Introduction
Nickel (Ni) compounds are classified as Group 1
carcinogens by the International Agency for Research on Cancer.
Previous studies reported that workers processing and refining
sulfidic nickel ores have increased risk of pulmonary and sinonasal
cancers (1–6). Previously, special AT-rich
sequence-binding protein (SATB2), a nuclear matrix-associated
protein, was identified as the only common gene upregulated in
BEAS-2B cells transformed by nickel, arsenic, vanadate, and
hexavalent chromium (7). It is a
member of the family of special AT-rich sequence-binding proteins
and is an embryonic gene important in regulating stem cell
differentiation and development (8).
SATB2 overexpression has been observed in numerous cancers, such as
colon, breast, ovarian, lung, and sinonasal carcinomas (9–12). SATB2
has been increasingly studied in cancer development and is
considered to be a biomarker for colon cancer (9,13). SATB2
is upstream of numerous prognostic markers of colorectal cancer
(CRC) such as acyl-CoA synthetase long-chain 6 (ACSL6) and Vav
guanine nucleotide exchange factor 3 (VAV3) (14). Numerous downstream targets of SATB2
such as Bcl-2, Bsp, c-Myc, Klf4, Hoxa2 and Nanog are also important
regulators of cell pluripotency and survival (15). Thus, it is considered that
inappropriate induction of SATB2 in normal mammalian cells may act
as a driver of cell transformation.
In fact, the critical role of SATB2 in Ni-induced
BEAS-2B cell transformation has been identified (16) and in the present study the upstream
regulators for SATB2 signaling were investigated. Runt-related
transcription factor 2 (RUNX2) is a master regulator of
osteogenesis and it has a number of overlapping functions with
SATB2 (17,18). The interaction between RUNX2 and SATB2
in osteogenesis has been identified in several previous studies
(19–26). RUNX2 was detected in a limited number
of non-osseous tissues, while increased expression of RUNX2 has
been described in the progression and metastasis of several human
cancers. Overexpression of RUNX2 was observed in prostate,
pancreatic, breast, thyroid, and bone cancers (27–30).
Increased expression of RUNX2 suppressed microRNA (miRNA or miR)-31
allowing SATB2 protein translation and initiation of
carcinogenesis. Moreover, genes located downstream of RUNX2, such
as matrix metalloproteinases (MMPs), vascular epithelial growth
factor (VEGF), bone sialoprotein (BSP) and c-Myc were also involved
in promoting cancer cell metastasis (31–34). Genes
such as SRY-box transcription factor 9 (SOX9), snail family
transcriptional repressor 2 (SNAI2), snail family transcription
repression 3 (SNAI3), twist family bHLH transcription factor 1
(TWIST1) and SMAD family member 3 (SMAD3) regulated by RUNX2
induced cell invasion (35,36). RUNX2 modulated SATB2 expression by
suppressing the expression of miRNAs that directly target SATB2
mRNA in both osteogenesis and carcinogenesis (22,23,26,37–41).
Expression of RUNX2 appears to be a critical upstream event leading
to the increase of SATB2 and transforming BEAS-2B cells.
miRNAs are small noncoding RNAs that are known to
mediate mRNA degradation and inhibit translation of proteins as
well as altering chromatin structure during early development,
differentiation, metabolic control and cell death (42–45). The
expression of miRNAs has been revealed to be globally suppressed in
tumors (46). Several studies have
reported that altered miRNA expression is involved in various
stages of Ni carcinogenesis (47).
SATB2 has been revealed to be negatively regulated by several
miRNAs that target its 3′-untranslated region (UTR) during
osteogenesis and carcinogenesis, including miR-211, miR-31,
miR-23a/27a/24-2 cluster, miR-499, miR-34c, miR-875, miR-599
(10,23,37,38,40,48–54).
miR-31 was identified as a negative regulator for SATB2 in
arsenic-induced BEAS-2B transformation (40). In fact, miR-31 was also targeted by
RUNX2 and miR-31 allowed RUNX2 to regulate and increase SATB2
during osteogeneses (21,22). The aim of the study was to investigate
whether RUNX2 is an upstream regulator for SATB2, mediating
Ni-induced BEAS-2B cell transformation by regulating the expression
level of miR-31. This carcinogenic pathway mirrors what occurs
during normal bone development.
Materials and methods
Cells and cell culture
BEAS-2B human bronchial epithelial cells were
obtained from the American Type Culture Collect (ATCC). Before the
start of the present study, the cells were examined at Genetica DNA
Laboratories and were revealed to be 100% authentic against a
reference BEAS-2B cell line (ATCC CRL-9609). Cells were cultured in
Dulbecco's modified Eagle's medium (DMEM, Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS;
Atlanta Biologicals, Inc.; R&D Systems, Inc.) and 1%
penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at
37°C in an atmosphere containing 5% CO2.
Ni-transformed (Ni-T) BEAS-2B cells were developed
by chronic exposure to 0.25 mM NiSO4 for 4 weeks
(7). Cells were seeded by single cell
suspension into soft agar and colonies were isolated and grown in
monolayer on culture dishes. Each clone was analyzed using
Affymetrix gene expression arrays and the clones with highest SATB2
expression were selected for the present study. The Ni-T cells were
maintained in DMEM supplemented with 10% FBS and 1% p/s at 37°C in
a humidified incubator containing 5% CO2.
Stable overexpression and
knockdown
Stable overexpression of miR-31 in Ni-T cells was
achieved using miR-31 construct (cat. no. HmiR0138-MR04-10), which
was purchased from GeneCopoeia, Inc. RUNX2 plasmid constructs
(clone ID OHU22872D) were acquired from Genscript. Short hairpin
(sh)-RUNX2 (cat. no. HSH021333-CU6; scrambled control ID
CSHCTR001-1-CU6.1) and miR-31 inhibitor plasmid (cat. no.
HmiR-AN0401-AM01; control ID CmiR-AN0001-AM01) were obtained from
GeneCopoeia, Inc. The sequence for sh-RUNX2 and sh-miR-31 were as
follows: sh-RUNX2-a: 5′-GCAGCACGCTATTAAATCCAA-3′; sh-RUNX2-b:
5′-GCAGAATGGATGAATCTGTTT-3′; sh-RUNX2-c: GCGACCATATTGAAATTCCTC-3′;
scrambled control: 5′-GCTTCGCGCCGTAGTCTTA-3′; sh-miR-31:
5′-AGGCAAGAUGCUGGCAUAGCU-3′. Each plasmid was purified by a Qiagen
QIAprep Spin Miniprep kit (cat. no. 27104) before transfection,
using the Lipofectamine™ LTX reagent with PLUS kit (cat. no.
15338030; Invitrogen; Thermo Fisher Scientific, Inc.) used
according to the manufacturer's instructions. Briefly, the cells
were plated in 2 ml growth medium in a 6-well plate and were
~70-80% confluent during transfection. For each transfection, 2.5
µg of purified plasmid was diluted in 250 µl Opti-MEM. Immediately
before transfection, 2.5 µl PLUS reagent was added to the diluted
plasmid. Then, 10 µl Lipofectamine™ LTX reagent was diluted in 250
µl Opti-MEM and then mixed with the plasmid and PLUS reagent. The
mixture was incubated for 5 min at room temperature before adding
into the well. The medium was replaced with fresh complete DMEM 24
h after transfection. The cells were divided onto 10 cm2
dishes 48 h after transfection and selection reagents (500 µg/ml
hygromycin or 0.5 µg/ml puromycin) were added. After selection for
2 weeks (hygromycin) or 3 days (puromycin), the pooled clones were
harvested for western blotting and RT-qPCR analysis.
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
Cells were lysed by TRI-reagent (Molecular Research
Center, Inc.) (1 ml/well of a 6-well plate). RNA was extracted
using 200 µl chloroform (Sigma-Aldrich; Merck KGaA) and centrifuged
at 13,000 × g for 10 min at 4°C, separating the RNA in the aqueous
phase. RNA was then precipitated by 500 µl isopropanol and washed
twice with 750 µl of 75% ethanol. RNA pellets were obtained by
centrifugation for 5 min at 13,000 × g at 4°C and air-dried for 10
min. RNA was solubilized in RNase-free water and heat-inactivated
at 65°C for 10 min.
Single-strand cDNA was generated from 250 ng RNA by
First Strand cDNA Synthesis kit (cat. no. E6300; New England
Biolabs, Inc.) using random primers according to the manufacturer's
instructions. The primer and RNA mix were denatured at 70°C for 5
min. Reaction enzyme and buffer were mixed at a ratio of 1:5 and
added into each sample for cDNA synthesis. Samples were incubated
at 25°C for 5 min, followed by cDNA synthesis reaction at 42°C for
1 h and then the enzymes were inactivated at 80°C for 5 min.
Quantitative PCR analysis was performed by SYBR green qPCR Master
Mix (Roche Diagnostics). A total of 4 ng cDNA product and master
mix were added into each well of a 384-well plate. The reaction was
performed on an ABI PRISM 7900HT system (Applied Biosystems; Thermo
Fisher Scientific, Inc.) and each sample was run in triplicate. The
thermocycling conditions were as follows: 95°C for 10 min, 30
cycles at 95°C for 15 sec and 60°C for 1 min, and 72°C for 10 min.
Relative gene expression level was quantified using the
2−ΔΔCq method (55) and
normalized to the expression of the house-keeping gene
glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
miRNA extraction was performed according to the
manufacturer's instructions using the miRVana RNA isolation kit
(cat. no. AM1560, Invitrogen; Thermo Fisher Scientific, Inc.) and
quantified by a NanoDrop spectrophotometer. cDNA was synthesized
using a TaqMan microRNA reverse transcription kit (cat. no.
4366596; Thermo Fisher Scientific, Inc.) for miR-31, according to
the manufacturer's instructions. The relative expression level was
normalized to the expression of U6. Finally, qPCR analysis was
conducted by TaqMan primers (Thermo Fisher Scientific, Inc.) on an
7900HT system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The thermocycling conditions were as follows: 95°C for 10 min, 40
cycles at 95°C for 15 sec and 60°C for 1 min. Primers were designed
using the Primer Expression Software (v3.0.1; Thermo Fisher
Scientific, Inc.) and the sequences were as follows: RUNX2 forward,
5′-GAGTCCTTCTGTGGCATGCA-3′ and reverse, 5′-TGGCCTGGTGGTGTCATTAG-3′;
SATB2 forward, 5′-TCTCCCCAAACACACCATCA-3′ and reverse,
5′-GCAGCTCCTCGTCCTTATATTCA-3′; SIRT6 forward,
5′-TTTTTCTCTCGTGGTCTCACTTTGT-3′ and reverse,
5′-ACGGAGGGCAGGTGTAACC-3′; BSP forward, 5′-AAAGTGAGAACGGGGAACCTT-3′
and reverse, 5′-GATGCAAAGCCAGAATGGAT-3′; VEGF forward,
5′-CTTGCCTTGCTGCTCTAC-3′ and reverse, 5′-TGGCTTGAAGATGTACTCG-3′;
GAPDH forward, 5′-TGCACCACCAACTGCTTAGC-3′ and reverse,
5′-GGCATGGACTGTGGTCATGA-3′; miR-31 promoter A forward,
5′-TGCTATCTGCAGTACTGTGCTGAGG-3′ and reverse,
5′-AGAGAGGCTGTGGTTAGTTCCTGCT-3′; miR-31 promoter B forward,
5′-ACTGCCTTGACTTCCTGCCTTGGTG-3′ and reverse,
5′-TAGCTAGGTCTGTACCCTGTCTCAG-3′.
Western blot analysis
Cells were washed with PBS and lysed by heating in
boiling buffer (1% SDS, 10 mM Tris-HCl pH 7.4, 1 mM sodium
orthovanadate). Samples were boiled at 100°C for 10 min and
sonicated on high for 15 min at 4°C. The cell debris was removed by
13,000 × g centrifugation for 20 min at 4°C and the supernatant was
collected. The protein concentration was analyzed using the
Bradford Protein Assay (Bio-Rad Laboratories, Inc.). A total of 5
different dilutions of bovine serum albumin (BSA) (Sigma-Aldrich;
Merck KGaA) were prepared in boiling buffer to create the standard
curve. The absorbance was measured at 595 nm on spectrophotometer
SpectraMax M2 (Molecular Devices, LLC). Samples (30 µg each) were
mixed with a 6X SDS sample buffer (375 mM Tris-HCl pH 6.8, 6% SDS,
4.8% glycerol, 9% 2-mercaptoethanol, 0.03% bromophenol blue) and
separated on a 10% SDS-PAGE. The proteins were then transferred
onto a 0.45-µm nitrocellulose membrane at 100 V for 60 min. After
blocking with 5% skimmed milk in TBS for 30 min at room
temperature, the membrane was incubated with primary antibody in 5%
milk/TBS solution overnight at 4°C. The primary antibodies used
were: Anti-RUNX2 (1:200; product no. sc101145; Santa Cruz
Biotechnology, Inc.) anti-SATB2 (1:100; product no. ab51502; Abcam)
and anti-β-actin (1:1,000; product no. 3700S; Cell Signaling
Technology, Inc.).
