Introduction
Chimeric antigen receptor (CAR) T cells are
individualized living drugs which combine the properties of T
lymphocytes with the specificity of antibodies. They represent
potent weapons to treat malignancies (1,2). CAR T
cells directed against CD19 (CD19.CAR T cells) have shown
remarkable clinical results in heavily pre-treated patients with
relapsed or refractory (r/r) lymphoid malignancies (3–6), including
acute lymphoblastic leukemia (ALL) (7,8), chronic
lymphocytic leukemia (CLL) (9,10) and
non-Hodgkin's lymphoma (NHL) (11,12). In
fact, several CAR T-cell products have been approved by the
regulatory authorities and have been adopted as standard of care
within the labelled indications (13–16).
However, antigen-positive as well as antigen-negative relapses and
resistance to treatment are commonly observed in ALL (17–19), CLL
(10) and NHL (20) patients following CD19.CAR T-cell
treatment. Considering that cancer therapy against only a single
target may facilitate the development of resistance, combining
antigen-specific CAR T cells with less specific anti-tumor agents
may overcome resistance to treatment, prevent disease relapse and
enhance anti-tumor responses in patients.
Exportin-1 (XPO1), also termed chromosome region
maintenance 1 (CRM1), is a nuclear export receptor involved in the
transportation of proteins such as histones, polymerases,
transcription factors and/or RNA from the nucleus into the cytosol
(21). Notably, the export of tumor
suppressor proteins (TSPs) depends on XPO1 (22,23).
Hematological as well as solid malignancies overexpress XPO1
(24–31) to limit nuclear TSP effects and evade
inherent tumor control. XPO1 overexpression has been observed in
aggressive diseases, and elevated XPO1 levels have been associated
with poor clinical outcome in numerous neoplasms (32–36). Thus,
downregulation of XPO1 constitutes an interesting therapeutic
strategy. Notably, inhibiting XPO1 by selective inhibitors of
nuclear export (SINEs) has been shown to restore and enhance the
function of TSPs (21), and
anti-tumor efficacy of SINEs has been demonstrated in hematological
malignancies including multiple myeloma (MM) (32), ALL (33), NHL (34,35), acute
myeloid leukemia (AML) (36) as well
as in solid tumors (24–26). The SINE compound selinexor was
approved for the treatment of adults with r/r MM by the U.S. Food
and Drug Administration (FDA) in September 2019, and by the
European Medicines Agency (EMA) in December 2019 (37). Moreover, selinexor is currently under
clinical evaluation for treatment of diffuse large B-cell lymphoma
(DLBCL; NCT02227251), r/r AML and myelodysplastic syndrome (MDS;
NCT03071276) as well as advanced liposarcoma (NCT02606461).
Given that protein transport regulation through XPO1
across the nuclear membrane is essential to normal cells (36), SINEs disturb normal immune homeostasis
resulting in side effects such as cytopenia. Selinexor can cross
the blood-brain-barrier (BBB) and cause central anorexia with
associated weight loss and malaise (36,38). The
second-generation SINE eltanexor has a reduced effect on
hematopoietic stem and progenitor (HSPCs) cells (39) and an approximately 30-fold lower
capacity to penetrate the BBB than selinexor (32). Therefore, eltanexor has a more
favorable side effect profile when compared to selinexor, while
maintaining anti-tumor efficacy. Eltanexor has shown potent
anti-lymphoblastic activity in pre-clinical patient-derived T- and
B-ALL xenograft models (32).
The aim of the present study was to investigate the
impact of the SINE compounds selinexor and eltanexor on tumor cells
as well as third-generation CAR T cells and to evaluate potential
combinatorial effects of eltanexor and CAR T cells.
Materials and methods
Peripheral bood mononuclear cell
(PBMCs)
Peripheral blood mononuclear cells (PBMCs) of seven
healthy donors (HDs) were collected at the Heidelberg University
Hospital, Heidelberg, Germany. Sample collection and analysis were
approved by the Ethics Committee of the University of Heidelberg
(S-254/2016) and all donors signed a written consent prior to
treatment. All experiments were performed in accordance with the
convention of Helsinki.
Cell lines
Burkitt lymphoma cell lines Daudi and Raji as well
as ALL cell lines Nalm-6 and Reh [German Collection of
Microorganisms and Cell Cultures (DSMZ)] were used as CD19-positive
CAR T-cell target cells. Chronic myelogenous leukemia (CML) cell
line K562 (DSMZ) was used as CD19-negative control cell line. 293T
cells were obtained from the American Type Culture Collection
(ATCC). Cells were cultured in RPMI-1640 (Thermo Fisher Scientific)
supplemented with 2 mM L-glutamine (Thermo Fisher Scientific) and
10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher
Scientific) at 37°C and 5% CO2.
Cell culturing
Due to the experimental design, co-culturing
experiments of CAR T cells (effector cells) with tumor cell lines
(Nalm-6, Daudi, Raji and Reh) and eltanexor were performed using
two different culture conditions: i) simultaneous co-culturing of
CAR T cells, target cells and eltanexor, or ii) pre-treatment of
target cells with eltanexor (0.05, 0.1 and 0.5 µM) and washing-out
of eltanexor prior to addition of CAR T cells.
CAR T-cell generation
Retrovirus generation and CD19 CAR
transfection
The third-generation retroviral vector
RV-SFG.CD19.CD28.4-1BB.CD3zeta used in a CD19.CAR T-cell clinical
trial conducted at the University Hospital Heidelberg (EudraCT
2016-004808-60; NCT03676504) comprising CD28 and 4-1BB (CD137) as
costimulatory domains (40) was used
for CAR T-cell manufacturing. Retroviral supernatant was generated
via transfection of 293T cells with three plasmids: i) plasmid
RV-SFG.CD19.CD28.4-1BB.CD3zeta (3.75 µg) containing the
CD19-specific CAR transgene, ii) packaging plasmid PegPam3
containing gag-pol (3.75 µg) and iii) the envelope plasmid RDF
plasmid containing env (2.5 µg). SFG.CD19.CD28.4-1BB.CD3zeta,
PegPam3 and RDF plasmids were kindly provided by Professor Malcolm
Brenner, Center for Cell and Gene Therapy, Houston, TX, USA.
Details of retrovirus generation and transfection have been
previously described (41,42).
CAR T-cell manufacturing
CAR T cells were manufactured as previously
described (41,42). In brief, on day 0, cryopreserved PBMCs
from HDs were thawed and seeded on anti-CD3- and anti-CD28 coated
24-well plates (Corning). On day 3, activated T cells (ATCs)
supplied with retroviral supernatant were transferred into 24-well
plates (Corning) previously coated with retronectin (Takara Bio).
Efficacy was evaluated on days 7, 10, 14 and 17 after transduction
using flow cytometry.
SINE compounds
Selinexor (KPT330) and eltanexor (KPT8602) (Selleck
Chemicals) were dissolved in DMSO to a stock concentration of 10
mmol/l.
CellTiter-Glo assay
Viability assay CellTiter-Glo (Promega, Fitchburg)
was used for cell number titration as well as subsequent compound
titration. After adding CellTiter-Glo buffer to the CellTiter-Glo
substrate (Promega) to reconstitute the lyophilized
enzyme/substrate mixture, CellTiter-Glo reagent was aliquoted and
stored at −20°C until use. CellTiter-Glo experiments including cell
number titration and SINE compound titration were performed
sequentially.
Cell number titration
To obtain cells growing in the logarithmic phase at
48 h, tumor cell lines (Daudi, Raji, Nalm-6, Reh), CAR T cells, and
non-transduced T cells (negative control) were added to 384-well
plates (Greiner Bio-One) and diluted 1:1.5 with RPMI-1640 medium
(tumor cell lines) or with complete medium (T cells).
Compound titration on tumor cell lines
and CAR T cells
To assess the effects of SINEs on tumor cells as
well as CAR T cells, effects of selinexor and eltanexor on cell
viability were analyzed.
The therapeutic window of SINEs was assessed via
compound titration: Following cell number titration, selinexor and
eltanexor or DMSO as control were added at concentrations from 10
to 0.001 µM (dilution of eltanexor and selinexor performed with
phosphate-buffered saline (PBS) at ratios of 1:3) to Daudi, Raji,
Nalm-6, Reh, CAR T cells as well as non-transduced T cells in
384-well plates (Greiner Bio-One) and the half-maximal inhibitory
concentrations (IC50) determined.
Tumor or CAR T cells with added SINEs were
cultivated for 48 h in 384-well plates (Greiner Bio-One). After
cultivation, 12 µl of CellTiter-Glo reagents, i.e., CellTiter-Glo
and substrate (Promega), were added into each well of the culturing
system. The mixture of the solution was incubated for 15–20 min at
room temperature (RT) and luminescence was recorded by the Ensight
Multimode Plate Reader (PerkinElmer). Relative viability of serial
dilutions was used to calculate the IC50.
Flow cytometric analysis
According to the location of expression of analyzed
markers, surface marker staining or intracellular staining (ICS)
was performed using the FoxP3 staining buffer set (cat. no.
130-093-142, Miltenyi Biotec) at 4°C for 6 h. For all staining
procedures, dead cells were excluded using the LIVE/DEAD fixable
near-infrared (IR) dead cell stain kit (Thermo Fisher Scientific).
Anti-human goat F(ab) IgG (H+L) PE antibody (cat. 109-116-088;
Dianova) was used to distinguish CD19-specific CAR T cells from
non-transduced T cells. After staining, all the samples were
measured on the flow cytometer LSRII (BD Biosciences) and data were
analyzed using FlowJo.
Surface marker staining
The following antibodies were used for surface
marker staining: Anti-human goat F(ab) IgG (H+L) PE antibody (cat.
no. 109-116-088) from Dianova; anti-CD3-PE eFluor 610 (cat. no.
61-0038-42), and anti-CD4-Alexa Fluor 700 (cat. no. 560049-42) from
eBioscience, San Diego; anti-CD8-PerCP (cat. no. 344708),
anti-CD10-APC (cat. no. 312210), anti-human CD223-APC (LAG-3, Cat.
369212), anti-PD-1-Alexa Fluor 488 (cat. no. 329935), anti-human
CD366 (Tim-3, Cat. 345007) all from Biolegend; anti-CD3-V500 (cat.
no. 561416) from BD Biosciences.
ICS for evaluation of cytokine release
by CAR T cells
For cytokine release assessment, CAR T cells with or
without the addition of different doses of eltanexor were
stimulated by incubation with CD19-positive target cells for 6 h in
96-well U-bottom microplates (Greiner BioOne) in the presence of
Brefeldin A (Biolegend). The cell mixture was fixated and
permeabilized using the FoxP3 staining buffer set (cat. no.
