2014. View Article : Google Scholar
|
|
15
|
Croessmann S, Formisano L, Kinch LN,
Gonzalez-Ericsson PI, Sudhan DR, Nagy RJ, Mathew A, Bernicker EH,
Cristofanilli M, He J, et al: Combined blockade of activating ERBB2
mutations and ER results in synthetic lethality of ER+/HER2 mutant
breast cancer. Clin Cancer Res. 25:277–289. 2019. View Article : Google Scholar
|
|
16
|
Sanchez-Vega F, Hechtman JF, Castel P,
Castel P, Ku GY, Tuvy Y, Won H, Fong CJ, Bouvier N, Nanjangud GJ,
et al: EGFR and MET amplifications determine response to HER2
inhibition in ERBB2-amplified esophagogastric cancer. Cancer
Discov. 9:199–209. 2019. View Article : Google Scholar
|
|
17
|
Ross JS, Fakih M, Ali SM, Elvin JA,
Schrock AB, Suh J, Vergilio JA, Ramkissoon S, Severson E, Daniel S,
et al: Targeting HER2 in colorectal cancer: The landscape of
amplification and short variant mutations in ERBB2 and ERBB3.
Cancer. 124:1358–1373. 2018. View Article : Google Scholar
|
|
18
|
Xiang L, Jiang W, Ye S, He T, Pei X, Li J,
Chan DW, Ngan HYS, Li F, Tao P, et al: ERBB2 mutation: A promising
target in non-squamous cervical cancer. Gynecol Oncol. 148:311–316.
2018. View Article : Google Scholar
|
|
19
|
Barceló F, Ortiz-Lombardia M, Martorell M,
Oliver M, Méndez C, Salas JA and Portugal J: DNA binding
characteristics of mithramycin and chromomycin analogues obtained
by combinatorial biosynthesis. Biochemistry. 49:10543–10552. 2010.
View Article : Google Scholar
|
|
20
|
Kennedy B and Torkelson JL: Long-term
follow-up of stage III testicular carcinoma treated with
mithramycin (Plicamycin). Med Pediatr Oncol. 24:327–328. 1995.
View Article : Google Scholar
|
|
21
|
Dutcher JP, Coletti D, Paietta E and
Wiernik PH: A pilot study of alpha-interferon and plicamycin for
accelerated phase of chronic myeloid leukemia. Leuk Res.
21:375–380. 1997. View Article : Google Scholar
|
|
22
|
Fernández-Guizán A, Mansilla S, Barceló F,
Barceló F, Vizcaíno C, Núñez LE, Morís F, González S and Portugal
J: The activity of a novel mithramycin analog is related to its
binding to DNA, cellular accumulation, and inhibition of Sp1-driven
gene transcription. Chem Biol Interact. 219:123–132. 2014.
View Article : Google Scholar
|
|
23
|
Choi ES, Nam JS, Jung JY, Cho NP and Cho
SD: Modulation of specificity protein 1 by mithramycin A as a novel
therapeutic strategy for cervical cancer. Sci Rep. 4:71622014.
View Article : Google Scholar
|
|
24
|
Rao M, Atay SM, Shukla V, Hong Y, Upham T,
Ripley RT, Hong JA, Zhang M, Reardon E, Fetsch P, et al:
Mithramycin depletes specificity protein 1 and activates p53 to
mediate senescence and apoptosis of malignant pleural mesothelioma
cells. Clin Cancer Res. 22:1197–1210. 2016. View Article : Google Scholar
|
|
25
|
Thorpe LM, Yuzugullu H and Zhao JJ: PI3K
in cancer: Divergent roles of isoforms, modes of activation and
therapeutic targeting. Nat Rev Cancer. 15:72015. View Article : Google Scholar
|
|
26
|
Wright AA, Howitt BE, Myers AP, Dahlberg
SE, Palescandolo E, Van Hummelen P, MacConaill LE, Shoni M, Wagle
N, Jones RT, et al: Oncogenic mutations in cervical cancer: Genomic
differences between adenocarcinomas and squamous cell carcinomas of
the cervix. Cancer. 119:3776–3783. 2013. View Article : Google Scholar
|
|
27
|
Benedet J, Pecorelli S, Ngan H and Hacker
NF: Staging classifications and clinical practice guidelines for
gynaecological cancers. Int J Gynecol Obstetr. 70:207–312. 2000.
