Introduction
Wnt signaling is implicated in various physiological
processes, including cell proliferation and differentiation,
metabolism, development, tissue homeostasis and tissue regeneration
(1–6).
In addition, the aberrant activation of Wnt signaling, which
contributes to cancer progression, invasion, metastasis and
recurrence, has been detected in various types of cancer, including
epidermoid, breast, prostate and lung malignancies, as well as in
glioblastoma (GBM) (7–9). Notably, Wnt3A has been reported to be
overexpressed in the human tumor microenvironment and in tumor
cells (10–13). Additionally, Wnt3A expression has been
reported to be associated with tumor grade (12,13). Wnt3A
is considered to function as an oncogenic factor through the
promotion of proliferation, invasion, metastasis and
chemo/radioresistance in a number of tumor types, including
epidermoid, breast, prostate and hepatocellular malignancies, as
well as in GBM (12–16).
In general, Wnt signaling is classified into the
canonical Wnt pathway (β-catenin-dependent) and the non-canonical
Wnt pathway (β-catenin-independent) (17,18). In
the canonical Wnt pathway, the binding of Wnt ligand to the
low-density lipoprotein receptor-related protein-5/6 and Frizzled
receptors results in the inhibition of the GSK-3β-mediated
phosphorylation of β-catenin. Subsequently, this promotes β-catenin
stabilization and translocation to the nucleus, where β-catenin
interacts with T-cell-specific factor (TCF)/lymphoid
enhancer-binding factor (LEF) and coactivators, in order to
activate Wnt target genes involved in the development of cancer
(19–23). In the non-canonical Wnt pathway, Wnt
signaling activates phospholipase C or small GTPases through the
binding of the Wnt ligand to Frizzled receptors and Ror/Ryk
co-receptors or Frizzled receptors, respectively, inducing a wide
range of cellular processes, including actin cytoskeleton
remodeling and cell motility (8,22,24). Accumulating evidence has indicated
that cancer cells acquire the ability to migrate and metastasize
through the non-canonical Wnt pathway (7,24–26).
The Warburg effect, a key metabolic hallmark of
cancer, is the phenomenon through which cancer cells produce
energy, predominantly through aerobic glycolysis and regardless of
the extracellular oxygen levels (27). Due to the reduced efficiency of
glycolysis in ATP production, cancer cells increase glucose uptake
for ATP production, followed by lactic acid fermentation in the
cytosol (28). This metabolic
alteration is required for the increased energy and biosynthetic
demands of tumor cells and facilitates their rapid growth (29,30).
β-catenin-dependent canonical Wnt signaling has been found to be
indirectly involved in the regulation of glycolysis though the
regulation of mitochondrial enzyme expression, including pyruvate
dehydrogenase kinase 1 and pyruvate carboxylase (31,32).
However, whether the β-catenin-independent non-canonical Wnt
signaling directly regulates the Warburg effect in cancer cells
remains unknown.
In glucose metabolism, phosphofructokinase 1 (PFK1),
which catalyzes the rate-limiting reaction of glycolysis, converts
fructose 6-phosphate and ATP to fructose 1,6-bisphosphate and ADP
(33). PFK1 has three isoforms, PFK1
platelet (PFKP), PFK1 liver (PFKL) and PFK1 muscle (PFKM), as
originally named, since PFKL is the most abundant isoform in the
liver, whereas PFKM and PFKP are the only isoforms present in
muscles and platelets in adults, respectively (33,34).
However, all three isoforms have been detected in the brain and
other tissues (35,36). Additionally, it has been previously
reported by the authors that all three isoforms are expressed in
GBM cells; PFKP is the prominent PFK1 isoform in GBM cells and has
been found to be overexpressed in human GBM specimens (37). AKT-mediated phosphorylation of PFKP at
S386 inhibits the proteasomal degradation of PFKP, resulting in the
increased PFKP expression and promoting aerobic glycolysis and
brain tumor growth (37). However,
the role of PFKP in Wnt signaling-induced tumor development remains
unknown.
