Introduction
From a genomic standpoint, colorectal cancer is not
a single disease, but a heterogeneous group of malignancies arising
within the colon and rectum. Colorectal cancer accounts for
approximately 10% of all annually diagnosed cancers and
cancer-related deaths worldwide. Currently, it is the world's
fourth deadliest cancer with almost 900,000 deaths annually
(1,2).
Metastasis to either liver, lung, brain or peritoneum is present in
about 90% of patients with stage IV disease. Twenty percent of CRC
patients have metastases at the time of diagnosis. About 40% of
patients with stage II–III succumb to recurrence in the next 5
years after surgical treatment (3).
Liver metastases are often detected only in advanced disease and
even when resection is combined with modern adjuvant systemic
regimens, it is curative in only 20% of patients, with 70%
developing recurrence. Owing to difficulties in the detection and
treatment of the metastatic spread of CRC, the research is based on
identifying high-risk patient cohorts and new biomarkers based on
the differences between metastatic vs. non-metastatic cells
(4).
Colon cancer progresses through a well-defined
series of transformations from normal colonic epithelial cells into
precursor adenoma lesions which eventually evolve to increasingly
more invasive and malignant stages. Generally, the defining
hallmark of metastasis is development of any secondary mass that is
no longer directly connected to the originating tumour. Metastases
are distinct and unique subsets of cells that emigrated from the
primary tumour and are molecularly, genetically, and biochemically
distinct from the cells remaining at the site of tumour origin
(5).
Metastatic cells are able to successfully
dissociate, disseminate, and colonize secondary sites, so they
acquire properties in addition to those necessary to become
neoplastic: Motility and invasion, ability to modulate the
secondary site or local microenvironments, plasticity, and ability
to colonize secondary tissues (5,6). The
generation of the tumour in a foreign organ is tightly bound to the
acquisition of a stem-like phenotype by cancer cells. Recent
findings suggest that cancer stem cells (CSCs) are a phenotypically
and functionally heterogeneous population, which is dynamic and is
able to adapt as a result of various extrinsic and intrinsic
cellular factors (7). Metastatic stem
cells use multiple phenotypes and behaviours and critically depend
on their interaction with the microenvironment to migrate, survive
in the circulation and thrive in a foreign organ (8).
A large study of CRC patients investigating the
molecular differences between primary tumours, lymph node
metastases and distant metastases using next-generation sequencing
(NGS) and immunohistochemistry showed that lymphatic and distant
metastases harbour different mutation profiles compared to their
primary tumour and between each other (9). The transcriptomes of primary CRC and
their metastatic lesions at both the gene and pathway levels were
compared and showed differences between them (10). Principles and differences with respect
to CRC organotropism, epithelial-mesenchymal transition,
angiogenesis and inflammation were also published (11). However, a complex summary of the
traits of metastatic colorectal cells through the lens of molecular
biology is missing.
This review therefore summarises biological
properties of metastatic colorectal cells, their genetic and
molecular determinants of metastatic competence and active
molecular pathways. A deeper understanding of molecular signatures
of metastatic cells can lead to finding new therapeutic targets and
drug regimens to cure metastatic dissemination.
Biology of metastatic colorectal cancer
Metastatic cells have the ability, the metastatic
competence, to fine-tune malignant properties of primary tumour
cancer cells by building upon an already tumourigenic program,
enhancing their own stem-like features and becoming predominant
during disease progression and metastatic dissemination.
According to the traditional model of progressive
acquisition of mutations during colon cancerogenesis, the
metastatic capacity/competence is acquired later in time during the
multi-step tumour progression process after accumulation of genetic
alterations. K-RAS, B-RAF, APC and P53 mutations in case of
CIN and mutations in the mismatch repair genes in case of MSI
result in the gradual transition of an adenoma into carcinoma
(12). Metastatic ability is thought
to be acquired in later stages of carcinogenesis. A limited number
of cells acquire the ability to move from the primary tumour into
the bloodstream or lymphatic system, to migrate to a distant
location in the body and to grow into tumours in the new location
(12).
On the other hand, even in 2003, a study from
Ramaswamy et al suggested a novel idea about rare cells with
an ability to metastasize within a primary tumour. Those authors
showed that the metastatic potential of human tumours is encoded in
the bulk of a primary tumour and the gene expression signatures of
primary tumours are predictive of distant recurrence (13) (Fig. 1).
Close genetic relationships between primary tumours and metastases
in a variety of cancer types indicate that, at least in certain
cases, the cells forming a metastatic colony derive from a dominant
clonal subpopulation of the primary tumour. These populations
manage to complete all of the steps required both for primary
tumour formation and the subsequent multi-step invasion of
metastasis cascade (14). The
completion of this cascade depends not only on genetic changes but
especially on specifically epigenetically organized programs that
complement the previously acquired genetic mutations (6).
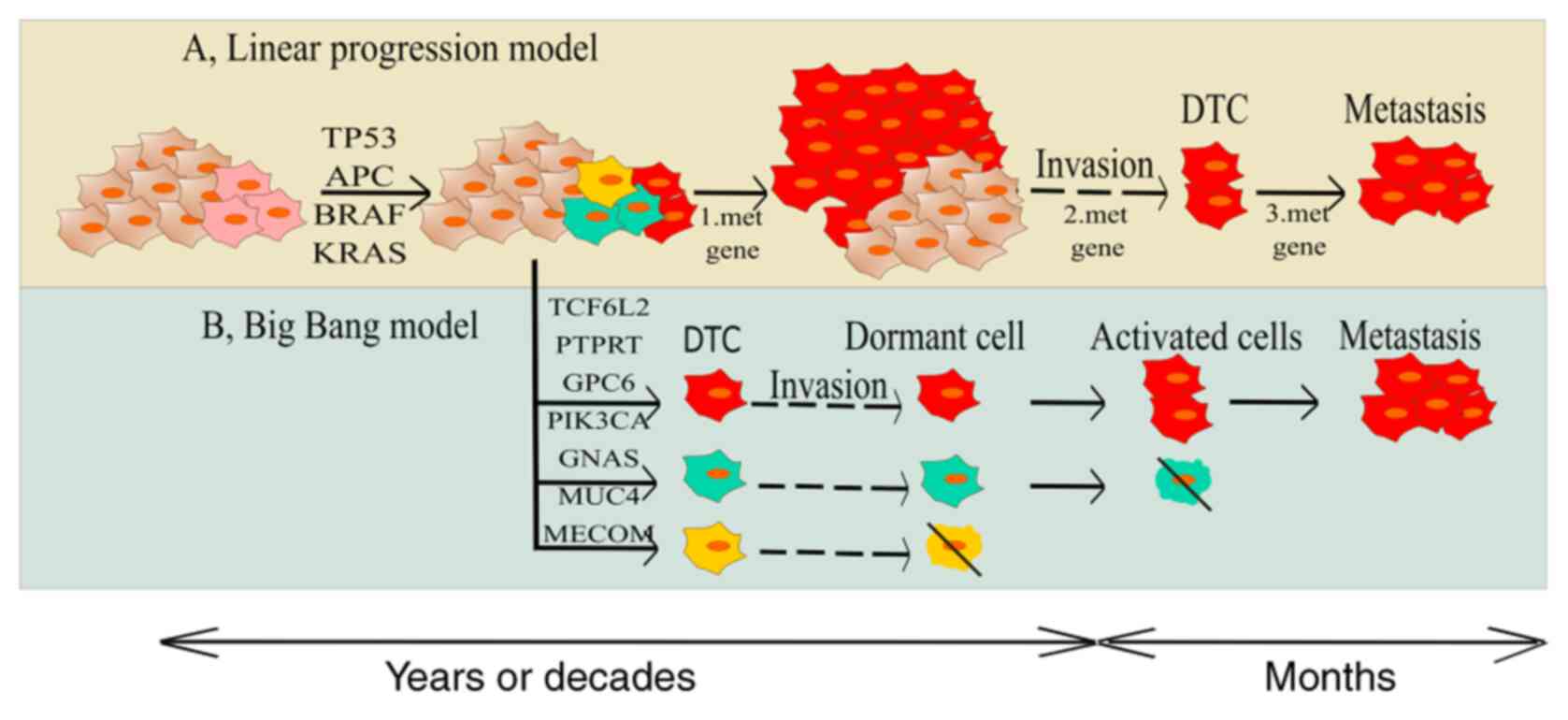 | Figure 1.Two models regarding gaining
metastatic competence. (A) Tranditional model is the concept of
transition of initiated cell to more aggressive state and gaining
of metastatic ability due to the accumulation of genetic and
epigenetic changes. (B) The second model explains that there are
some clones in tumour bulk with metastatic potential already
present in early stages of carcinogenesis and their individual gene
signature is predictive for the invasiveness and distant
recurrence. APC, adenomatous polyposis coli; CIN, chromosome
instability; DTC, dormant tumour cells; MSI, microsatellite
instability; SMAD4, mothers against decapentaplegic homolog 4;
PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic
subunit alpha; TCF7L2, transcription factor 7-like 2; AMER1, APC
membrane recruitment protein 1; PTPRT, Receptor-type
tyrosine-protein phosphatase T; GNAS, heterotrimeric G-protein
alpha subunit Gs-α; FXR1, fragile X mental retardation
syndrome-related protein 1; MUC4, Mucin 4; GPC6, Glypican-6; MECOM,
MDS1 and EVI1 complex locus protein EVI1. |
The ‘Big Bang’ theory of tumour evolution says that
after transformation some cancer cells grow as a single expansion
giving rise to effective subclones constituting intratumoural
heterogeneity (ITH) (15). Genomic
profiling of 349 individual samples from 15 colorectal tumours
showed an absence of selective steps, uniformly high ITH and
subclone mixing in distant regions. The most detectable ITH
originates from early private alterations and not from later clonal
expansions (16). Recently, large NGS
studies of paired colorectal cancer and their metastases were
performed showing that the primary tumours' vs. paired metastases'
genomic divergence is low and their invasive and metastatic
potential were acquired early, even during the time the tumour
could not be diagnosed (17). An
important finding is that the vast majority (90%) of primary
tumours exhibits subclonal selection consistent with the metastatic
clone gaining a selective growth advantage. On the other hand,
subclonal selection was detected only in 33% of patients with
early-stage CRC (17).
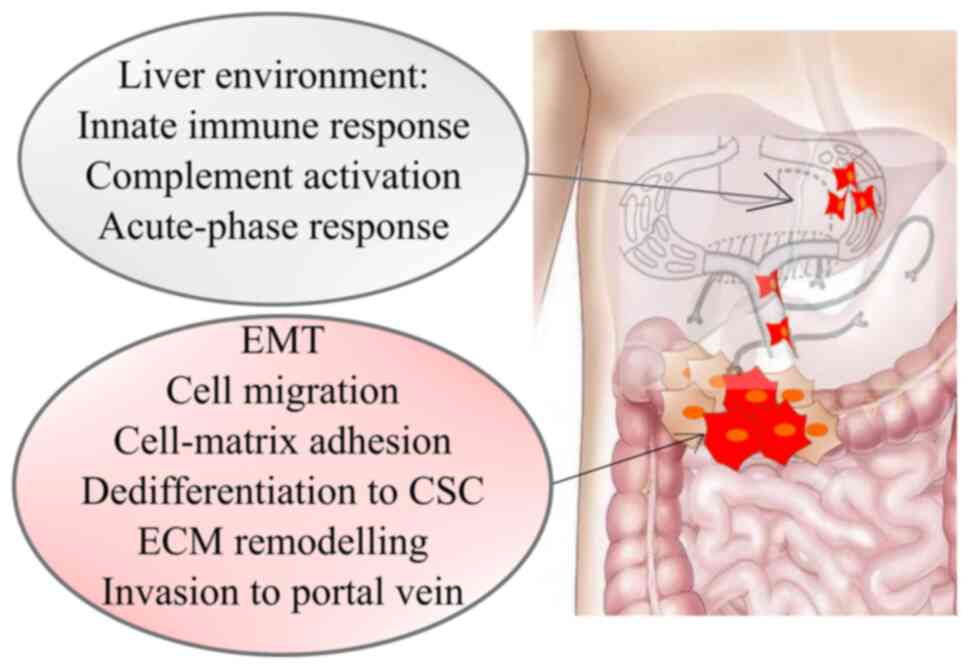
Pattern of colorectal cancer metastasis
Colorectal cancer shows sequential organ-specific
colonization, with the first site of metastasis being usually the
liver and secondarily the lung. Focusing on these most frequent
metastatic sites, 70% of colorectal cancer metastasizes to the
liver, and 47.5% of the patients presented with metastases confined
to the lungs, distant lymph nodes (16%), and peritoneum (15%)
(18). When it comes to metastases
arising from the colon and proximal parts of the rectum, the portal
system guides the blood flow directly to the liver (Fig. 2). In hematogenous spread platelets and
neutrophils help CTC by protecting them from elimination by NK
cells. The entry of colorectal carcinoma cells into the hepatic
microvasculature can initiate a pro-inflammatory cascade that
results in Kupffer as well as stellate cells being triggered to
secrete chemokines that upregulate vascular adhesion receptors,
thereby enabling the adhesion of CTC in the microvasculature of the
liver (19). Neutrophils support
metastatic spread by forming an extracellular trap for CTC in the
bloodstream helping them adhere to endothelial cells and
extravasate. The blood group antigens sLea and sLex may play an
important role in the attachment (11). The primary tumour itself can actively
support preparing of premetastatic niche by recruiting VEGFR-1
expressing haematopoietic progenitor cells. The pro-tumour
microenvironment is composed of inflammatory and immune cells
involving cancer-associated fibroblasts (CAFs), neutrophils and
macrophages and environmental conditions such as hypoxia, soluble
factors, signalling molecules, and ECM components. At the early
stages, the proinflammatory TAMs subtype 1 are active and work to
eliminate malignant cells. At later stages, macrophages switch to
immunosuppressive TAMs subtype 2 creating the microenvironment
permissive for tumour growth by secretion of ECM-degrading
components (MMP1, 7, 9, 12) (11).
The metastatic cascade ends with the invasion of colorectal cancer
cells, as well as the adaptation and colonization of the hepatic
parenchyma (11).
Lung metastases usually appeared together with liver
metastases (73% in colon cancer/60% in rectal cancer) (20,21). CRC
cells disseminate through portal circulation to the liver, and from
there to the lungs. Metastatic cells flow to the lungs directly by
lymphatic system or from the distal rectum via systemic circulation
through the hemorrhoidal veins. Metastases of bones and the nervous
system in CRC patients occur more frequently in the patients with
lung metastases, but are sporadic in patients with liver metastases
(20). This indicated that the lungs
are an important waypoint towards further metastatic dissemination
(20). A study on the onset of growth
of lung metastases revealed that some CRC patients with isolated
synchronous liver metastasis already have lung metastases even when
the metastatic sites are considered to be limited to the liver
(22). Compared to other distal
metastases, lung metastases grow slowly and have better overall
survival (21).
Expression analysis of CRC metastasis revealed 22
specific genes related to liver metastasis and they were strongly
associated with: i) Cell migration, adhesion, proliferation (cell
adhesion/focal adhesion/chemokine signalling pathway/PI3K-AKT
signalling pathway/APOH/ F5/CXCL14); and ii) immune response
(innate immune response/complement activation/acute-phase
response/SERPIN A1/CXCL14). CXCL14 may be a favourable prediction
factor that is involved in liver metastasis of colon carcinoma
(23). Signalling network analyses
have indicated that the PI3K-AKT pathway is highly activated in
liver metastases from colorectal cancer compared with matched
primary tumours.
Testing of gene signature in the metastatic process
of CRC lung metastasis revealed APC, TP53 and KRAS
being the most commonly mutated genes. Mutations in EGFR, GNAQ,
KIT, MET and PTPN11 genes were associated with an early
pulmonary recurrence. The two strongest affected pathways were the
RAS signalling pathway (EGFR, KIT, MET and PTPN11) and the RAP1
signalling pathway (EGFR, GNAQ, KIT and MET) (24).
CRC metastasizes within the abdominal cavity too,
resulting in non-hematogenous metastases in ovaries. All parts of
the gastrointestinal system share a mutual lymphatic drain, flowing
to the left subclavian vein. Moreover, metastases may spread
through the peritoneal fluid within the peritoneal cavity (25).
Functional traits of metastatic cells
Metastasis formation is a multi-step process
involving invasion of primary tumour cells through the basement
membrane and intravasation into nearby lymphatics or blood vessels.