The membrane was washed with TBST (Tris-buffered
saline with 0.1% Tween-20 detergent) for 5 min 3 times before
horseradish peroxidase (HRP)-labeled secondary antibody incubation
for 1 h at room temperature. The secondary antibodies used were as
follows: HRP-conjugated anti-mouse (1:2,000 dilution; cat. no.
sc-516102; Santa Cruz Biotechnology, Inc.); or HPR-conjugated
anti-rabbit (1:2,000 dilution; cat. no. sc-2357; Santa Cruz
Biotechnology, Inc.). The protein bands were visualized by the
chemiluminescence after incubating the membrane in an
enhanced-chemiluminescence (ECL) reagent (Thermo Fisher Scientific,
Inc.) for 2 min at room temperature and imaging films were
developed by an OPTIMAX X-Ray Film Processor (Merry X-Ray
Corporation). To control for sample loading and protein transfer,
β-actin was probed as an internal reference control. Quantification
of immunodetected protein bands was performed by ImageJ software
(V1.53e, National Institutes of Health).
Soft agar assay
Approximately 5,000 cells were mixed in a top layer
of 0.35% 2-hydroxyethylagarose (Type VII low gelling temperature;
Sigma-Aldrich; Merck KGaA)/DMEM upon a bottom layer of 0.5%
2-hydroxethylagarose/DMEM in a 6-well plate. A total of 200 cells
for each sample were seeded onto a 10 cm2 dish and
cultured for two weeks to assess plating efficiency. The colonies
in the plates were stained by 5% Giemsa solution (40% methanol/60%
glycerol) for ~4 h at room temperature (Thermo Fisher Scientific,
Inc.) and the number of colonies were determined. The cells in
agarose were humidified with 200 µl DMEM for each well twice a week
and cultured for four weeks until individual colonies were visible
under the light microscope at a magnification of ×20. Each well was
stained with 0.1% INT/BCIP solution (Roche Diagnostics) in 0.1 M
Tris/0.05 M MgCl2/0.1 M NaCl at 4°C overnight, and
images were captured by a Bio-Rad Molecular Imager Gel-Doc XR+
system and analyzed on the Image Lab software (v2.0.1 build 18;
Bio-Rad Laboratories, Inc.). The number of colonies were counted by
ImageJ software using defined particle size of 6-infinity pixel
units and circularity of 0.30–1.00.
Scratch test
Cells were plated onto gridded plates (2×2 mm grids
on the bottom) (Nalge Nunc International). Upon reaching 90–100%
confluency, cells were washed with PBS twice and a single scratch
was produced across the monolayer between the grids by a 200-µl
pipette tip held perpendicular to the plate bottom. The plates were
gently washed with PBS to remove detached cells and cells were then
maintained in serum-free medium to inhibit proliferation that might
interfere with measurement of cell migration. For the CADD522
group, cells were cultured in medium containing 50 µM CADD522
during this experiment. CADD522 is a small molecule inhibitor for
RUNX2, and it was kindly provided by Dr. Antonino Passaniti from
University of Maryland School of Medicine (Baltimore, MD, USA).
Images were captured every 24 h after the scratch using a Nikon
digital DS-Fil1-U3 camera linked with a microscope by bright field
light at a magnification of ×20 and controlled by NIS-Elements F3.2
software on a Nikon Eclipse TS100 microscope (Nikon Corporation).
The wound healing rate was estimated by calculating the percentage
of the area of the wound covered or migrated during 48 h in the
image (wound area at 0 h-wound area at 48 h)/wound area at 0 h
×100%) measured by ImageJ software (v1.53e).
Invasion assay
The Matrigel-coated invasion chambers (Corning,
Inc.) were placed in a 24-well plate and balanced with low serum
medium (0.1% FBS/DMEM) at 37°C for 3 h. The medium was carefully
removed, and the lower wells were filled with 1 ml 10% FBS/DMEM.
Cells were washed with PBS twice and 5×104 cells were
suspended in 400 µl of 0.1% FBS/DMEM and seeded into each upper
chamber. For the CADD522 group, cells were pretreated with 50 µM
CADD522 for 24 h at 37°C and the medium in the chamber contained 50
µM CADD522. After culturing for 24 h, Transwell chambers were
washed with PBS and fixed by 10% formalin for 5 min and methanol
for 20 min at room temperature. The membranes in the bottom
chambers were stained with 10% Giemsa overnight at room temperature
and washed with H2O three times on the following day.
Non-invasive cells were inside of the chamber and were gently
removed by wet cotton swabs. The images were captured with a bright
field light microscope at a magnification of ×20 controlled by
NIS-Elements F3.2 software on a Nikon Eclipse TS100 microscope.
Three fields were randomly selected for each chamber membrane and
the number of invading cells were determined by ImageJ software
(v1.53e).
Cell viability and cytotoxicity
assay
Cell viability and cytotoxicity were assessed by the
CellTiter-Fluor Cell Viability Assay kit and CytoTox-Fluor
Cytotoxicity Assay kit (cat. nos. G6080 and G9260, respectively;
both from Promega Corporation) used according to the manufacturer's
instructions. Cells were seeded onto a 96-well plate and then
treated with CADD522 at the indicated time-point and doses (25, 50,
100, 150 and 300 µg/ml for 48 h, respectively). Each treatment was
repeated 4 times. GF-AFC substrate and bis-AAF-R110 substrate were
diluted (200X) in the indicated assay buffers and mixed with
culture media at a ratio of 1:5. A total of 60 µl mixture was added
into each well. Plates were then incubated in the dark for 1 h at
room temperature. Cell viability was measured at
400Ex/505Em nm, and cell toxicity was
assessed at 485Ex/520Em nm on a fluorescence
plate.
RNA stability assay
Actinomycin D can form a stable complex with DNA and
block DNA-dependent RNA polymerase activity to inhibit new mRNA
synthesis, allowing measurements of mRNA decay (56). Briefly, approximately 1×105
cells were seeded on a 6-well plate. For the CADD522 treatment
group, cells were treated with CADD522 at 50 µg/ml for 24 h at 37°C
before collection. Cells were then treated with 10 µg/ml of
actinomycin D (Invitrogen; Thermo Fisher Scientific, Inc.) for
indicated time intervals (0, 8, 16 and 24 h) at 37°C. Cells were
then washed twice with PBS and collected by TRI-reagent and
subjected to RNA extraction, cDNA synthesis with random primers and
RT-qPCR analysis for SATB2 mRNA expression.
ChIP assay
Chromatin immunoprecipitation (ChIP) was used to
confirm the binding of RUNX2 in the promoter region of miR-31. Ni-T
cells were treated with 50 µM CADD522 for 48 h and were crosslinked
with 1% formaldehyde (Sigma-Aldrich; Merck KGaA) in DMEM for 10 min
at room temperature. The crosslink reaction was stopped by treating
cells with 250 mM glycine for 5 min and were washed twice with cold
PBS supplemented with 1 mM PMSF and protease inhibitors (1 tablet
in 10 ml buffer; cOmplete mini EDTA-free protease inhibitor
cocktail; MilliporeSigma). The ChIP Assay kit (cat. no. 17-295;
MilliporeSigma) was used for immunoprecipitation of DNA-protein
complexes and reverse crosslinked DNA. Briefly, cells were
collected and suspended in SDS lysis buffer provided in the kit and
followed by sonication at 60 kHz to shear DNA between 200 and 1,000
base pairs. The chromatin size was verified on a 2% agarose gel.
Once the sonication conditions were optimized (36 cycles at 40%
power), samples were then diluted in ChIP dilution buffer. The
chromatin complex was then immunoprecipitated with anti-RUNX2
antibody (1:50; cat. no. ab236639; Abcam) or anti-IgG (1:50; cat.
no. 12-370; Sigma-Aldrich; Merck KGaA) as a control overnight at
4°C. The following day, Protein A Agarose/Salmon Sperm DNA (50%
Slurry) was added at 4°C and incubated for 1 h. The beads were then
collected by centrifugation at 200 × g at 4°C for 1 min to remove
unbound non-specific DNA. The beads were washed in the following
order: Low salt, high salt, LiCl, and TE buffer. The RUNX2 protein
was then eluted from the antibody by elution buffer (1% SDS, 0.1M
NaHCO3) and the protein-DNA crosslinks were reversed by heating at
65°C for 4 h. Samples were then treated with 10 µl of 0.5 M
ethylenediaminetetraacetic acid (EDTA), 20 µl of 1 M Tris-HCl, 2 µl
of 10 mg/ml Proteinase K, and 5 µl of 10 mg/ml RNase A and
incubated at 45°C for 1 h to digest proteins and RNAs. DNA was then
recovered by phenol/chloroform extraction and ethanol
precipitation. To increase recovery of DNA, glycogen and sodium
acetate were added. DNA was eluted in TE buffer and the
concentration of DNA in the samples was analyzed by PicoGreen (cat.
no. P7589, Invitrogen; Thermo Fisher Scientific, Inc.). For qPCR
analysis, approximately 0.2 ng of DNA was used per well on 384-well
plates. The Ct values were determined using the percent input
method [100*2^ (Adjusted input-Ct (IP)] to calculate
enrichment.
Bioinformatics
To identify the target genes for miRNA or regulator
miRNAs for a particular gene, the potential interaction of the 3′
UTR of the genes with miRNAs was searched and confirmed by miRNA
target prediction databases TargetScanHuman v7.2 (www.targetscan.org). The promoter of miR-31 was
identified by searching the FANTOM5 data website (http://fantom.gsc.riken.jp), which is based on
prediction of the promoters of protein-coding genes, and the
specific promoter sequence upstream from the transcription start
site (TSS) of miR-31 host gene MIR31HG (RefSeq NR_027054) was
obtained at Ensembl website (http://www.ensembl.org/index.html).
Statistical analysis
Experiments were performed at least three times
unless otherwise indicated and statistical significance of the
study was assessed using the mean ± standard deviation (SD) by the
two-tailed, unpaired t-test to compare the means of two groups or a
one-way ANOVA and post-hoc Tukey's test for comparison of the means
of more than two groups using GraphPad Prism 8.0 software (GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Ni-T BEAS-2B cells exhibit enhanced
anchorage-independent growth, migration and invasion, as well as
increased SATB2 and RUNX2
Ni-induced cell transformation has been documented
in numerous cell studies (7,57–63). Ni-T
human lung epithelial cells were obtained following four weeks of
chronic exposure to 0.25 mM NiSO4 and isolated from
colonies that grew in soft agar (7).