130-093-142, Miltenyi Biotec). Cells were incubated for 30 min at
RT in the dark for fixation with fixation/permeabilization solution
(fixation/permeabilization solution 1: Fixation/permeabilization
solution 2=1:4) and 15 min at RT for permeabilization with the
FoxP3 permeabilization buffer. ICS was performed with
anti-interferon (IFN)-γ-Alexa Fluor 488 (cat. no. 502515;
Biolegend) and anti-tumor necrosis factor (TNF)-α-BV421 (cat. no.
562783; BD Biosciences).
ICS for evaluation of
phosphorylated-STAT3
For staining of phosphorylated-STAT3, CAR T cells
were fixed by incubation with fixation buffer (Biolegend) for 15
min at RT, before they were permeabilized with True-Phos perm
buffer (Biolegend) at −20°C overnight. The anti-STAT3
phosphorylated (Tyr705) antibody (Biolegend) was added to stain
phosphorylated-STAT3.
Flow cytometric analysis used to
evaluate cytotoxicity of CAR T cells towards Nalm-6 cells
As Nalm-6 cells have a low Chromium 51
(51Cr) ∆release (∆release=maximum release-spontaneous
release), Cr release (mentioned in a section below) is an
inadequate method to assess the cytotoxicity of CAR T cells towards
Nalm-6 cells (Fig. S1).
Consequently, flow cytometric analysis was used to evaluate
cytotoxicity of CAR T cells towards Nalm-6 cells. After either
simultaneous co-culturing (CAR T cells, Nalm-6 cells and eltanexor)
or culturing of CAR T cells with pre-treated Nalm-6 cells, cells
were collected and stained with the following antibodies:
Anti-human goat F(ab) IgG (H+L) PE antibody (Dianova), anti-CD3-PE
eFluor 610 (eBioscience), anti-human CD223 (LAG-3), anti-PD-1-Alexa
Fluor 488, anti-human CD366 (Tim-3) (Biolegend) and flow cytometry
was performed.
Chromium 51 release assay
51Cr release assay to address
functionality of CAR T cells towards Daudi, Raji or Reh cells was
performed as previously described (43,44).
Effector to target cell ratios of 10:1, 5:1, 2.5:1 and 1:1 were
used.
Pre-treating tumor cell lines with
eltanexor and CAR T cells
Tumor cells were labeled with 51Cr
(Hartmann Analytic) for 2 h in a humidified incubator at 37°C and
5% CO2. Subsequently, CAR T cells were added and
co-culturing was performed for 4 h at 37°C and 5% CO2 in
a 96-well U-bottom microplate (Greiner Bio-One). The supernatant
was collected to perform radioactive activity measurement as
previously described (41,42).
Simultaneous co-culturing of tumor
cell lines, CAR T cells and eltanexor
Either Daudi, Raji or Reh cells were labeled with
51Cr for 2 h in a humidified incubator at 37°C and 5%
CO2. After labeling, the cells were co-cultured with CAR
T cells, followed by the addition of eltanexor at different
concentrations (0.05, 0.1 and 0.5 µM). The negative control
contained DMSO instead of eltanexor. The cells and eltanexor were
cultured in 96-well U-bottom microplates (Greiner Bio-One) for 4 h
at 37°C and 5% CO2. The supernatant was collected to
perform radioactive activity measurement as previously described
(41,42).
Western blot analysis
One million CD19 CAR T cells, non-transduced T cells
and tumor cells, respectively, with or without the addition of
eltanexor were lysed in 200 µl radio-immunoprecipitation assay
buffer (RIPA buffer; Thermo Fisher Scientific) after the addition
of complete protease inhibitor (Sigma-Aldrich; Merck KGaA) at RT
for 10 min followed by centrifugation for 10 min at 12.000 × g at
4°C. Protein-containing supernatants were collected and 20 µg
protein was loaded on a 4–12% SDS-PAGE gel. Separated proteins were
immediately blotted onto nitrocellulose membranes. Prior to
incubation with a primary antibody at a dilution of 1:200
(anti-exportin-1/CRM1, anti-phosphorylated-STAT3 (only for CAR T
cells) or 1:500 [anti-beta (β) actin (as internal reference),
anti-total STAT3 (only for CAR T cells)] at 4°C overnight, the
membranes were blocked for 1 h at RT with 5% milk in Tris-buffered
saline with Tween-20 (TBST). Appropriate horseradish
peroxidase-conjugated secondary antibodies (anti-mouse IgG or
anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology,
Frankfurt) were used at a dilution of 1:2,000. Proteins were
visualized in an Amersham Imager 600 (GE Healthcare).
Quantification of the of protein bands was performed using software
ImageJ.
Statistical analysis
Statistical analysis was performed with GraphPad
Prism 6 (GraphPad Software Inc.). P-values were calculated using
the parametric two-way t-test between two groups, and the one-way
analysis of variance (ANOVA) with Bonferroni's multiple comparison
test for three or four groups. P<0.05 was considered
statistically significant. When not otherwise indicated, results
were represented as mean ± standard deviation (SD). IC50s were
presented as mean ± standard error of the mean (SEM). Graphs and
tables were designed using GraphPad Prism 6.
Results
Sensitivity of tumor cells towards
selinexor and eltanexor and measuring of XPO1 protein levels
In order to improve CAR T cell efficacy and overcome
refractory disease, we evaluated the combination of CAR T cells
with the SINE compounds selinexor and eltanexor.
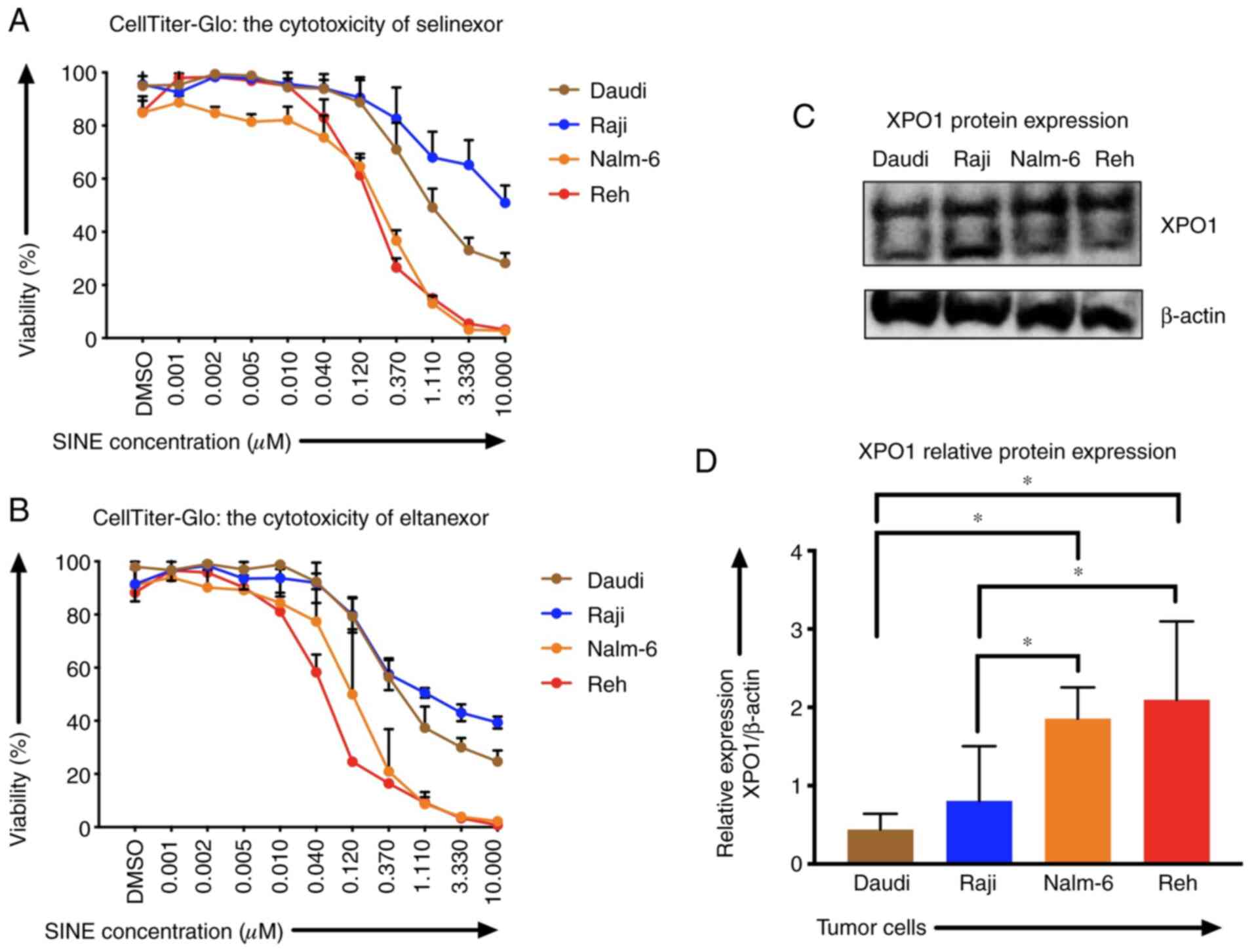
Sensitivities of Reh, Nalm-6, Daudi and Raji cells
to SINEs were analyzed assessing IC50 of selinexor and eltanexor.
Selinexor and eltanexor effectively inhibited viabilities of Reh
[IC50: Selinexor: 0.16±0.01 µM (Fig.
1A), eltanexor: 0.05±0.01 µM (Fig.
1B)] and Nalm-6 cells [IC50: Selinexor: 0.30±0.02 µM (Fig. 1A), eltanexor: 0.14±0.03 µM (Fig. 1B)]. Daudi cells showed medium
sensitivity [selinexor: 0.60±0.09 µM (Fig. 1A), eltanexor: 0.30±0.03 µM (Fig. 1B)], whereas Raji cells exhibited the
lowest sensitivity to SINEs [IC50s: Selinexor: 1.33±1.16 µM
(Fig. 1A), eltanexor: 0.23±0.03 µM
(Fig. 1B)].
Sensitivity of ALL and NHL tumor cells towards
selinexor and eltanexor was associated with the protein levels of
the SINE target CRM1/XPO1: ALL cells Reh (2.10±0.01) and Nalm-6
(1.86±0.01), that had shown the highest sensitivity to SINEs,
displayed higher relative XPO1 protein levels compared to the NHL
cells Daudi (0.44±0.01) and Raji (0.81±0.01) (Fig. 1C and D) (P<0.05).
Tumor cells were more sensitive to eltanexor,
suggesting a superior toxicity profile of eltanexor compared to
selinexor (32,39). Consequently, eltanexor was chosen as
SINE compound to perform further experiments.