These tumour cells must then survive transport to distant organ
sites (typically liver in CRC) where they extravasate and colonize
the secondary organ site to establish micro-metastases, which
proliferate to expand to macrometastases (26).
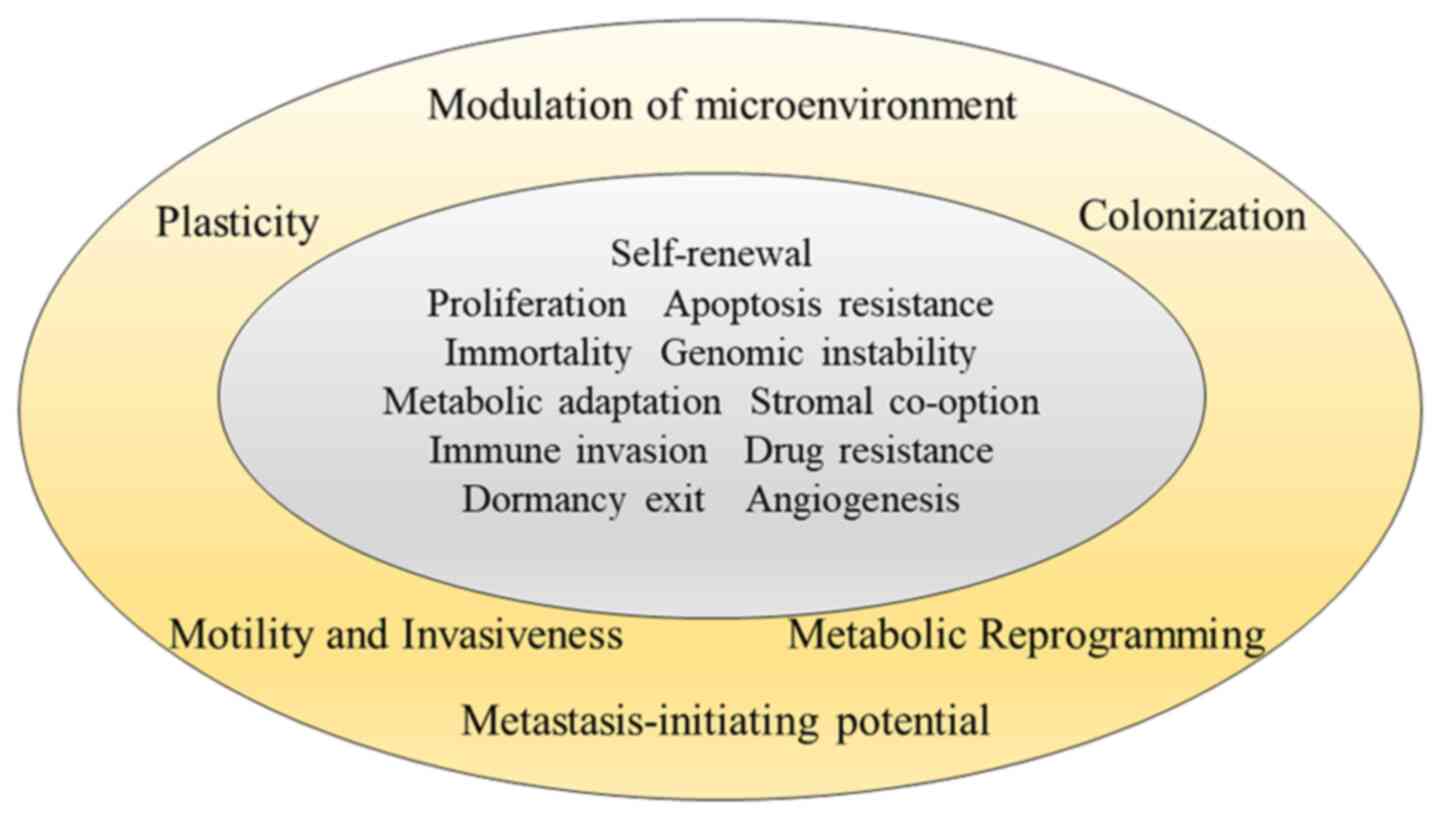
Any hallmarks of metastasis are superimposed upon
the hallmarks of cancer itself published by Hanahan and Weinberg in
2000 and 2011 (27,28). The traits of metastatic cells contain
motility and invasion, ability to modulate the secondary site or
local microenvironments, plasticity, and ability to colonize
secondary tissues (5,6) (Fig.
3).
Motility and invasion
To manifest their properties and live without
connection to a primary tumour, the cells must possess the ability
to move and penetrate through a basement membrane. Cellular
movement requires coordinated cell-cell and cell-matrix adhesion,
matrix degradation, and cytoskeletal activity. During motility and
invasion, the cells organize adhesive, proteolytic, and motility
components into specialized structures: Invadopodia (5).
One of the first steps in invasion in CRC is tumour
budding (TB), which represents an infiltrating growth pattern at
the invasive front. It promotes progression and dissemination of
tumour cells by penetrating the vascular and lymphatic vessels. The
balance between pro-tumour (budding) and anti-tumour (immune
response or certain inflammatory cell types) factors at the
invasive front of colorectal cancer may be decisive in determining
tumour progression in CRC (29).
The invasive front in CRC tumours and interactions
within it represent a critical interface encompassing a dynamic
process of de-differentiation of colorectal carcinoma cells.
Histopathological analyses of invasive cells suggest their internal
complexity, with invading cells at the leading edge paving the way
for subsequent cells to which they remain attached via cell-cell
junctions (30). The leading cells at
the invasive fronts exhibit certain mesenchymal traits during
collective migration (31–33). In comparison to epithelial CRC cells,
leading cells in the invasive front possess increased motility,
invasiveness and the ability to degrade components of the
extracellular matrix. Such invading leaders release proteases that
degrade the extracellular matrix that would otherwise impede the
forward progress of the cohort as a whole. Moreover, such leader
cells may also possess the motility to enable the forward motion of
the cohort as a whole (6).
Dedifferentiation of originally epithelial
colorectal cancer cells to those with invasive potential and
tumour-initiating capability is triggered by induction of
epithelial-mesenchymal transition (EMT) programs (34–36). WNT
signalling has a pivotal role in colon cancer initiation and
consequent downstream EMT activation and also underlies the onset
of migrating CSCs at the invasive front of the primary lesion which
locally invade the tumour microenvironment and eventually form
distant metastases (34). The
dedifferentiation process may be an alternative mechanism in
acquisition of CSC-like properties in human colorectal cancer
cells. External stimulation with TGF-β and the induction of TWIST1
converted the epithelial CRC into undifferentiated CSCs, leading to
a significant increment of stem cell properties in human colorectal
cancer (37).
EMT is characterised by a switch of production of
E-cadherin (epithelial) to N-cadherin. E-cadherin reduction is
regulated by two groups of transcription factors: i) Direct
repressors of E-cadherin including SNAI1 and 2, ZEB1, ZEB2,
E12/E47, Brachyury, and AP4; ii) indirect repressors: Twist1,2,
FOXC2, TCF4, SOX2, OCT4, NANOG, PROX1, SIX1, PRRX1, HMGA1, and
FRA-1 that regulate the transcription of E-cadherin at different
levels including activation of direct repressors (11,38–47). With
respect to the clinical significance in CRC, the repressors AP4,
SOX2, and OCT4 have been associated with liver metastasis (11,40,41). In
addition, 85% of CRC patients show moderate to strong expression of
the repressor Twist1 which is associated with nodal invasion and
poor outcome. Upregulation of SNAI2 significantly correlates with
strong Vimentin expression, and both SNAI2 and Vimentin expression
is associated with lymph node metastasis and poor prognosis
(11,48).
In particular, RAS signalling has been reported to
play a crucial role in EMT inducing a decrease of E-cadherin
expression and an increase of Vimentin expression initiation
(49,50). KRAS mutation alone is not able
to modify the epithelial morphology of CRC cells but requires
cooperation with TGF-β growth factor to accomplish the cell
transformation (51).
Recent studies revealed new potential molecules
connected to CRC invasion. Wan et al revealed that MEIS2
serves a role as a promoter of metastasis in CRC. In vitro
and in vivo experiments revealed that knockdown of MEIS2
significantly suppressed CRC migration, invasion and EMT (52). Considerable evidence indicates that
S100A4 expression by cancer cells alters their adhesive properties,
possibly by remodelling the ECM and promoting the redeployment of
adhesion-mediating molecules. Additionally, the induction of S100A4
may be linked to the downregulation of E-cadherin and cytoskeletal
dysregulation (53).
Extracellular matrices are remodelled by proteolytic
enzymes that contribute to matrix degradation and facilitate tumour
cell invasion, e.g., serine proteinases (plasmin, plasminogen
activator, seprase, hepsin), cysteine proteinases (cathepsins B and
K), aspartyl proteinases (cathepsins D and E), and metal-dependent
proteinases of the matrix metalloproteinase and a disintegrin and
ADAM families (5).
Plasticity
Plasticity is the fundamental trait of metastatic
cells (54) due to: i) The dynamics
of epithelial-to-mesenchymal transition and the reverse process,
ii) the redundancy of mechanisms to accomplish all steps in
metastatic cascade (55), and iii)
cell dedifferentiation and gaining of stemness potential early in
the metastatic process compared to the dormancy exit and resumption
of proliferation during metastasis formation in distant organs
(54,56,57).
Chromatin remodelling complexes such as Polycomb and NuRD
(Nucleosome Remodeling Deacetylase) regulate the transcription of
EMT-related transcription factors (58,59).
Migrating cancer cells display intermediate phenotypes featuring
both epithelial and mesenchymal (E/M) characteristics. These hybrid
E/M cancer cells have been the focus of much attention as they are
likely to be metastable and as such very efficient in causing
metastasis, while cells with fixed epithelial or mesenchymal states
lose their plasticity and associated stem cell capabilities
(60). A study by Liu et al
reported that mesenchymal-like CSCs exhibited high invasiveness and
were quiescent, whereas epithelial-like CSCs were highly
proliferative and less invasive (61). During colonization of distant organs,
disseminated cells have to undergo MET to gain back their
proliferative potential. The ability of cancer cells to revert back
from EMT-induced phenotypes is critical for metastasis formation in
distant organs and full mesenchymal transformation may result in
the irreversible loss of MET capacity (62,63).
Another form of plasticity is the fact that
metastatic cells evolve more mechanisms on how to accomplish steps
of the metastatic process. This redundancy provides a clear
competitive advantage to cells and the loss of the ability to adapt
(i.e., terminal differentiation) can block the ability to
metastasize (55).
Colonization
Colonization is dependent on a combination of tumour
cell- and tissue-specific factors. Intravasating CRC cells interact
with pre-metastatic niches that are permissive for proliferation
and colonization of secondary sites. The interaction of
intravasating tumour cells with organ-specific microenvironment can
lead to the formation of metastasis in colonised site as summarized
in a later subsection (Modulation of microenvironment).
Comparing CRC to other types of cancer, its latency
period between dissemination and colonization can be estimated
approximately in between, since it is longer than that of lung
cancer, but shorter than that of prostate cancer (64).
Metastasis-initiating ability
Critical hallmark of progression through the
invasion-metastasis cascade is a metastasis-initiating ability,
since disseminated tumour cells must function as founders of new
metastatic colonies. They often exhibit long-term self-renewal
capacity, quiescence and resistance to chemotherapy, which belong
to the main traits of CSCs. CSCs have the ability to differentiate
into multiple cell types found in particular metastatic samples,
but represent a small fraction of the tumour population.
Disseminated cells invading into the circulation are usually known
as circulating tumour cells (CTC). CTCs consist of cells acquiring
the right combination of motility, invasiveness, and resistance to
anoikis (apoptosis caused by lack of attachment to neighbouring
cells or extracellular matrix). There is a stochastic model saying
CSCs form a small, randomly determined fraction of the CTCs, but
they preferentially survive and initiate secondary tumours. On the
other hand, a dynamic model hypothesis posits that CSCs undergo
substantial changes in phenotype driven by responses to their
microenvironment and become CTCs. After finding a target tissue,
the cells extravasate, invade the target microenvironment and
re-establish their stem-like properties (65).
CMS classification of primary tumours and
its relation to their metastasis
A classification of a primary tumour before 2015 was
based on non-overlapping genomic phenotypes of microsatellite
instability (MSI) and chromosomal instability (CIN). The marked
interconnectivity between independent gene expression classifiers
gave rise to a classification system of CMS 1–4 groups in 2015,
which do not only reflect cancer cell phenotypes nor
microenvironment features present in bulk tumour tissue samples.
Based on the distinct molecular and clinicopathologic hallmarks
such as mutation status of KRAS, BRAF, TP53 and MSI status,
each of CMS classification groups have distinct treatment pathways
(66).
Previous findings in melanoma suggested that genetic
instability appears to be necessary for the development of
metastases (67). The observance of
activated DNA repair pathways in metastases suggests that a similar
metastatic program may be at play in CRC. Comparison of the
transcriptomes of primary CRCs and their metastatic lesions at both
the gene and pathway levels are in concordance to a study by
Guinney et al (66) reporting
that genetically unstable subtypes such as CMS1 and CMS3 are almost
non-existent among metastases of CRC (10). CMS classification results show that
metastases are more likely to be CMS2 in reference to CMS4 compared
to primary tumours. Although CMS4 has previously been associated
with advanced stages (III, IV) of disease, CMS2 was not previously
associated with advanced disease and is characterized by epithelial
differentiation and strong upregulation of MYC and WNT signalling
(10,66). In relation to the CMS taxonomy of CRC,
tumour buds (invasive protrusions), not the tumour bulk are more
related to mesenchymal phenotype and CMS4 class (29). In addition, a large CRC patient cohort
analysed by IHC showed that a greater number of tumour buds were
found in CMS4 compared to CMS2 and CMS3 tumours and was connected
with KRAS and BRAF mutations (51,68).
The CMS classification framework was originally
developed from microarray-based gene expression profiles or RNA
sequencing of fresh-frozen primary CRC samples mostly (>90%)
from patients with non-metastatic disease (66,69). Some
authors opposed that most CRC tumours do not have one unique and
clonal CMS assignment and present an indefinite heterogeneity, and
only some subclones are able to generate metastasis. The CMS
classification should be a starting point to deepen our knowledge
about CRC biology and the driver genes are important for metastatic
progression, but it does not currently provide a rationale for
therapy selection in metastatic CRC (mCRC) (69).
Identification of metastatic CRC
markers
Colorectal cancer stem cells are closely linked to
tumour metastasis, drug resistance and recurrence after primary
treatment. Not all CSCs in primary tumour are metastatic, and
metastases are produced from a specific subpopulation of CSCs,
known as migrating cancer stem cells (70). Current knowledge of normal and tumour
tissues indicates that CSCs are rarely defined by a single marker
but by a combination of multiple molecular markers. On the other
hand, several studies have linked a high surface expression of some
of following markers (Table I) with
the tumour degree of differentiation, depth of invasion, clinical
stage and metastatic status in CRC (70–80).
 | Table I.Cancer stem cell markers associated
with metastases in CRC. |
Table I.