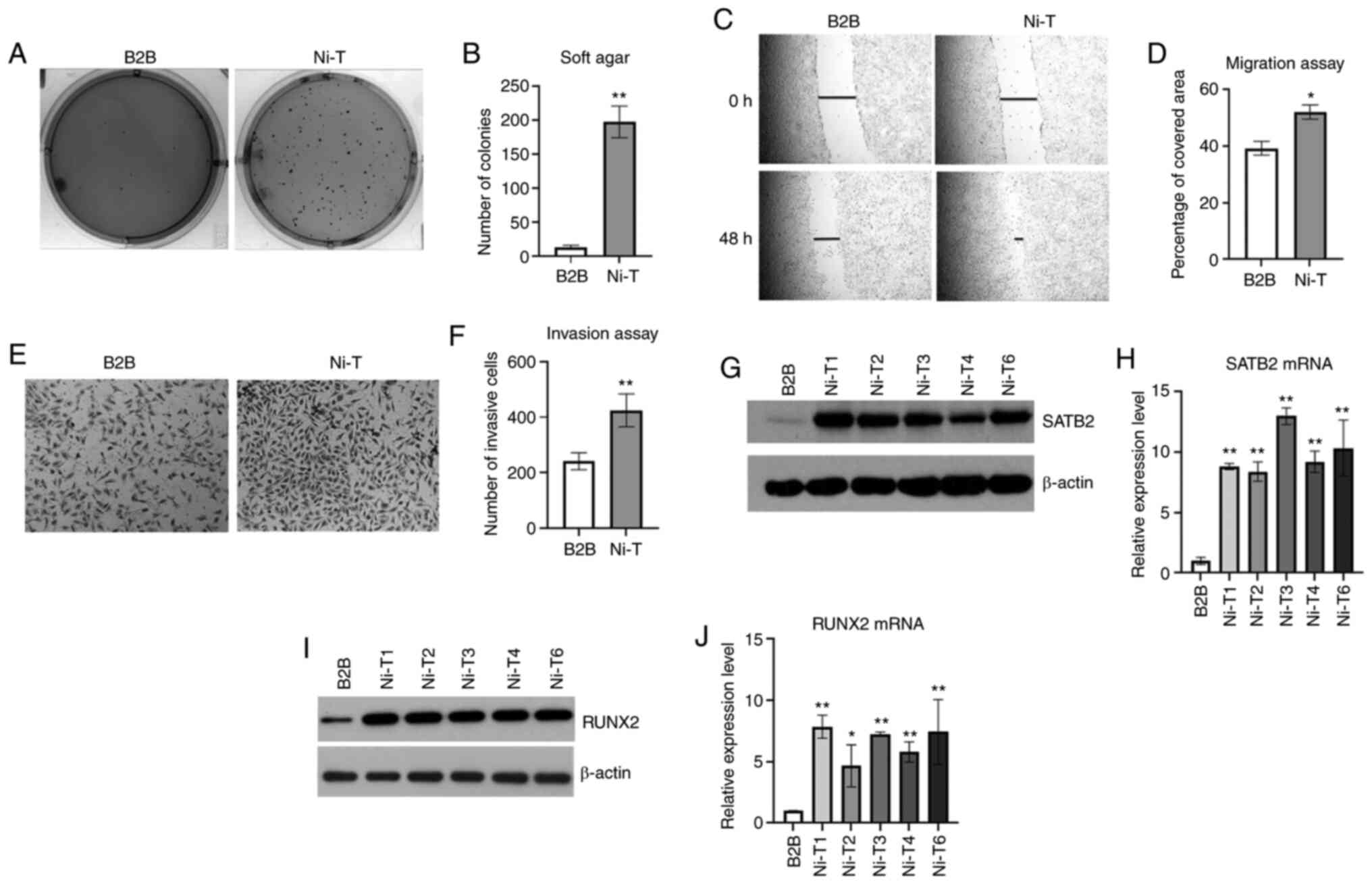
As demonstrated in Fig. 1A and B,
Ni-T cells exhibited increased colony growth in agar compared with
normal BEAS-2B cells, indicating the acquisition of
anchorage-independent growth. Cellular anchorage-independent growth
in the soft agar matrix implies malignant transformation (64). Cell migration and invasion are key
processes for cancer metastasis and are also hallmarks of
malignancy (65). Cell migration
ability was determined by the scratch test. Cell invasion was
assessed by culturing cells in invasion chambers coated with
Matrigel matrix, which only allows cells with metastatic ability to
invade and transfer (66). As
revealed in Fig. 1C-F, Ni-T cells
demonstrated higher wound-healing capacity and increased cell
invasion compared with normal BEAS-2B cells.
In previous studies, SATB2 was indicated to be an
important mediator of Ni-induced cell transformation (7,16). It was
reported that knockdown of SATB2 in Ni-T cells reduced
anchorage-independent growth, cell migration, and tumor formation
in nude mice (16). SATB2 mRNA and
protein levels were significantly increased in Ni-T cells (Fig. 1G and H). To further study the
mechanisms of SATB2 induction, RUNX2 levels were measured, as the
latter is an upstream regulator for SATB2. RUNX2 was revealed as a
master regulator for osteogenesis in mesenchymal stem cells
(67). It is not commonly expressed
in normal mammalian cells, but it has been revealed to be increased
in numerous cancers (68). RUNX2
indirectly regulates SATB2 expression by inhibiting miRNAs in both
osteogenesis and cancers. The mRNA and protein levels of RUNX2 were
examined in Ni-T cells. As revealed in Fig. 1I and J, the expression of RUNX2 was
increased as compared with control BEAS-2B cells, indicating a
possible role of RUNX2 in Ni-mediated induction of SATB2.
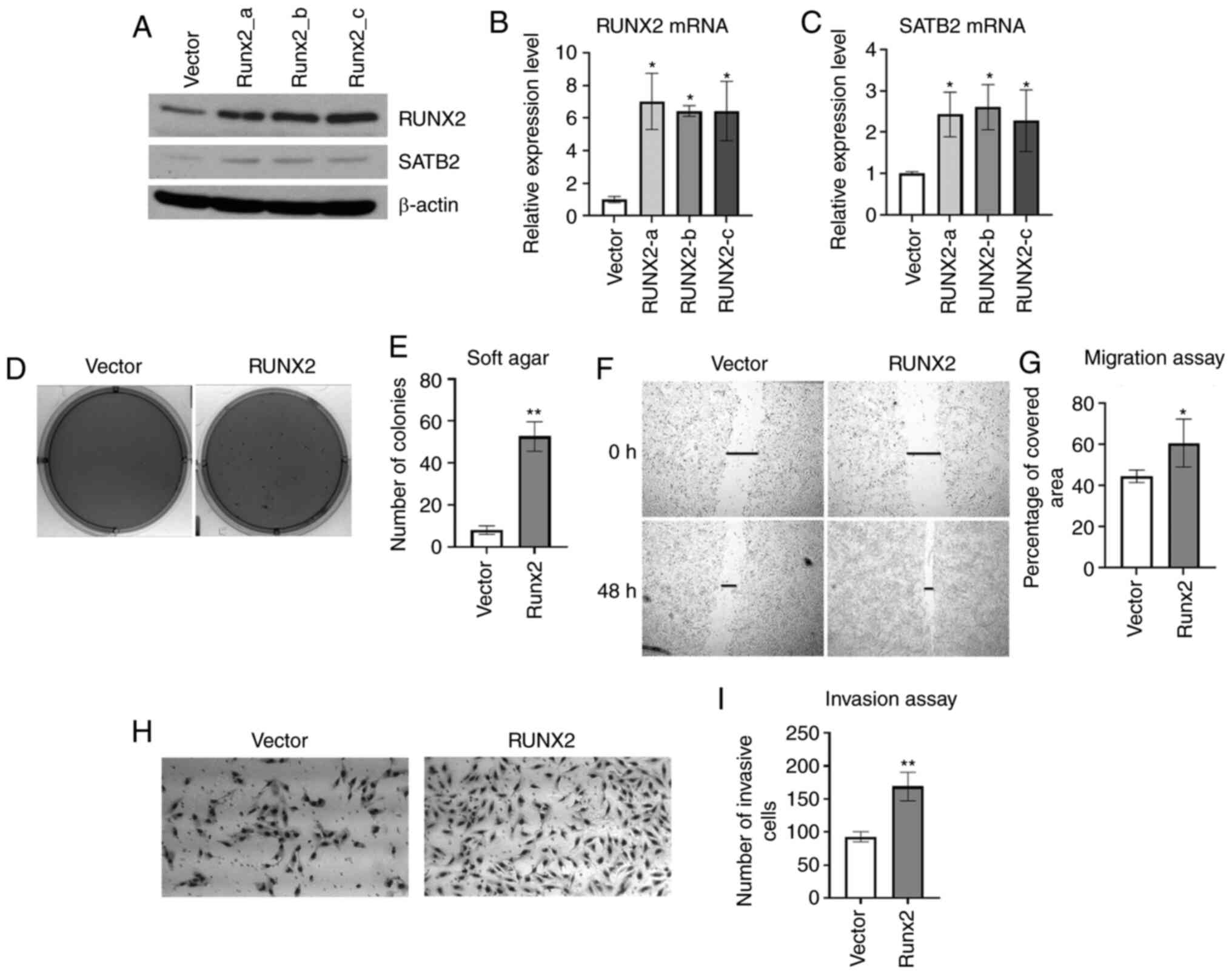
Overexpression of RUNX2 promotes
cancer hallmarks in BEAS-2B cells
RUNX2 appears to be an upstream regulator of SATB2.
To investigate whether SATB2 can be induced by RUNX2 in BEAS-2B
cells, RUNX2 was stably overexpressed in BEAS-2B cells (Fig. 2A and B). The mRNA and protein levels
of SATB2 were increased by expressing RUNX2 (Fig. 2A and C). Overexpression of SATB2 in
BEAS-2B cells increased anchorage-independent growth (16). RUNX2 is an oncogene that plays an
important role in the progression and metastasis of several human
cancers (27–29,69).
Cancer hallmarks were examined in BEAS-2B cells stably expressing
RUNX2. To assess whether this was sufficient to promote
transformation, BEAS-2B cells overexpressing RUNX2 that were
cultured in soft agar demonstrated increased colony growth compared
with an empty vector control (Fig. 2D and
E). Overexpression of RUNX2 in BEAS-2B cells also promoted
acceleration of wound healing and cell invasion compared with an
empty vector control (Fig. 2F-I).
These findings indicated that RUNX2 induced cancer hallmarks in
BEAS-2B cells by inducing SATB2.
Inhibition of RUNX2 activity in
Ni-transformed BEAS-2B cells eliminates cancer hallmarks
Previous studies have reported the role of RUNX2 in
lung cancer and inhibition of RUNX2 has been revealed to suppress
tumorigenesis in lung cancer cells (70–72). A
previous study using computer-assisted drug design (CADD) screening
detected a novel inhibitor of RUNX2 transcriptional activity termed
CADD522 (73–75). This inhibitor was used in Ni-T cells
to investigate whether inhibition of RUNX2 activity can suppress
cancer-related properties. Cell viability and cytotoxicity were
assessed by treating BEAS-2B cells for 24 h at 0–150 µM of CADD522.
There was no loss of cell viability or increased cytotoxicity at 50
µM of CADD522 for 24 h (Fig. 3A and
B). Since BEAS-2B cells tolerated a dose of 50 µM, this dose
was selected for further studies. To confirm that RUNX2 activity
can be suppressed by CADD522, Ni-T cells were treated with 50 µM
CADD522 for 24 h. RUNX2 mRNA and protein levels were not
significantly reduced in treated BEAS-2B or Ni-T cells (Fig. 3C and D). However downstream target
genes of RUNX2, VEGF and BSP were significantly inhibited by 50 µM
CADD522 treatment for 24 h in Ni-T cells (Fig. 3E-F). In addition, SIRT6 (sirtuin 6),
that was negatively regulated by RUNX2, was increased by CADD522
(Fig. 3G), indicating that CADD522
repressed RUNX2 trans-activity. SATB2 mRNA and protein were also
found reduced in Ni-T cells treated with 50 µM CADD522 (Fig. 3H and I).