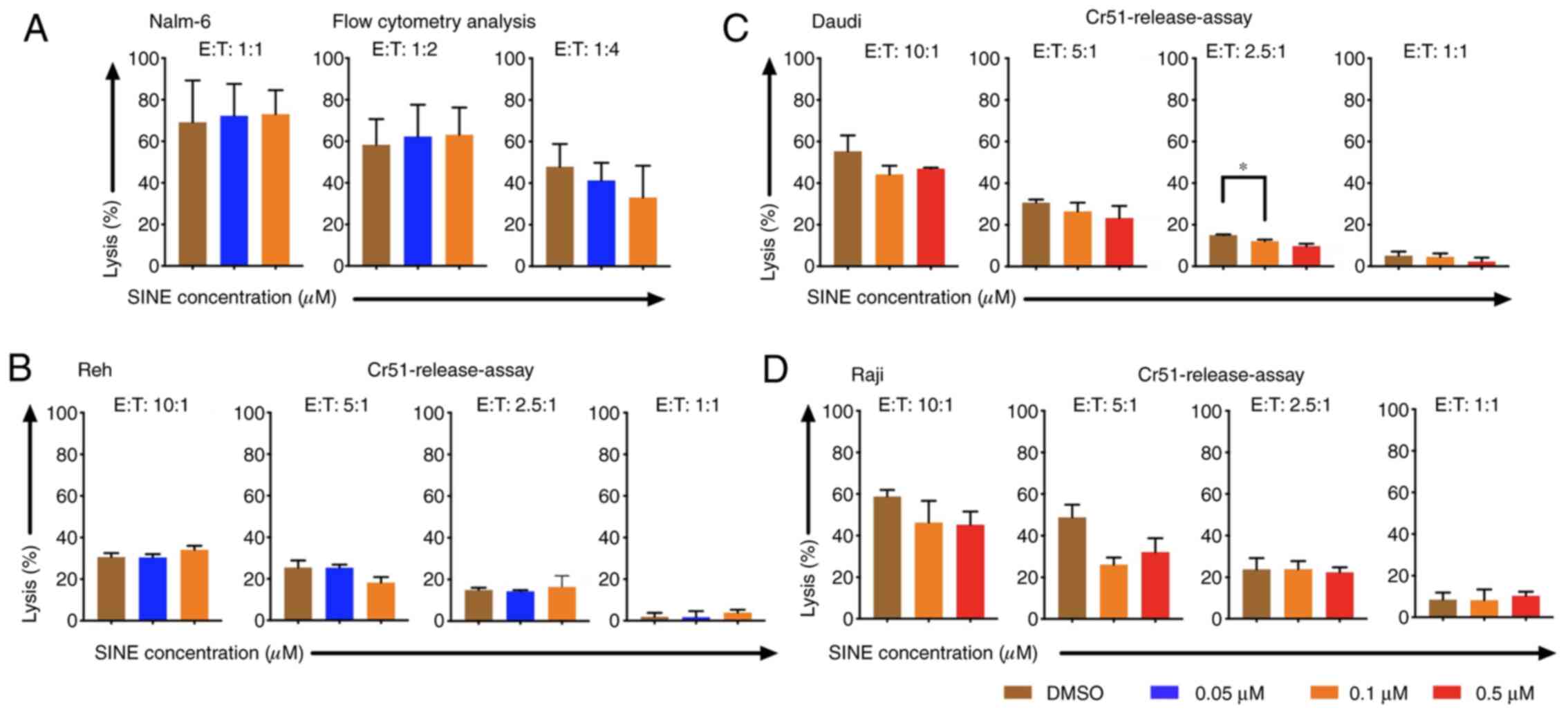
Cytotoxicity of CAR T cells was
abrogated when CAR T cells and tumor cells were cultivated
concomitantly with eltanexor
Cytotoxicity of CAR T cells towards tumor cells was
addressed when they were cultivated simultaneously with target
cells and eltanexor. Nalm-6, Reh, Daudi and Raji cells were
co-cultured with eltanexor at their respective IC50 (0.05 and 0.1
µM for Nalm-6 and Reh; 0.1 and 0.5 µM for Daudi and Raji) as well
as CAR T cells. Simultaneous co-culturing decreased cytotoxicity of
CAR T cells towards Nalm-6 (Fig. 2A,
assessed via flow cytometry), Reh (Fig.
2B, assessed via 51Cr release assay) (0.05 µM
eltanexor used), Daudi (Fig. 2C,
assessed via 51Cr release assay) as well as Raji
(Fig. 2D, assessed via
51Cr release assay) (0.1 µM eltanexor used) cells
compared to DMSO [Nalm-6: 1:1 ratio: 72.3 vs. 69.2%; 1:2 ratio:
62.3 vs. 58.3%; 1:4 ratio: 41.3 vs. 47.8% (Fig. 2A); Reh: 10:1 ratio: 30.4±1.6 vs.
30.7±1.9%, 5:1 ratio: 25.4±1.5 vs. 25.5±3.4%, 1:1 ratio: 1.7±3.0
vs. 1.8±1.8% (Fig. 2B). Daudi cells:
10:1 ratio: 44.2±4.2 vs. 55.2±7.6%, 5:1 ratio: 26.5±4.2 vs.
30.7±1.4%, 2.5:1 ratio: 12.2±0.7 vs. 15.0±0.3% (P=0.0088), 1:1
ratio: 4.6±1.6 vs. 5.1±2.0% (Fig. 2C)
and Raji cells: 10:1 ratio: 46.3±10.5 vs. 58.8±3.2%, 5:1 ratio:
26.1±3.4 vs. 48.8±6.1%, 2.5:1 ratio: 23.8±5.4 vs. 23.8±3.9%, 1:1
ratio: 8.2±5.p =2% vs. 8.5±3.4%) (Fig.
2D)].
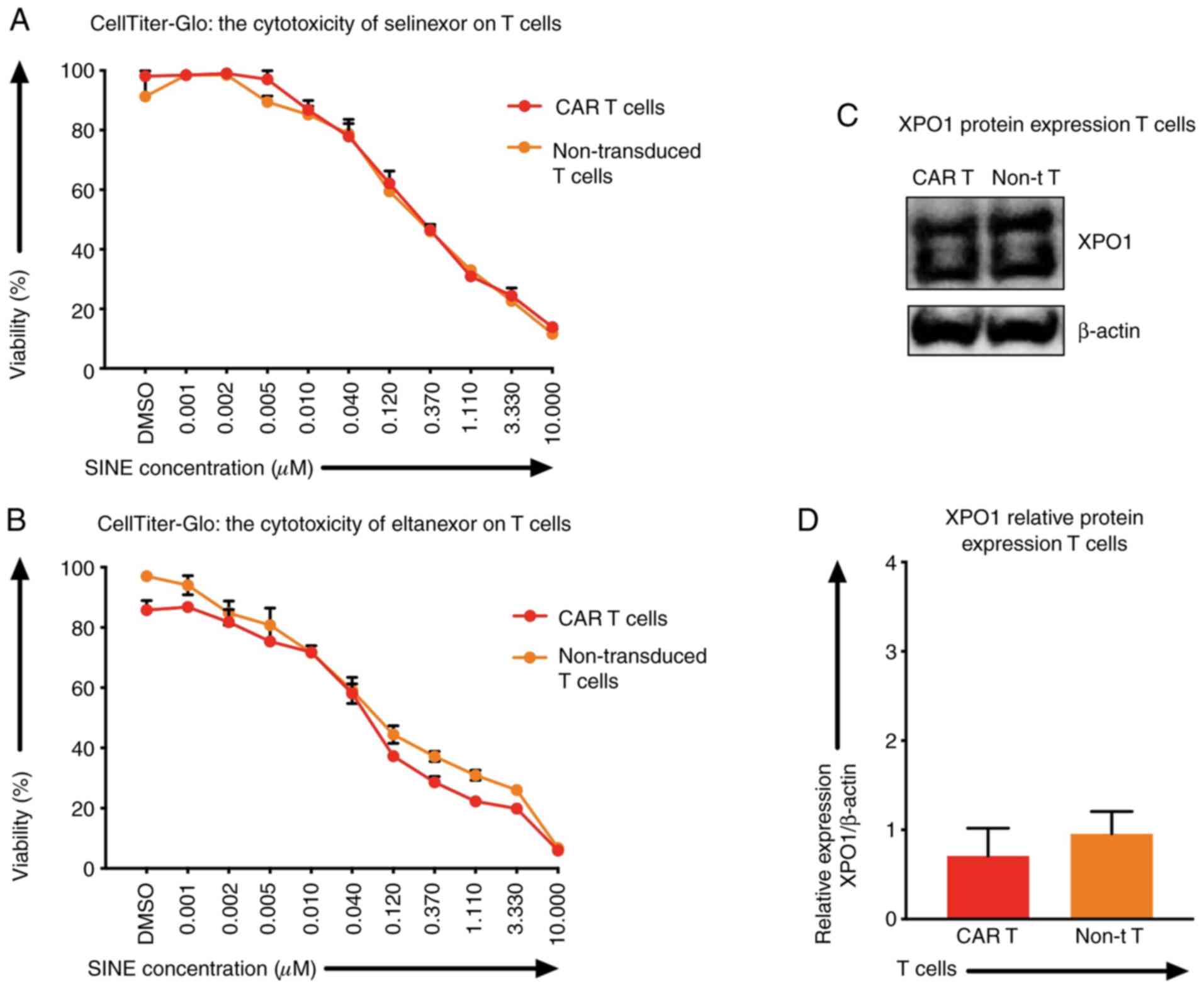
XPO1 protein levels and the
sensitivity of T cells towards SINEs
Besides tumor cells, selinexor and eltanexor also
inhibited viabilities of T cells, i.e., CAR T cells [IC50:
Selinexor (0.20±0.04 µM) (Fig. 3A,
red line), eltanexor (0.06±0.02 µM) (Fig.
3B, red line)] and non-transduced T cells [IC50s: Selinexor
(0.28±0.08 µM) (Fig. 3A, orange
line), eltanexor (0.04±0.05 µM) (Fig.
3B, orange line)]. The protein levels of XPO1 in CAR T cells
and non-transduced T cells were quantified by western blot
analysis: The relative expression of XPO1 protein of CAR T cells
was 0.71±0.01 and of non-transduced T cells 0.96±0.01 (Fig. 3C and D). Differences of XPO1 protein
levels and differences of sensitivity of CAR T cells and
non-transduced T cells towards selinexor or eltanexor were not
statistically significant (Fig.
3C).
Effects of eltanexor on CAR T cells
were pronounced when CAR T cells were cultivated simultaneously
with Daudi cells and eltanexor
Lower cytokine secretion of TNF-α and IFN-γ by CD4-
and CD8-positive CAR T cells after stimulation with Daudi cells and
eltanexor (0.1 and 0.5 µM) was observed when compared to the DMSO
control: CD4 TNF-α+: 39.2±5.5%, 38.0±3.0% vs. 46.9±2.3%;
CD4+ IFN-γ+: 14.8±11.8%, 15.6±10.4% vs.