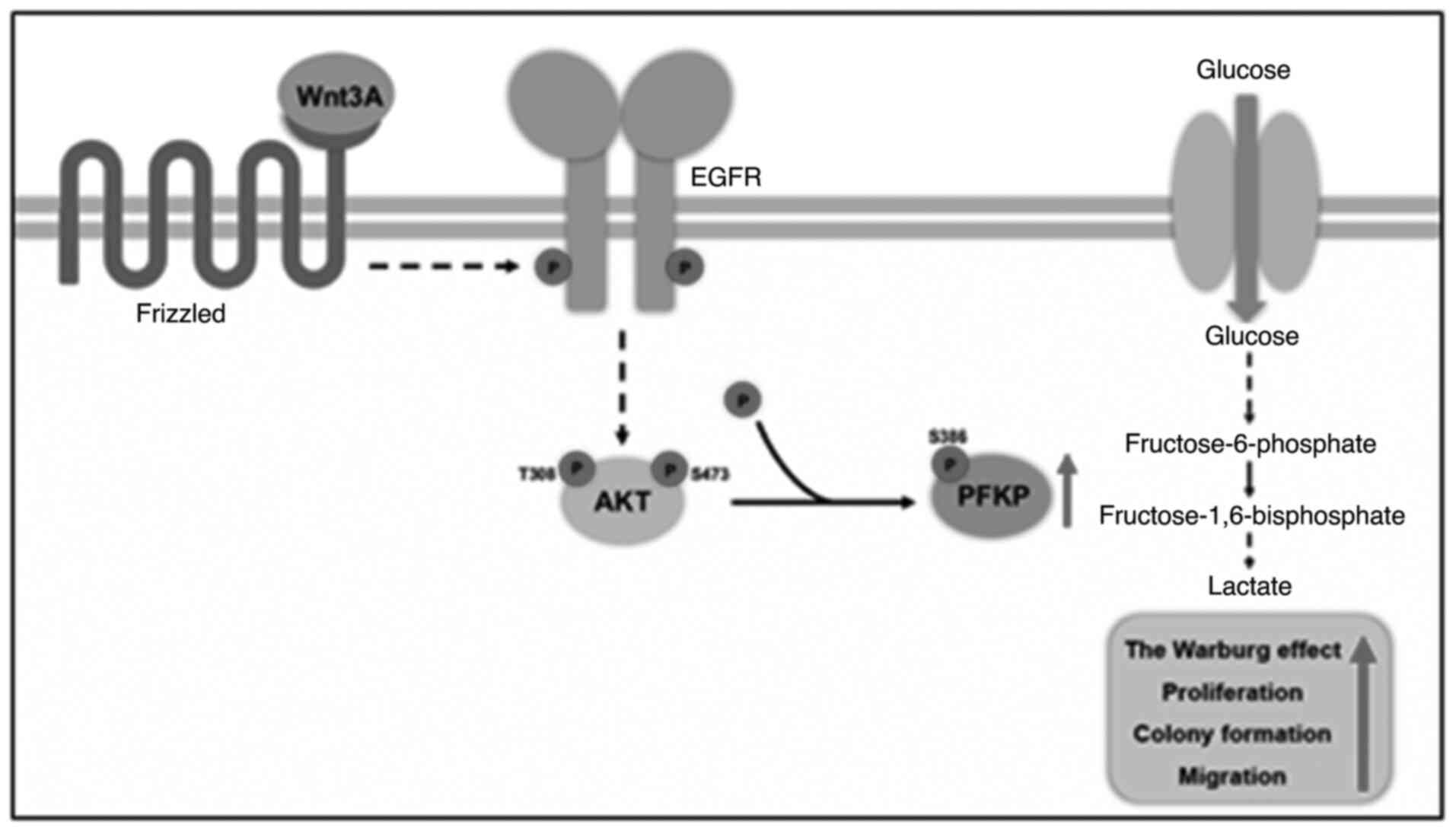
In the present study, it was demonstrated that Wnt
signaling transactivated epidermal growth factor receptor (EGFR) in
order to induce PI3K/AKT activation, resulting in PFKP
phosphorylation at S386 and stabilizing PFKP protein, thereby
promoting the Warburg effect, as well as cell proliferation, colony
formation and cancer cell migration.