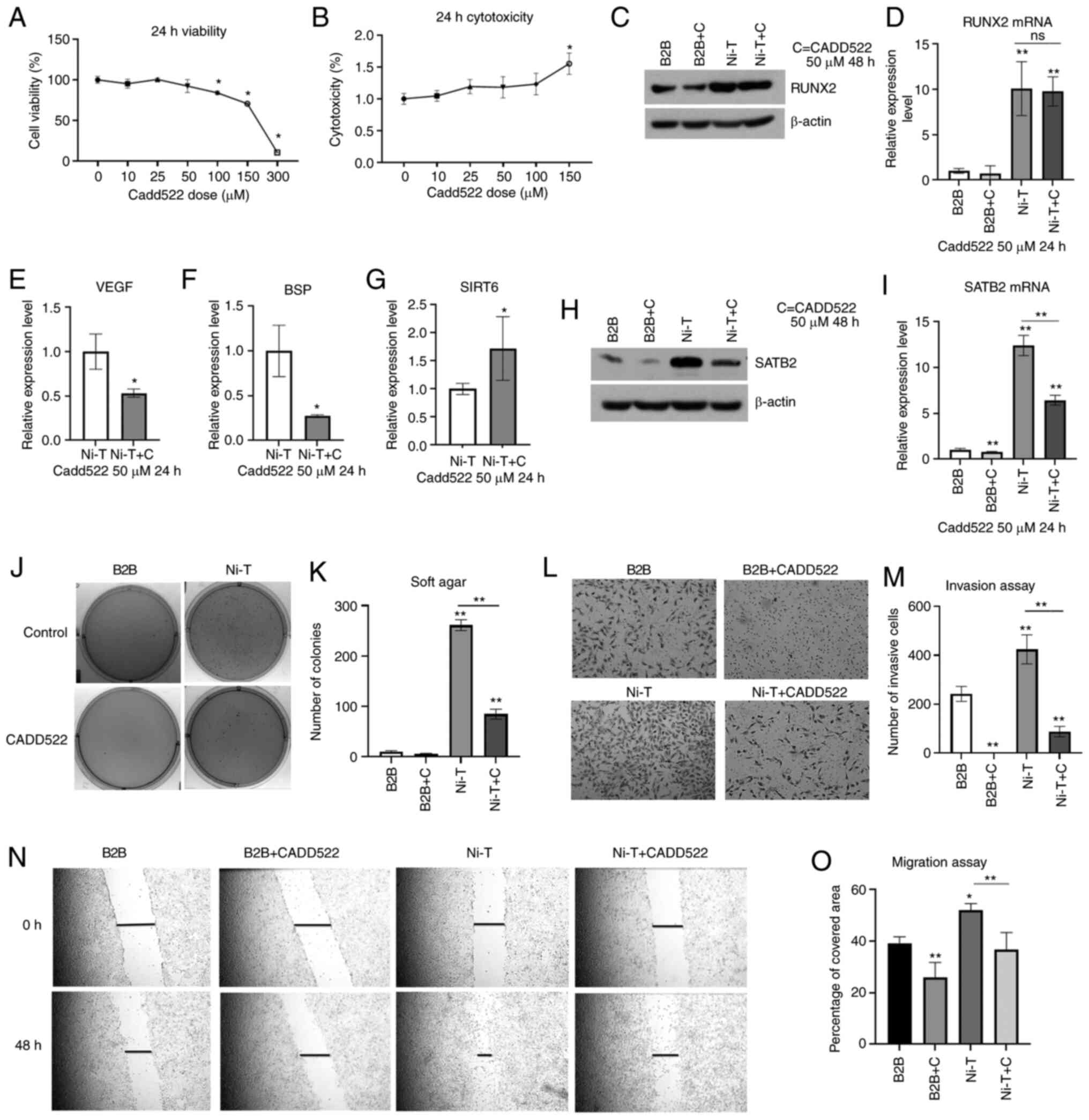 | Figure 3.Inhibition of RUNX2 activity in Ni-T
BEAS-2B cells reverses cancer hallmarks. (A and B) Cell viability
and cytotoxicity assays in BEAS-2B cells treated with CADD522 for
24 h. (C and D) The expression levels of RUNX2 protein and mRNA in
CADD522-treated BEAS-2B cells and Ni-T cells by western blotting
and RT-qPCR assays. (E-G) The expression level of RUNX2 downstream
genes, VEGF, BSP, and SIRT6 mRNA in Ni-T cells treated with 50 µM
CADD522 by RT-qPCR. (H and I) The expression level of SATB2 protein
and mRNA in BEAS-2B cells and Ni-T cells treated with 50 µM CADD522
by western blotting and RT-qPCR assays. (J and K) Soft agar assay
of BEAS-2B cells and Ni-T cells treated with CADD522 at 50 µM and
compared with that treated with control. (L and M) The invasion of
BEAS-2B cells and Ni-T cells treated with CADD522 at 50 µM was
measured by Transwell assay and images were captured at a
magnification of ×20. (N and O) Cell migration ability in BEAS-2B
cells Ni-T cells with inhibited RUNX2 activity was measured by
scratch wound healing assays, and images were captured at a
magnification of ×20. Statistical significance was calculated by
the GraphPad Prism software using ANOVA for comparisons among
groups. *P<0.05 and **P<0.01. RUNX2, runt-related
transcription factor 2; Ni-T, Ni-transformed; RT-qPCR, reverse
transcription-quantitative PCR; VEGF, vascular epithelial growth
factor; BSP, bone sialoprotein; SIRT6, sirtuin 6; SATB2, AT-rich
sequence-binding protein 2. |
The anchorage independent growth of cells was
analyzed by culturing Ni-T cells in soft agar containing 50 µM
CADD522 for 4 weeks. The colony number and size were significantly
reduced in CADD522-treated Ni-T cells compared with control
(Fig. 3J and K). The metastatic
potential of Ni-T treated with CADD522 was assessed in cell
invasion and migration assays. As revealed in Fig. 3L and M, CADD522-treated Ni-T cells
demonstrated reduced wound healing rate and invasion as compared
with the control.
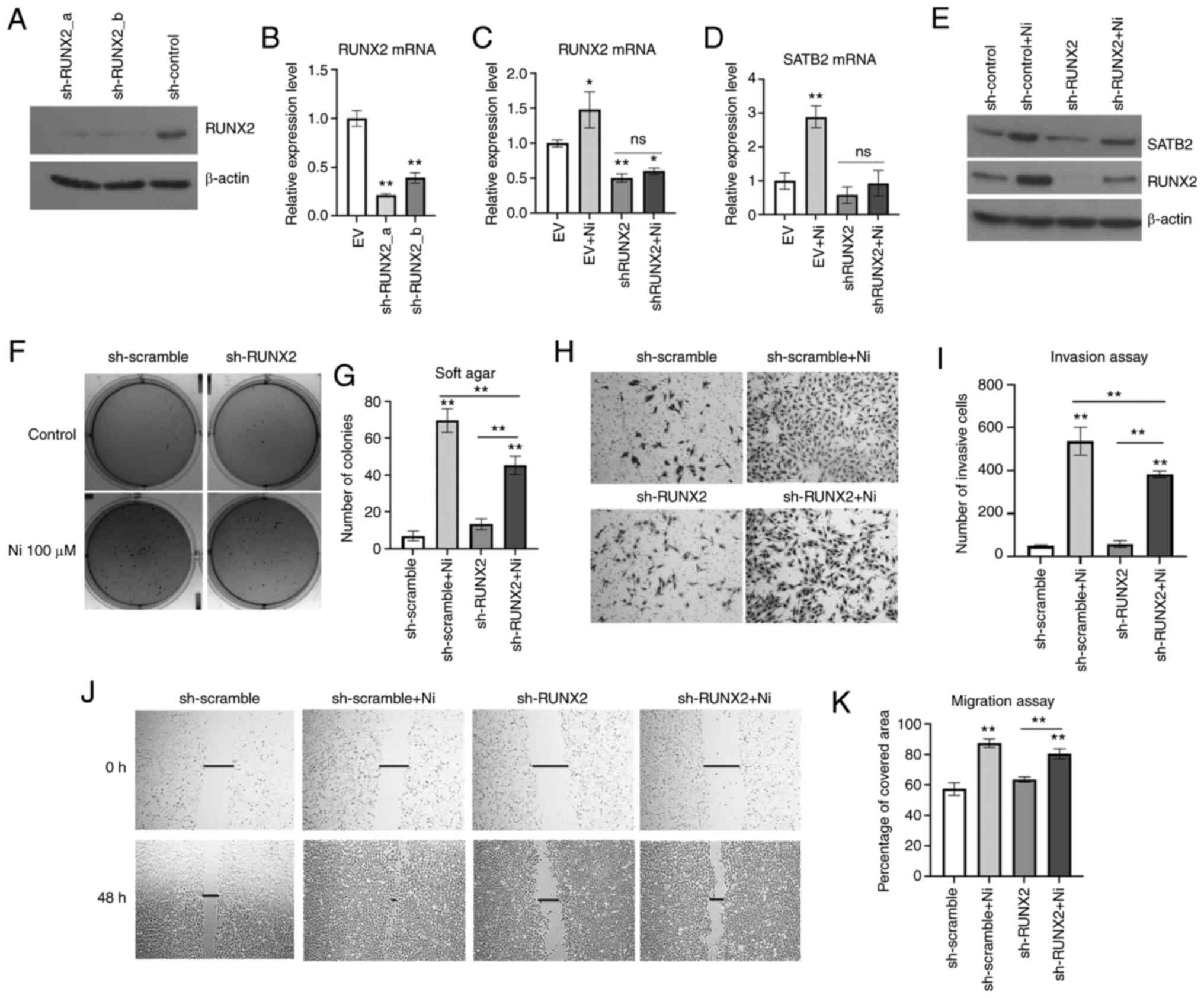
Knockdown of RUNX2 prevents Ni-induced
transformation in BEAS-2B cells
To further study the role of RUNX2 in Ni-induced
BEAS-2B transformation, RUNX2 was knocked down in normal BEAS-2B
cells by shRNA stable transfection prior to a chronic exposure to
Ni (Fig. 4A and B). NiCl2
and NiSO4 are two common soluble forms of Ni compounds
used in cell transformation studies (7,57–63). The RUNX2-knockdown cells experienced
massive cell death after treatment of 0.25 mM NiCl2 for
2 weeks. Thus, a lower dose and a longer treatment time were tried
with this compound. Cells were treated with 100 µM NiCl2
for 6 consecutive weeks to induce cell transformation. As
demonstrated in Fig. 4C-E, the
scramble control cells demonstrated significantly higher levels of
RUNX2 compared with shRUNX2-transfected cells. Chronic exposure to
Ni induced both RUNX2 and SATB2 level in control cells, while in
RUNX2-knockdown cells, Ni failed to increase the levels of RUNX2 or
SATB2 compared with controls. These results indicated that
inhibition of RUNX2 decreased the induction of SATB2 by Ni chronic
exposure.
Ni-induced cancer hallmarks were investigated in
BEAS-2B cells harboring knockdown of RUNX2. As revealed in Fig. 4F and G, Ni-treated BEAS-2B cells with
knockdown of RUNX2 demonstrated significantly reduced
anchorage-independent growth compared with the control. Although
knockdown of RUNX2 only slightly reduced cell migration of BEAS-2B
cells chronically treated with Ni (Fig.
4H and I), Ni-treated cells with knockdown of RUNX2
demonstrated a reduced invasiveness (Fig.
4J and K). These findings indicated that RUNX2 was essential in
maintaining the cancer hallmarks of Ni-T cells, which was likely
dependent on SATB2.
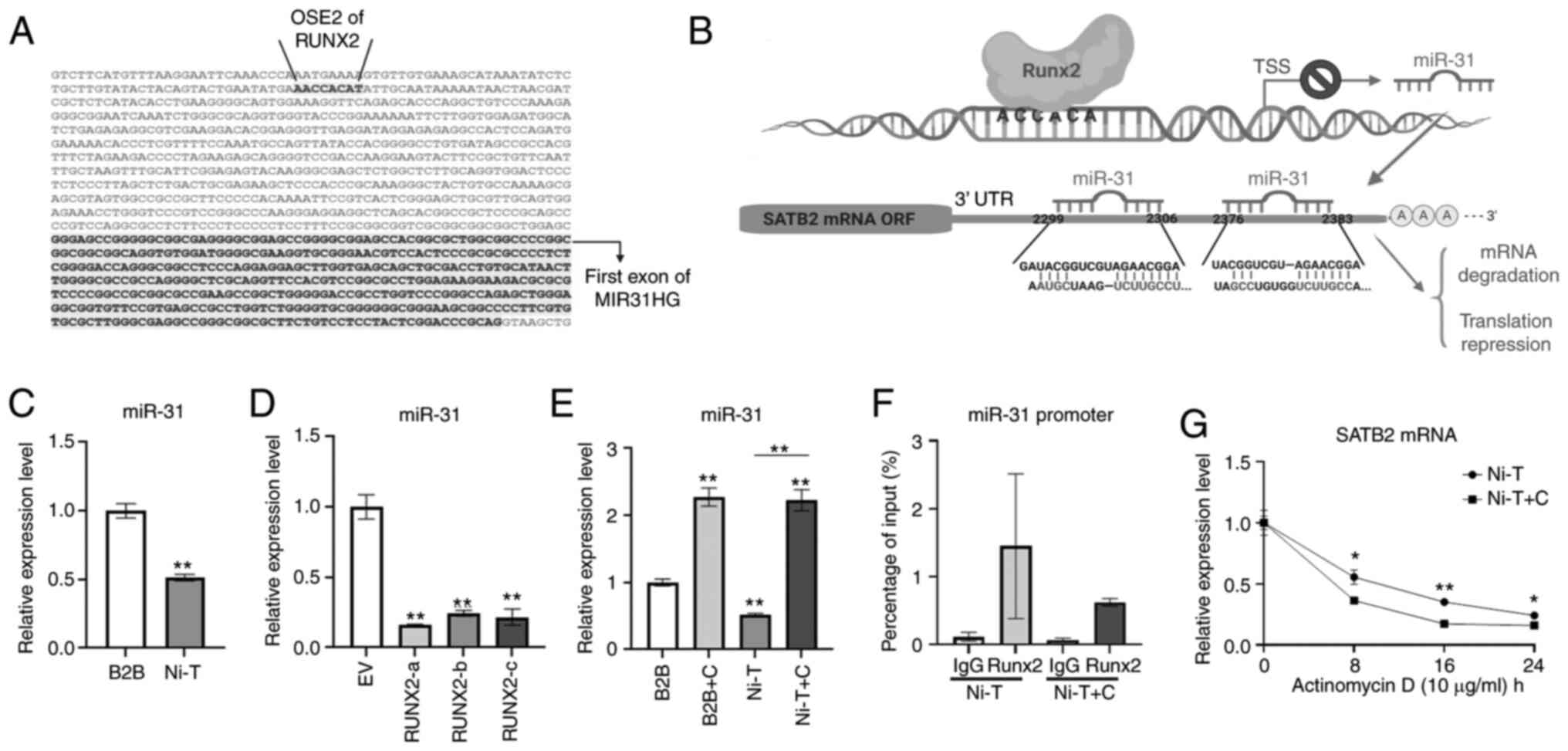
miR-31 connects RUNX2 and SATB2 in
Ni-induced cell transformation
SATB2 is maintained at low levels in most mature
organisms, and it is regulated by post-transcriptional
modifications and several miRNAs (48,76).
miR-31 is a negative regulator for SATB2 (22,38,40,41).