19.4±12.2%; CD8+ TNF-α+: 25.7±2.7%
(P=0.0007), 25.5±3.1% (P=0.0012) vs. 39.6±2.6%; CD8
IFN-γ+: 31.3±8.3%, 21.1±14.1% vs. 36.6±7.5% (Fig. 4A).
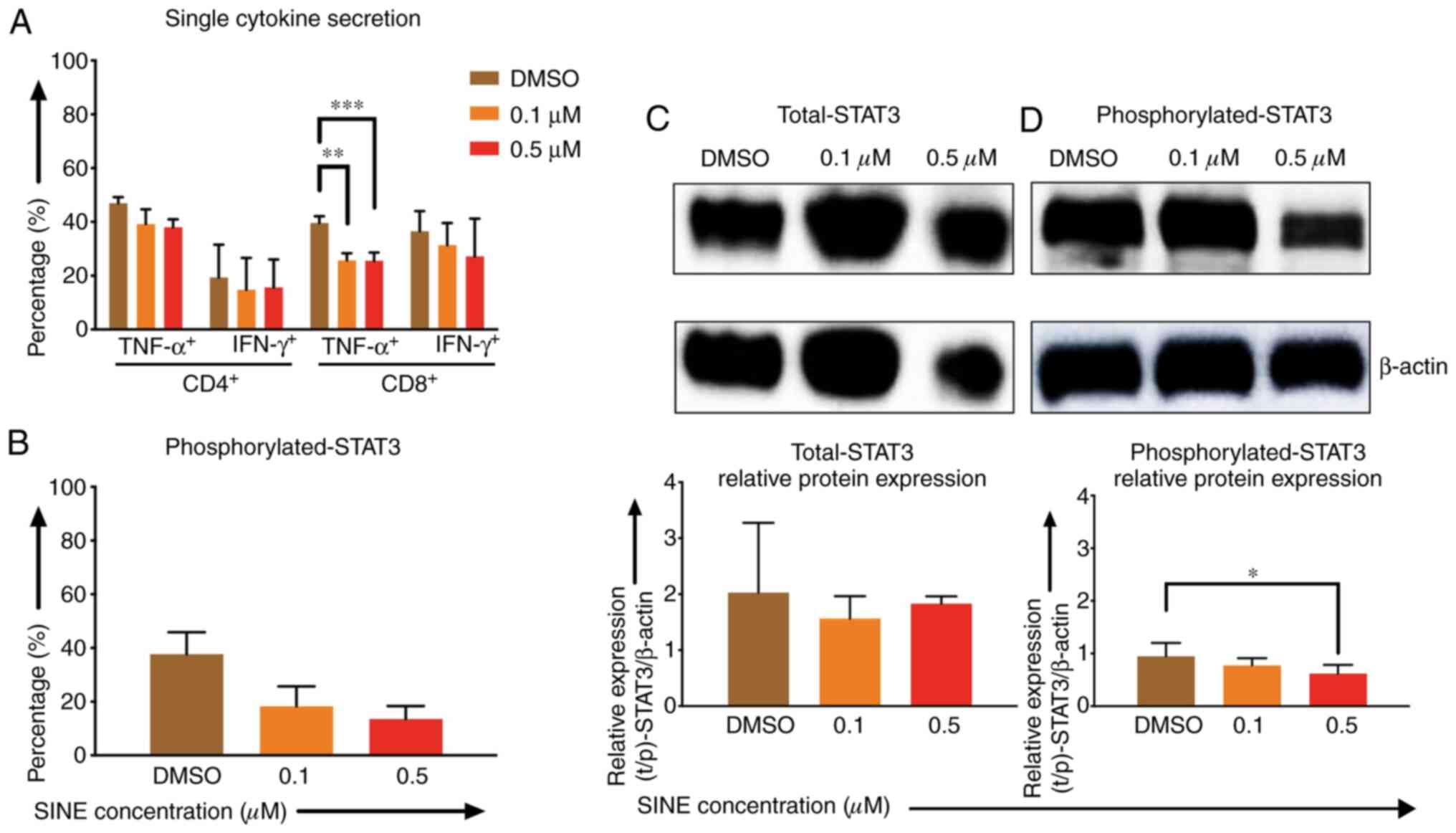 | Figure 4.Effects of SINEs when eltanexor, CAR
T cells and Daudi cells were cultivated simultaneously. Daudi cells
were cultivated simultaneously with CAR T cells and eltanexor (0.1
µM: Orange, 0.5 µM: Red) or with DMSO as control (brown) for 6 h
(A, assessed via flow cytometry analysis) or 4 h (B, assessed via
flow cytometry analysis, C and D via western blot analysis).
Cytokine release (IFN-γ and TNF-α) of CD4- and CD8-positive CAR T
cells was assessed and decreased cytokine secretion after
stimulation of CAR T cells with Daudi cells compared to the DMSO
control was observed. (A) Phosphorylated-STAT3 level of CAR T cells
was evaluated. Decreased phosphorylated-STAT3 of CAR T cells
cultured with eltanexor was observed when compared to the DMSO
control. (B) Evaluation of total STAT3 and phosphorylated-STAT3
protein levels of CAR T cells was performed after protein isolation
via western blot analysis on STAT3 of CAR T cells; β-actin was used
as the internal reference. Raw total STAT3 protein level (C, upper
panel), relative STAT3 expression (C, lower panel), raw
phosphorylated-STAT3 protein level (D, upper panel) and relative
phosphorylated-STAT3 expression (D, lower panel) were assessed. The
protein levels of phosphorylated-STAT3 compared to total STAT3 were
decreased. Experiments were performed in triplicate. Mean values
were calculated for each group; results are presented as mean ±
standard deviation (SD). *P<0.05, **P<0.005, ***P<0.0005
indicates statistical significance. |
Phosphorylated-STAT3 (p-STAT3) levels of CAR T cells
when co-cultured with eltanexor were assessed via flow cytometry:
p-STAT3 decreased significantly when eltanexor was used within the
culture [0.1 and 0.5 µM eltanexor vs. DMSO control: 18.3±7.4%
(P=0.0244), 13.5±4.8% (P=0.0094) vs. 37.1±8.1% (Fig. 4B)]. To further verify the decrease of
p-STAT3 in the cytoplasm of CAR T cells, difference in protein
levels of total STAT3 and phosphorylated STAT3 was quantified by
western blot analysis. Total STAT3 protein levels showed no
difference (Fig. 4C). However,
compared to the DMSO control, 0.1 and 0.5 µM of eltanexor
demonstrated a decreased phosphorylated STAT3 protein expression in
the cytoplasm: 0.1 and 0.5 µM vs. DMSO: 0.8±0.1% (P=0.2932),
0.4±0.2% (P=0.0281), vs. 0.9±0.3% (Fig.
4D).
Cytotoxicity of CAR T cells was
improved when tumor cells were pre-treated with eltanexor
Pre-sensitizing of tumor cells with eltanexor before
CAR T-cell exposure was addressed: Nalm-6, Reh, Daudi as well as
Raji cells were pre-treated with eltanexor (0.05 and 0.1 µM for
Nalm-6 and Reh; 0.1 and 0.5 µM for Daudi and Raji) and eltanexor
was removed by additional washing with culturing medium before CAR
T cells were added. Pre-treated Nalm-6 cells were cultivated with
CAR T cells for 24 h. Assessment of cytotoxicity of CAR T cells was
performed via flow cytometry as 51Cr release assay was
not adequate to evaluate the toxicity of CAR T cells towards Nalm-6
cells (Fig. S1). Toxicity of CAR T
cells on Reh, Daudi and Raji cells was assessed via 51Cr
release assay. Pre-treatment with 0.05 µM eltanexor significantly
increased cytotoxicity of CAR T cells towards Nalm-6 and Reh cells
as compared to DMSO [Fig. 5A and B:
Nalm-6: 1:1 ratio: 75.1±2.5 vs. 59.3±4.1% (P=0.0025); 1:2 ratio:
66.7±2.7 vs. 45.8±0.9% (P<0.0001), 1:4 ratio: 54.5±3.3 vs.
33.3±1.7% (P=0.0004); Reh: 10:1 ratio: 52.4±8.0 vs. 40.3±2.4%
(P=0.0163), 5:1 ratio: 44.3±5.4 vs. 36.1±1.5% (P=0.0472), 2.5:1
ratio: 38.3±4.3 vs. 27.8±1.8% (P=0.0130), 1:1 ratio: 31.6±2.9 vs.
16.9±4.5% (P=0.0025)]. The increase of cytotoxicity of CAR T cells
was also observed when Daudi cells pretreated with 0.1 µM eltanexor
were used as target cells [10:1 ratio: 76.3±5.3 vs. 69.9±2.3%
(P=0.0278), 5:1 ratio: 70.3±5.2% vs. 59.7±4.0% (P=0.0007), 2.5:1
ratio: 61.0±2.3% vs. 43.8±2.1% (P<0.0001), 1:1 ratio: 31.6±1.6%
vs. 17.7±2.0% (P<0.0001) (Fig.
5C)]. However, improvement of toxicity was not observed for CAR
T cells towards pre-treated Raji cells with 0.1 or 0.5 µM eltanexor
(Fig. 5D) that had previously shown
the lowest sensitivity to SINEs (Fig.
1A). However, pre-treating tumor cells with high concentrations
of eltanexor reversed the cytotoxicity of CAR T cells [Reh, 0.1 µM
eltanexor vs. DMSO: 5:1 ratio: 25.3±2.3 vs. 36.1±1.5% (P=0.0159);
Daudi, 0.5 µM eltanexor vs. DMSO: 10:1 ratio: 59.3±4.0 vs.
69.9±2.3% (P=0.0008), 5:1 ratio: 52.4±2.0 vs. 59.7±4.0% (P=0.0121),
2.5:1 ratio: 38.4±2.6 vs. 43.8±2.1% (P=0.0023) (Fig. 5B and C)]. This effect of reversing
cytotoxicity of CAR T cells was also observed for Nalm-6 cells
pre-treated with high concentrations of eltanexor, although this
was not statistically significant [0.1 µM eltanexor vs. DMSO: 1:1
ratio: 51.5±3.6 vs. 59.3±4.1% (P=0.0533) (Fig. 5A)].