Materials and methods
Reagents and antibodies
The following reagents and antibodies were purchased
from the indicated companies: Rabbit polyclonal antibodies
recognizing PFKP (cat. no. 12746, 1:1,000 for western blot
analysis), phosphorylated (p-)AKT (pT308, cat. no. 4056, 1:1,000
for western blot analysis), p-AKT (pS473, cat. no. 4060, 1:1,000
for western blot analysis) and AKT (cat. no. 9272, 1:1,000 for
western blot analysis) from Cell Signaling Technology, Inc.; mouse
monoclonal antibodies for PFKL (A-6, cat. no. sc-393713, 1:500 for
western blot analysis), PFKM (cat. no. sc-67028, 1:1,000 for
western blot analysis), PFK2 (cat. no. sc-377416, 1:500 for western
blot analysis), lactate dehydrogenase A (LDHA) (cat. no. sc-137243,
1:500 for western blot analysis) and β-catenin (E-5, cat. no.
sc-7963, 1:200 for western blot analysis) from Santa Cruz
Biotechnology, Inc.; mouse monoclonal antibody for tubulin (clone
B-5-1-2, T6074, 1:5,000 for western blot analysis) from
MilliporeSigma; rabbit polyclonal antibodies for EGFR (pY869, cat.
no. 11229, 1:1,000 for western blot analysis) and PFKP [pS386,
1:1,000 for western blot analysis; customized as previously
described (37)] from Signalway
Antibody LLC; mouse monoclonal EGFR antibody (cat. no. 610016,
1:1,000 for western blot analysis) from Abcam; human recombinant
EGF (E9644) and cycloheximide (CHX; 66-81-9) from MilliporeSigma;
Wnt3A (5036-WN) from R&D Systems, Inc.; AG1478 (658552) from
Calbiotech, Inc.; LY294002 (L-7988) from LC Laboratories; MK-2206
(S1078) from Selleck Chemicals; DAPI and Alexa Fluor 488 goat
anti-rabbit antibody (A11008, 1:10,000 for immunofluorescence) from
Molecular Probes; Thermo Fisher Scientific, Inc.; HyFect
transfection reagents (E2650) from Denville Scientific, Inc.
Cell culture
A431 (21555) human epidermoid carcinoma cells and
MDA-MB-231 (30026) human breast carcinoma cells were purchased from
the Korean Cell Line Bank (KCLB). LN18 and LN229 GBM cells were
kindly provided by Dr Hyunggee Kim (Korea University, Seoul,
Korea). U251 GBM cells were kindly provided by Dr Kyu Heo (Dongnam
Institute of Radiological and Medical Sciences, Busan, Korea). The
human epidermal squamous carcinoma cell lines, SCC12 and SCC13,
were kindly provided by Dr Tae-Jin Yoon (Gyeongsang National
University and Hospital, Jinju, Korea). These cells were maintained
in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10%
bovine calf serum (Capricorn Scientific GmbH) and 1%
penicillin/streptomycin (Capricorn Scientific GmbH).