In a previous study, it was demonstrated that arsenic induced SATB2
level by inhibiting miR-31 in BEAS-2B cells (40). miR-31 could directly target SATB2 at
the 3′ UTR to diminish SATB2 mRNA stability and translation
(22,41). miR-31 has also been revealed to be
downstream of RUNX2 during osteogenic differentiation (22). It connects RUNX2 and SATB2 by acting
as a direct target of RUNX2 and an upregulator of SATB2 in
osteogenesis and carcinogenesis. A search of the promoter of miR-31
on Ensembl genome browser revealed a core binding sequence for
RUNX2 upstream of TSS of miR-31 (Fig.
5A). An illustration of RUNX2 binding at the miR-31 promoter
region and inhibiting its transcription is demonstrated in Fig. 5B. Furthermore, miR-31 complementary
base pairing was revealed with the sequence on SATB2 mRNA 3′ UTR
region to mediate its mRNA degradation and translation repression
and two target sites on 3′ UTR of SATB2 mRNA were identified by
TargetScan.
The expression level of miR-31 in Ni-T cells was
reduced compared with normal BEAS-2B cells (Fig. 5C). To confirm whether miR-31 was
regulated by RUNX2, BEAS-2B cells overexpressing RUNX2 and
CADD522-treated Ni-T cells were subjected to miRNA extraction and
qPCR analysis for the expression level of miR-31. Overexpression of
RUNX2 in BEAS-2B cells reduced the expression level miR-31 compared
with the vector control (Fig. 5D),
while increased miR-31 expression levels were observed in Ni-T
cells and BEAS-2B cells with inhibited RUNX2 activity (Fig. 5E). The physical binding of RUNX2 to
the core binding site on miR-31 promoter was demonstrated by Deng
et al (22). To further
investigate the mechanism by which CADD522 induced the expression
level of miR-31, the binding of RUNX2 at the core binding site of
miR-31 promotor region was examined by ChIP in Ni-T cells. A primer
that spans the binding site in the promoter of miR-31 was used for
qPCR analysis and another primer spanning an unrelated promoter
region was used as a negative control. The binding of RUNX2 to
miR-31 promoter was confirmed. Upon treatment with CADD522, the
binding of RUNX2 was slightly reduced as compared with control
(Fig. 5F), indicating that CADD522
alleviated the inhibitory effect by RUNX2 on miR-31 promoter and
thereby increased the expression level of miR-31.
It is known that miRNAs target downstream mRNAs by
promoting mRNA degradation and reducing translation (77). Since CADD52 induced expression of
miR-31, an inhibition of DNA transcription by actinomycin D in
CADD522-treated Ni-T cells was conducted to assess the stability of
SATB2 mRNA. CADD522-treated Ni-T cells had less SATB2 mRNA
stability compared with the control (Fig.
5G), indicating that increased SATB2 mRNA degradation was the
operative event. These findings indicated that RUNX2 increased
SATB2 by suppressing the expression of miR-31, since RUNX2 directly
targeted miR-31 promoter to negatively regulate its expression, and
miR-31 was hypothesized to contribute to decreased stability of
SATB2 mRNA in Ni-T cells.
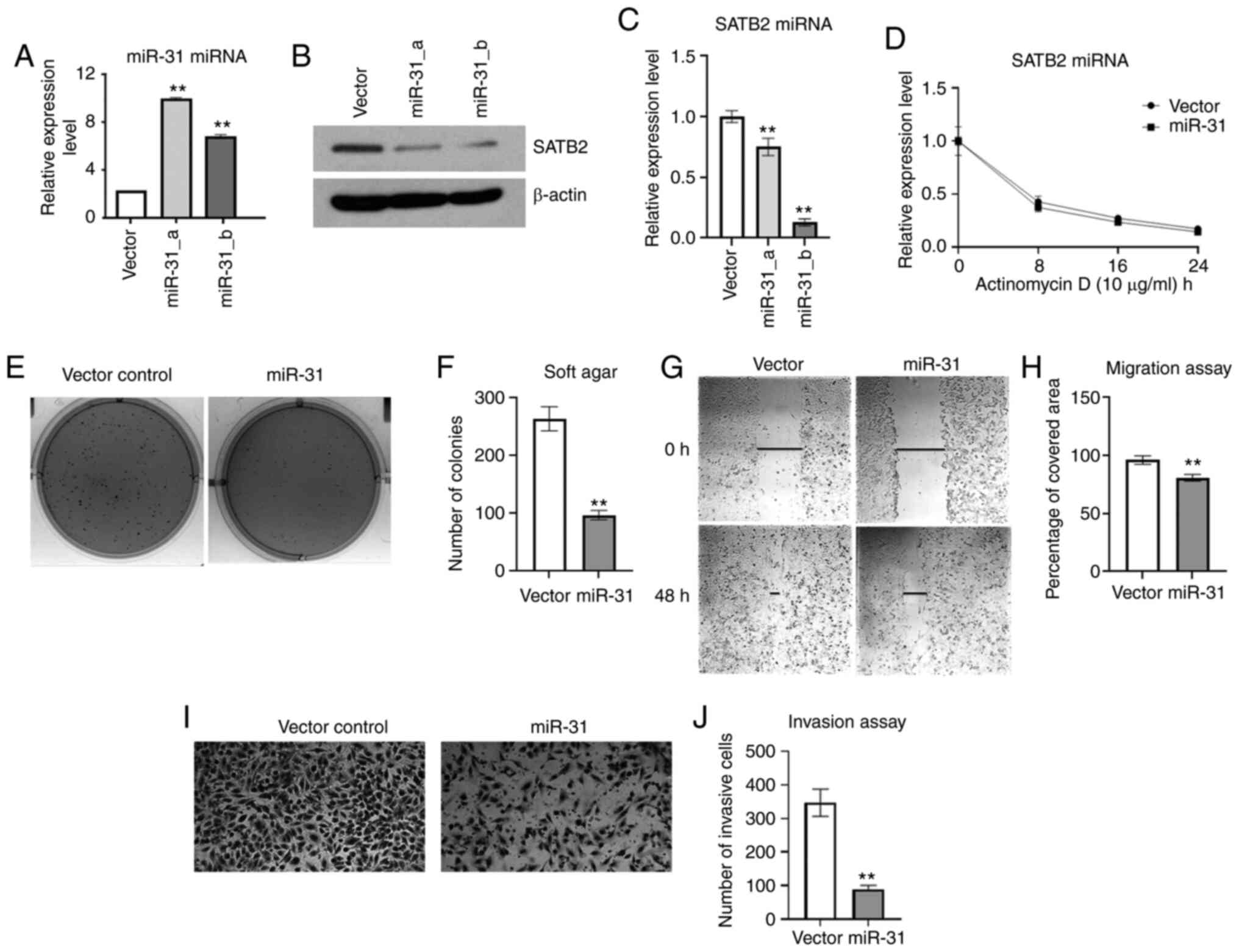
Overexpression of miR-31 represses
Ni-induced cancer hallmarks
miRNAs target downstream mRNAs by base-paring with a
complementary sequence on their 3′ UTR, while the levels of target
mRNAs are not necessarily affected by miRNAs. Genes not affected by
the binding of targeting miRNAs are termed tuning genes, and those
that can be turned off by the binding of miRNAs are termed switch
genes (44,78). To investigate whether miR-31 regulates
the level of SATB2 in Ni-T cells, miR-31 was ectopically expressed
(Fig. 6A). SATB2 protein and mRNA
expression levels were measured by western blotting and qPCR. As
revealed in Fig. 6B and C,
ectopically expressed miR-31 in Ni-T cells suppressed the protein
and mRNA levels of SATB2. However, unlike CADD522 which promoted
SATB2 mRNA degradation, the stability of SATB2 mRNA was not
significantly impaired in miR-31 overexpressed cells compared with
vector control (Fig. 6D). This may be
due to suppressed transcription of miR-31 under the treatment of
actinomycin D, failing to show the direct link between miR-31 and
SATB2.
Since exogenous expression of miR-31 in Ni-T cells
reduced SATB2 levels, and knockdown of SATB2 was revealed to reduce
cell transformation (16), it was
hypothesized that cell transformation and metastasis would also be
inhibited in Ni-T cells stably expressing exogenous miR-31. The
colony number and size in soft agar were significantly decreased in
miR-31-overexpressing cells compared with the vector control as
demonstrated in Fig. 6E and F. To
investigate whether the expression of miR-31 in Ni-T cells also
suppressed cellular metastasis, both migration and invasion were
analyzed using the scratch test and invasion assay, respectively.
As demonstrated in Fig. 6G and H, the
wound healing rate was reduced in Ni-T cells ectopically expressing
miR-31 compared with the control, indicating a decreased cell
migration ability. The number of invasive cells was also
significantly reduced in miR-31-overexpressed cells (Fig. 6I and J). Collectively, overexpression
of miR-31 reduced cell transformation and metastatic potential in
Ni-T cells, indicating that miR-31 could be a critical regulator in
Ni-induced cell transformation by its effects on SATB2
expression.
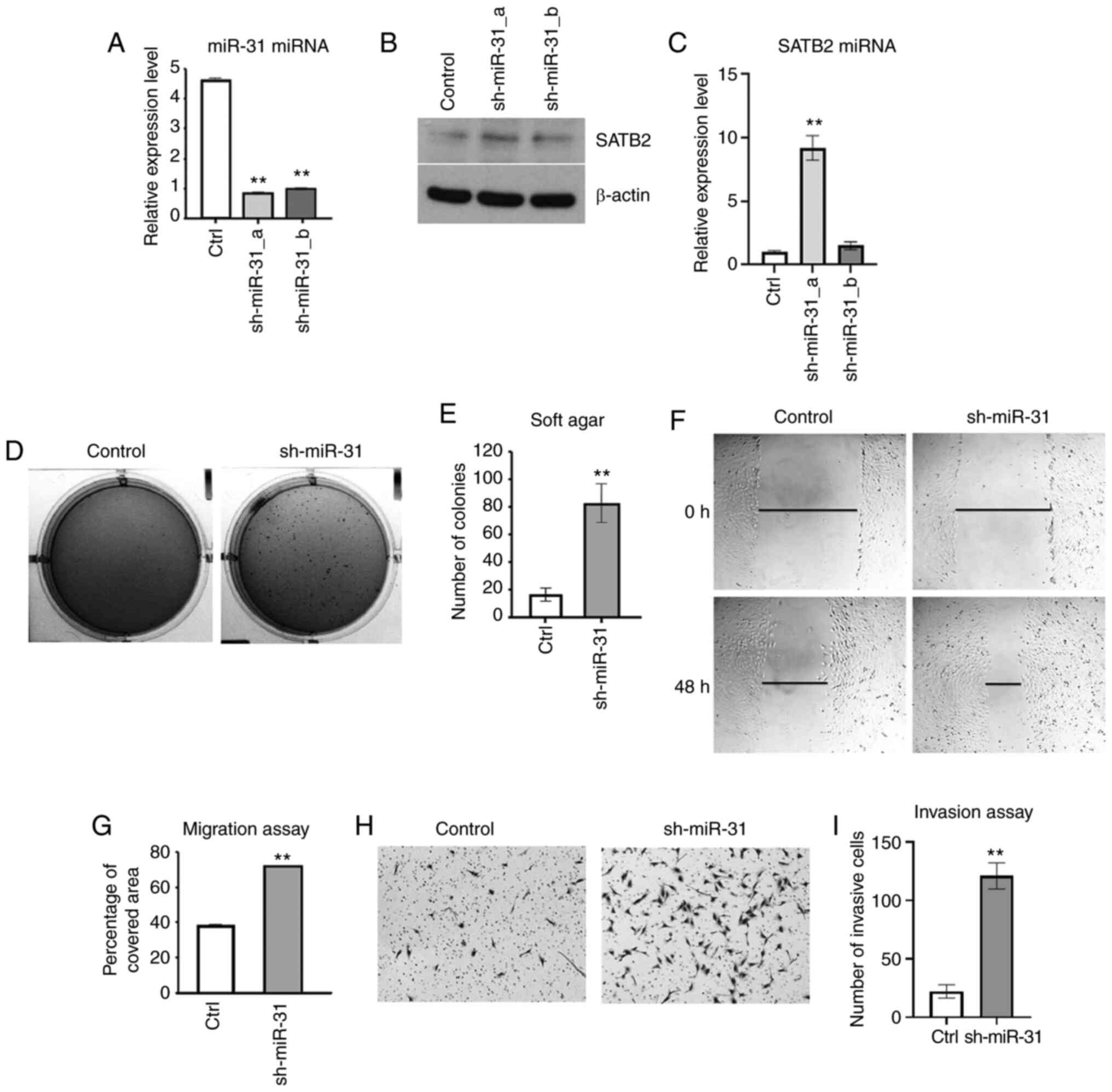
Inhibition of miR-31 increases cancer
hallmarks in BEAS-2B cells
To further examine the role of miR-31 in cell
transformation, miR-31 was knocked down in BEAS-2B cells by shRNA
(Fig. 7A). The mRNA and protein
levels of SATB2 were increased in one of the clones of
miR-31-knockdown BEAS-2B cells (Fig. 7B
and C). Inhibition of miR-31 level in BEAS-2B cells
significantly promoted anchorage-independent growth as compared
with the scramble shRNA-transfected controls (Fig. 7D and E). The cell metastasis was
investigated in BEAS-2B cells with knockdown of miR-31. Cells with
reduced expression of miR-31 exhibited increased migration compared
with the scramble shRNA control (Fig. 7F
and G). Furthermore, knockdown of miR-31 effectively increased
the invasiveness as compared with the control (Fig. 7H and I). Collectively, these results
indicated that miR-31 played an important role in inhibiting cell
transformation and metastasis in BEAS-2B cells.