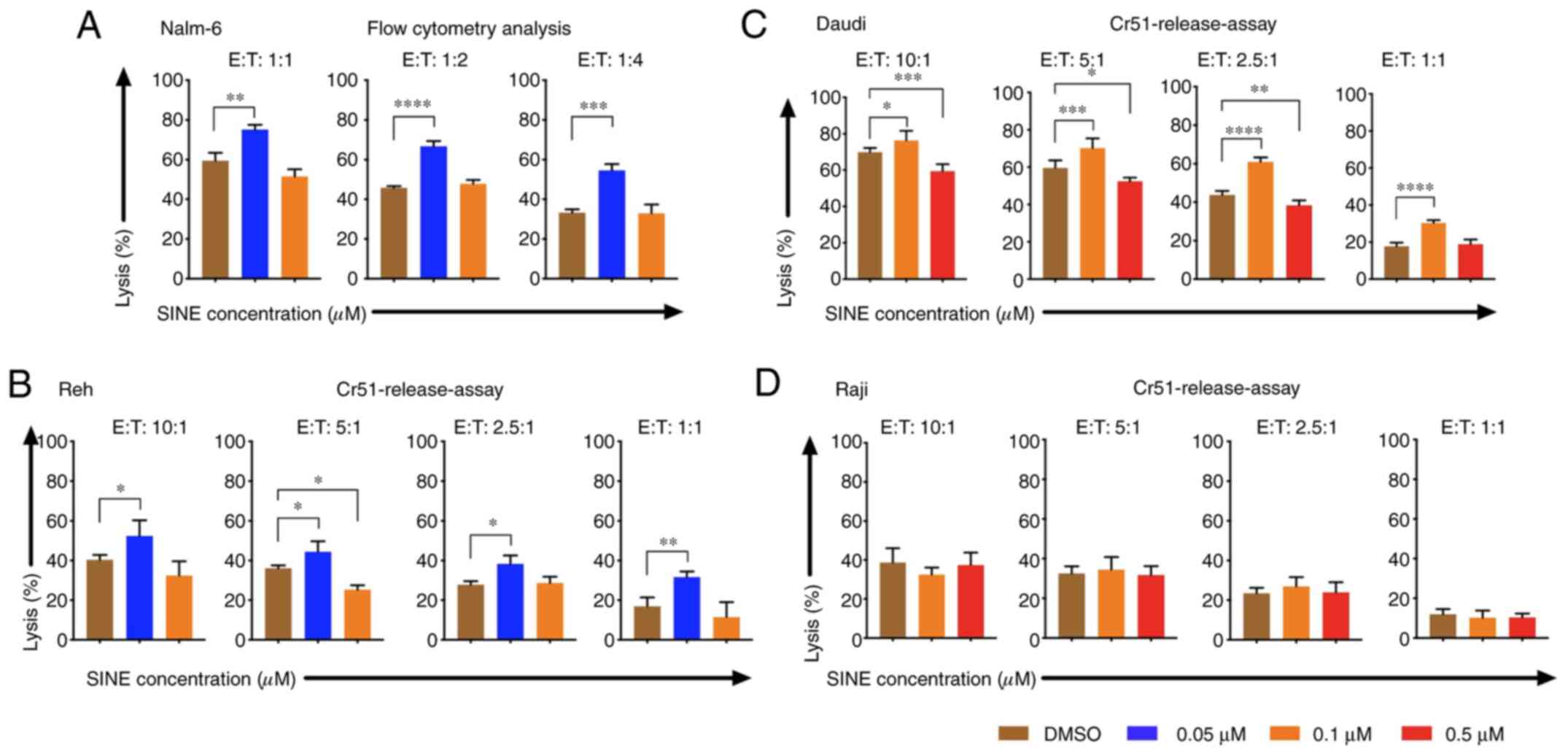 | Figure 5.Cytotoxicity of CAR T cells when
target cells were pre-treated with the SINE compound eltanexor.
Nalm-6 (A) Reh (B) Daudi (C) and Raji (D) cells were cultivated
with eltanexor at different concentrations (0.05 µM: Blue, 0.1 µM:
Orange, 0.5 µM: Red) or with DMSO as control (brown) for 24 h.
After washing with medium, CAR T cells were added. Cytotoxicity of
CAR T cells on Nalm-6 cells was assessed via flow cytometry
following co-cultivation for 24 h. Reh, Daudi and Raji cells were
co-cultured for 4 h with CAR T cells prior to evaluation of
cytotoxicity of CAR T cells via chromium release. Increased
cytotoxicity of CAR T cells was observed for Nalm-6, Reh and Daudi
cells when low concentrations of eltanexor pre-treatment were used
(0.05 µM on both Nalm-6 and Reh, 0.1 µM on Daudi) as compared to
the DMSO control (brown). Higher concentrations of eltanexor (0.1
µM on Reh, 0.5 µM on Daudi) abrogated the lytic effects of CAR T
cells. Improvement of lytic capacity after pretreatment with
eltanexor was not observed for Raji cells. Experiments were
performed in triplicate. Results are presented as mean ± standard
deviation (SD). *P<0.05, **P<0.005, ***P<0.0005,
****P<0.00005 indicate statistical significance. E: Effector
cells, i.e., CAR T cells; T: Target cells. i.e., Nalm-6, Reh, Daudi
and Raji cells. |
Cytokine release levels of CAR T cells
increased when tumor cells were pre-treated with eltanexor
CD4- and CD8-positive CAR T cells displayed higher
cytokine (TNF-α and IFN-γ) secretion levels after stimulation of
CAR T cells with Nalm-6 and Daudi cells when target cells had been
pre-treated with eltanexor [(Nalm-6: 0.05 µM eltanexor vs. DMSO):
CD4 TNF-α+: 66.7±2.0 vs. 40.9±5.9% (P=0.0157), CD4
IFN-γ+: 29.6±0.6 vs. 20.8±0.9% (P=0.0093), CD8
TNF-α+: 43.3±1.3 vs. 29.0±6.6% (P=0.1272), CD8
IFN-γ+: 53.1±0.2 vs. 46.3±0.8% (P=0.0056) (Fig. 6A, upper panel); Daudi (0.1 µM
eltanexor vs. DMSO): CD4 TNF-α+: 38.8±1.9 vs. 32.1±1.0%
(P=0.1805), CD4 IFN-γ+: 20.7±0.2 vs. 16.6±1.0%
(P=0.0299), CD8 TNF-α+: 28.0±0.7 vs. 22.3±0.8%
(P=0.0467), CD8 IFN-γ+: 46.2±0.4 vs. 39.4±1.1%
(P=0.0167) (Fig. 6C, upper panel)].
Although a trend towards an increase in multi-cytokine release
(IFN-γ and TNF-α double positive) was also observed after
stimulation of CAR T cells with pre-treated Nalm-6 (0.05 µM
eltanexor) and Daudi (0.1 µM eltanexor) cells, this was not
statistically significant (Fig. 6A and
C, lower panel). An increase in secretion of single cytokine or
multiple cytokines was also observed for Reh cells pre-treated with
0.1 µM eltanexor, but this was without statistical significance
when compared to DMSO (Fig. 6B).
Improved cytokine-secretion was not observed for CAR T cells
stimulated with Raji cells (Fig.
6D).
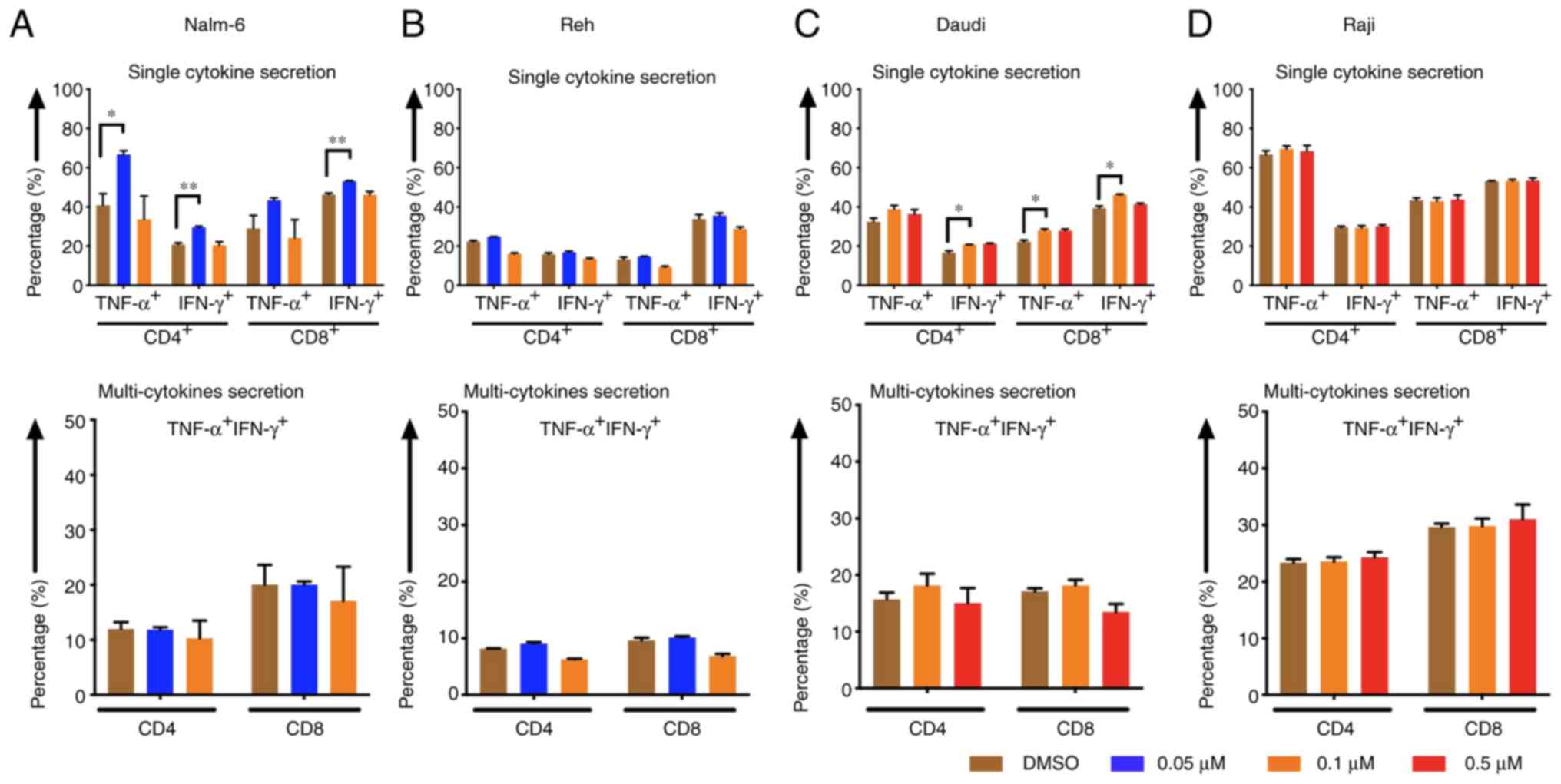 | Figure 6.Cytokine release of CAR T cells after
pre-treatment of tumor cells with the SINE compound eltanexor.