Cancer stem cell markers associated
with metastases in CRC.
| Metastatic
marker | Localisation | Interaction
with | Results in | In CRC patients
correlate with | (Refs.) |
|---|
| LGR5 | Membrane
receptor | R-spondin,
IQGAP1-Rac1 and WNT signalling | Proliferation,
changes in actin cytoskeletal structure and cell adhesion | TNM staging, lymph
node mts, vascular invasion, OS | (71, 81–83) |
| CD26 (dipeptidyl
peptidase IV) | Membrane
receptor | CXCR4, CD45,
adenosine deaminase, fibronectin, collagen | Motility and
invasive ability in vitro, mts formation in vivo,
modulation of chemokine activity | Distant mts
formation, advanced tumour staging, OS | (34,72,
73,84) |
| CD44v6 | Membrane
receptor | HGF/c-MET, MYC,
STAT3, WNT signalling, stabilization of Cys/Glu exchange | EMT and resistance
to anoikis, upregulation of MDR genes, protection against
ROS | Poor prognosis,
resistance to anti-cancer therapy-together with LGR5 liver mts | (74,85) |
| CD110
(thrombopoietin receptor) | Membrane
receptor | Lysine degradation,
c-MYC, WNT signalling | Shift in redox
status, chromatin remodelling Self-renewal and metabolic
re-programming in CD110+ TICs. Liver mts in vivo | Grading, vascular
invasion, synchronous or metachronous liver mts | (87,151) |
| CDCP1 (CUB
domain-containing protein 1) | Membrane
receptor | Enhancer of Src
activation | Reduction of
cell-cell adhesion, raise of cell migration in vitro. Lung
mts in vivo | Grading, vascular
invasion, synchronous or metachronous lung mts | (75,87) |
| Notch1 | Cytoplasm | TGFβ | It creates TME of
poorly differentiated tumour and drives metastasis via
TGFβ-dependent neutrophil recruitment | Grading, LV
invasion and metastasis, peritumoural budding | (76,77) |
| ALDH1A1 | Cytoplasm | Synthesis of
retinoic acid, activation of Akt, c-Myc, RARβ- | Chemoresistance,
clonogenicity, tumourigenicity, stem cell potential Regulates ROS
and synthesis of carboxyl acids | Grading, LV
invasion and metastasis, peritumoural budding Poorly differentiated
or RCRC. More ALDH1A1 in liver mts vs. paired tumours | (76,78) |
Commonly used CRC cell lines show that a percentage
of stem cells from intestinal origin (as indicated by LGR5
expression from 1–22.5% and frequency of EpCAM+ cells)
is high in fully differentiated carcinoma cells. Sphere-derived
cells showed enhanced frequencies (2- to 3-fold) of LGR5 positivity
compared to the original cell lines (79). LGR5+, E-cadherin high,
EpCAM high and CD26 high are frequently associated with
sphere-derived cells. These results are highlighted by a recent
report showing that LGR5+ cells are more important for
the process of metastasis than for primary tumour growth (81).
LGR5 is a membrane receptor of intestinal stem cells
acting like local enhancer of the WNT/β-catenin signalling pathway.
LGR5 expression is higher in colon carcinomas than in adenomas. It
correlates with TNM staging, lymph node metastases, vascular
invasion and significantly lower overall survival of patients
(82,83).
Pang et al (36) demonstrated that a subpopulation of
colorectal CSCs expressing CD26 has both tumour-initiating and
metastatic capacities. Orthotopic implantation of
CD133+/CD26+ cells isolated from primary CRCs
of a patient with hepatic metastasis led to metastasis formation in
the liver, following orthotopic tumour formation in vivo,
whereas their CD26-counterparts led to no metastasis. In
vitro evaluations revealed that CD26 knockdown by siRNA reduced
the migratory and invasive capacities of the CD26+ cells
(36). The presence of
CD26+/CD326− marker in CTC tested in a CRC
patient blood sample was higher in advanced Dukes' stages and was
significantly associated with poor survival and high recurrence
rates (84). Examination of
paraffin-embedded tissues in 143 patients with CRC revealed
CD26+ cells have a significant clinical impact on the
prediction of distant metastasis development in colorectal cancer
(72).
Notably, Todaro et al (85) phenotypically identified colorectal
CSCs with metastatic capacity based on the expression of CD44v6.
CD44v6+ cells were able to induce tumour growth in the
gut, lung, and liver after orthotopic injection into mice, whereas
their negative counterparts grew locally without forming distant
metastases (85). Interestingly,
while there was a substantial overlap between CD44v6+
and CD26+ cells, CD44v6+/CD26−
cells showed considerable metastatic potential in the orthotopic
model (85), indicating that there is
phenotypic heterogeneity even within metastatic CSCs. CD44 is
involved in HGF/MET signalling and many other oncogenic mediators,
including MYC and STAT3, which promote CRC metastasis via anoikis
resistance and enhancing EMT properties.
Zhang et al showed that the CRC cell line
HCT116 also contains CD133+/CXCR4+ cells,
which have a significantly higher metastatic capacity than
CD133+/CXCR4− cells (86).
Previous findings suggested that the formation of
metastases in certain favoured target organs would be attributable
in part to the diversity within metastatic CSCs. Gao et al
reported that only CRCs expressing CD110, a specific receptor for
thrombopoietin, were able to colonize the liver after orthotopic
implantation in immunocompromised mice, while CRCs expressing CUB
domain-containing protein 1 (CDCP1) were associated with the
development of lung metastasis (87).
They also confirmed that knockdown of either CD110 or CDCP1 by
siRNA reduced the liver or lung metastasis burden, respectively,
but had no discernible effect on primary tumour growth. In
addition, authors of that study showed that CD110 and CDCP1 would
be involved in integral parts of the metastatic process, including
in vivo extravasation.
Accumulating evidence suggests that alternation or
even loss of differentiation control may result in complete
dedifferentiation as well as acquisition of properties typical for
stem cells which contribute to metastasis initiation traits.
Besides known Yamanaka factors (SOX2, MYC, KLF4, OCT4,
NANOG), other tissue-specific cell fate determinants essential
for metastasis initiation responsible for metastatic relapse should
be identified. The decrease of differentiation via genetic
manipulation of either of two distinct differentiation-promoting
transcription factors (Smad4 or Cdx2) in mouse model was associated
with BRAF-driven serrated tumour development and restored stem cell
activity in BRAFV600E intestine. In human patients
reduced levels of differentiation in normal tissue are associated
with increased susceptibility to serrated colon tumours (88).
The stem cell markers Notch1 and ALDH1 correlate
with lymph node metastasis, advanced stage, and tumour recurrence
and represent an independent prognostic factor in colorectal
carcinoma (76). ALDH1+
cells, particularly those displaying high WNT activity, are able to
initiate and maintain colon tumours when inoculated into
immunodeficient mice (82). In
relation to stem cell properties, much emphasis has been placed on
ALDH1A1 and ALDH1A3 isoforms. Overexpression of ALDH1A3 isoform was
detected in chemoresistant colorectal carcinoma cells derived from
human cell line HT-29 which formed distant metastases in mouse
lungs spontaneously, although the parental HT-29 colorectal cell
line possesses no metastatic ability (89).
Gene signature of metastatic CRC cells
Gene signature of metastatic cells able to survive
the complete metastatic cascade is derived from that of primary
tumour's subclones and in CRC is based on either a chromosomal
instability (CIN, 80–85% of all CRC cases) or microsatellite
instability and/or CpG island methylation phenotype.
Defects in mismatch repair in tumours with MSI
affect MLH1, MSH2, MSH6, PMS1 and PMS2 genes
(90,91). Their proteins form heterodimers that
repair DNA damage. The most common and relevant heterodimers in
colorectal carcinogenesis are MLH1/PMS2 and MSH2/MSH6.
Immunohistochemical staining of these proteins in primary tumours
and paired metastatic lesions revealed identical results in 77%
(92) to 100% of the cases (93).
The CIN pathway is characterized by imbalance in the
number of chromosomes, their segregation, telomere dysfunction and
DNA damage response, which affect the main proliferation and cell
cycle check-points: APC, KRAS, PI3K and TP53 proteins. APC
mutations cause translocation of β-catenin to the nucleus and drive
the transcription of genes implicated in tumourigenesis and
invasion, whereas mutations in KRAS and PI3K lead to
constant activation of MAP kinase, thus increasing cell
proliferation. Finally, loss-of-function mutations in TP53
encoding p53, the main cell-cycle checkpoint, cause an uncontrolled
entry in the cell cycle (91,94). These canonical drivers are shared by
primary colorectal tumours but also their paired metastases and are
known as metastatic-associated early driver genes (17). Specific combination of early driver
genes may confer metastatic competence. Data are in concordance
with CMS classification, where the main early driver mutations
consist of APC, KRAS, TP53, PIK3CA genes in CMS2-4 tumours
(95). The study of Fumagalli et
al (96) triggered the APC,
KRAS, TP53 and SMAD4 gene in normal primary colon
organoids which resulted in the seeding of metastatic cells after
xenotransplantation. SMAD4 acts as a metastasis suppressor by
interacting to block the functionality of transcription factors
that promote metastatic cancer progression (97). In accordance with the organoid study
(96), another group confirmed that
oncogenic mutated KRAS combined with APC and TP53 deficiency can
trigger metastatic cascade in a mouse model (98). Although the amount of canonical driver
genes is low, there are many combinations of mutations that
collectively disrupt key signalling pathways (WNT, TP53, TGFβ, EGFP
and cellular adhesion) enabling dissemination and outgrowth in
distant organs.
Early CRC driver genes were supplemented with
additional gene/genes from a candidate metastasis driver in
numerous CRC samples: TCF7L2, AMER1, PTPRT, PIK3CA, GNAS, SRC,
FXR1, MUC4, GPC6 and MECOM (99–107),
which seemed to be specific to metastases (Fig. 4). Mutations in coding regions of
BRCA1 and 2, FLCN, HNF1A, PTEN, RNF43 leading to
progression and metastases were also reported (95). Collectively, the early driver genes
plus an additional candidate metastasis driver showed statistically
significant enrichment in metastatic vs. early-stage CRCs (18%
compared to 5.6%, respectively, q=2.9×10-20) (17). All are connected to basic signalling
pathways activated in CRC.
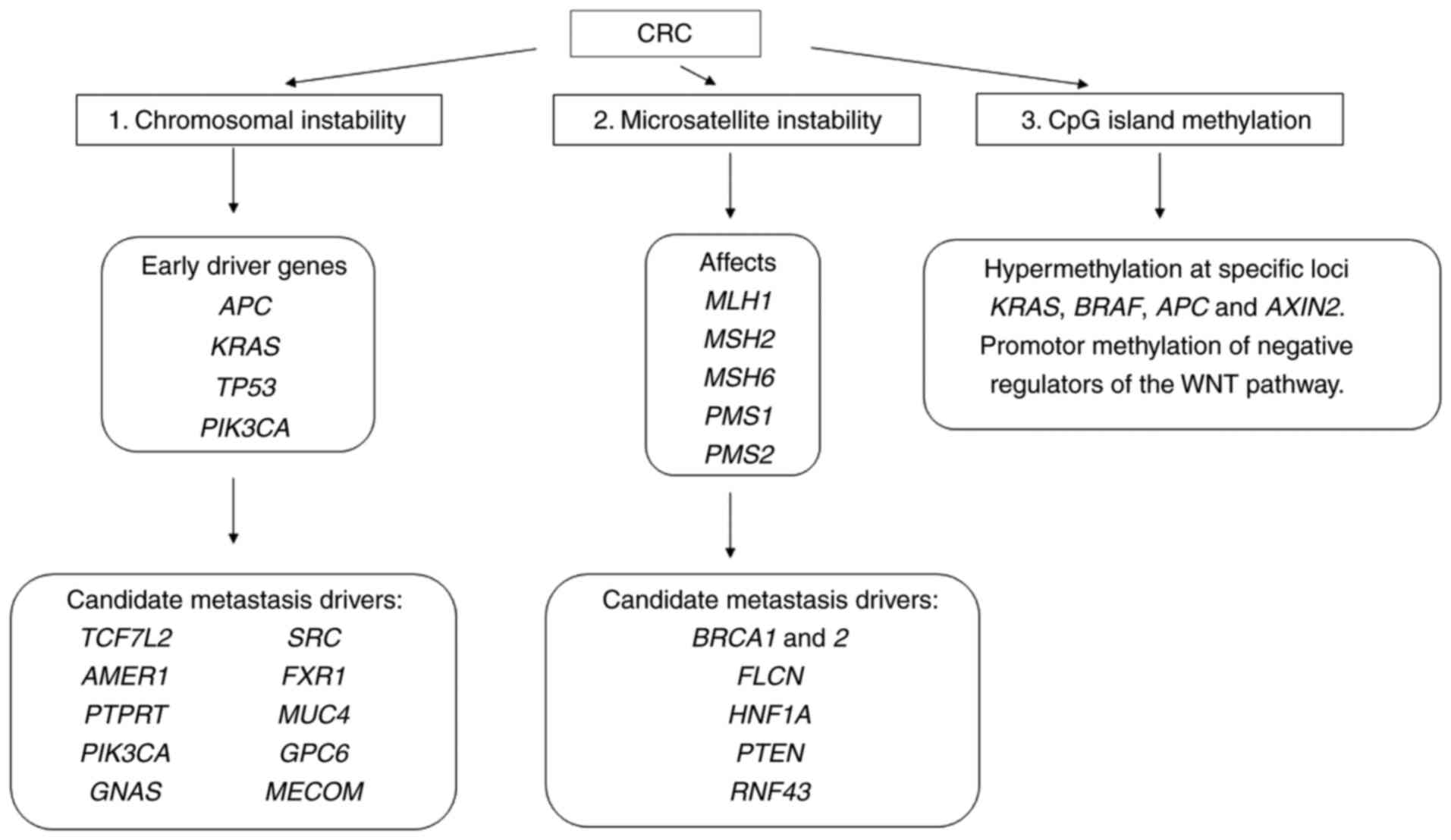 | Figure 4.Genetic alterations associated with
CRC metastasis. Primary CRC tumours are divided into three
subgroups based on genomic phenotypes of chromosomal instability
(CIN) and microsatellite instability (MSI) or epigenetic phenotype.
Genetic alterations of early CRC driver genes required for
transformation of an adenoma into carcinoma are supplemented with
genetic/epigenetic changes in the candidate metastasis drivers to
gain full metastatic competence. AMER1, APC membrane recruitment
protein 1; APC, adenomatous polyposis coli; BRCA1 and 2, breast
cancer type 1 and 2 susceptibility protein; FLCN, Folliculin; FXR1,
f ragile X mental retardation syndrome-related protein 1; GNAS,
heterotrimeric G-protein alpha subunit Gs,α; GPC6,
Glypican-6; HNF1A, hepatocyte nuclear factor 1 homeobox A; MECOM,
MDS1 and EVI1 complex locus protein EVI1; MSH2, 6, MutS homolog 2,
6; MLH1, MutL homolog 1; MUC4, Mucin 4; PI3K, Phosphatidylinositol
3-kinase; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase
catalytic subunit alpha; PTPRT, receptor-type tyrosine-protein
phosphatase T; SMAD4, Mothers against decapentaplegic homolog 4;
PTEN, phosphatase and tensin homolog; TCF7L2, transcription factor
7-like 2, TP53, cellular tumour antigen p53. |
Transcription factor 7-like 2
(TCF7L2)-negative CRC cell lines exhibited morphological
changes, enhanced migration, invasion, and collagen adhesion.
Stimulation of the WNT signalling pathway leads to the association
of β-catenin with BCL9, translocation to the nucleus, and
association with TCF7L2, which in turn results in the activation of
WNT target genes (99).
AMER1 acts as scaffold for β-catenin degradation,
assembling the destruction complex at the plasma membrane by
recruiting β-catenin, APC, and Axin/Conductin, and therefore acting
as a key negative regulator of WNT signalling (100).
Gain-of-function mutations in PIK3CA
upregulate the downstream AKT-mTOR signalling pathway thereby
promoting cancer cell growth and proliferation.
By stimulating adenylyl cyclases, GNAS activation
leads to intracellular accumulation of cyclic adenosine
monophosphate (cAMP), a central second messenger which in turn
activates protein kinase A (PKA). Similarly, 10% of CRC patients
harbour gene amplification of GNAS, and another 45% have
mutations in PKA subunits. There are indications that GNAS-PKA
signalling is promoting growth and is connected to RAS (101).
SRC is non-receptor tyrosine kinase and plays a role
in signalling through a variety of membrane-bound receptors. EGF
and VEGFR-1-induced increase in SRC kinase activity has been shown
to stimulate the movement of carcinoma cells into basement
membranes. The multiple effectors of SRC include the PI3K-AKT,
RAS-RAF-MAPK, STAT3-STAT5B, and p130 pathways (102).
Fragile X-related gene 1 (FXR1) was also
elevated in the plasma of colorectal cancer patients and acted as
an oncogene to promote proliferation, invasion and migration of
cancer cells (103). Functionally,
this is likely due to FXR1 participation in an RNP complex that
regulates translation in metastasis. FXR1 was also found to
destabilize p21 mRNA in cancer cells, and deletion of FXR1
in these cells rescues cell cycle control (104).
Increased expression of Mucin4 (MUC4) is
associated with poor survival of early-stage (I and II) CRC. The
formation of the tetrameric MUC4-ErbB2-ErbB3-NRG complex leads to
the hyperphosphorylation of ErbB2. This phosphorylation enables the
downstream activation of the PI3K-AKT and RAS-ERK pathways, which
induce a loss of cell polarity in epithelial tumour cells as an
early stem of invasion (105).