Discussion
The present study proposed a signaling pathway,
RUNX2/miR-31/SATB2, that drives Ni-induced BEAS-2B cell
transformation. Expression of SATB2 and RUNX2 were both increased
in Ni-transformed BEAS-2B cells (Ni-T) and miR-31 levels were
reduced. Ectopic expression of RUNX2 in BEAS-2B cells promoted
cancer hallmarks, while an inhibition of RUNX2 in Ni-T cells or
knockdown of RUNX2 in BEAS-2B cells prior to Ni treatment
significantly prevented the induction of SATB2 as well as
carcinogenesis. These results indicated an important role of RUNX2
in Ni-induced cell transformation, which is likely mediated by
inducing the expression of SATB2. miR-31 was demonstrated to be an
upstream negative regulator of SATB2 in arsenic-transformed BEAS-2B
cells (40). miR-31 is targeted by
RUNX2 at the promoter and its expression level was revealed to be
regulated by RUNX2 in BEAS-2B cells. Knockdown of miR-31 in normal
BEAS-2B cells increased SATB2 level and cancer hallmarks, while
overexpression of miR-31 in Ni-T cells reversed transformation by
decreasing SATB2. Different doses and treatment times for soluble
Ni-induced cell transformation have been reported in numerous
studies (7,57–63),
however a dose-response effect of soluble Ni on cell growth,
migration and invasion has not been well explored in the same
detail and could be studied in the future. Furthermore, a
dose-response relationship between soluble Ni exposure and
expression levels of these components will be explored in future
studies. Collectively, these results indicated that long-term
soluble NiSO4 exposure induced SATB2 expression through
the induction of RUNX2 and inhibition of miR-31 which is a
mechanism for Ni-induced BEAS-2B cell transformation.
Ni-induced cell transformation occurs through
several mechanisms, including the hypoxia-inducible signaling
pathway, epigenetic changes including DNA methylation, histone
acetylation and miRNA regulation, tumor suppressor gene loss and
oncogene activation (79).
Hypoxia-inducible factor (HIF) is a heterodimeric transcriptional
factor that consists of 2 subunits, HIF-1α and HIF-1β, both of
which are required for HIF-1 to function; HIF-1β is integral in
HIF-1 heterodimer formation, while HIF-1α is the key regulatory
subunit and is responsible for HIF-1 transcriptional activity
(80). Previous studies demonstrated
that Ni could replace iron in the HIF prolyl hydroxylases and thus
inhibit the association of HIF-1α with Von-Hippel-Lindau (VHL) E3
ubiquitin ligase, which in turn stabilizes HIF protein (81–83). RUNX2
was also revealed to cause accumulation of HIF-1α protein in
chondrocytes since it stabilizes HIF-1α by competition with pVHL at
the oxygen-dependent degradation domain (ODDD) of HIF-1α and
prevents HIF-1α ubiquitination (84).
Another study confirmed the physical binding of the Runt domain of
RUNX2 with HIF-1α and indicated that RUNX2 and HIF-1α interaction
induced the expression of the downstream gene vascular epithelial
growth factor (VAGF) (85). Recently
SATB2 was also revealed to regulate HIF-1a expression under hypoxia
signaling and mediate cell stemness, proliferation and cell
survival in oral squamous cell carcinoma (86). These findings indicated that both
RUNX2 and SATB2 may also be involved in the mechanism of the
Ni-induced hypoxia signaling pathway.
Carcinogenic mechanisms induced by Ni can ultimately
lead to the activation of proto-oncogenes or inactivation of tumor
suppressor genes such as p53 (87–89). SATB2
was reported to be an oncogene in numerous cancers (48). SATB2 was indicated as a diagnostic
marker for CRC since SATB2 stained positively in 71–97% of primary
and metastatic CRC biopsies (90). A
reduced expression level of SATB2 in colon cancer was associated
with poor prognosis, while increased SATB2 expression enhanced
therapeutic sensitivity (90–94). A previous study demonstrated that
SATB2 induction and subsequent transformation of colon epithelial
cells (CRL-1831) was mediated through Wnt/β-catenin/T-cell factor
(TCF)/lymphoid enhancer factor (LEF) pathway (13). Numerous downstream genes of the
pathway have been associated with carcinogenesis, including c-Myc
and VEGF (95). In fact, these genes
are also downstream of RUNX2 (96,97). RUNX2
has been linked with SATB2 by numerous osteo-specific markers and
miRNAs during bone development and carcinogenesis (19,20,22–25,39).
RUNX2 was indicated to be a novel prognostic marker
in non-small cell lung cancer (NSCLC) since its expression was
revealed to be tightly related with tumor size, stage, and lymph
node metastasis (72). The present
study focused on the role of RUNX2 in Ni-T cells and confirmed that
RUNX2 is an upstream regulator of SATB2, and it is important in
mediating cell transformation induced by Ni. Inhibition of RUNX2
was achieved by two methods in the present study, a small molecule
inhibitor and gene knockdown. The advantage of small molecule drugs
is functioning rapidly, continuously, and often reversibly across
cell types and species (98) From a
clinical point of view, it has the potential for therapeutic
development, and the results of the present study could possibly
add meaning to it. A concern in the use of small molecule
inhibitors of RUNX2 would be its off-target effects. A previous
study has demonstrated that CADD522 is specific for the RUNX family
and highly specific in targeting RUNX2 transcriptional activity
(75). Stable knockdown of RUNX2 by
shRNA in normal BEAS-2B cells was carried out to provide further
evidence that RUNX2 is critical in maintaining cancer hallmarks
during Ni-induced transformation. Although SATB2 is considered a
tumor suppressor for NSCLC (99),
SATB2 levels increased in Ni-T BEAS-2B cells. In addition,
inhibition of the expression level of SATB2 in Ni-T cells reduced
cell proliferation and anchorage-independent growth (100). SATB2 was also induced by arsenic in
BEAS-2B transformed cells, and this induction was demonstrated to
be achieved by downregulated miR-31, which was required to induce
malignant transformation of BEAS-2B cells (40).
RUNX2 acts as an upstream regulator of miR-31 by
directly targeting its promoter region in bone marrow mesenchymal
stem cells (22). The expression
level of miR-31 was negatively regulated by RUNX2 following
Ni-transformation, and the binding of RUNX2 at miR-31 promoter was
demonstrated. miR-31 is a negative regulator of SATB2 since
overexpression of miR-31 suppressed SATB2 level in Ni-T cells,
while inhibition of miR-31 restored SATB2 to normal low levels.
miRNAs function by binding to the 3′ UTR of target mRNAs and
subsequently mediate mRNA de-capping, triggering degradation, as
well as reducing mRNA translation (101). SATB2 mRNA was directly targeted by
miR-31 in CRC and breast cancer cells (38,53). In
the present study, it was observed that miR-31 affected SATB2
levels, and further studies on the mechanism by which miR-31
regulates SATB2 in this pathway are required.
Two target sequences that miR-31 bind to the 3′ UTR
of SATB2 mRNA were identified. Inhibition of RUNX2 transcriptional
activity by CADD522 increased miR-31 levels, reducing the half-life
of SATB2 mRNA, indicating that miR-31 promoted SATB2 mRNA
degradation. miR-31 is a highly evolutionarily conserved miRNA and
its expression was revealed to be disrupted in the progression of
numerous cancers and diseases such as psoriasis and systemic lupus
erythematosus (102). The
transcriptional regulation of miR-31 is suppressed in breast,
prostate, liver, leukemia, and melanoma cancers, and this is partly
mediated by hypermethylation of a CpG island in miR-31 promoter
(103–107). In fact, DNA methylation is the most
prevalent epigenetic event in Ni-induced lung cancer (81,108–112).
Ni elicited global hypermethylation and suppressed key tumor
suppressor genes to induce senescence as part of its carcinogenic
pathway (113). Ni was also revealed
to alter the expression levels of numerous ncRNAs by regulating the
activity of DNA methyltransferases (DMNTs) (47). The level of miR-31 was reduced in Ni-T
cells, allowing SATB2 protein to be expressed. In addition,
RUNX2/miR-31 may not be the only upstream signaling for
SATB2-mediated carcinogenesis induced by Ni since RUNX2 and SATB2
are regulated by other miRNAs such as miR-23a/27a/24-2 in cancer
cells (23).
The present study described a signaling pathway
leading to increased SATB2 in Ni-induced BEAS-2B transformation. It
demonstrated that induced expression of RUNX2 by Ni-suppressed
miR-31 expression by silencing its promoter, thereby allowing SATB2
mRNA translation, and increasing the protein level of SATB2,
leading to cell transformation. Since SATB2 is a common gene
significantly induced by different heavy metals in BEAS-2B cells
(7), it would be of interest to
investigate whether this is a shared pathway for other
metal-induced lung carcinogenesis in future studies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Institutes of Health grants (grant nos. ES000260, ES023174,
ES029359, ES030583 CA229234 and ES026138).
Availability of data and materials
All datasets generated or analyzed during this study
are available from the corresponding author upon reasonable
request.
Author's contributions
MC, YZ and AJ contributed to the conception and
design of the study. AJ performed preliminary experiments to
establish the methodology to assess RUNX2 and miR-31 expression in
cells. QYC generated miR-31 overexpression and knockdown clones,
confirmed that these clones had authentic overexpression and
knockdown and conducted cell transformation assays for these
clones. YZ performed all of the remaining experiments that are
reported in the study. YZ, QYC and NR analyzed the data and
produced the final images of the study using the software mentioned
in the study. HS and NR provided technical support and data
interpretation for the study. YZ prepared the first draft of the
manuscript. YZ and MC revised the manuscript according to the
comments from the reviewers and editors. HS also assisted in
confirming and providing detailed methods of the study. MC
supervised the preparation of the manuscript and was the PI on NIH
grants that financially supported this publication. All authors
have read and approved the submitted manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huvinen M and Pukkala E: Cancer incidence
among Finnish ferrochromium and stainless steel production workers
in 1967–2011: A cohort study. BMJ Open. 3:e0038192013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grimsrud TK and Andersen A: Evidence of
carcinogenicity in humans of water-soluble nickel salts. J Occup
Med Toxicol. 5:72010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Andersen A, Berge SR, Engeland A and
Norseth T: Exposure to nickel compounds and smoking in relation to
incidence of lung and nasal cancer among nickel refinery workers.
Occup Environ Med. 53:708–713. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seilkop SK and Oller AR: Respiratory
cancer risks associated with low-level nickel exposure: An
integrated assessment based on animal, epidemiological, and
mechanistic data. Regul Toxicol Pharmacol. 37:173–190. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anttila A, Pukkala E, Aitio A, Rantanen T
and Karjalainen S: Update of cancer incidence among workers at a
copper/nickel smelter and nickel refinery. Int Arch Occup Environ
Health. 71:245–250. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moulin JJ, Clavel T, Roy D, Dananche B,
Marquis N, Fevotte J and Fontana JM: Risk of lung cancer in workers
producing stainless steel and metallic alloys. Int Arch Occup
Environ Health. 73:171–180. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Clancy HA, Sun H, Passantino L, Kluz T,
Munoz A, Zavadil J and Costa M: Gene expression changes in human
lung cells exposed to arsenic, chromium, nickel or vanadium
indicate the first steps in cancer. Metallomics. 4:784–793. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Savarese F, Davila A, Nechanitzky R, De La
Rosa-Velazquez I, Pereira CF, Engelke R, Takahashi K, Jenuwein T,
Kohwi-Shigematsu T, Fisher AG and Grosschedl R: Satb1 and Satb2
regulate embryonic stem cell differentiation and Nanog expression.