Nalm-6 (A) Reh (B) Daudi (C) and Raji (D) cells were cultivated
with eltanexor at different concentrations (0.05 µM: Blue, 0.1 µM:
Orange, 0.5 µM: Red) or with DMSO as control (brown). After washing
with culturing medium, CAR T cells were incubated with tumor cells
for 6 h. Single cytokine release (IFN-γ and TNF-α) of CD4- and
CD8-positive CAR T cells was assessed. Higher cytokine secretion
levels were detected after stimulation of CAR T cells with Nalm-6
(pre-treated with 0.05 µM eltanexor, blue, A, upper panel) and
Daudi cells (pre-treated with 0.1 µM eltanexor, orange, C, upper
panel) when compared to the DMSO control (brown, A and C, upper
panel). The increase was also observable when multi-cytokine
secretion (IFN-γ and TNF-α double positive, A and C, lower panel)
of CD4- and CD8-positive CAR T cells was measured, although this
was not statistically significant. No alternation of cytokine
secretion of CAR T cells towards pre-treated Raji cells
(pre-treated with 0.1 and 0.5 µM eltanexor, D) was detected.
Experiments were performed in triplicate. Results are presented as
mean ± standard deviation (SD). *P<0.05 and **P<0.005
indicate statistical significance. |
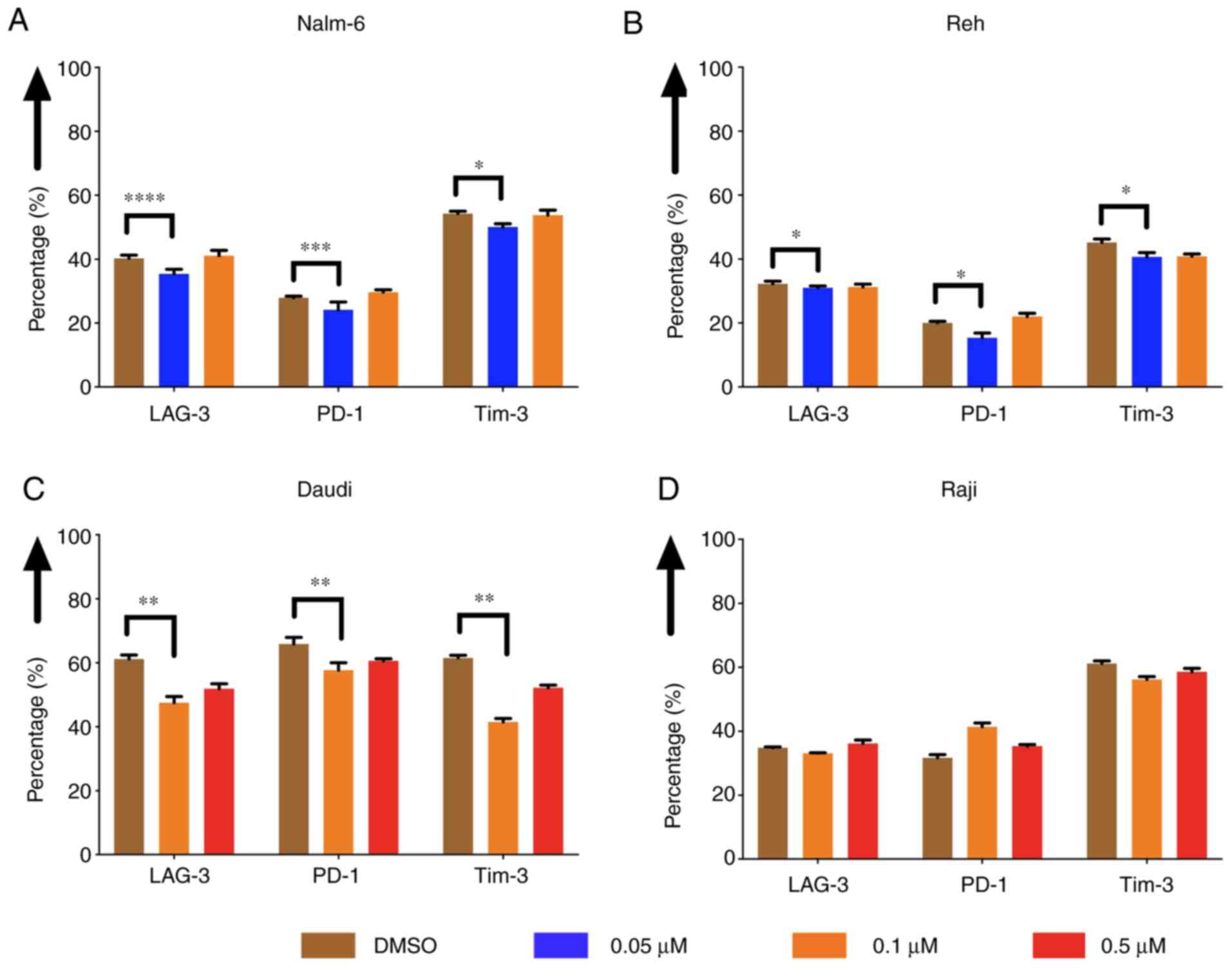
The expression of exhaustion markers
of CAR T cells was decreased when tumor cells were pre-treated with
eltanexor
The expression of exhaustion markers of CAR T cells,
such as LAG-3, PD-1 and Tim-3, was evaluated after co-culturing CAR
T cells with pre-treated Nalm-6, Reh, Daudi and Raji cells. The
expression was decreased when Nalm-6 and Reh had been pre-treated
with eltanexor (0.05 µM) compared to DMSO [Nalm-6: LAG-3: 35.7±1.4
vs. 40.3±1.0% (P<0.0001), PD-1: 24.1±2.5 vs. 27.9±0.5%
(P=0.0004), Tim-3: 50.1±1.0 vs. 54.3±0.8% (P=0.0225) (Fig. 7A); Reh: LAG-3: 31.0±0.6 vs. 32.3±0.8%
(P=0.0335), PD-1: 15.4±1.5% vs. 20.0±0.5% (P=0.0317), Tim-3:
40.7±1.3 vs. 45.7±1.2% (P=0.0100) (Fig.
7B)]. Exhaustion marker expression on CAR T cells co-cultured
with pre-treated Daudi cells (0.1 µM eltanexor) compared to DMSO
was also decreased [LAG-3: 47.5±2.0 vs. 61.2±1.2% (P=0.0030), PD-1:
57.7±2.3 vs. 65.9±2.0% (P=0.1585), Tim-3: 41.5±1.1 vs. 61.6±0.8%
(P=0.0060) (Fig. 7C)]. No difference
in exhaustion marker expression was observed for CAR T cells
co-cultured with pre-treated Raji cells (0.1 µM) (Fig. 7D). The decrease in exhaustion markers
was not observed when CAR T cells were cultivated simultaneously
with Daudi cells and eltanexor (Fig.
S2).
Discussion
In patients with lymphoid malignancies CAR T cells
have mediated high response rates. However, relapses and resistance
after treatment with CAR T-cell therapy (10,45)
constitute a challenge; to address this, several approaches are
under investigation. To improve persistence and efficacy of CAR T
cells, CAR T-cell production can be enhanced: For example, the
PI3Kδ inhibitor idelalisib rendered enhanced in vivo
function to CAR T cells that were manufactured from T-lymphocytes
of CLL patients (42), a starting
T-cell population with limited CAR T-cell responses due to
functional characteristics of terminally differentiated lymphocytes
(46). In addition, armored CAR T
cells have been developed. Due to additional genetic modifications,
these advanced CAR T cells intrinsically express additional
costimulatory ligands or cytokines to augment CAR T-cell response
(47). Furthermore, approaches that
combine different mechanisms to target malignancies are of
considerable interest in order to prevent escape of malignant cells
from CAR T-cell treatment. Combination of CAR T cells with
PD-1/PD-L1 inhibitors has been shown to enhance CAR T-cell efficacy
and improve the clinical outcome of treated patients (48,49). CAR T
cells combined with reactive oxygen species (ROS) accelerators were
able to overcome tumor microenvironment-mediated treatment
resistance (43). In combination with
ibrutinib, CAR T-cell proliferation and antitumor efficacy in a
human xenograft model were enhanced (50) while occurrence of CAR T-cell toxicity,
i.e., cytokine release syndrome (CRS), was reduced (51). Recent clinical studies confirmed these
data rendering superior clinical responses to CLL patients treated
concomitantly with ibrutinib and CAR T cells (52).
Due to their general anti-malignant effect, SINEs
constitute interesting combination partners for CAR T-cell therapy.
XPO1 promotes cell deregulation exporting TSPs involved in
apoptotic-inhibition from the nucleus (53) and SINEs can disrupt this process and
regain tumor control (33,35,53). In
this study, we evaluated the potential of SINEs in combination with
third-generation CD19.CAR T cells.
The approved SINE compound selinexor as well as the
second-generation SINE eltanexor mediated robust in vitro
growth-inhibition of CD19-positive tumor cell lines. These data are
in accordance with previous reports demonstrating that selinexor at
a concentration up to 0.22 µM induced apoptosis in isolated MM
cells (27). Eltanexor has been shown
to mediate apoptosis in primary CLL cells and significantly
inhibited proliferation of DLBCL cell lines (54). Moreover, eltanexor at a concentration
of 0.15 µM has shown to induce apoptosis in AML cell lines but has
displayed a better tolerability when compared to selinexor
(55). With regards to the superior
toxicity profile of eltanexor over selinexor as demonstrated also
by others (39), we performed
experiments addressing the combinatorial approach of SINEs and CAR
T cells with eltanexor.
Our data demonstrate that sensitivity of tumor cells
to SINEs correlated with the XPO1 protein levels in Nalm-6 and Reh
cells. Besides confirming the anti-tumor efficacy of SINEs on
malignant cells, the impact of SINEs on T cells, i.e., also CAR T
cells, was assessed. It is known that in T cells XPO1-inhibition
affects transcription factors that are crucial for T-cell
functionality, e.g., NFATc1, p100 and p65 (subunits of NF-κB),
cIAP1, stat1 and STAT3 (36,55,56). We
confirmed this by observing that eltanexor decreased the levels of
phosphorylated STAT3. The reduction of phosphorylated STAT3 in the
cytoplasm with unaltered levels of total STAT3 suggests that
retention of STAT3 within the nucleus may impair anti-tumor
function of CAR T cells. This is in line with previous findings
that demonstrated that the XPO1 inhibitor leptomycin B decreased
the levels of phosphorylated STAT3 in the cytoplasm by limiting the
transport through the nuclear membrane and accumulating the
inactive STAT3 conformation within the nucleus (57). In addition, CLL patients achieving a
complete response after CAR T-cell treatment had higher activation
levels of the IL-6/STAT3 pathway when compared to non-responding
patients, suggesting that decrease of phosphorylated-STAT3 is
associated with poor clinical outcomes (58). Moreover, a novel gene-edited CAR
containing a JAK-STAT3 signaling domain mediated superior
anti-tumor effects (59).