View Article : Google Scholar
|
|
28
|
Gioia G, Werner B, Nydam D and Moroni P:
Validation of a mycoplasma molecular diagnostic test and
distribution of mycoplasma species in bovine milk among New York
State dairy farms. J Dairy Sci. 99:4668–4677. 2016. View Article : Google Scholar
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Dong P, Xiong Y, Watari H, Hanley SJ,
Konno Y, Ihira K, Yamada T, Kudo M, Yue J and Sakuragi N: MiR-137
and miR-34a directly target Snail and inhibit EMT, invasion and
sphere-forming ability of ovarian cancer cells. J Exp Clin Cancer
Res. 35:1322016. View Article : Google Scholar
|
|
31
|
Ihira K, Dong P, Xiong Y, Watari H, Konno
Y, Hanley SJ, Noguchi M, Hirata N, Suizu F, Yamada T, et al: EZH2
inhibition suppresses endometrial cancer progression via
miR-361/Twist axis. Oncotarget. 8:13509–13520. 2017. View Article : Google Scholar
|
|
32
|
Hsiao YH, Hsieh MJ, Yang SF, Chen SP, Tsai
WC and Chen PN: Phloretin suppresses metastasis by targeting
protease and inhibits cancer stemness and angiogenesis in human
cervical cancer cells. Phytomedicine. 62:1529642019. View Article : Google Scholar
|
|
33
|
Hu Y, Ma Y, Liu J, Cai Y, Zhang M and Fang
X: LINC01128 expedites cervical cancer progression by regulating
miR-383-5p/SFN axis. BMC Cancer. 19:1–11. 2019. View Article : Google Scholar
|
|
34
|
Lv Y, Cang W, Li Q, Liao X, Zhan M, Deng
H, Li S, Jin W, Pang Z, Qiu X, et al: Erlotinib overcomes
paclitaxel-resistant cancer stem cells by blocking the
EGFR-CREB/GRβ-IL-6 axis in MUC1-positive cervical cancer.
Oncogenesis. 8:702019. View Article : Google Scholar
|
|
35
|
Han SA, Jang JH, Won KY, Lim SJ and Song
JY: Prognostic value of putative cancer stem cell markers (CD24,
CD44, CD133, and ALDH1) in human papillary thyroid carcinoma.
Pathol Res Pract. 213:956–963. 2017. View Article : Google Scholar
|
|
36
|
Mizukami T, Kamachi H, Mitsuhashi T,
Tsuruga Y, Hatanaka Y, Kamiyama T, Matsuno Y and Taketomi A:
Immunohistochemical analysis of cancer stem cell markers in
pancreatic adenocarcinoma patients after neoadjuvant
chemoradiotherapy. BMC Cancer. 14:6872014. View Article : Google Scholar
|
|
37
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar
|
|
38
|
Siegel PM, Ryan ED, Cardiff RD and Muller
WJ: Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3
are involved in the induction of mammary tumors in transgenic mice:
Implications for human breast cancer. EMBO J. 18:2149–2164. 1999.
View Article : Google Scholar
|
|
39
|
Zhou BP, Hu MC, Miller SA, Yu Z, Xia W,
Lin SY and Hung MC: HER-2/neu blocks tumor necrosis factor-induced
apoptosis via the Akt/NF-kappaB pathway. J Biol Chem.
275:8027–8031. 2000. View Article : Google Scholar
|
|
40
|
Prigent SA and Gullick WJ: Identification
of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and
SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 13:2831–2841.
1994. View Article : Google Scholar
|
|
41
|
Vasudevan KM, Barbie DA, Davies MA,
Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P,
Kim SY, et al: AKT-independent signaling downstream of oncogenic
PIK3CA mutations in human cancer. Cancer Cell. 16:21–32. 2009.
View Article : Google Scholar
|
|
42
|
Bonazzoli E, Cocco E, Lopez S, Bellone S,
Zammataro L, Bianchi A, Manzano A, Yadav G, Manara P, Perrone E, et
al: PI3K oncogenic mutations mediate resistance to afatinib in
HER2/neu overexpressing gynecological cancers. Gynecol Oncol.
153:158–164. 2019. View Article : Google Scholar
|
|
43
|
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto
T, Kato S and Miyazono K: Modulation of microRNA processing by p53.
Nature. 460:529–533. 2009. View Article : Google Scholar
|
|
44
|
Dong P, Xiong Y, Hanley SJ, Yue J and
Watari H: Musashi-2, a novel oncoprotein promoting cervical cancer
cell growth and invasion, is negatively regulated by p53-induced
miR-143 and miR-107 activation. J Exp Clin Cancer Res. 36:1502017.