Genes Dev. 23:2625–2638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Magnusson K, de Wit M, Brennan DJ, Johnson
LB, McGee SF, Lundberg E, Naicker K, Klinger R, Kampf C, Asplund A,
et al: SATB2 in combination with cytokeratin 20 identifies over 95%
of all colorectal carcinomas. Am J Surg Pathol. 35:937–948. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang G, Cui Y, Yu X, Wu Z, Ding G and Cao
L: miR-211 suppresses hepatocellular carcinoma by downregulating
SATB2. Oncotarget. 6:9457–9466. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cartularo L, Kluz T, Cohen L, Shen SS and
Costa M: Molecular mechanisms of malignant transformation by low
dose cadmium in normal human bronchial epithelial cells. PLoS One.
11:e01550022016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fukuhara M, Agnarsdottir M, Edqvist PH,
Coter A and Ponten F: SATB2 is expressed in Merkel cell carcinoma.
Arch Dermatol Res. 308:449–454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu W, Ma Y, Shankar S and Srivastava RK:
SATB2/β-catenin/TCF-LEF pathway induces cellular transformation by
generating cancer stem cells in colorectal cancer. Sci Rep.
7:109392017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bae T, Rho K, Choi JW, Horimoto K, Kim W
and Kim S: Identification of upstream regulators for prognostic
expression signature genes in colorectal cancer. BMC Syst Biol.
7:862013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu W, Ma Y, Shankar S and Srivastava RK:
Role of SATB2 in human pancreatic cancer: Implications in
transformation and a promising biomarker. Oncotarget.
7:57783–57797. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu F, Jordan A, Kluz T, Shen S, Sun H,
Cartularo LA and Costa M: SATB2 expression increased
anchorage-independent growth and cell migration in human bronchial
epithelial cells. Toxicol Appl Pharmacol. 293:30–36. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao X, Qu Z, Tickner J, Xu J, Dai K and
Zhang X: The role of SATB2 in skeletogenesis and human disease.
Cytokine Growth Factor Rev. 25:35–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Komori T: Regulation of skeletal
development by the Runx family of transcription factors. J Cell
Biochem. 95:445–453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakashima K, Zhou X, Kunkel G, Zhang Z,
Deng JM, Behringer RR and de Crombrugghe B: The novel zinc
finger-containing transcription factor osterix is required for
osteoblast differentiation and bone formation. Cell. 108:17–29.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang W, Li Y, Osimiri L and Zhang C:
Osteoblast-specific transcription factor Osterix (Osx) is an
upstream regulator of Satb2 during bone formation. J Biol Chem.
286:32995–33002. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu L, Xu Y, Qu H, Yu Y, Li W, Zhao Y and
Qiu G: Decrease of miR-31 induced by TNF-α inhibitor activates
SATB2/RUNX2 pathway and promotes osteogenic differentiation in
ethanol-induced osteonecrosis. J Cell Physiol. 234:4314–4326. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deng Y, Wu S, Zhou H, Bi X, Wang Y, Hu Y,
Gu P and Fan X: Effects of a miR-31, Runx2, and Satb2 regulatory
loop on the osteogenic differentiation of bone mesenchymal stem
cells. Stem Cells Dev. 22:2278–2286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hassan MQ, Gordon JA, Beloti MM, Croce CM,
van Wijnen AJ, Stein JL, Stein GS and Lian JB: A network connecting
Runx2, SATB2, and the miR-23a~27a~24-2 cluster regulates the
osteoblast differentiation program. Proc Natl Acad Sci USA.
107:19879–19884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dobreva G, Chahrour M, Dautzenberg M,
Chirivella L, Kanzler B, Farinas I, Karsenty G and Grosschedl R:
SATB2 is a multifunctional determinant of craniofacial patterning
and osteoblast differentiation. Cell. 125:971–986. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dowrey T, Schwager EE, Duong J, Merkuri F,
Zarate YA and Fish JL: Satb2 regulates proliferation and nuclear
integrity of pre-osteoblasts. Bone. 127:488–498. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang J, Tu Q, Grosschedl R, Kim MS,
Griffin T, Drissi H, Yang P and Chen J: Roles of SATB2 in
osteogenic differentiation and bone regeneration. Tissue Eng Part
A. 17:1767–1776. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kayed H, Jiang X, Keleg S, Jesnowski R,
Giese T, Berger MR, Esposito I, Lohr M, Friess H and Kleeff J:
Regulation and functional role of the Runt-related transcription
factor-2 in pancreatic cancer. Br J Cancer. 97:1106–1115. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pratap J, Lian JB, Javed A, Barnes GL, van
Wijnen AJ, Stein JL and Stein GS: Regulatory roles of Runx2 in
metastatic tumor and cancer cell interactions with bone. Cancer
Metastasis Rev. 25:589–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Niu DF, Kondo T, Nakazawa T, Oishi N,
Kawasaki T, Mochizuki K, Yamane T and Katoh R: Transcription factor
Runx2 is a regulator of epithelial-mesenchymal transition and
invasion in thyroid carcinomas. Lab Invest. 92:1181–1190. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thomas DM, Johnson SA, Sims NA, Trivett
MK, Slavin JL, Rubin BP, Waring P, McArthur GA, Walkley CR,
Holloway AJ, et al: Terminal osteoblast differentiation, mediated
by runx2 and p27KIP1, is disrupted in osteosarcoma. J Cell Biol.
167:925–934. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Akech J, Wixted JJ, Bedard K, van der Deen
M, Hussain S, Guise TA, van Wijnen AJ, Stein JL, Languino LR,
Altieri DC, et al: Runx2 association with progression of prostate
cancer in patients: Mechanisms mediating bone osteolysis and
osteoblastic metastatic lesions. Oncogene. 29:811–821. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun X, Wei L, Chen Q and Terek RM: HDAC4
represses vascular endothelial growth factor expression in
chondrosarcoma by modulating RUNX2 activity. J Biol Chem.
284:21881–21890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Papachristou DJ, Papachristou GI,
Papaefthimiou OA, Agnantis NJ, Basdra EK and Papavassiliou AG: The
MAPK-AP-1/-Runx2 signalling axes are implicated in chondrosarcoma
pathobiology either independently or via up-regulation of VEGF.
Histopathology. 47:565–574. 2005.PubMed/NCBI
|
|
34
|
Inman CK and Shore P: The osteoblast
transcription factor Runx2 is expressed in mammary epithelial cells
and mediates osteopontin expression. J Biol Chem. 278:48684–48689.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baniwal SK, Khalid O, Gabet Y, Shah RR,
Purcell DJ, Mav D, Kohn-Gabet AE, Shi Y, Coetzee GA and Frenkel B:
Runx2 transcriptome of prostate cancer cells: Insights into
invasiveness and bone metastasis. Mol Cancer. 9:2582010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuen HF, Kwok WK, Chan KK, Chua CW, Chan
YP, Chu YY, Wong YC, Wang X and Chan KW: TWIST modulates prostate
cancer cell-mediated bone cell activity and is upregulated by
osteogenic induction. Carcinogenesis. 29:1509–1518. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aprelikova O, Yu X, Palla J, Wei BR, John
S, Yi M, Stephens R, Simpson RM, Risinger JI, Jazaeri A and
Niederhuber J: The role of miR-31 and its target gene SATB2 in
cancer-associated fibroblasts. Cell Cycle. 9:4387–4398. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang MH, Yu J, Chen N, Wang XY, Liu XY,
Wang S and Ding YQ: Elevated microRNA-31 expression regulates
colorectal cancer progression by repressing its target gene SATB2.
PLoS One. 8:e853532013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Xie RL, Croce CM, Stein JL, Lian
JB, van Wijnen AJ and Stein GS: A program of microRNAs controls
osteogenic lineage progression by targeting transcription factor
Runx2. Proc Natl Acad Sci USA. 108:9863–9868. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen QY, Li J, Sun H, Wu F, Zhu Y, Kluz T,
Jordan A, DesMarais T, Zhang X, Murphy A and Costa M: Role of
miR-31 and SATB2 in arsenic-induced malignant BEAS-2B cell
transformation. Mol Carcinog. 57:968–977. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ge J, Guo S, Fu Y, Zhou P, Zhang P, Du Y,
Li M, Cheng J and Jiang H: Dental follicle cells participate in
tooth eruption via the RUNX2-miR-31-SATB2 Loop. J Dent Res.
94:936–944. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Z, Hassan MQ, Volinia S, van Wijnen AJ,
Stein JL, Croce CM, Lian JB and Stein GS: A microRNA signature for
a BMP2-induced osteoblast lineage commitment program. Proc Natl
Acad Sci USA. 105:13906–13911. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Z, Hassan MQ, Jafferji M, Aqeilan RI,
Garzon R, Croce CM, van Wijnen AJ, Stein JL, Stein GS and Lian JB:
Biological functions of miR-29b contribute to positive regulation
of osteoblast differentiation. J Biol Chem. 284:15676–15684. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pawlicki JM and Steitz JA: Nuclear
networking fashions pre-messenger RNA and primary microRNA
transcripts for function. Trends Cell Biol. 20:52–61. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu Y, Chen QY, Li AH and Costa M: The
role of non-coding RNAs involved in nickel-induced lung
carcinogenic mechanisms. Inorganics. 7:812019. View Article : Google Scholar
|
|
48
|
Chen QY, Des Marais T and Costa M:
Deregulation of SATB2 in carcinogenesis with emphasis on
miRNA-mediated control. Carcinogenesis. 40:393–402. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tian W, Wang G, Liu Y, Huang Z, Zhang C,
Ning K, Yu C, Shen Y, Wang M, Li Y, et al: The miR-599 promotes
non-small cell lung cancer cell invasion via SATB2. Biochem Biophys
Res Commun. 485:35–40. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
El Bezawy R, Cominetti D, Fenderico N,
Zuco V, Beretta GL, Dugo M, Arrighetti N, Stucchi C, Rancati T,
Valdagni R, et al: miR-875-5p counteracts epithelial-to-mesenchymal
transition and enhances radiation response in prostate cancer
through repression of the EGFR-ZEB1 axis. Cancer Lett. 395:53–62.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gu J, Wang G, Liu H and Xiong C: SATB2
targeted by methylated miR-34c-5p suppresses proliferation and
metastasis attenuating the epithelial-mesenchymal transition in
colorectal cancer. Cell Prolif. 51:e124552018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sun X, Liu S, Chen P, Fu D, Hou Y, Hu J,
Liu Z, Jiang Y, Cao X, Cheng C, et al: miR-449a inhibits colorectal
cancer progression by targeting SATB2. Oncotarget. 8:100975–100988.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Luo LJ, Yang F, Ding JJ, Yan DL, Wang DD,
Yang SJ, Ding L, Li J, Chen D, Ma R, et al: miR-31 inhibits
migration and invasion by targeting SATB2 in triple negative breast
cancer. Gene. 594:47–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gong Y, Xu F, Zhang L, Qian Y, Chen J,
Huang H and Yu Y: MicroRNA expression signature for Satb2-induced
osteogenic differentiation in bone marrow stromal cells. Mol Cell
Biochem. 387:227–239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ratnadiwakara M and Änkö ML: mRNA
stability assay using transcription inhibition by actinomycin D in
mouse pluripotent stem cells. Bio-Protocol. 8:e30722018. View Article : Google Scholar
|
|
57
|
Costa M: Molecular mechanisms of nickel
carcinogenesis. Annu Rev Pharmacol Toxicol. 31:321–337. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen QY and Costa M: A comprehensive
review of metal-induced cellular transformation studies. Toxicol
Appl Pharmacol. 331:33–40. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Huang H, Zhu J, Li Y, Zhang L, Gu J, Xie
Q, Jin H, Che X, Li J, Huang C, et al: Upregulation of SQSTM1/p62
contributes to nickel-induced malignant transformation of human
bronchial epithelial cells. Autophagy. 12:1687–1703. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pang Y, Li W, Ma R, Ji W, Wang Q, Li D,
Xiao Y, Wei Q, Lai Y, Yang P, et al: Development of human cell
models for assessing the carcinogenic potential of chemicals.
Toxicol Appl Pharmacol. 232:478–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rani AS, Qu DQ, Sidhu MK, Panagakos F,
Shah V, Klein KM, Brown N, Pathak S and Kumar S: Transformation of
immortal, non-tumorigenic osteoblast-like human osteosarcoma cells
to the tumorigenic phenotype by nickel sulfate. Carcinogenesis.
14:947–953. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang L, Fan J, Hitron JA, Son YO, Wise JT,
Roy RV, Kim D, Dai J, Pratheeshkumar P, Zhang Z and Shi X: Cancer
stem-like cells accumulated in nickel-induced malignant
transformation. Toxicol Sci. 151:376–387. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang Q, Salnikow K, Kluz T, Chen LC, Su
WC and Costa M: Inhibition and reversal of nickel-induced
transformation by the histone deacetylase inhibitor trichostatin A.