DNA constructs, mutagenesis, and
transfection
PCR-amplified human PFKP was cloned into the
pcDNA3.1/hygro(+)-Flag vector. pcDNA3.1/hygro(+)-Flag PFKP S386A
was created using the QuikChange site-directed mutagenesis kit
(Stratagene; Agilent Technologies, Inc.). The pGIPZ shRNA vector
(RHS4348) was purchased from GE Dharmacon, Inc. shRNA-resistant (r)
PFKP contained the a448c, g450c, c453t and c456g mutations. The
following pGIPZ shRNAs were used: Control shRNA oligonucleotide,
5-GCTTCTAACACCGGAGGTCTT-3; PFKP shRNA oligonucleotide,
5-AGGAACGGCCAGATCGATA-3; and β-catenin shRNA oligonucleotide,
5-TTACCACTCAGAGAAGGAG-3. Cells were plated at a density of
4×105/60-mm dish 18 h prior to transfection and were
directly transfected with the constructed vectors (1 µg/6-well
plate) using HyFect transfection reagent (Denville Scientific,
Inc.) according to the manufacturer's instructions. Transfected
cells were stabilized for 1 day, and then the cells were selected
for 1 week using 200 µg/ml hygromycin (400053; EMD Millipore) and 2
µg/ml puromycin (540222; EMD Millipore).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was prepared from tumor cells using an
RNeasy Mini kit (Qiagen, Inc.) according to the manufacturer's
instructions, and cDNA was synthesized from 2 µg of total RNA by
reverse transcriptase (Superscript II Preamplification System,
Gibco; Thermo Fisher Scientific, Inc.). Quantitative PCR (qPCR) was
performed on an ABI Prism 7500 sequence detection system using a
SYBR®-Green PCR Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.) and following the manufacturer's
protocols. The ABI 7500 sequence detector was programmed with the
following PCR conditions: 40 cycles of 15-sec denaturation at 95°C
and 1-min amplification at 60°C. All reactions were run in
triplicate and normalized to the housekeeping gene β-actin. The
evaluation of relative differences of PCR results was calculated
using the 2−ΔΔCq method (38). The following primer pairs were used
for RT-qPCR: Human PFKP forward, 5′-CGGAAGTTCCTGGAGCACCTCTC-3′ and
reverse, 5′-AAGTACACCTTGGCCCCCACGTA-3′; human PFKL forward,
5′-GGCATTTATGTGGGTGCCAAAGTC-3′ and reverse,
5′-CAGTTGGCCTGCTTGATGTTCTCA-3′; human PFKM forward,
5′-GAGTGACTTGTTGAGTGACCTCCAGAAA-3′ and reverse,
5′-CACAATGTTCAGGTAGCTGGACTTCG-3′; human PFKPB1 forward,
5′-AGAAGGGGCTCATCCATACCC-3′ and reverse,
5′-CTCTCGTCGATACTGGCCTAA-3′; human PFKFB2 forward,
5′-AGTCCTACGACTTCTTTCGGC-3′ and reverse,
5′-TCTCCTCAGTGAGATACGCCT-3′; human PFKFB3 forward,
5′-ATTGCGGTTTTCGATGCCAC-3′ and reverse, 5′-GCCACAACTGTAGGGTCGT-3′;
human PFKFB4 forward, 5′-CAACATCGTGCAAGTGAAACTG-3′ and reverse,
5′-GACTCGTAGGAGTTCTCATAGCA-3′; and human β-actin forward,
5′-ATGGATGATGATATCGCCGCGC-3′ and reverse,
5′-GCAGCACGGGGTGCTCCTCG-3′.
Western blot analysis
Protein extraction from the cultured cancer cells
was performed using a lysis buffer [50 mM Tris-HCl (pH 7.5), 0.1%
SDS, 1% Triton X-100, 150 mM NaCl, 1 mM DTT, 0.5 mM EDTA, 100 µM
sodium orthovanadate, 100 µM sodium pyrophosphate, 1 mM sodium
fluoride, and proteinase inhibitor cocktail]. The cell extracts
were centrifuged at 12,000 × g (at 4°C for 15 min) and protein
concentrations of cell lysates were determined using the DC protein
assay kit (5000116, Bio-Rad Laboratories, Inc.). Equal amounts of
lysates (2 mg/ml protein) were resolved by 8–10% SDS-PAGE and were
then transferred to a nitrocellulose membrane. The membrane was
blocked with 5% skim milk in TBST at room temperature for 1 h and
then incubated with the indicated antibodies (as indicated above in
Reagents and antibodies) at 4°C overnight. The blots were then
incubated with horseradish peroxidase-conjugated secondary
antibodies (anti-rabbit (NA934V; Cytiva) or anti-mouse (NA931V;
Cytiva) at a dilution of 1:3,000) at room temperature for 2 h and
were visualized using a chemiluminescence technique (Amersham
Biosciences). Band intensity was quantified using ImageJ 1.53e
software (National Institutes of Health). Each experiment was
repeated at least three times.