View Article : Google Scholar
|
|
45
|
Cousin S, Khalifa E, Crombe A, Laizet Y,
Lucchesi C, Toulmonde M, Le Moulec S, Auzanneau C, Soubeyran I and
Italiano A: Targeting ERBB2 mutations in solid tumors: Biological
and clinical implications. J Hematol Oncol. 11:862018. View Article : Google Scholar
|
|
46
|
Breyer J, Wirtz RM, Laible M, Schlombs K,
Erben P, Kriegmair MC, Stoehr R, Eidt S, Denzinger S, Burger M, et
al: ESR1, ERBB2, and Ki67 mRNA expression predicts stage and grade
of non-muscle-invasive bladder carcinoma (NMIBC). Virchows Arch.
469:547–552. 2016. View Article : Google Scholar
|
|
47
|
Ursini-Siegel J, Schade B, Cardiff RD and
Muller WJ: Insights from transgenic mouse models of ERBB2-induced
breast cancer. Nat Rev Cancer. 7:389–397. 2007. View Article : Google Scholar
|
|
48
|
Di Cosimo S and Baselga J: Targeted
therapies in breast cancer: Where are we now? Eur J Cancer.
44:2781–2790. 2008. View Article : Google Scholar
|
|
49
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View Article : Google Scholar
|
|
50
|
Olayioye MA, Neve RM, Lane HA and Hynes
NE: The ErbB signaling network: Receptor heterodimerization in
development and cancer. EMBO J. 19:3159–3167. 2000. View Article : Google Scholar
|
|
51
|
Knuefermann C, Lu Y, Liu B, Jin W, Liang
K, Wu L, Schmidt M, Mills GB, Mendelsohn J and Fan Z:
HER2/PI-3K/Akt activation leads to a multidrug resistance in human
breast adenocarcinoma cells. Oncogene. 22:3205–3212. 2003.
View Article : Google Scholar
|
|
52
|
Hurtado A, Holmes KA, Geistlinger TR,
Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S and
Carroll JS: Regulation of ERBB2 by oestrogen receptor-PAX2
determines response to tamoxifen. Nature. 456:663–666. 2008.
View Article : Google Scholar
|
|
53
|
Demidov ON, Kek C, Shreeram S, Timofeev O,
Fornace AJ, Appella E and Bulavin DV: The role of the MKK6/p38 MAPK
pathway in Wip1-dependent regulation of ErbB2-driven mammary gland
tumorigenesis. Oncogene. 26:2502–2506. 2007. View Article : Google Scholar
|
|
54
|
Daly JM, Jannot CB, Beerli RR, Graus-Porta
D, Maurer FG and Hynes NE: Neu differentiation factor induces ErbB2
down-regulation and apoptosis of ErbB2-overexpressing breast tumor
cells. Cancer Res. 57:3804–3811. 1997.
|
|
55
|
Wang Y, Zhang X, Chao Z, Kung HF, Lin MC,
Dress A, Wardle F, Jiang BH and Lai L: MiR-34a modulates ErbB2 in
breast cancer. Cell Biol Int. 41:93–101. 2017. View Article : Google Scholar
|
|
56
|
He XH, Zhu W, Yuan P, Jiang S, Li D, Zhang
HW and Liu MF: miR-155 downregulates ErbB2 and suppresses
ErbB2-induced malignant transformation of breast epithelial cells.
Oncogene. 35:6015–6025. 2016. View Article : Google Scholar
|
|
57
|
Malek A, Núñez LE, Magistri M, Brambilla
L, Jovic S, Carbone GM, Morís F and Catapano CV: Modulation of the
activity of Sp transcription factors by mithramycin analogues as a
new strategy for treatment of metastatic prostate cancer. PLoS One.
7:e351302012. View Article : Google Scholar
|
|
58
|
Wang L, Guan X, Zhang J, Jia Z, Wei D, Li
Q, Yao J and Xie K: Targeted inhibition of Sp1-mediated
transcription for antiangiogenic therapy of metastatic human
gastric cancer in orthotopic nude mouse models. Int J Oncol.
33:161–167. 2008.
|
|
59
|
Haupt S, Raghu D and Haupt Y: Mutant p53
drives cancer by subverting multiple tumor suppression pathways.
Front Oncol. 6:122016. View Article : Google Scholar
|