Glypican-6 (GPC6) has been found to be
frequently mutated and aberrantly methylated in colorectal cancer.
In breast cancer progression, GPC6 effectively upregulates WNT5A
signalling, which, in turn, inhibits JNK and p38 MAPK signalling,
thereby promoting metastasis and invasion. In CRC patient samples,
glypicans modulate WNT/β-catenin signal pathways (106).
MECOM encodes positive regulatory domain zinc
finger protein 3 (also named as PRDM3 or EVI-1) involved in
downstream signalling pathway of TGF-β.
PTPRT seems to be a specific driver of the
metastatic process, because its association with APC, KRAS, TP53
and SMAD4 is almost exclusively present in patients with
metastases. Loss of PTPRT in CRC results in increased STAT3
activation and cellular survival. If the observations are
confirmed, PTPRT mutations could be predictive biomarkers
for STAT3 pathway inhibitors (17,107).
In the case of CMS1 tumours the early driver genes
involve two DNA repair genes MSH6 (MutS homolog 6, DNA
repair gene) and ATM (ATM serine/threonine kinase); negative
regulator of WNT signalling RNF43 (ring finger protein 43);
transforming growth factor β receptor II; BRAF; PTEN
(Phosphatase and tensin homolog) mutations and genes of
microsatellite instability (95).
Metastatic cells are derived from the primary
tumour's subclones, where epigenetic changes are accumulated and
the EMT program is triggered. Hypermethylation at specific loci
KRAS and BRAF leads to upregulation of the oncogenes,
and promotor methylation of negative regulators of the WNT pathway
lead to invasion (98). Loss of
E-cadherin expression due to hypermethylation results in decreased
cell-cell adhesion, tumour progression and increased invasion.
EMT-promoting signals epigenetically modify the repression of
epithelial genes and consequently drive the transition of cells in
more mesenchymal-like states (59,108).
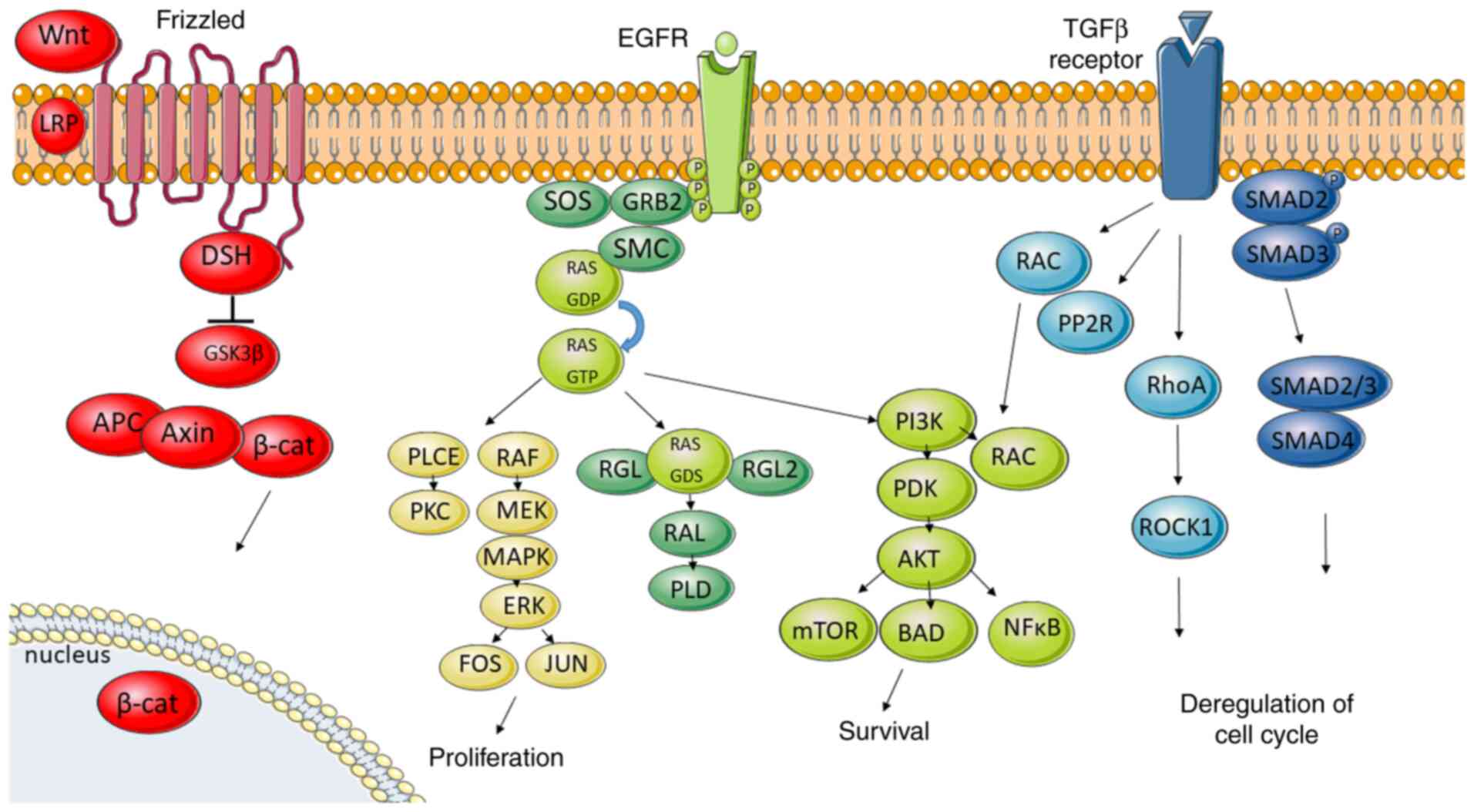
Signalling pathways involved in metastatic
CRC
Genetic changes in the driver genes together with
the abovementioned candidate metastatic driver/s are reflected in a
limited amount of signalling pathways, critical for cell survival
and differentiation. The most important signalling pathways
involved in metastatic colorectal cancer are WNT, EGFR, TGF-β and
HGF/c-MET signalling pathways (Fig.
5).
The WNT pathway plays an important role in stem
cell differentiation and cellular growth, but in CRC this pathway
is associated in several ways with metastasis. Concerning the
metastatic process, the WNT pathway is connected with ‘β-catenin
paradox’ in the case of CRC. The initiating event in many CRCs is
represented by the constitutive activation of canonical WNT
signalling through loss of function mutations of APC or gain of
function of WNT agonists such as β-catenin (109). Co-activation of the Frizzled and LRP
receptors prevents formation of the β-catenin destruction complex
(consists of PP2A, GSK3β, CK1α, APC, Axin1/2). This leads to
transport of β-catenin to the nucleus, where it interacts with
members of the TCF/LEF family of transcription factors and
modulates the cellular functions ranging from stemness to
proliferation (110). However, the
nuclear localisation of β-catenin is not distributed through the
tumour. IHC analysis revealed that cells in the centre of the
tumour mass are characterised by normal, membrane-bound and
cytoplasmic β-catenin staining. However, cells of invasive front of
CRC showed marked nuclear β-catenin accumulation in the proximity
of stromal microenvironment (111).
This ‘β-catenin paradox’ is only achieved in colon cancer cells
located at the invasive front exposed to stromal signals capable of
further promoting the nuclear translocation of β-catenin from the
cytoplasm (112,113). The same phenomenon, i.e., nuclear
β-catenin staining, only in less differentiated cells located in
closer proximity to the microenvironment unlike the rest of tumour
mass, was previously described in colorectal metastasis (109). WNT pathway plays a role also in
weakening of tight junctions, leading to reduced cellular adhesion
and thus favours migration and metastasis (114).
The TGF-β pathway is fundamental in growth,
differentiation or apoptosis. Activation starts by binding TGF-β to
TGF-β receptor 2, which recruits TGF-β receptor 1. Then
receptor-associated SMAD2 and SMAD3 are phosphorylated, thus
allowing them to bind to SMAD4. The complex is translocated into
the nucleus to regulate the transcription of the target genes
(105). Chromosomal changes
involving TGF-β are connected to the CIN pathway in CRC. Loss of
18q is one of the main genomic aberrations related to the TGF-β
pathway in colorectal cancer (115).
Chromosome 18q encodes the tumour suppressor genes SMAD2 and
SMAD4, the loss of which leads to an ability to evade
apoptosis and deregulation of the cell cycle. SMAD proteins act in
the transcription of EMT factors. Loss of 17q-TP53 can drive tumour
progression by allowing excessive proliferation (82,116).
Activation of the TGF-β pathway turns on also several non-SMAD
signalling pathways including MAPK, PI3K, Notch and WNT signalling
(105).
Binding of growth factor to EGFR receptor induces
activation of its intrinsic kinase activity recruiting Src homology
2 proteins. Adaptor protein GRB2 mobilises SOS to the membrane.
Next, SOS activates GDP/GTP exchange which activates transition of
RAF to the membrane. Consequently, this activates mitogen-activated
protein kinase kinases 1 and 2 (MEK1 and MEK2) and subsequently
activates extracellular signal-regulated kinases 1 and 2 (ERK1 and
ERK2). Phosphorylated ERK then translocates to the nucleus and
activates transcription factors enhancing the expression of
c-FOS, c-JUN and MYC genes (25), thus promoting cell survival,
proliferation, invasion, and migration (117,118).
Moreover, the activation of MEK1 in the RAS-RAF-MEK cascade allows
the enrolment of downstream effectors EGR1 and FRA-1 which can
promote the expression of SNAI1 and SNAI2, which in turn,
downregulate E-cadherin expression (119). In EMT, the pathways that regulate
actomyosin and cytoskeleton dynamics drive plasticity and KRAS
mutations can determine the mode and effectiveness of migration by
means of RhoA and Rac1 signalling (51,120,121).
GRB2 also recruits the phosphoinositide 3-kinase
(PI3K), another main messenger of the EGFR signalling pathway.
Alterations in MAPK and PI3K pathways are involved in cell
proliferation and survival. The PI3K/AKT pathway propagates signals
from growth factors, cytokines, and oncogenic-mediators to
contribute to CRC pathogenesis. PI3K activation leads to the
production of the second messenger
phosphatidylinositol-3,4,5-triphosphate which recruits a subset of
signalling proteins, including phosphoinositide-dependent kinase 1
(PDK1) and AKT/protein kinase B (PKB) (27). AKT/PKB regulates several cellular
processes involved in cell survival and cell cycle progression such
as activating the survival factor NFκ and mTOR (122). As for cell cycle progression and
cell growth, several targets of AKT are involved in protein
synthesis, glycogen metabolism, and cell cycle regulation (27). Loss of PTEN, which downregulates the
PI3K pathway, was present in CRC primary tumours and their matched
liver metastases and was significantly related to an increased
death risk and with poor overall survival (123). BRAF mutation V600E is a poor
prognostic factor in metastatic cancer (124). Nevertheless, this mutation is a
promising target for personalized medicine, and the combination of
specific BRAF inhibitors with other MAPK/PI3K pathway inhibitors
has been shown to be more effective for treating mCRC (91,125).
Recent evidence has shown the importance of mTORC1 and 2 pathways
for mCRC. SMAD4 interacts with RICTOR to suppress mTORC2
functionality and therefore the loss of SMAD4 function results in
oncogenic activation of the mTORC2 pathway, leading to enhancement
in metastatic colon cancer progression. Overactivation of mTORC1
can promote tumour formation, proliferation, and metastasis, while
mTORC2 can regulate the expression of mTORC1 through the mTORC2/AKT
pathway (126,127).
Hepatocyte growth factor (HGF) binds to cMET
receptor tyrosine kinase, which is hyper-activated in primary CRC
tumour development and promotes metastasis. A number of
transcription factors that directly facilitate cMET expression,
promote metastasis in in vivo models, and whose expression
is elevated in metastatic patient samples have been identified
(128). FOXC2 directly targets cMET
to promote metastasis in vivo (129). The transcription factor
metastasis-associated in colon cancer-1 (MACC1) (130) directly binds to the cMET promoter to
enhance transcription in an HGF-dependent manner. Findings have
demonstrated that targeting MACC1 genetically or chemically reduces
metastases in animal models (131,132).
Signalling through cMET is not only controlled by receptor
expression but also by structural components that facilitate HGF
ligand binding. CD44v6 acts as coreceptor and together with cMET
together promotes the growth of ex vivo adenoma organoid
cultures. Blocking CD44v6 or MET limited migration in vitro
and reduced the metastatic spread of patient-derived tumour cells
in animal models (26).
Metastasis connected to the localisation of
CRC
Gene expression studies of right- and left-side
colon biopsies revealed distinct expression profiles, and the
researchers noted higher transcriptional activity in the descending
colon (133).
Right-sided CRC (RCRC) patients tend to have
advanced and larger tumours than left-sided CRC patients, where the
tumours are often poorly differentiated with worse prognosis. Those
tumour types usually metastasize to the peritoneal region (134). In RCRC, mutations in the DNA
mismatch repair pathway are commonly observed and are detected in
more advanced stages because of their flat histology. The greater
proportions of the ‘microsatellite unstable/immune’ CMS1 and the
‘metabolic’ CMS3 subtypes are found in right-sided colon cancers
(135). In the RCRC, the MSI or BRAF
mutations are predominantly activated.
Left-sided CRC (LCRC) occurs at a younger age with
metastasis to lung and liver. This type of carcinogenesis is led by
chromosomal instability pathway-related mutations, such as KRAS,
APC, PIK3CA, p53 mutations and they demonstrate polypoid-like
morphology. While RCRC patients with RAS mutation are
resistant to anti-EGFR therapy, they benefit more from adjuvant
chemotherapies such as 5-fluorouracil-based regimens. LCRC patients
show promising results with targeted therapies such as anti-EGFR
therapy and have a better prognosis. Right-sided mCRC tumours that
are RAS and BRAF wild-type and non-bulky may respond
to combination chemotherapy with anti-EGFR antibodies, but not to
anti-EGFR therapy alone (136).
Transverse colon tumours have mutation profiles that more closely
resemble left-sided tumours, keeping right-sided tumours distinct
(137). Mucinous adenocarcinomas,
another type of CRC, are commonly observed in RCRC and MSI-high
tumours, and are characterized by excessive mucin excretion. They
also have faster progression compared to adenomatous polyps
(138). There is evidence that the
immune system and the microbial communities vary along the length
of the colon (139).
Modulation of microenvironment
Distant tissues are normally a hostile environment
for the newly arriving tumour cells. Metastatic cells are commonly
attacked by the immune system, which is responsible for preventing
the formation of more than 80% of metastases (140). Immune cells such as T cells,
macrophages, natural killer cells and neutrophils infiltrate
tumours and destroy tumour cells as part of immunosurveillance. The
communication of tumour cells with the cellular compartment is
recruiting new cells into the tumour microenvironment (TME) and is
involved with mobilization of immune/inflammatory cells,
restructuring of other tissues, altering metabolism of surrounding
stroma and interruption of anti-tumour actions of the immune system
(5). Tumour cells and the cells of
TME can ‘programme’ immune cells into tumour-permissive or
tumour-promoting phenotypes. EMT transcription factors, including
SNAI1, ZEB1 and TGF-β have been shown to suppress the functioning
of immune system (28).
CRC tumours with microsatellite instability have a
high mutational burden that creates many neoantigens that are
loaded on the MHC of antigen-presenting cells and recognized as
foreign by T cells (141). Thus, a
microenvironment of MSI tumours is rich in tumour-infiltrating
lymphocytes (TIL) in comparison with MSS (142) and the adaptive immune system plays
an important role in suppressing tumour progression (143,144).
The strong activation of tumour-directed immune cells triggers the
feedback expression of immune checkpoint blockade receptors and
ligands, such as surface protein PD-1 and transmembrane protein
PD-L1, on tumour cells, TILs and tumour-associated macrophages
(145). This explains the reason for
patients with MSI mCRC constituting a rare group (5% of all cases)
responsive to PD-1 blockade. However, the vast majority of mCRC
patients (95%) are microsatellite stable (MSS) with low tumour
burden and a lack of immune cell infiltration, and are completely
refractory to checkpoint blockade therapy, including PD-1 or PD-L1
inhibitors (141,146,147).
In mCRC, PD-L1-positive expression in tumour cells ranges between
22 and 38% in MSI and 13 and 67% in MSS (148). The expression of PD-L1 in MSI
tumours is localized in polarized macrophages (CD163+)
at the invasive front and in the stroma, but not in tumour cells.