Despite identifying STAT3 as a relevant SINE target,
further studies extending the analysis to other relevant proteins
and transcription factors in CAR T cells are required to define CAR
T-cell impairment by SINEs. Our study is further limited by the
fact that the effect of SINEs on CAR T cells was only assessed
under artificial two-dimensional cell culture conditions, which do
not reflect the dynamic activity of SINEs in tumor cells and in CAR
T cells. Further evaluations in more complex culture conditions are
required to clarify the mechanisms of the combination of SINEs and
CAR T cells.
With regard to a potential combinatory approach,
optimal synergistic effects of SINEs with CAR T cells should render
effective inhibition of target tumor cells without affecting CAR T
cells. Given that in this study SINEs impaired CAR T-cell function
already at low concentrations, concomitant administration of SINEs
and CAR T cells does not seem advisable. However, applying a
pre-treatment strategy to protect the CAR T cells from SINEs seems
to be promising. In fact, when used sequentially, pre-treatment
with eltanexor mediated enhanced anti-tumor cytotoxicity of CAR T
cells. The increase in secretion of cytokines and the decrease in
expression of exhaustion markers on CAR T cells were consistent
with improved cytotoxicity, which may-at least partially-explain
the mechanism of enhancement.
According to our data, Reh and Nalm-6 were more
sensitive than NHL cells Daudi and Raji to SINE treatment.
Accordingly, we chose 0.05 and 0.1 µM as SINE concentrations for
the ALL cell group (Reh and Nalm-6 cells), and higher
concentrations of 0.1 and 0.5 µM to treat the NHL group (Daudi and
Raji cells). In fact, in both groups a lower SINE concentration
(0.05 µM for ALL; 0.1 µM for NHL) was more effective, increasing
CAR T-cell cytotoxicity and enhancing CAR T-cell cytokine release.
Consequently, pre-sensitizing tumor cells with SINEs may display a
window of effect-concentration, whereby higher SINE concentrations
may be associated with severe damage and killing of tumor cells
resulting in loss of targets for CAR T cells and consequently
decreased cytokine release. In contrast, lower SINE concentrations
may pre-sensitize the tumor cells only (without completely killing
them), preparing them for treatment with CAR T cells. Taken
together, this finding is clinically promising and supports the
combination approach of SINEs and CAR T cells as a low dose of
SINEs displaying a favourable toxicity profile may be sufficient to
enhance CAR T-cell function.
In summary, this study has focused on the intrinsic
toxicity of SINEs against malignant cells. The toxicity of SINEs,
however, also affected CAR T cells and significantly impaired their
function, thereby limiting the potential for the combination and
concomitant application of SINEs and CAR T cells. Nonetheless,
pre-sensitizing tumor cells with eltanexor was shown to be an
effective strategy to improve anti-malignant effects. Therefore,
sequential use of SINEs and CAR T cells is a potential option to
improve the efficacy of CAR T-cell treatment and should be
addressed in further trials.
Supplementary Material
Supporting Data
Acknowledgements
We thank Amy Publicover for editing the text. We are
grateful to Ulrike Gern and Stefanie Mechler as technicians
supporting this study.
Funding
MLS was supported by the Olympia Morata Program of
the Medical Faculty of Heidelberg. SW was supported by the China
Scholarship Council.
Availability of data and materials
The datasets used and/or analyzed within this study
are available from the corresponding author on reasonable request.
Original flow cytometry analysis data are displayed within the
supplementary material (Figs.
S3–S12).
Authors' contributions
SW, MLS, MS and LS designed the study; SW and HY
performed the experiments; SW analyzed the data and wrote the
primary manuscript; MLS, LS, TS and MS revised the manuscript
critically for important intellectual content; SW, LW, MLS, LS, BN,
WG, SS, MN, CK, AS and CMT discussed and contributed to the
experimental design. SW and MLS are responsible for confirming the
authenticity of all the raw data. All authors reviewed the
manuscript. All authors approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
MS received funding for collaborative research from
Apogenix, Hexal and Novartis, travel grants from Hexal and Kite,
financial support for educational activities and conferences from
Bluebird Bio, Kite and Novartis, is an advisory board member for
MSD and (co-)PI of clinical trials of MSD, GSK, Kite and BMS, as
well as co-founder and shareholder of TolerogenixX, Ltd. AS
received travel grants from Hexal and Jazz Pharmaceuticals,
research grant from Therakos/Mallinckrodt and is co-founder of
TolerogenixX, Ltd. AS is a part-time employee of TolerogenixX, Ltd.
LS is a full-time employee of Takeda (current address: Oncology
Business Unit, Takeda Pharma Vertrieb GmbH & Co. KG, Berlin,
Germany). LW is a full-time employee of TolerogenixX, Ltd. CMT:
Bayer AG (research support); Pfizer, Janssen-Cilag GmbH (advisory
board member). Pfizer, Daiichi Sankyo, BiolineRx (grants and/or
provision of investigational medicinal products). SW, BN, WG, SS,
MN, HY, CK, TS and MLS have no conflict of interest to declare.
References
|
1
|
Mohanty R, Chowdhury CR, Arega S, Sen P,
Ganguly P and Ganguly N: CAR T cell therapy: A new era for cancer
treatment (Review). Oncol Rep. 42:2183–2195. 2019.PubMed/NCBI
|
|
2
|
Ma CC, Wang ZL, Xu T, He ZY and Wie YQ:
The approved gene therapy drugs worldwide: From 1998 to 2019.
Biotechnol Adv. 40:1075022020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schubert ML, Hückelhoven A, Hoffmann JM,
Schmitt A, Wuchter P, Sellner L, Hofmann S, Ho AD, Dreger P and
Schmitt M: Chimeric antigen receptor T cell therapy targeting
CD19-positive leukemia and lymphoma in the context of stem cell
transplantation. Hum Gene Ther. 27:758–771. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sadelain M, Riviere I and Riddell S:
Therapeutic T cell engineering. Nature. 545:423–431. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
June CH and Sadelain M: Chimeric antigen
receptor therapy. N Engl J Med. 379:64–73. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maude SL, Laetsch TW, Buechner J, Rives S,
Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers
GD, et al: Tisagenlecleucel in children and young adults with
B-cell lymphoblastic leukemia. N Engl J Med. 378:439–448. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davila ML, Riviere I, Wang X, Bartido S,
Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska
M, et al: Efficacy and toxicity management of 19-28z CAR T cell
therapy in B cell acute lymphoblastic leukemia. Sci Transl Med.
6:224ra2252014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park JH, Riviere I, Gonen M, Wang X,
Sénéchal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et
al: Long-term follow-up of CD19 CAR therapy in acute lymphoblastic
leukemia. N Engl J Med. 378:449–459. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Porter DL, Hwang WT, Frey NV, Lacey SF,
Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al:
Chimeric antigen receptor T cells persist and induce sustained
remissions in relapsed refractory chronic lymphocytic leukemia. Sci
Transl Med. 7:303ra1392015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Turtle CJ, Hay KA, Hanafi LA, Li D,
Cherian S, Chen X, Wood B, Lozanski A, Byrd JC, Heimfeld S, et al:
Durable molecular remissions in chronic lymphocytic leukemia
treated with CD19-specific chimeric antigen receptor-modified T
cells after failure of ibrutinib. J Clin Oncol. 35:3010–3020. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schuster SJ, Svoboda J, Chong EA, Nasta
SD, Mato AR, Anak O, Brogdon JL, Pruteanu-Malinici I, Bhoj V,
Landsburg D, et al: Chimeric antigen receptor T cells in refractory
B-cell lymphomas. N Engl J Med. 377:2545–2554. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Neelapu SS, Locke FL, Bartlett NL, Lekakis
LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T,
Lin Y, et al: Axicabtagene ciloleucel CAR T-cell therapy in
refractory large B-cell lymphoma. N Engl J Med. 377:2531–2544.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Grupp S, Hu ZH, Zhang Y, Keating A,
Pulsipher MA, Philips C, Margossian SP, Rosenthal J, Salzberg D,
Schiff DE, et al: Tisagenlecleucel Chimeric Antigen Receptor (CAR)
T-Cell Therapy for Relapsed/Refractory Children and young adults
with Acute Lymphoblastic Leukemia (ALL): Real World Experience from
the Center for International Blood and Marrow Transplant Research
(CIBMTR) and Cellular Therapy (CT) Registry. Blood. 134:26192019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jaglowski S, Hu ZH, Zhang Y, Kamdar M,
Ghosh M, Lulla P, Sasine J, Perales MA, Hematti P, Nikiforow S, et
al: Tisagenlecleucel Chimeric Antigen Receptor (CAR) T-Cell Therapy
for Adults with Diffuse Large B-Cell Lymphoma (DLBCL): Real World
Experience from the Center for International Blood & Marrow
Transplant Research (CIBMTR) Cellular Therapy (CT) Registry. Blood.
134:7662019. View Article : Google Scholar
|
|
15
|
Nastoupil LJ, Jain MD, Feng L, Spiegel JY,
Ghobadi A, Lin Y, Dahiya S, Lunning M, Lekakis L, Reagan P, et al:
Standard-of-Care Axicabtagene Ciloleucel for relapsed or refractory
large B-cell lymphoma: Results From the US Lymphoma CAR T
consortium. J Clin Oncol. 38:3119–3128. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang M, Munoz J, Goy A, Locke FL, Jacobson
CA, Hill BT, Timmerman JM, Holmes H, Jaglowski S, Flinn IW, et al:
KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell
lymphoma. N Engl J Med. 382:1331–1342. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sotillo E, Barrett DM, Black KL, Bagashev
A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et
al: Convergence of acquired mutations and alternative splicing of
CD19 enables resistance to CART-19 immunotherapy. Cancer Discov.
5:1282–1295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fischer J, Paret C, El Malki K, Alt F,
Wingerter A, Neu MA, Kron B, Russo A, Lehmann N, Roth L, et al:
CD19 isoforms enabling resistance to CART-19 immunotherapy are
expressed in B-ALL patients at initial diagnosis. J Immunother.
40:187–195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee DW, Kochenderfer JN, Stetler-Stevenson
M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M,
Shah NN, et al: T cells expressing CD19 chimeric antigen receptors
for acute lymphoblastic leukaemia in children and young adults: A
phase 1 dose-escalation trial. Lancet. 385:517–528. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shah NN and Fry TJ: Mechanisms of
resistance to CAR T cell therapy. Nat Rev Clin Oncol. 16:372–385.
2019.PubMed/NCBI
|
|
21
|
Cook A, Bono F, Jinek M and Conti E:
Structural biology of nucleocytoplasmic transport. Annu Rev
Biochem. 76:647–671. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao Y, Dong Y, Lin F, Zhao H, Shen Z, Chen
P, Sun YJ, Tang LN and Zheng SE: The expression of CRM1 is
associated with prognosis in human osteosarcoma. Oncol Rep.