Toxicol Appl Pharmacol. 192:201–211. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Borowicz S, Van Scoyk M, Avasarala S,
Karuppusamy Rathinam MK, Tauler J, Bikkavilli RK and Winn RA: The
soft agar colony formation assay. J Vis Exp. e519982014.PubMed/NCBI
|
|
65
|
Jiang WG, Sanders AJ, Katoh M, Ungefroren
H, Gieseler F, Prince M, Thompson SK, Zollo M, Spano D, Dhawan P,
et al: Tissue invasion and metastasis: Molecular, biological and
clinical perspectives. Semin Cancer Biol. 35 Suppl:S244–S275. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sahai E: Mechanisms of cancer cell
invasion. Curr Opin Genet Dev. 15:87–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Komori T, Yagi H, Nomura S, Yamaguchi A,
Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al:
Targeted disruption of Cbfa1 results in a complete lack of bone
formation owing to maturational arrest of osteoblasts. Cell.
89:755–764. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Valenti MT, Serafini P, Innamorati G, Gili
A, Cheri S, Bassi C and Dalle Carbonare L: Runx2 expression: A
mesenchymal stem marker for cancer. Oncol Lett. 12:4167–4172. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Thomas DM, Carty SA, Piscopo DM, Lee JS,
Wang WF, Forrester WC and Hinds PW: The retinoblastoma protein acts
as a transcriptional coactivator required for osteogenic
differentiation. Mol Cell. 8:303–316. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bai X, Meng L, Sun H, Li Z, Zhang X and
Hua S: MicroRNA-196b Inhibits cell growth and metastasis of lung
cancer cells by targeting Runx2. Cell Physiol Biochem. 43:757–767.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Herreno AM, Ramirez AC, Chaparro VP,
Fernandez MJ, Canas A, Morantes CF, Moreno OM, Bruges RE, Mejia JA,
Bustos FJ, et al: Role of RUNX2 transcription factor in epithelial
mesenchymal transition in non-small cell lung cancer lung cancer:
Epigenetic control of the RUNX2 P1 promoter. Tumour Biol.
41:10104283198510142019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li H, Zhou RJ, Zhang GQ and Xu JP:
Clinical significance of RUNX2 expression in patients with nonsmall
cell lung cancer: A 5-year follow-up study. Tumour Biol.
34:1807–1812. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Underwood KF, D'Souza DR, Mochin-Peters M,
Pierce AD, Kommineni S, Choe M, Bennett J, Gnatt A, Habtemariam B,
MacKerell AD Jr and Passaniti A: Regulation of RUNX2 transcription
factor-DNA interactions and cell proliferation by vitamin D3
(cholecalciferol) prohormone activity. J Bone Miner Res.
27:913–925. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Underwood KF, Mochin MT, Brusgard JL, Choe
M, Gnatt A and Passaniti A: A quantitative assay to study protein:
DNA interactions, discover transcriptional regulators of gene
expression, and identify novel anti-tumor agents. J Vis Exp.
78:e505122013.PubMed/NCBI
|
|
75
|
Kim MS, Gernapudi R, Choi EY, Lapidus RG
and Passaniti A: Characterization of CADD522, a small molecule that
inhibits RUNX2-DNA binding and exhibits antitumor activity.
Oncotarget. 8:70916–70940. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Dobreva G, Dambacher J and Grosschedl R:
SUMO modification of a novel MAR-binding protein, SATB2, modulates
immunoglobulin mu gene expression. Genes Dev. 17:3048–3061. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
O'Brien J, Hayder H, Zayed Y and Peng C:
Overview of MicroRNA biogenesis, mechanisms of actions, and
circulation. Front Endocrinol (Lausanne). 9:4022018. View Article : Google Scholar
|
|
78
|
Flynt AS and Lai EC: Biological principles
of microRNA-mediated regulation: Shared themes amid diversity. Nat
Rev Genet. 9:831–842. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Cameron KS, Buchner V and Tchounwou PB:
Exploring the molecular mechanisms of nickel-induced genotoxicity
and carcinogenicity: A literature review. Rev Environ Health.
26:81–92. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lee JW, Bae SH, Jeong JW, Kim SH and Kim
KW: Hypoxia-inducible factor (HIF-1)alpha: Its protein stability
and biological functions. Exp Mol Med. 36:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li Q, Chen H, Huang X and Costa M: Effects
of 12 metal ions on iron regulatory protein 1 (IRP-1) and
hypoxia-inducible factor-1 alpha (HIF-1alpha) and HIF-regulated
genes. Toxicol Appl Pharmacol. 213:245–255. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Davidson TL, Chen H, Di Toro DM, D'Angelo
G and Costa M: Soluble nickel inhibits HIF-prolyl-hydroxylases
creating persistent hypoxic signaling in A549 cells. Mol Carcinog.
45:479–489. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lee SH, Che X, Jeong JH, Choi JY, Lee YJ,
Lee YH, Bae SC and Lee YM: Runx2 protein stabilizes
hypoxia-inducible factor-1α through competition with von
Hippel-Lindau protein (pVHL) and stimulates angiogenesis in growth
plate hypertrophic chondrocytes. J Biol Chem. 287:14760–14771.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kwon TG, Zhao X, Yang Q, Li Y, Ge C, Zhao
G and Franceschi RT: Physical and functional interactions between
Runx2 and HIF-1α induce vascular endothelial growth factor gene
expression. J Cell Biochem. 112:3582–3593. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Dong W, Chen Y, Qian N, Sima G, Zhang J,
Guo Z and Wang C: SATB2 knockdown decreases hypoxia-induced
autophagy and stemness in oral squamous cell carcinoma. Oncol Lett.
20:794–802. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ali T, Mushtaq I, Maryam S, Farhan A, Saba
K, Jan MI, Sultan A, Anees M, Duygu B, Hamera S, et al: Interplay
of N acetyl cysteine and melatonin in regulating oxidative
stress-induced cardiac hypertrophic factors and microRNAs. Arch
Biochem Biophys. 661:56–65. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Clemens F, Verma R, Ramnath J and Landolph
JR: Amplification of the Ect2 proto-oncogene and over-expression of
Ect2 mRNA and protein in nickel compound and
methylcholanthrene-transformed 10T1/2 mouse fibroblast cell lines.
Toxicol Appl Pharmacol. 206:138–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chiocca SM, Sterner DA, Biggart NW and
Murphy EC Jr: Nickel mutagenesis: Alteration of the MuSVts110
thermosensitive splicing phenotype by a nickel-induced duplication
of the 3′ splice site. Mol Carcinog. 4:61–71. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhang YJ, Chen JW, He XS, Zhang HZ, Ling
YH, Wen JH, Deng WH, Li P, Yun JP, Xie D and Cai MY: SATB2 is a
promising biomarker for identifying a colorectal origin for liver
metastatic adenocarcinomas. EBioMedicine. 28:62–69. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Dragomir A, de Wit M, Johansson C, Uhlen M
and Ponten F: The role of SATB2 as a diagnostic marker for tumors
of colorectal origin: Results of a pathology-based clinical
prospective study. Am J Clin Pathol. 141:630–638. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Eberhard J, Gaber A, Wangefjord S, Nodin
B, Uhlen M, Ericson Lindquist K and Jirstrom K: A cohort study of
the prognostic and treatment predictive value of SATB2 expression
in colorectal cancer. Br J Cancer. 106:931–938. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lin F, Shi J, Zhu S, Chen Z, Li A, Chen T,
Wang HL and Liu H: Cadherin-17 and SATB2 are sensitive and specific
immunomarkers for medullary carcinoma of the large intestine. Arch
Pathol Lab Med. 138:1015–1026. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Moh M, Krings G, Ates D, Aysal A, Kim GE
and Rabban JT: SATB2 Expression distinguishes ovarian metastases of
colorectal and appendiceal origin from primary ovarian tumors of
mucinous or endometrioid type. Am J Surg Pathol. 40:419–432. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yu W, Ma Y, Ochoa AC, Shankar S and
Srivastava RK: Cellular transformation of human mammary epithelial
cells by SATB2. Stem Cell Res. 19:139–147. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Shin MH, He Y, Marrogi E, Piperdi S, Ren
L, Khanna C, Gorlick R, Liu C and Huang J: A RUNX2-Mediated
epigenetic regulation of the survival of p53 defective cancer
cells. PLoS Genet. 12:e10058842016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Tang W, Yang F, Li Y, de Crombrugghe B,
Jiao H, Xiao G and Zhang C: Transcriptional regulation of Vascular
Endothelial Growth Factor (VEGF) by osteoblast-specific
transcription factor Osterix (Osx) in osteoblasts. J Biol Chem.
287:1671–1678. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Efe JA and Ding S: The evolving biology of
small molecules: Controlling cell fate and identity. Philos Trans R
Soc Lond B Biol Sci. 366:2208–2221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ma YN, Zhang HY, Fei LR, Zhang MY, Wang
CC, Luo Y and Han YC: SATB2 suppresses non-small cell lung cancer
invasiveness by G9a. Clin Exp Med. 18:37–44. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wei MM and Zhou GB: Long non-coding RNAs
and their roles in non-small-cell lung cancer. Genomics Proteomics
Bioinformatics. 14:280–288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Valencia-Sanchez MA, Liu J, Hannon GJ and
Parker R: Control of translation and mRNA degradation by miRNAs and
siRNAs. Genes Dev. 20:515–524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Stepicheva NA and Song JL: Function and
regulation of microRNA-31 in development and disease. Mol Reprod
Dev. 83:654–674. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Asangani IA, Harms PW, Dodson L, Pandhi M,
Kunju LP, Maher CA, Fullen DR, Johnson TM, Giordano TJ, Palanisamy
N and Chinnaiyan AM: Genetic and epigenetic loss of microRNA-31
leads to feed-forward expression of EZH2 in melanoma. Oncotarget.
3:1011–1025. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Augoff K, Das M, Bialkowska K, McCue B,
Plow EF and Sossey-Alaoui K: miR-31 is a broad regulator of
β1-integrin expression and function in cancer cells. Mol Cancer
Res. 9:1500–1508. 2011.PubMed/NCBI
|
|
105
|
Yamagishi M, Nakano K, Miyake A, Yamochi
T, Kagami Y, Tsutsumi A, Matsuda Y, Sato-Otsubo A, Muto S,
Utsunomiya A, et al: Polycomb-mediated loss of miR-31 activates
NIK-dependent NF-κB pathway in adult T cell leukemia and other
cancers. Cancer Cell. 21:121–135. 2012.PubMed/NCBI
|
|
106
|
Ling H, Fabbri M and Calin GA: MicroRNAs
and other non-coding RNAs as targets for anticancer drug
development. Nat Rev Drug Discov. 12:847–865. 2013.PubMed/NCBI
|
|
107
|
Kim HS, Lee KS, Bae HJ, Eun JW, Shen Q,
Park SJ, Shin WC, Yang HD, Park M, Park WS, et al: MicroRNA-31
functions as a tumor suppressor by regulating cell cycle and
epithelial-mesenchymal transition regulatory proteins in liver
cancer. Oncotarget. 6:8089–8102. 2015.PubMed/NCBI
|
|
108
|
Sakai T, Toguchida J, Ohtani N, Yandell
DW, Rapaport JM and Dryja TP: Allele-specific hypermethylation of
the retinoblastoma tumor-suppressor gene. Am J Hum Genet.
48:880–888. 1991.PubMed/NCBI
|
|
109
|
Lee YW, Klein CB, Kargacin B, Salnikow K,
Kitahara J, Dowjat K, Zhitkovich A, Christie NT and Costa M:
Carcinogenic nickel silences gene expression by chromatin
condensation and DNA methylation: A new model for epigenetic
carcinogens. Mol Cell Biol. 15:2547–2557. 1995.PubMed/NCBI
|
|
110
|
Ohtani-Fujita N, Fujita T, Aoike A,
Osifchin NE, Robbins PD and Sakai T: CpG methylation inactivates
the promoter activity of the human retinoblastoma tumor-suppressor
gene. Oncogene. 8:1063–1067. 1993.PubMed/NCBI
|
|
111
|
Sutcliffe JS, Nelson DL, Zhang F, Pieretti
M, Caskey CT, Saxe D and Warren ST: DNA methylation represses FMR-1
transcription in fragile X syndrome. Hum Mol Genet. 1:397–400.
1992.PubMed/NCBI
|
|
112
|
Greger V, Debus N, Lohmann D, Hopping W,
Passarge E and Horsthemke B: Frequency and parental origin of
hypermethylated RB1 alleles in retinoblastoma. Hum Genet.
94:491–496. 1994.PubMed/NCBI
|
|
113
|
Hansen RS, Gartler SM, Scott CR, Chen SH
and Laird CD: Methylation analysis of CGG sites in the CpG island
of the human FMR1 gene. Hum Mol Genet. 1:571–578. 1992.PubMed/NCBI
|