The quantification of T cells and cytotoxic T cells (CD3 and CD8)
in mCRC tumours (immune score) shows that a higher immune score
correlates with a decreased likelihood of metastasis (149) and can also predict the overall
survival (150). However, it
excludes regulatory T cells and inflammatory T cells
(IL-17+), which have potentially important roles in CRC
immunosuppression (151,152).
Moreover, carcinoma cells can transform fibroblasts
located at metastatic sites into CAFs known to promote metastasis
(153). They do so by producing ECM
niche components Periostin and Tenascin C (154). In colorectal cancer, the release of
TGF-β stimulates CAFs to secrete IL-11, which recruits carcinoma
cells to activate STAT3 signalling, therefore further strengthening
the ability of metastatic cells to survive in the liver (155).
Based on an analysis of 100 mCRC patients in which
NGS data yielding immune signatures were integrated with TME,
clinical scores, and metabolic pathway inferences, Gastrointestinal
Immune-Signature mCRC was classified into three distinct
immune-metabolic clusters: Inflamed-stromal-dependent (IM Cluster
1), inflamed-non-stromal-dependent (IM Cluster 2), and
non-inflamed/cold (IM Cluster 3) (152).
Metabolic reprogramming in metastatic
CRC
A typical hallmark of cancer cells is their energy
metabolism switch from oxidative phosphorylation to anaerobic
glycolysis. Changes in cellular metabolism may precede the
acquisition of driver mutations ultimately leading to colonocyte
transformation. Oncogenic mutations and loss of tumour suppressor
genes further reprogram CRC cells to upregulate glycolysis,
glutaminolysis, one-carbon metabolism, and fatty acid synthesis
(156). For example, the activation
of RAS and MYC oncogenes leads to upregulation of the oxidative
phosphorylation system, which is caused by MAPK activation followed
by mitochondrial biogenesis induced by PGC-1β expression (157).
The expression profile of metabolic genes in IM
Cluster 1 suggests a predominance of the aerobic glycolytic pathway
over oxidative mitochondrial TCA (Krebs) cycle with
lactate-mediated TME acidification. IM Cluster II is associated
with immune (T, NK and B cells) and myeloid-monocytes with
enrichment for immune checkpoint genes and features of EMT. The
cluster underwent a distinctive metabolic reprogramming with
enhanced TCA oxidative phosphorylation and glutaminolysis. IM
Cluster 3 is characterized by the absence of myeloid-derived
suppressor cells, T-regulation and CAF markers. Glycolytic tumour
cell metabolism limits glucose availability in the TME, which,
coupled with high lactate excretion and extracellular acidification
rate, can potentially dampen CD8+ T-cell differentiation
and function (158).
Upregulation of glycolytic genes and glycolytic
capacity was also detected in mCRC. Specifically, glucose
transporter 1 and 3 (GLUT1 and GLUT3) overexpression was associated
with metastasis and poor survival in colorectal cancer patients. In
detail, hypoxia-inducible factor-1α (HIF-1α) is activated in
cultured HCT116 colon cancer cells under hypoxic conditions as well
as in tumour budding cells of CRC (159). Hypoxia and HIF-1α upregulate cancer
cell expression of GLUT1 and induce glycogen metabolizing enzymes
(160,161). Additionally, GLUT3 promotes
invasiveness and stemness in a Yes-associated protein
(YAP)-dependent manner. Activation of YAP in turn transactivated
GLUT3 and regulated a group of glycolytic genes. Importantly, a
high-fat high-sucrose diet promoted tumour metastasis, whereas the
inhibition of either GLUT3 or YAP effectively reduced the
metastatic burden. Activation of the GLUT3-YAP signalling pathway
acts as a master activator to reprogram cancer metabolism and
thereby promotes metastasis (162).
Reservoirs of nutrients and oxygen vary between
different host organs. The lungs are rich in glucose and oxygen
supplies, which may grant easier colonization of metastatic cells
using aerobic glycolysis (163) or
oxidative phosphorylation (164). In
stark contrast, the liver has lower levels of oxygen and irregular
glucose availability which indicates that metastatic cells in this
organ need to urgently adapt to such metabolic stresses (56). In addition, metastatic cells may use
other sources of energy than primary tumour cells. Indeed, aerobic
glycolysis observed in primary tumours is often replaced by other
types of energy production in metastasis (165). In other organs with low oxygen
tension, metastatic cells are able to use the creatine cycle to
scavenge ATP or activate β-oxidation (56). According to Bu et al colon
cancer-derived liver metastases upregulate Aldolase-B, an enzyme
for fructose metabolism that utilizes fructose as a source of
energy. Targeting Aldolase-B or its upstream regulator GATA6 or
reduction of fructose decreases liver metastatic growth (166). A detachment of tumour cells from the
extracellular matrix during metastasis also reduces glucose uptake
(164).
Moreover, metastatic colorectal CD110+
cancer cells in liver can utilize thrombopoietin-mediated
activation of lysine degradation. The mechanism of action is based
on lysine catabolism leading to activated WNT signalling and a
shift in redox status (167).
Effect of microRNAs on the metastatic
process of CRC
Dysregulation and aberrant expression of microRNAs
(miRNAs) has been found in various types of cancer including
colorectal cancer (up to 35 miRNAs) (165). This makes them potential biomarkers
for predicting prognosis, characterisation of the tumour and a tool
for diagnosis. Dysregulation can be explained by various genetic
alterations and epigenetic modifications such as methylation
(168).
MiRNAs can have an opposite effect on tumour
progression by targeting oncogenes while acting as tumour
suppressors. While being downregulated, they can serve as
biomarkers for early diagnosis. MiR-19 (169), miR-885-5p (170) and miR-155 (171) were associated with induction of
migration and invasion of CRC cells. MiR-21 augments invasion and
migration by downregulating tumour suppressor gene PDCD4
(172).
A study by Loo et al (173) has also underlined the necessity of
CRC cells to get energy from the extracellular environment to
overcome metabolic stress in liver. In that study, colon cancer
cells, by downregulating miR-483 and miR-551, derepressed and
secreted creatine kinase brain type into the extracellular space.
The cancer cells benefit from elevated levels of creatine in the
liver, which is converted into phosphocreatine to serve as an ATP
source for growth functions in metastatic cells (174).
Post-transcriptional regulation of gene expression
by EMT-related miRNAs showed a great impact on promoting the
epithelial or mesenchymal phenotype of CRC cells targeting specific
mRNA (173). Some miRNAs can
regulate genes involved in EMT thereby regulating the early steps
of metastasis formation in CRC, for example, miR-31-5p targets and
inhibits c-MET, a mediator of EMT (175). Other miRNAs that are target
mediators of EMT are miR-34 which targets c-MET, SNAI1 and
β-catenin (176), miR-302 which
targets AP4, SNAI1 and vimentin (177), and miR-15a functioning similarly to
miR302 and targeting AP4 (178).
Altered expression of miRNA affects the expression of Cadherin-1
and EMT transcription factors. For example, the expression of
miR-508 negatively correlates with stemness and EMT-associated gene
expression and positively correlates with patient survival in
colorectal cancer (179).
Members of the miR-200 family (miR-200a, miR-200b,
miR-200c, miR-141, and miR-429) promote epithelial phenotype
preventing the translation of ZEB1 and ZEB2 mRNA (180). This, in turn, acts in a negative
feedback loop downregulating the miR-200 family expression
(62). Moreover, ZEB2 is also
identified as a direct target of miR-132, miR-192, and miR-335.
Downregulation of these miRNAs is usually associated with the
acquisition of an aggressive mesenchymal phenotype leading to
distant metastasis and a poor prognosis (181,182).
MiR-34a/b/c is another caretaker of the epithelial phenotype
through the downregulation of SNAI1, SNAI2 and ZEB1 (183). Suppression of miR-34a/b/c causes
upregulation of SNAI1 resulting in the enhanced expression of EMT
markers, mesenchymal features, and improved cell invasion and
motility (51). Targeting of this
overexpressed miRNA associated with metastasis can improve the
overall survival of CRC patients.
Epigenetic changes in metastatic CRC
Proto-oncogenes and tumour suppressors can be
affected not only by genetic but also by epigenetic alternations.
Inactivation of tumour suppressor genes can affect apoptosis,
invasion and cell proliferation (184). For example, a tumour suppressor gene
termed N-MYC downstream-regulated gene 1 in a highly
metastatic cell line SW620 is silenced by reduced H4 acetylation
(185).
Aberrant DNA methylation has also been extensively
demonstrated in CRC and occurs early in the adenoma to carcinoma
sequence, as the hypermethylation in the gene promoters
transcriptionally silences the tumour suppressor genes (9,186). The
CpG island methylator phenotype pathway in CRC is characterized by
significant changes in promoter methylation (179). Loss of E-cadherin expression is
caused by hypermethylation and results in decreased cell-cell
adhesion, tumour progression and increased invasion (187).
Changes in metabolic flux impact epigenetics in
both normal and cancer cells because metabolites serve as essential
cofactors for chromatin remodelling enzymes responsible for
epigenetic alterations (188,189).
S-adenosyl methionine (SAM), generated by the methionine cycle,
serves as the methyl donor for histone methyltransferases and
DNA-methyltransferases (188). Thus,
changes in intracellular SAM levels directly affect histone
methylation associated with active gene transcription (190). On the other hand, CRC also exhibits
global DNA hypomethylation outside of CpG islands (191). Metabolites generated in the citric
acid cycle and electron transfer chain serve as cofactors for DNA
and histone demethylation. Alpha ketoglutarate is required for
activity of the TET (Ten-eleven translocation methylcytosine
dioxygenase 1) family of DNA demethylases and the Jumonji C family
of histone demethylases (148,188).
A variety of chromatin remodelling complexes play a
central role in the transcriptional regulation of EMT-related
transcription factors and microRNAs by determining the
accessibility of regulatory DNA elements and positioning of
nucleosomes (58,59). In addition, post-translational histone
modifications modulate chromatin folding, influence the recruitment
of regulatory proteins and control gene expression (192). Accordingly, contextual EMT-promoting
signals epigenetically modify the repression of epithelial genes
and consequently drive the transition of cells in more
mesenchymal-like states. These are epigenetically sustained unless
the presence of EMT-promoting signals is discontinued leading to
reversion to more epithelial phenotypes (59,108).
However, the heterogeneity of metastatic cancer cells represents a
difficulty in specifying epigenetic background of these cells.
Conclusion
This review has demonstrated a growing
understanding of biological and molecular traits of metastatic
colorectal cells and describes molecular drivers and enhancers of
metastasis in CRC. A central core property of metastatic cells is
their cellular plasticity, which underlies almost all other
hallmarks: Motility, invasiveness, the ability to degrade
components of the extracellular matrix, ability to colonise
distinct organs and the metastasis-initiating ability. Disseminated
cancer cells display intermediate phenotypes featuring both
epithelial and mesenchymal characteristics depending on changing
conditions within the metastatic cascade. The ability to modulate
the local microenvironment is based on strong activation of
tumour-directed immune cells and blockade of their receptors and
ligands. After the process of intravasation, the metastatic cells
inclined from invasive properties back to their proliferative
potential and ability to differentiate into more cell types to form
macrometastasis.
The genetic and molecular determinants of
metastatic competence and active molecular pathways mimic the
changing metastatic traits. Nevertheless, knowledge of the dynamics
of transcriptional and metabolic changes in disseminated cells, CTC
and intravasated cells on the RNA, protein and pathway level is
limited by the current technical possibilities. However, the
genomic landscape of mCRC together with expression profiles have
led to the identification of driver genetic events in metastatic
development. The effect of mutations on early driver genes
including APC, KRAS, TP53, PIK3CA in CMS 2–4 tumour
(95) is increased by one or more
candidate metastasis drivers: TCF7L2, AMER1, PTPRT, PIK3CA,
GNAS, SRC, FXR1, MUC4, GPC6 and MECOM (17). They encode regulators of signalling
pathways: WNT signalling pathway (TCF7L2, AMER1), activator of cAMP
and protein kinase A (GNAS), PI3K-Akt, and AKT-mTOR signalling
pathway (PIK3CA), PI3K-Akt, Ras-Raf-MAPK, STAT3/STAT5B, and p130
pathways (SRC), the PI3K-Akt and Ras-ERK pathways (Mucin4), JNK and
p38 MAPK signalling (Glypican 4) and TGF-β pathway (MECOM). The
overproduction, abrogation or incorrect location of proteins
encoded by these genes and their combination dysregulate many
pivotal signalling pathways enabling the dissemination and
outgrowth in distant organs. In addition, epigenetic mechanisms and
metabolic reprogramming play significant roles in driving tumour
progression towards the metastatic process.
Acknowledgements
We would like to thank Martin Benej, PhD, for
language editing.
Funding
This study was supported by VEGA (grant nos.
2/0128/17, 2/0050/19 and 2/0124/17); by the Ministry of Health of
the Slovak Republic under the contract 2019/60-BMCSAV and by
funding from the European Union's Horizon 2020 Research and
Innovation Strategies to Programme under grant agreement no. 857381
(project VISION).
Availability of data and materials
Not applicable.
Authors' contributions
MP, TF, SB, SS, LK, and MM contributed to this
paper with conception of the study, literature review and analysis.