21:229–235. 2009.PubMed/NCBI
|
|
23
|
van der Watt PJ, Zemanay W, Govender D,
Hendricks DT, Parker MI and Leaner VD: Elevated expression of the
nuclear export protein, CRM1 (exportin 1), associates with human
oesophageal squamous cell carcinoma. Oncol Rep. 32:730–738. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Noske A, Weichert W, Niesporek S, Roske A,
Buckendahl AC, Koch I, Sehouli J, Dietel M and Denkert C:
Expression of the nuclear export protein chromosomal region
maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian
cancer. Cancer. 112:1733–1743. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shen A, Wang Y, Zhao Y, Zou L, Sun L and
Cheng C: Expression of CRM1 in human gliomas and its significance
in p27 expression and clinical prognosis. Neurosurgery. 65:153–160.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van der Watt PJ, Maske CP, Hendricks DT,
Parker MI, Denny L, Govender D, Birrer MJ and Leaner VD: The
Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed
in cervical cancer and are critical for cancer cell survival and
proliferation. Int J Cancer. 124:1829–1840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tai YT, Landesman Y, Acharya C, Calle Y,
Zhong MY, Cea M, Tannenbaum D, Cagnetta A, Reagan M, Munshi AA, et
al: CRM1 inhibition induces tumor cell cytotoxicity and impairs
osteoclastogenesis in multiple myeloma: Molecular mechanisms and
therapeutic implications. Leukemia. 28:155–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kojima K, Kornblau SM, Ruvolo V, Dilip A,
Duvvuri S, Davis RE, Zhang M, Wang Z, Coombes KR, Zhang N, et al:
Prognostic impact and targeting of CRM1 in acute myeloid leukemia.
Blood. 121:4166–4174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo B, Huang L, Gu Y, Li C, Lu H, Chen G,
Peng Z and Feng Z: Expression of exportin-1 in diffuse large B-cell
lymphoma: Immunohistochemistry and TCGA analyses. Int J Clin Exp
Pathol. 11:5547–5560. 2018.PubMed/NCBI
|
|
30
|
Lapalombella R, Sun Q, Williams K,
Tangeman L, Jha S, Zhong Y, Goettl V, Mahoney E, Berglund C, Gupta
S, et al: Selective inhibitors of nuclear export show that
CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood.
120:4621–4634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang K, Wang M, Tamayo AT, Shacham S,
Kauffman M, Lee J, Zhang L, Ou Z, Li C, Sun L, et al: Novel
selective inhibitors of nuclear export CRM1 antagonists for therapy
in mantle cell lymphoma. Exp Hematol. 41:67–78.e4. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bahlis NJ, Sutherland H, White D, Sebag M,
Lentzsch S, Kotb R, Venner CP, Gasparetto C, Del Col A, Neri P, et
al: Selinexor plus low-dose bortezomib and dexamethasone for
patients with relapsed or refractory multiple myeloma. Blood.
132:2546–2554. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vercruysse T, De Bie J, Neggers JE,
Jacquemyn M, Vanstreels E, Schmid-Burgk JL, Hornung V, Baloglu E,
Landesman Y, Senapedis W, et al: The second-generation exportin-1
inhibitor KPT-8602 demonstrates potent activity against acute
lymphoblastic leukemia. Clin Cancer Res. 23:2528–2541. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ming M, Wu W, Xie B, Sukhanova M, Wang W,
Kadri S, Sharma S, Lee J, Shacham S, Landesman Y, et al: XPO1
inhibitor selinexor overcomes intrinsic ibrutinib resistance in
mantle cell lymphoma via nuclear retention of IκB. Mol Cancer Ther.
17:2564–2574. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuruvilla J, Savona M, Baz R, Mau-Sorensen
PM, Gabrail N, Garzon R, Stone R, Wang M, Savoie L, Martin P, et
al: Selective inhibition of nuclear export with selinexor in
patients with non-Hodgkin lymphoma. Blood. 129:3175–3183. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gravina GL, Senapedis W, McCauley D,
Baloglu E, Shacham S and Festuccia C: Nucleo-cytoplasmic transport
as a therapeutic target of cancer. J Hematol Oncol. 7:852014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chari A, Vogl DT, Gavriatopoulou M, Nooka
AK, Yee AJ, Huff CA, Moreau P, Dingli D, Cole C, Lonial S, et al:
Oral selinexor-dexamethasone for triple-class refractory multiple
myeloma. N Engl J Med. 381:727–738. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Machlus KR, Wu SK, Vijey P, Soussou TS,
Liu ZJ, Shacham E, Unger TJ, Kashyap T, Klebanov B, Sola-Visner M,
et al: Selinexor-induced thrombocytopenia results from inhibition
of thrombopoietin signaling in early megakaryopoiesis. Blood.
130:1132–1143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Etchin J, Berezovskaya A, Conway AS,
Galinsky IA, Stone RM, Baloglu E, Senapedis W, Landesman Y,
Kauffman M, Shacham S, et al: KPT-8602, a second-generation
inhibitor of XPO1-mediated nuclear export, is well tolerated and
highly active against AML blasts and leukemia-initiating cells.
Leukemia. 31:143–150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schubert ML, Schmitt A, Sellner L, Neuber
B, Kunz J, Wuchter P, Kunz A, Gern U, Michels B, Hofmann S, et al:
Treatment of patients with relapsed or refractory CD19+ lymphoid
disease with T lymphocytes transduced by RV-SFG.CD19.CD28.4-1BBzeta
retroviral vector: A unicentre phase I/II clinical trial protocol.
BMJ Open. 9:e0266442019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hoffmann JM, Schubert ML, Wang L,
Huckelhoven A, Sellner L, Stock S, Schmitt A, Kleist C, Gern U,
Loskog A, et al: Differences in expansion potential of naive
chimeric antigen receptor T cells from healthy donors and untreated
chronic lymphocytic leukemia patients. Front Immunol. 8:19562018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stock S, Ubelhart R, Schubert ML, Fan F,
He B, Hoffmann JM, Wang L, Wang S, Gong W, Neuber B, et al:
Idelalisib for optimized CD19-specific chimeric antigen receptor T
cells in chronic lymphocytic leukemia patients. Int J Cancer.
145:1312–1324. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yoo HJ, Liu Y, Wang L, Schubert ML,
Hoffmann JM, Wang S, Neuber B, Huckelhoven-Krauss A, Gern U,
Schmitt A, et al: Tumor-specific reactive oxygen species
accelerators improve chimeric antigen receptor t cell therapy in B
cell malignancies. Int J Mol Sci. 20:24692019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang L, Gong W, Wang S, Neuber B, Sellner
L, Schubert ML, Huckelhoven-Krauss A, Kunz A, Gern U, Michels B, et
al: Improvement of in vitro potency assays by a resting step for
clinical-grade chimeric antigen receptor engineered T cells.
Cytotherapy. 21:566–578. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Armitage JO, Gascoyne RD, Lunning MA and
Cavalli F: Non-hodgkin lymphoma. Lancet. 390:298–310. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Riches JC, Davies JK, McClanahan F, Fatah
R, Iqbal S, Agrawal S, Ramsay AG and Gribben JG: T cells from CLL
patients exhibit features of T-cell exhaustion but retain capacity
for cytokine production. Blood. 121:1612–1621. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schubert ML, Hoffmann JM, Dreger P,
Muller-Tidow C and Schmitt M: Chimeric antigen receptor transduced
T cells: Tuning up for the next generation. Int J Cancer.
142:1738–1747. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gargett T, Yu W, Dotti G, Yvon ES, Christo
SN, Hayball JD, Lewis ID, Brenner MK and Brown MP: GD2-specific CAR
T cells undergo potent activation and deletion following antigen
encounter but can be protected from activation-induced cell death
by PD-1 blockade. Mol Ther. 24:1135–1149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao Y, Lu W, Sun R, Jin X, Cheng L, He X,
Wang L, Yuan T, Lyu C and Zhao M: Anti-CD19 chimeric antigen
receptor T cells in combination with nivolumab are safe and
effective against relapsed/refractory B-cell Non-Hodgkin lymphoma.
Front Oncol. 9:7672019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fraietta JA, Beckwith KA, Patel PR, Ruella
M, Zheng Z, Barrett DM, Lacey SF, Melenhorst JJ, McGettigan SE,
Cook DR, et al: Ibrutinib enhances chimeric antigen receptor T-cell
engraftment and efficacy in leukemia. Blood. 127:1117–1127. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ruella M, Kenderian SS, Shestova O,
Klichinsky M, Melenhorst JJ, Wasik MA, Lacey SF, June CH and Gill
S: Kinase inhibitor ibrutinib to prevent cytokine-release syndrome
after anti-cd19 chimeric antigen receptor t cells for b-cell
neoplasms. Leukemia. 31:246–248. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gauthier J, Hirayama AV, Purushe J, Hay
KA, Lymp J, Li DH, Yeung CCS, Sheih A, Pender BS, Hawkins RM, et
al: Feasibility and efficacy of CD19-targeted CAR-T cells with
concurrent ibrutinib for CLL after ibrutinib failure. Blood.
135:1650–1660. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tan DS, Bedard PL, Kuruvilla J, Siu LL and
Razak AR: Promising SINEs for embargoing nuclear-cytoplasmic export
as an anticancer strategy. Cancer Discov. 4:527–537. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hing ZA, Fung HY, Ranganathan P, Mitchell
S, El-Gamal D, Woyach JA, Williams K, Goettl VM, Smith J, Yu X, et
al: Next-generation XPO1 inhibitor shows improved efficacy and in
vivo tolerability in hematological malignancies. Leukemia.
30:2364–2372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Xu D, Grishin NV and Chook YM: Nesdb: A
database of NES-containing CRM1 cargoes. Mol Biol Cell.
23:3673–3676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tyler PM, Servos MM, de Vries RC, Klebanov
B, Kashyap T, Sacham S, Landesman Y, Dougan M and Dougan SK:
Clinical dosing regimen of selinexor maintains normal immune
homeostasis and T-cell effector function in mice: Implications for
combination with immunotherapy. Mol Cancer Ther. 16:428–439. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bhattacharya S and Schindler C: Regulation
of STAT3 nuclear export. J Clin Invest. 111:553–559. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fraietta JA, Lacey SF, Orlando EJ,
Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y,
O'Connor RS, Hwang WT, et al: Determinants of response and
resistance to CD19 chimeric antigen receptor (CAR) T cell therapy
of chronic lymphocytic leukemia. Nat Med. 24:563–571. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kagoya Y, Tanaka S, Guo T, Anczurowski M,
Wang CH, Saso K, Butler MO, Minden MD and Hirano N: A novel
chimeric antigen receptor containing a JAK-STAT signaling domain
mediates superior antitumor effects. Nat Med. 24:352–359. 2018.
View Article : Google Scholar : PubMed/NCBI
|