MP prepared the figures. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ADAM
|
A disintegrin and
metalloproteinase
|
|
AKT1 (PKB)
|
protein kinase B
|
|
ALDH1A1, 3
|
aldehyde dehydrogenase 1 isoforms A1,
A3
|
|
AMER1
|
APC membrane recruitment protein
1
|
|
AP4
|
murine transcription factor activator
protein 4
|
|
APC
|
adenomatous polyposis coli
|
|
APOH
|
apolipoprotein H
|
|
ATM
|
ATM serine/threonine kinase
|
|
BRAF
|
serine/threonine-protein kinase
|
|
CAF
|
carcinoma-associated fibroblast
|
|
cAMP
|
cyclic adenosine monophosphate
|
|
CD133
|
transmembrane glycoprotein
prominin-1
|
|
CD26
|
dipeptidyl peptidase-4
|
|
CD44
|
homing cell adhesion molecule
|
|
CDCP1
|
CUB domain-containing protein 1
|
|
CDX2
|
homeobox transcription factor 2
|
|
CIN
|
chromosomal instability
|
|
CK1α
|
casein kinase 1α
|
|
CMS
|
classification of molecular
subtypes
|
|
CRC
|
colorectal cancer
|
|
CSC
|
cancer stem cell
|
|
CTC
|
circulating tumour cell
|
|
CXCL14
|
C-X-C motif chemokine ligand 14
|
|
CXCR4
|
C-X-C chemokine receptor type 4
|
|
ECM
|
extracellular matrix
|
|
EGFR
|
epidermal growth factor receptor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
EpCAM
|
epithelial cell adhesion molecule
|
|
ErbB2
|
(NEU, HER2) receptor tyrosine
kinase
|
|
ErbB3
|
(HER3) receptor tyrosine kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
ERK1, 2
|
extracellular signal-regulated
kinases 1, 2
|
|
F5
|
factor five, protein of the
coagulation system
|
|
FOXC2
|
forkhead box protein C2
|
|
FRA-1
|
Fos-related antigen 1
|
|
FXR1-Fragile
|
X mental retardation syndrome-related
protein 1
|
|
GATA
|
transcription factor
|
|
GDP/GTP
|
guanosine diphosphate/guanosine
triphosphate
|
|
GLUT1, 3
|
Glucose transporter 1, 3
|
|
GNAS
|
heterotrimeric g-protein α subunit
Gs-α
|
|
GPC6
|
glypican-6
|
|
GRB2
|
growth factor receptor-bound protein
2
|
|
GSK3β
|
glycogen synthase kinase 3β
|
|
HGF
|
hepatocyte growth factor
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
HMGA1
|
high-mobility group gene
|
|
ITH
|
intratumoural heterogeneity
|
|
JNK
|
c-Jun N-terminal kinase
|
|
Klf4
|
kruppel-like factor 4
|
|
LCRC
|
left-sided colorectal carcinoma
|
|
LEF 1
|
lymphoid enhancer-binding factor
1
|
|
LGR5
|
leucine-rich repeat-containing
G-protein coupled receptor 5
|
|
LRP
|
low-density lipoprotein-related
protein
|
|
MACC1
|
metastasis-associated in colon
cancer-1
|
|
MAPK
|
mitogen-activated protein kinase
|
|
mCRC
|
metastatic CRC
|
|
MECOM
|
MDS1 and EVI1 complex locus protein
EVI1
|
|
MEIS2
|
Homeobox 2
|
|
MEK
|
mitogen-activated protein kinase
kinase
|
|
MEK1, 2
|
mitogen-activated protein kinases 1,
2
|
|
cMET (HGFR)
|
hepatocyte growth factor receptor
|
|
MLH1
|
MutL homolog 1
|
|
MMP
|
matrix metalloproteinase
|
|
MSH2, 6
|
MutS homolog 2, 6
|
|
MSI
|
microsatellite instability
|
|
MSS
|
microsatellite stability
|
|
mTOR
|
mammalian target of rapamycin
|
|
mTORC1, 2
|
mammalian target of rapamycin complex
1, 2
|
|
MUC4
|
mucin 4 glycoprotein
|
|
NANOG
|
homeobox protein, transcriptional
factor
|
|
NFκB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
NGS
|
next-generation sequencing
|
|
NK
|
natural killer
|
|
Notch1
|
Notch homolog 1, transmembrane
receptor
|
|
NRG
|
neuroregulin
|
|
OCT4
|
octamer-binding transcription factor
4PKB-Protein kinase B
|
|
PD-1
|
programmed cell death protein 1
|
|
PDCD4
|
programmed cell death protein 4
|
|
PDK1
|
phosphoinositide-dependent
kinase-1
|
|
PD-L1
|
programmed death-ligand 1
|
|
PGC-1β
|
peroxisome proliferator-activated
receptor γ coactivator 1-β
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
PIK3CA
|
phosphatidylinositol-4,5-bisphosphate
3-kinase, catalytic subunit α
|
|
PIP2
|
phosphatidylinositol-4,5-bisphosphate
|
|
PIP3
|
phosphatidylinositol-3,4,5-triphosphate
|
|
PKA
|
protein kinase A
|
|
PKB
|
protein kinase B
|
|
PMS1, 2
|
PMS1 protein homolog 1,2
|
|
PP2A
|
protein phosphatase 2A
|
|
PROX1
|
Prospero homeobox protein 1
|
|
PRRX1
|
paired related homeobox 1
|
|
PTEN
|
phosphatase and tensin homolog
|
|
PTPRT
|
receptor-type tyrosine-protein
phosphatase T
|
|
RAC1
|
Ras-related C3 botulinum toxin
substrate 1
|
|
RCRC
|
right-sided colorectal carcinoma
|
|
RhoA
|
member of family of GTPases
|
|
RICTOR
|
rapamycin-insensitive companion of
mammalian target of rapamycin
|
|
S100A4
|
S100 calcium-binding protein A4
|
|
SAM
|
S-adenosyl methionine
|
|
SIX1
|
Homeobox protein
|
|
sLeA
|
sialyl Lewis A
|
|
sLeX
|
sialyl Lewis X
|
|
SMAD2, 4
|
Mothers against decapentaplegic
homolog 2, 4
|
|
SNAI1
|
(Snail) zinc finger protein 1
|
|
SNAI2
|
(Slug) zinc finger protein 2
|
|
SOS
|
son of sevenless
|
|
SOX2
|
SRY (sex determining region Y)-box
2
|
|
SRC
|
Proto-oncogene tyrosine-protein
kinase
|
|
STAT3, 5
|
Signal transducer and activator of
transcription 3, 5
|
|
SW620
|
colorectal carcinoma cell line
|
|
TAM
|
tumour-associated macrophage
|
|
TCA
|
citric acid (Krebs) cycle
|
|
TCF4 (TCF7L2)
|
transcription factor 7-like 2
|
|
TGF-β
|
transforming growth factor-β
|
|
TIL
|
tumour-infiltrating lymphocytes
|
|
TME
|
tumour microenvironment
|
|
TP53
|
cellular tumour antigen p53
|
|
TWIST1,2
|
TWIST-related protein 1, 2
|
|
VEGFR-1
|
vascular endothelial growth factor
receptor 1
|
|
WNT5A
|
WNT family member 5A
|
|
YAP
|
Yes-associated protein
|
|
ZEB1, 2
|
Zinc finger E-box-binding homeobox 1,
2
|
References
|
1
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qiu M, Hu J, Yang D, Cosgrove DP and Xu R:
Pattern of distant metastases in colorectal cancer: A SEER based
study. Oncotarget. 6:38658–38666. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zarour LR, Anand S, Billingsley KG, Bisson
WH, Cercek A, Clarke MF, Coussens LM, Gast CE, Geltzeiler CB,
Hansen L, et al: Colorectal cancer liver metastasis: Evolving
paradigms and future directions. Cell Mol Gastroenterol Hepatol.
3:163–173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Welch DR and Hurst DR: Defining the
hallmarks of metastasis. Cancer Res. 79:3011–3027. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lambert AW, Pattabiraman DR and Weinberg
RA: Emerging biological principles of metastasis. Cell.
168:670–691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirata A, Hatano Y, Niwa M, Hara A and
Tomita H: Heterogeneity of colon cancer stem cells. Adv Exp Med
Biol. 1139:115–126. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tauriello DV, Calon A, Lonardo E and
Batlle E: Determinants of metastatic competency in colorectal
cancer. Mol Oncol. 11:97–119. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Puccini A, Xiu J, Goldberg RM, Grothey A,
Shields AF, Salem ME, Seeber A, Battaglin F, Berger MD, El-Deiry
WS, et al: Molecular differences between lymph nodes (LNs) and
distant metastases (mets) in colorectal cancer (CRC). J Clin Oncol.
37 (Suppl 15):S31302019. View Article : Google Scholar
|
|
10
|
Kamal Y, Schmit SL, Hoehn HJ, Amos CI and
Frost HR: Transcriptomic differences between primary colorectal
adenocarcinomas and distant metastases reveal metastatic colorectal
cancer subtypes. Cancer Res. 79:4227–4241. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pretzsch E, Bösch F, Neumann J, Ganschow
P, Bazhin A, Guba M, Werner J and Angele M: Mechanisms of
metastasis in colorectal cancer and metastatic organotropism:
Hematogenous versus peritoneal spread. J Oncol. 2019:74071902019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tariq K and Ghias K: Colorectal cancer
carcinogenesis: A review of mechanisms. Cancer Biol Med.
13:120–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ramaswamy S, Ross KN, Lander ES and Golub
TR: A molecular signature of metastasis in primary solid tumors.
Nat Genet. 33:49–54. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Naxerova K and Jain RK: Using tumour
phylogenetics to identify the roots of metastasis in humans. Nat
Rev Clin Oncol. 12:258–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meacham CE and Morrison SJ: Tumour
heterogeneity and cancer cell plasticity. Nature. 501:328–337.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sottoriva A, Kang H, Ma Z, Graham TA,
Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D and
Curtis C: A big bang model of human colorectal tumor growth. Nat
Genet. 47:209–216. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hu Z, Ding J, Ma Z, Sun R, Seoane JA,
Scott Shaffer J, Suarez CJ, Berghoff AS, Cremolini C, Falcone A, et
al: Quantitative evidence for early metastatic seeding in
colorectal cancer. Nat Genet. 51:1113–1122. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Holch JW, Ricard I, Stintzing S, Modest DP
and Heinemann V: The relevance of primary tumour location in
patients with metastatic colorectal cancer: A meta-analysis of
first-line clinical trials. Eur J Cancer. 70:87–98. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Auguste P, Fallavollita L, Wang N, Burnier
J, Bikfalvi A and Brodt P: The host inflammatory response promotes
liver metastasis by increasing tumor cell arrest and extravasation.
Am J Pathol. 170:1781–1792. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Riihimäki M, Hemminki A, Sundquist J and
Hemminki K: Patterns of metastasis in colon and rectal cancer. Sci
Rep. 6:297652016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Prasanna T, Craft PS, Chua YJ, Karapetis
CS, Gibbs P, Wong R, Tie J, Roder DM, Price TJ, Padbury R, et al:
The outcome of patients (pts) with metastatic colorectal cancer
(mCRC) based on site of metastases (mets) and the impact of
molecular markers and site of primary cancer on metastatic pattern.
J Clin Oncol. 35 (Suppl 15):S35602017. View Article : Google Scholar
|
|
22
|
Sadahiro S, Suzuki S, Ishikawa K, Nakamura
T, Tanaka Y, Ishizu K, Yasuda S, Makuuchi H and Murayama C:
Estimation of the time of pulmonary metastasis in colorectal cancer
patients with isolated synchronous liver metastasis. Jpn J Clin
Oncol. 35:18–22. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Wang D, Zhang C, Zhang Z, Chen X,
Lian J, Liu J, Wang G, Yuan W, Sun Z, et al: Identification of
liver metastasis-associated genes in human colon carcinoma by mRNA
profiling. Chin J Cancer Res. 30:633–646. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schweiger T, Liebmann-Reindl S, Glueck O,
Starlinger P, Laengle J, Birner P, Klepetko W, Pils D, Streubel B
and Hoetzenecker K: Mutational profile of colorectal cancer lung
metastases and paired primary tumors by targeted next generation
sequencing: Implications on clinical outcome after surgery. J
Thorac Dis. 10:6147–6157. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hugen N, van de Velde CJH, de Wilt JHW and
Nagtegaal ID: Metastatic pattern in colorectal cancer is strongly
influenced by histological subtype. Ann Oncol. 25:651–657. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Andres SF, Williams KN and Rustgi AK: The
molecular basis of metastatic colorectal cancer. Curr Colorectal
Cancer Rep. 14:69–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanahan D and Weinber RA: The hallmark of
cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanahan D and Weinber RA: The hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
De Smedt L, Palmans S, Andel D, Govaere O,
Boeckx B, Smeets D, Galle E, Wouters J, Barras D, Suffiotti M, et
al: Expression profiling of budding cells in colorectal cancer
reveals an EMT-like phenotype and molecular subtype switching. Br J
Cancer. 116:58–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheung KJ, Gabrielson E, Werb Z and Ewald
AJ: Collective invasion in breast cancer requires a conserved basal
epithelial program. Cell. 155:1639–1651. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Revenu C and Gilmour D: EMT 2.0: Shaping
epithelia through collective migration. Curr Opin Genet Dev.
19:338–342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Westcott JM, Prechtl AM, Maine EA, Dang
TT, Esparza MA, Sun H, Zhou Y, Xie Y and Pearson GW: An
epigenetically distinct breast cancer cell subpopulation promotes
collective invasion. J Clin Invest. 125:1927–1943. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ye X, Tam WL, Shibue T, Kaygusuz Y,
Reinhardt F, Ng Eaton E and Weinberg RA: Distinct EMT programs
control normal mammary stem cells and tumour-initiating cells.
Nature. 525:256–260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brabletz T, Hlubek F, Spaderna S,
Schmalhofer O, Hiendlmeyer E, Jung A and Kirchner T: Invasion and
metastasis in colorectal cancer: Epithelial-mesenchymal transition,
mesenchymal-epithelial transition, stem cells and beta-catenin.
Cells Tissues Organs. 179:56–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fan F, Samuel S, Evans KW, Lu J, Xia L,
Zhou Y, Sceusi E, Tozzi F, Ye XC, Mani SA and Ellis LM:
Overexpression of snail induces epithelial-mesenchymal transition
and a cancer stem cell-like phenotype in human colorectal cancer
cells. Cancer Med. 1:5–16. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pang R, Law WL, Chu AC, Poon JT, Lam CS,
Chow AK, Ng L, Cheung LW, Lan XR, Lan HY, et al: A subpopulation of
CD26+ cancer stem cells with metastatic capacity in
human colorectal cancer. Cell Stem Cell. 6:603–615. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakano M, Kikushige Y, Miyawaki K,
Kunisaki Y, Mizuno S, Takenaka K, Tamura S, Okumura Y, Ito M,
Ariyama H, et al: Dedifferentiation process driven by TGF-beta
signaling enhances stem cell properties in human colorectal cancer.
Oncogene. 38:780–793. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Watanabe T, Kobunai T, Yamamoto Y, Matsuda
K, Ishihara S, Nozawa K, Iinuma H, Kanazawa T, Tanaka T, Konishi T,
et al: Gene expression of mesenchyme forkhead 1 (FOXC2)
significantly correlates with the degree of lymph node metastasis
in colorectal cancer. Int J Surg. 96:207–216. 2011. View Article : Google Scholar
|
|
39
|
Sánchez-Tilló E, de Barrios O, Siles L,
Cuatrecasas M, Castells A and Postigo A: β-catenin/TCF4 complex
induces the epithelial-to-mesenchymal transition (EMT)-activator
ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA.
108:19204–19209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han X, Fang X, Lou X, Hua D, Ding W, Foltz
G, Hood L, Yuan Y and Lin B: Silencing SOX2 induced
mesenchymal-epithelial transition and its expression predicts liver
and lymph node metastasis of CRC patients. PLoS One. 7:e413352012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dai X, Ge J, Wang X, Qian X, Zhang C and
Li X: OCT4 regulates epithelial-mesenchymal transition and its
knockdown inhibits colorectal cancer cell migration and invasion.
Oncol Rep. 29:155–160. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Meng HM, Zheng P, Wang XY, Liu C, Sui HM,
Wu SJ, Zhou J, Ding YQ and Li J: Over-expression of Nanog predicts
tumor progression and poor prognosis in colorectal cancer. Cancer
Biol Ther. 9:295–302. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lu MH, Huang CC, Pan MR, Chen HH and Hung
WC: Prospero homeobox 1 promotes epithelial-mesenchymal transition
in colon cancer cells by inhibiting E-cadherin via miR-9. Clin
Cancer Res. 18:6416–6425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ono H, Imoto I, Kozakiet K, Tsuda H,
Matsui T, Kurasawa Y, Muramatsu T, Sugihara K and Inazawa J: SIX1
promotes epithelial-mesenchymal transition in colorectal cancer
through ZEB1 activation. Oncogene. 31:4923–4934. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takahashi Y, Sawada G, Kurashige J, Uchi
R, Matsumura T, Ueo H, Takano Y, Akiyoshi S, Eguchi H, Sudo T, et
al: Paired related homoeobox 1, a new EMT inducer, is involved in
metastasis and poor prognosis in colorectal cancer. Br J Cancer.
109:307–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Belton A, Gabrovsky A, Bae YK, Reeves R,
Iacobuzio-Donahue C, Huso DL and Resar LM: HMGA1 induces intestinal
polyposis in transgenic mice and drives tumor progression and stem
cell properties in colon cancer cells. PLoS One. 7:e300342012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Diesch J, Sanij E, Gilan O, Love C, Tran
H, Fleming NI, Ellul J, Amalia M, Haviv I, Pearson RB, et al:
Widespread FRA1-dependent control of mesenchymal
transdifferentiation programs in colorectal cancer cells. PLoS One.
9:e889502014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Toiyama Y, Yasuda H, Saigusa S, Tanaka K,
Inoue Y, Goel A and Kusunoki M: Increased expression of Slug and
Vimentin as novel predictive biomarkers for lymph node metastasis
and poor prognosis in colorectal cancer. Carcinogenesis.
34:2548–2557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Janda E, Lehmann K, Killisch I, Jechlinger
M, Herzig M, Downward J, Beug H and Grünert S: Ras and TGF[beta]
cooperatively regulate epithelial cell plasticity and metastasis:
Dissection of Ras signaling pathways. J Cell Biol. 156:299–313.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gotzmann J, Mikula M, Eger A,
Schulte-Hermann R, Foisner R, Beug H and Mikulitz W: Molecular
aspects of epithelial cell plasticity: Implications for local tumor
invasion and metastasis. Mutat Res. 566:9–20. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Maffeis V, Nicolè L and Cappellesso R:
RAS, cellular plasticity, and tumor budding in colorectal cancer.
Front Oncol. 9:12552019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wan Z, Chai R, Yuan H, Chen B, Dong Q,
Zheng B, Mou X, Pan W, Tu Y, Yang Q, et al: MEIS2 promotes cell
migration and invasion in colorectal cancer. Oncol Rep. 42:213–223.
2019.PubMed/NCBI
|
|
53
|
Sugai T, Yamada N, Eizuka M, Sugimoto R,
Uesugi N, Osakabe M, Ishida K, Otsuka K, Sasaki A and Matsumoto T:
Vascular invasion and stromal S100A4 expression at the invasive
front of colorectal cancer are novel determinants and tumor
prognostic markers. J Cancer. 8:1552–1561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Varga J and Greten FR: Cell plasticity in
epithelial homeostasis and tumorigenesis. Nat Cell Biol.
19:1133–1141. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ishay-Ronen D, Diepenbruck M, Kalathur
RKR, Sugiyama N, Tiede S, Ivanek R, Bantug G, Morini MF, Wang J,
Hess C and Christofori G: Gain fat-lose metastasis: Converting
invasive breast cancer cells into adipocytes inhibits cancer
metastasis. Cancer Cell. 35:17–32.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Celià-Terrassa T and Kang Y: Distinctive
properties of metastasis-initiating cells. Genes Dev. 30:892–908.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tam WL and Weinberg RA: The epigenetics of
epithelial-mesenchymal plasticity in cancer. Nat Med. 19:1438–1449.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mohd-Sarip A, Teeuwssen M, Bot AG, De
Herdt MJ, Willems SM, Baatenburg de Jong RJ, Looijenga LHJ,
Zatreanu D, Bezstarosti K, van Riet J, et al: DOC1-dependent
recruitment of NURD reveals antagonism with SWI/SNF during
epithelial-mesenchymal transition in oral cancer cells. Cell Rep.
20:61–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu
Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al:
Breast cancer stem cells transition between epithelial and
mesenchymal states reflective of their normal counterparts. Stem
Cell Report. 2:78–91. 2013. View Article : Google Scholar
|
|
62
|
Biddle A, Gammon L, Liang X, Costea DE and
Mackenzie IC: Phenotypic plasticity determines cancer stem cell
therapeutic resistance in oral squamous cell carcinoma.
EBioMedicine. 4:138–145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Katsuno Y, Meyer DS, Zhang Z, Shokat KM,
Akhurst RJ, Miyazono K and Derynck R: Chronic TGF-β exposure drives
stabilized EMT, tumor stemness, and cancer drug resistance with
vulnerability to bitopic mTOR inhibition. Sci Signal.
12:eaau85442019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Obenauf AC and Massagué J: Surviving at a
distance: Organ specific metastasis. Trends Cancer. 1:76–91. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Agnoletto C, Corrà F, Minotti L,
Baldassari F, Crudele F, Cook WJJ, Di Leva G, d'Adamo AP, Gasparini
P and Volinia S: Heterogeneity in circulating tumor cells: The
relevance of the stem-cell subset. Cancers (Basel). 11:4832019.
View Article : Google Scholar
|
|
66
|
Guinney J, Dienstmann R, Wang X, de
Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda
G, Angelino P, et al: The consensus molecular subtypes of
colorectal cancer. Nat Med. 21:1350–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kauffmann A, Rosselli F, Lazar V,
Winnepenninckx V, Mansuet-Lupo A, Dessen P, van den Oord JJ, Spatz
A and Sarasin A: High expression of DNA repair pathways is
associated with metastasis in melanoma patients. Oncogene.
27:565–573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Trinh A, Lädrach C, Dawson HE, Ten Hoorn
S, Kuppen PJ, Reimers MS, Koopman M, Punt CJ, Lugli A, Vermeulen L
and Zlobec I: Tumour budding is associated with the mesenchymal
colon cancer subtype and RAS/RAF mutations: A study of 1320
colorectal cancers with consensus molecular subgroup. Br J Cancer.
119:1244–1251. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sveen A, Cremolini C and Dienstmann R:
Predictive modeling in colorectal cancer: Time to move beyond
consensus molecular subtypes. Ann Oncol. 30:1682–1685. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhou Y, Xia L, Wang H, Oyang L, Su M, Liu
Q, Lin J, Tan S, Tian Y, Liao Q and Cao D: Cancer stem cells in
progression of colorectal cancer. Oncotarget. 9:33403–33415. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Carmon KS, Gong X, Yi J, Wu L, Thomas A,
Moore CM, Masuho I, Timson DJ, Martemyanov KA and Liu QJ: LGR5
receptor promotes cell-cell adhesion in stem cells and colon cancer
cells via the IQGAP1-Rac1 pathway. J Biol Chem. 292:14989–15001.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lam CS, Cheung AH, Wong SK, Wan TM, Ng L,
Chow AK, Cheng NS, Pak RC, Li HS, Man JH, et al: Prognostic
significance of CD26 in patients with colorectal cancer. PLoS One.
9:e985822014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mortier A, Gouwy M, Van Damme J, Proost P
and Struyf S: CD26/dipeptidylpeptidase IV-chemokine interactions:
Double-edged regulation of inflammation and tumor biology. J Leukoc
Biol. 99:955–969. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ma L, Dong L and Chang P: CD44v6 engages
in colorectal cancer progression. Cell Death Dis. 10:302019.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liu H, Ong SE, Badu-Nkansah K, Schindler
J, White FM and Hynes RO: CUB-domain-containing protein 1 (CDCP1)
activates Src to promote melanoma metastasis. Proc Natl Acad Sci
USA. 108:1379–1384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mohamed SY, Kaf RM, Ahmed MM, Elwan A,
Ashour HR and Ibrahim A: The prognostic value of cancer stem cell
markers (Notch1, ALDH1, and CD44) in primary colorectal carcinoma.
J Gastrointest Cancer. 50:824–837. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jackstadt R, van Hooff SR, Leach JD,
Cortes-Lavaud X, Lohuis JO, Ridgway RA, Wouters VM, Roper J,
Kendall TJ, Roxburgh CS, et al: Epithelial NOTCH signaling rewires
the tumor microenvironment of colorectal cancer to drive
poor-prognosis subtypes and metastasis. Cancer Cell. 36:319–336.e7.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Van der Waals LM, Borel Rinkes IH and
Kranenburg O: ALDH1A1 expression is associated with poor
differentiation, ‘right-sidedness’ and poor survival in human
colorectal cancer. PLoS One. 13:e02055362018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Vázquez-Iglesias L, Barcia-Castro L,
Rodríguez-Quiroga M, Páez de la Cadena M, Rodríguez-Berrocal J and
Cordero OJ: Surface expression marker profile in colon cancer cell
lines and sphere-derived cells suggests complexity in
CD26+ cancer stem cells subsets. Biol Open.
8:bio0416732019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Dotse E and Bian Y: Isolation of
colorectal cancer stem-like cells. Cytotechnology. 68:609–619.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
De Sousa E, Melo F, Wang X, Jansen M,
Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van
Leersum R, Bijlsma MF, et al: Poor-prognosis colon cancer is
defined by a molecularly distinct subtype and develops from
serrated precursor lesions. Nat Med. 19:614–618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Testa U, Pelosi E and Castelli G:
Colorectal cancer: Genetic abnormalities, tumor progression, tumor
heterogeneity, clonal evolution and tumor-initiating cells. Med Sci
(Basel). 6:312018.
|
|
83
|
Fedyanin M, Popova A, Polyanskaya E and
Tjulandin S: Role of stem cells in colorectal cancer progression
and prognostic and predictive characteristics of stem cell markers
in colorectal cancer. Curr Stem Cell Res Ther. 12:19–30. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lieto E, Galizia G, Orditura M, Romano C,
Zamboli A, Castellano P, Mabilia A, Auricchio A, De Vita F and
Gemei M: CD26-positive/CD326-negative circulating cancer cells as
prognostic markers for colorectal cancer recurrence. Oncol Lett.
9:542–550. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Todaro M, Gaggianesi M, Catalano V,
Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S,
Cocorullo G, et al: CD44v6 is a marker of constitutive and
reprogrammed cancer stem cells driving colon cancer metastasis.
Cell Stem Cell. 14:342–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhang SS, Han ZP, Jing YY, Tao SF, Li TJ,
Wang H, Wang Y, Li R, Yang Y, Zhao X, et al: CD133(+)CXCR4(+) colon
cancer cells exhibit metastatic potential and predict poor
prognosis of patients. BMC Med. 10:852012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Gao W, Chen L, Ma Z, Du Z, Zhao Z, Hu Z
and Li Q: Isolation and phenotypic characterization of colorectal
cancer stem cells with organ-specific metastatic potential.
Gastroenterology. 145:636–646.e5. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tong K, Pellón-Cárdenas O, Sirihorachai
VR, Warder BN, Kothari OA, Perekatt AO, Fokas EE, Fullem RL, Zhou
A, Thackray JK, et al: Degree of tissue differentiation dictates
susceptibility to BRAF-driven colorectal cancer. Cell Rep.
21:3833–3845. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Durinikova E, Kozovska Z, Poturnajova M,
Plava J, Cierna Z, Babelova A, Bohovic R, Schmidtova S, Tomas M,
Kucerova L and Matuskova M: ALDH1A3 upregulation and spontaneous
metastasis formation is associated with acquired chemoresistance in
colorectal cancer cells. BMC Cancer. 18:8482018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Boland CR and Goel A: Microsatellite
instability in colorectal cancer. Gastroenterology.
138:2073–2087.e3. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mármol I, Sánchez-de-Diego C, Pradilla
Dieste A, Cerrada E and Rodriguez Yoldi MJ: Colorectal carcinoma: A
general overview and future perspectives in colorectal cancer. Int
J Mol Sci. 18:1972017. View Article : Google Scholar
|
|
92
|
Jung J, Kang Y, Lee YJ, Kim E, Ahn B, Lee
E, Kim JY, Lee JH, Lee Y, Kim CH and Chae YS: Comparison of the
mismatch repair system between primary and metastatic colorectal
cancers using immunohistochemistry. J Pathol Transl Med.
51:129–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen W, Swanson BJ and Frankel WL:
Molecular genetics of microsatellite-unstable colorectal cancer for
pathologists. Diagn Pathol. 12:242017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Pino MS and Chung DC: The chromosomal
instability pathway in colon cancer. Gastroenterology.
138:2059–2072. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Thanki K, Nicholls ME, Gajjar A, Senagore
AJ, Qiu S, Szabo C, Hellmich MR and Chao C: Consensus molecular
subtypes of colorectal cancer and their clinical implications. Int
Biol Biomed J. 3:105–111. 2017.PubMed/NCBI
|
|
96
|
Fumagalli A, Drost J, Suijkerbuijk SJ, van
Boxtel R, de Ligt J, Offerhaus GJ, Begthel H, Beerling E, Tan EH,
Sansom OJ, et al: Genetic dissection of colorectal cancer
progression by orthotopic transplantation of engineered cancer
organoids. Proc Natl Acad Sci USA. 114:E2357–E2364. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhao M, Mishra L and Deng CX: The role of
TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. 14:111–123. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Boutin AT, Liao WT, Wang M, Hwang SS,
Karpinets TV, Cheung H, Chu GC, Jiang S, Hu J, Chang K, et al:
Oncogenic KRAS drives invasion and maintains metastases in
colorectal cancer. Genes Dev. 31:370–382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wenzel J, Rose K, Haghighi EB, Lamprecht
C, Rauen G, Freihen V, Kesselring R, Boerries M and Hecht A: Loss
of the nuclear Wnt pathway effector TCF7L2 promotes migration and
invasion of human colorectal cancer cells. Oncogene. 39:3893–3909.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Major MB, Camp ND, Berndt JD, Yi X,
Goldenberg SJ, Hubbert C, Biechele TL, Gingras AC, Zheng N, Maccoss
MJ, et al: Wilms tumor suppressor WTX negatively regulates
WNT/beta-catenin signaling. Science. 316:1043–1046. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Steffen DJ, Amornphimoltham P, Valera JLC,
Taylor S, Hunter T, Tamayo P and Gutkind JS: GNAS-PKA Oncosignaling
Network in Colorectal Cancer. Pharmacology. 31:lb5272017.
|
|
102
|
Lieu C and Kopetz S: The SRC family of
protein tyrosine kinases: A new and promising target for colorectal
cancer therapy. Clin Colorectal Cancer. 9:89–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jin X, Zhai B, Fang T, Guo X and Xu L:
FXR1 is elevated in colorectal cancer and acts as an oncogene.
Tumor Biol. 37:2683–2690. 2016. View Article : Google Scholar
|
|
104
|
Gumireddy K, Li A, Yan J, Setoyama T,
Johannes GJ, Ørom UA, Tchou J, Liu Q, Zhang L, Speicher DW, et al:
Identification of a long non-coding RNA-associated RNP complex
regulating metastasis at the translational step. EMBO J.
32:2672–2684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Xia P, Choi AH, Deng Z, Yang Y, Zhao J,
Wang Y, Hardwidge PR and Zhu G: Cell membrane-anchored MUC4
promotes tumorigenicity in epithelial carcinomas. Oncotarget.
8:14147–14157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
De Robertis M, Arigoni M, Loiacono L,
Riccardo F, Calogero RA, Feodorova Y, Tashkova D, Belovejdov V,
Sarafian V, Cavallo F and Signori E: Novel insights into Notum and
glypicans regulation in colorectal cancer. Oncotarget.
6:41237–41257. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lui VW, Peyser ND, Ng PK, Hritz J, Zeng Y,
Lu Y, Li H, Wang L, Gilbert BR, General IJ, et al: Frequent
mutation of receptor protein tyrosine phosphatases provides a
mechanism for STAT3 hyperactivation in head and neck cancer. Proc
Natl Acad Sci USA. 111:1114–1119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Brabletz T, Jung A, Reu S, Porzner M,
Hlubek F, Kunz-Schughart LA, Knuechel R and Kirchner T: Variable
beta-catenin expression in colorectal cancers indicates tumor
progression driven by the tumor environment. Proc Natl Acad Sci
USA. 98:10356–10361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Fodde R, Smits R and Clevers H: APC,
signal transduction and genetic instability in colorectal cancer.
Nat Rev Cancer. 1:55–67. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gregorieff A and Clevers H: Wnt signaling
in the intestinal epithelium: From endoderm to cancer. Genes Dev.
19:877–890. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Brabletz T, Jung A, Hermann K, Günther K,
Hohenberger W and Kirchner T: Nuclear overexpression of the
oncoprotein beta-catenin in colorectal cancer is localized
predominantly at the invasion front. Pathol Res Pract. 194:701–704.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Le NH, Franken P and Fodde R:
Tumour-stroma interactions in colorectal cancer: Converging on
beta-catenin activation and cancer stemness. Br J Cancer.
98:1886–1893. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Teeuwssen M and Fodde R: Cell
heterogeneity and phenotypic plasticity in metastasis formation:
The case of colon cancer. Cancers (Basel). 11:13682019. View Article : Google Scholar
|
|
114
|
Brocardo M and Henderson B: APC shuttling
to the membrane, nucleus and beyond. Trends Cell Boil. 18:587–596.
2008. View Article : Google Scholar
|
|
115
|
Sarli L, Bottarelli L, Bader G, Iusco D,
Pizzi S, Costi R, D'Adda T, Bertolani M, Roncoroni L and Bordi C:
Association between recurrence of sporadic colorectal cancer, high
level of microsatellite instability, and loss of heterozygosity at
chromosome 18q. Dis Colon Rectum. 47:1467–1482. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Munro AJ, Lain S and Lane DP: P53
abnormalities and outcomes in colorectal cancer: A systematic
review. Br J Cancer. 92:434–444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of Ras mutations in cancer. Cancer Res.
72:2457–2467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Makrodouli E, Oikonomou E, Koc M, Andera
L, Sasazuki T, Shirasawa S and Pintzas A: BRAF and RAS oncogenes
regulate Rho GTPase pathways to mediate migration and invasion
properties in human colon cancer cells: A comparative study. Mol
Cancer. 10:1182011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lemieux E, Bergeron S, Durand V, Asselin
C, Saucier C and Rivard N: Constitutively active MEK1 is sufficient
to induce epithelial-to-mesenchymal transition in intestinal
epithelial cells and to promote tumor invasionand metastasis. Int J
Cancer. 125:1575–1586. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yamazaki D, Kurisu S and Takenawa T:
Involvement of Rac and Rho signaling in cancer cell motility in 3D
substrates. Oncogene. 28:1570–1583. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Sanz-Moreno V and Marshall CJ: The
plasticity of cytoskeletal dynamics underlying neoplastic cell
migration. Curr Opin Cell Biol. 22:690–696. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Koveitypour Z, Panahi F, Vakilian M,
Peymani M, Seyed Forootan F, Nasr Esfahani MH and Ghaedi K:
Signaling pathways involved in colorectal cancer progression. Cell
Biosci. 9:972019. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Atreya CE, Sangale Z, Xu N, Matli MR,
Tikishvili E, Welbourn W, Stone S, Shokat KM and Warren RS: PTEN
expression is consistent in colorectal cancer primaries and
metastases and associates with patient survival. Cancer Med.
2:496–506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Day F, Muranyi A, Singh S, Shanmugam K,
Williams D, Byrne D, Pham K, Palmieri M, Tie J, Grogan T, et al: A
mutant BRAF V600E-specific immunohistochemical assay: Correlation
with molecular mutation status and clinical outcome in colorectal
cancer. Target Oncol. 10:99–109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Yaeger R, Cercek A, O'Reilly EM, Reidy DL,
Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, et
al: Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant
metastatic colorectal cancer patients. Clin Cancer Res.
21:1313–1320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Wong CK, Lambert AW, Ozturk S, Papageorgis
P, Lopez D, Shen N, Sen Z, Abdolmaleky HM, Győrffy B, Feng H and
Thiagalingam S: Targeting RICTOR sensitizes SMAD4-negative colon
cancer to irinotecan. Mol Cancer Res. 18:414–423. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zou Z, Tao T, Li H and Zhu X: mTOR
signaling pathway and mTOR inhibitors in cancer: Progress and
challenges. Cell Biosci. 10:312020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Demkova L and Kucerova L: Role of the
HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol
Cancer. 17:262018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Cui YM, Jiao HL, Ye YP, Chen CM, Wang JX,
Tang N, Li TT, Lin J, Qi L, Wu P, et al: FOXC2 promotes colorectal
cancer metastasis by directly targeting MET. Oncogene.
34:4379–4390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Stein U, Walther W, Arlt F, Schwabe H,
Smith J, Fichtner I, Birchmeier W and Schlag PM: MACC1, a newly
identified key regulator of HGF-MET signaling, predicts colon
cancer metastasis. Nat Med. 15:59–67. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Pichorner A, Sack U, Kobelt D, Kelch I,
Arlt F, Smith J, Walther W, Schlag PM and Stein U: In vivo imaging
of colorectal cancer growth and metastasis by targeting MACC1 with
shRNA in xenografted mice. Clin Exp Metastasis. 29:573–583. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Juneja M, Kobelt D, Walther W, Voss C,
Smith J, Specker E, Neuenschwander M, Gohlke B, Dahlmann M,
Radetzki S, et al: Statin and rottlerin small-molecule inhibitors
restrict colon cancer progression and metastasis via MACC1. PLoS
Biol. 15:e20007842017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Glebov OK, Rodriguez LM, Nakahara K,
Jenkins J, Cliatt J, Humbyrd CJ, DeNobile J, Soballe P, Simon R,
Wright G, et al: Distinguishing right from left colon by the
pattern of gene expression. Cancer Epidemiol Biomark Prev.
12:755–762. 2003.
|
|
134
|
Baran B, Mert Ozupek N, Yerli Tetik N,
Acar E, Bekcioglu O and Baskin Y: Difference between left-sided and
right-sided colorectal cancer: A focused review of literature.
Gastroenterol Res. 11:264–273. 2018. View Article : Google Scholar
|
|
135
|
Lee MS, Menter DG and Kopetz S: Right
versus left colon cancer biology: Integrating the consensus
molecular subtypes. J Natl Compr Canc Netw. 15:411–419. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Weinberg BA: Anti-EGFR therapy in
right-sided metastatic colorectal cancer: Right or wrong? J Natl
Compr Canc Netw. 16:1547–1548. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Loree JM, Pereira AAL, Lam M, Willauer AN,
Raghav K, Dasari A, Morris VK, Advani S, Menter DG, Eng C, et al:
Classifying colorectal cancer by tumor location rather than
sidedness highlights a continuum in mutation profiles and consensus
molecular subtypes. Clin Cancer Res. 24:1062–1072. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Hugen N, Brown G, Glynne-Jones R, de Wilt
JH and Nagtegaal ID: Advances in the care of patients with mucinous
colorectal cancer. Nat Rev Clin Oncol. 13:361–369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Esterházy D, Canesso MC, Mesin L, Muller
PA, de Castro TB, Lockhart A, ElJalby M, Faria AM and Mucida D:
Compartmentalized gut lymph node drainage dictates adaptive immune
responses. Nature. 569:126–130. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Piskounova E, Agathocleous M, Murphy MM,
Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ
and Morrison SJ: Oxidative stress inhibits distant metastasis by
human melanoma cells. Nature. 527:186–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Llosa NJ, Cruise M, Tam A, Wicks EC,
Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS,
et al: The vigorous immune microenvironment of microsatellite
instable colon cancer is balanced by multiple counter-inhibitory
checkpoints. Cancer Discov. 5:43–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Deschoolmeester V, Baay M, Van Marck E,
Weyler J, Vermeulen P, Lardon F and Vermorken JB: Tumor
infiltrating lymphocytes: An intriguing player in the survival of
colorectal cancer patients. BMC Immunol. 11:192010. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Salama P, Phillips M, Grieu F, Morris M,
Zeps N, Joseph D, Platell C and Iacopetta B: Tumor-infiltrating
FOXP3+ T regulatory cells show strong prognostic
significance in colorectal cancer. J Clin Oncol. 27:186–192. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Schwitalle Y, Kloor M, Eiermann S,
Linnebacher M, Kienle P, Knaebel HP, Tariverdian M, Benner A and
von Knebel Doeberitz M: Immune response against frameshift-induced
neopeptides in HNPCC patients and healthy HNPCC mutation carriers.
Gastroenterology. 134:988–997. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Han Y, Liu D and Li L: PD-1/PD-L1 pathway:
Current researches in cancer. Am J Cancer Res. 10:727–742.
2020.PubMed/NCBI
|
|
146
|
Kreidieh M, Mukherji D, Temraz S and
Shamseddine A: Expanding the scope of immunotherapy in colorectal
cancer: Current clinical approaches and future directions. Biomed
Res Int. 2020:90372172020. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Kroemer G, Galluzzi L, Zitvogel L and
Fridman WH: Colorectal cancer: The first neoplasia found to be
under immunosurveillance and the last one to respond to
immunotherapy? Oncoimmunology. 4:e10585972015. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 blockade in tumors with mismatch-repair DEfiCiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Mlecnik B, Bindea G, Kirilovsky A, Angell
HK, Obenauf AC, Tosolini M, Church SE, Maby P, Vasaturo A, Angelova
M, et al: The tumor microenvironment and Immunoscore are critical
determinants of dissemination to distant metastasis. Sci Trans Med.
8:327ra262016. View Article : Google Scholar
|
|
150
|
Mlecnik B, Van den Eynde M, Bindea G,
Church SE, Vasaturo A, Fredriksen T, Lafontaine L, Haicheur N,
Marliot F, Debetancourt D, et al: Comprehensive intrametastatic
immune quantification and major impact of immunoscore on survival.
J Natl Cancer Inst. 110:97–108. 2018. View Article : Google Scholar
|
|
151
|
Girardin A, McCall J, Black MA, Edwards F,
Phillips V, Taylor ES, Reeve AE and Kemp RA: Inflammatory and
regulatory T cells contribute to a unique immune microenvironment
in tumor tissue of colorectal cancer patients. Int J Cancer.
132:1842–1850. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Pedrosa L, Esposito F, Thomson TM and
Mauriel J: The tumor microenvironment in colorectal cancer therapy.
Cancers (Basel). 11:11722019. View Article : Google Scholar
|
|
153
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Malanchi I, Santamaria-Martínez A, Susanto
E, Peng H, Lehr HA, Delaloye JF and Huelsken J: Interactions
between cancer stem cells and their niche govern metastatic
colonization. Nature. 481:85–89. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Calon A, Espinet E, Palomo-Ponce S,
Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung
P, Zhang XH, et al: Dependency of colorectal cancer on a
TGF-β-driven program in stromal cells for metastasis initiation.
Cancer Cell. 22:571–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Brown RE, Short SP and Williams CS:
Colorectal cancer and metabolism. Curr Colorectal Cancer Rep.
14:226–241. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Gureev AP, Shaforostova EA and Popov VN:
Regulation of mitochondrial biogenesis as a way for active
longevity: Interaction between the Nrf2 and PGC-1α signaling
pathways. Front Genet. 10:4352019. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Chang CH, Qiu J, O'Sullivan D, Buck MD,
Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ,
et al: Metabolic competition in the tumor microenvironment is a
driver of cancer progression. Cell. 162:1229–1241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Vyas M, Patel N, Nagarajan A, Wajapeyee N,
Jain D and Zhan X: Hypoxia induced HIF-1α expression promotes
angiogenesis, tumor budding cell survival and cell proliferation
arrest in high-grade tumor budding colorectal carcinomas. Int J
Clin Exp Patho. 9:13047–13055. 2016.
|
|
160
|
Ancey PB, Contat C and Meylan E: Glucose
transporters in cancer-from tumor cells to the tumor
microenvironment. FEBS J. 285:2926–2943. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Ritterson Lew C, Guin S and Theodorescu D:
Targeting glycogen metabolism in bladder cancer. Nat Rev Urol.
12:383–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kuo CC, Ling HH, Chiang MC, Chung CH, Lee
WY, Chu CY, Wu YC, Chen CH, Lai YW, Tsai IL, et al: Metastatic
colorectal cancer rewrites metabolic program through a
Glut3-YAP-dependent signaling circuit. Theranostics. 9:2526–2540.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
DeBerardinis RJ: Is cancer a disease of
abnormal cellular metabolism? New angles on an old idea. Genet Med.
10:767–777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Weber GF: Metabolism in cancer metastasis.
Int Jour Cancer. 138:2061–2066. 2016. View Article : Google Scholar
|
|
165
|
Corté H, Manceau G, Blons H and
Laurent-Puig P: MicroRNA and colorectal cancer. Dig liver dis.
44:195–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Bu P, Chen KY, Xiang K, Johnson C, Crown
SB, Rakhilin N, Ai Y, Wang L, Xi R, Astapova I, et al: Aldolase
B-mediated fructose metabolism drives metabolic reprogramming of
colon cancer liver metastasis. Cell Metab. 27:1249–1262.e4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Wu Z, Wei D, Gao W, Xu Y, Hu Z, Ma Z, Gao
C, Zhu X and Li Q: TPO-induced metabolic reprogramming drives liver
metastasis of colorectal cancer CD110+ tumor-initiating
cells. Cell Stem Cells. 17:47–59. 2015. View Article : Google Scholar
|
|
168
|
Calin GA, Sevignani C, Dumitru CD, Hyslop
T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M
and Croce CM: Human microRNA genes are frequently located at
fragile sites and genomic regions involved in cancers. Proc Natl
Acad Sci USA. 101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Cellura D, Pickard K, Quaratino S, Parker
H, Strefford JC, Thomas GJ, Mitter R, Mirnezami AH and Peake NJ:
miR-19-mediated inhibition of transglutaminase-2 leads to enhanced
invasion and metastasis in colorectal cancer. Mol Cancer Res.
13:1095–1105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Lam CS, Ng L, Chow AK, Wan TM, Yau S,
Cheng NS, Wong SK, Man JH, Lo OS, Foo DC, et al: Identification of
microRNA 885–5p as a novel regulator of tumor metastasis by
targeting CPEB2 in colorectal cancer. Oncotarget. 8:26858–26870.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Shibuya H, Iinuma H, Shimada R, Horiuchi A
and Watanabe T: Clinicopathological and prognostic value of
microRNA-21 and microRNA-155 in colorectal cancer. Oncology.
79:313–320. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Asangani IA, Rasheed SA, Nikolova DA,
Leupold JH, Colburn NH, Post S and Allgayer H: MicroRNA-21 (miR-21)
post-transcriptionally downregulates tumor suppressor Pdcd4 and
stimulates invasion, intravasation and metastasis in colorectal
cancer. Oncogene. 27:2128–2136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Loo JM, Scherl A, Nguyen A, Man FY,
Weinberg E, Zeng Z, Saltz L, Paty PB and Tavazoie SF: Extracellular
metabolic energetics can promote cancer progression. Cell.
160:393–406. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Chi Y and Zhou D: MicroRNAs in colorectal
carcinoma-from pathogenesis to therapy. J Exp Clin Cancer Res.
35:432016. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Heublein S, Albertsmeier M, Pfeifer D,
Loehrs L, Bazhin AV, Kirchner T, Werner J, Neumann J and Angele MK:
Association of differential miRNA expression with hepatic vs.
peritoneal metastatic spread in colorectal cancer. BMC Cancer.
18:2012018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Siemens H, Neumann J, Jackstadt R,
Mansmann U, Horst D, Kirchner T and Hermeking H: Detection of
miR-34a promoter methylation in combination with elevated
expression of c-Met and β-catenin predicts distant metastasis of
colon cancer. Clin Cancer Res. 19:710–720. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Ma YS, Lv ZW, Yu F, Chang ZY, Cong XL,
Zhong XM, Lu GX, Zhu J and Fu D: MicroRNA-302a/d inhibits the
self-renewal capability and cell cycle entry of liver cancer stem
cells by targeting the E2F7/AKT axis. J Exp Clin Cancer Res.
37:2522018. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Shi L, Jackstadt R, Siemens H, Li H,
Kirchner T and Hermeking H: p53-induced miR-15a/16-1 and AP4 form a
double-negative feedback loop to regulate epithelial-mesenchymal
transition and metastasis in colorectal cancer. Cancer Res.
74:532–542. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Yan TT, Ren LL, Shen CQ, Wang ZH, Yu YN,
Liang Q, Tang JY, Chen YX, Sun DF, Zgodzinski W, et al: miR-508
defines the stem-like/mesenchymal subtype in colorectal cancer.
Cancer Res. 78:1751–1765. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Hur K, Toiyama Y, Takahashi M, Balaguer F,
Nagasaka T, Koike J, Hemmi H, Koi M, Boland CR and Goel A:
MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT)
in human colorectal cancer metastasis. Gut. 62:1315–1326. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Sun Z, Zhang Z, Liu Z, Qiu B, Liu K and
Dong G: MicroRNA-335 inhibits invasion and metastasis of colorectal
cancer by targeting ZEB2. Med Oncol. 31:9822014. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Geng L, Chaudhuri A, Talmon G, Wisecarver
JL, Are C, Brattain M and Wang J: MicroRNA-192 suppresses liver
metastasis of colon cancer. Oncogene. 33:5332–5340. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Hahn S, Jackstadt R, Siemens H, Hünten S
and Hermeking H: SNAIL and miR-34a feed-forward regulation of
ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J.
32:3079–3095. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Miranda E, Destro A, Malesci A, Balladore
E, Bianchi P, Baryshnikova E, Franchi G, Morenghi E, Laghi L,
Gennari L and Roncalli M: Genetic and epigenetic changes in primary
metastatic and nonmetastatic colorectal cancer. Br J Cancer.
95:1101–1107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Li Z, Chen Y, Ren WU, Hu S, Tan Z, Wang Y,
Chen Y, Zhang J, Wu J, Li T, et al: Transcriptome alterations in
liver metastases of colorectal cancer after acquired resistance to
cetuximab. Cancer Genom Proteom. 16:207–219. 2019. View Article : Google Scholar
|
|
186
|
Markowitz SD and Bertagnolli MM: Molecular
origins of cancer: Molecular basis of colorectal cancer. N Engl J
Med. 361:2449–2460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Garinis GA, Menounos PG, Spanakis NE,
Papadopoulos K, Karavitis G, Parassi I, Christeli E, Patrinos GP,
Manolis EN and Peros G: Hypermethylation-associated transcriptional
silencing of E-cadherin in primary sporadic colorectal carcinomas.
J Pathol. 198:442–449. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Ryall JG, Cliff T, Dalton S and Sartorelli
V: Metabolic reprogramming of stem cell epigenetics. Cell Stem
Cell. 17:651–662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Kim JA and Yeom YI: Metabolic signaling to
epigenetic alterations in cancer. Biomol Ther (Seoul). 26:69–80.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Zee BM, Levin RS, Xu B, LeRoy G, Wingreen
NS and Garcia BA: In vivo residue-specific histone methylation
dynamics. J Biol Chem. 285:3341–3350. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Tse JWT, Jenkins LJ, Chionh F and
Mariadason JM: Aberrant DNA methylation in colorectal cancer: What
should we target? Trends Cancer. 3:698–712. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Zentner GE and Henikoff S: Regulation of
nucleosome dynamics by histone modifications. Nat Struct Mol Biol.
20:259–266. 2013. View Article : Google Scholar : PubMed/NCBI
|