Immunofluorescence analysis
A431 cells were fixed with 4% paraformaldehyde for
20 min and permeabilized using 0.5% Triton X-100 at room
temperature for 10 min. The cells were blocked with 5% normal serum
at 37°C for 30 min and incubated with an anti-PFKP antibody at a
dilution of 1:100 at 4°C overnight, subsequently, an Alexa Fluor
dye-conjugated secondary antibody at 37°C for 1 h, and in the end,
stained with DAPI at room temperature for 10 min. The cells were
examined using a deconvolution microscope (Zeiss AG) with a 63-Å
oil-immersion objective. Axio Vision software from Zeiss AG was
used to deconvolute the Z-series images.
Measurement of glucose consumption and
lactate production
The A431 cells were seeded in culture dishes, and
the medium was changed after 6 h with non-serum DMEM. The cells
were then incubated at 37°C for 48 h, and the culture medium was
collected for the measurement of glucose and lactate
concentrations. Glucose levels were determined using a glucose (GO)
assay kit (GAGP20, Sigma-Aldrich; Merck KGaA). Glucose consumption
was calculated as the difference in glucose concentration between
the collected culture medium and DMEM. The absorbance was recorded
at 540 nm at room temperature in a 96-well plate. Lactate levels
were determined using a lactate assay kit (1200051002, Eton
Bioscience, Inc.). The absorbance was recorded at 570 nm using a
microplate reader (SpectraMax® ABS; Molecular Devices,
LLC) at room temperature in a 96-well plate. All results were
normalized to the final cell number.
Cell proliferation assay
A total of 2×104 A431 cells were plated
and incubated for 5 days after seeding in DMEM with 0.5% bovine
calf serum. The cells were trypsinized and counted using a
hemocytometer following staining with trypan blue at room
temperature for 1 min (EBT-001; NanoEnTek, Inc.). The data
represent the mean ± SD of three independent experiments.
Measurement of PFK activity
PFK activity was assessed in A431 cells with the use
of the PFK activity colorimetric assay kit (K776-100, BioVision,
Inc.). The reaction was performed using cell lysate (3 µg) in 100
µl of reaction buffer, which was prepared according to the
instructions provided with the kit. The absorbance was recorded at
450 nm using a microplate reader (SpectraMax® ABS) at
37°C in a 96-well plate.
Measurement of LDH activity
LDH activity was assessed in A431 cells using a
lactate dehydrogenase activity colorimetric assay kit (K726-500,
BioVision, Inc.). The reaction was performed using cell lysate (1
µg) in 100 µl of reaction buffer, which was prepared according to
the instructions provided with the kit. The absorbance was recorded
at 450 nm using a microplate reader (SpectraMax® ABS) at
37°C in a 96-well plate.
Wound healing assay
A431 cells (1.5×105 cells/well) were
seeded into 12-well plates and cultured at 100% confluence,
followed by starvation in serum-free DMEM for 24 h. A sterile 200
µl pipette tip was used to create a scratch in the monolayer in the
middle of the well. At the 0 and 18-h incubation at 37°C time
points, cells were photographed using a light microscope (×200
magnification) and the migration distance was measured using ImageJ
1.53e software (National Institutes of Health). The data represent
the mean ± SD of three independent experiments.
Colony formation assay
A431 cells (1×103 cells/well) were seeded
into 6-well plates and cultured in DMEM supplemented with 10%
bovine calf serum with or without Wnt3A (20 ng/ml). Colony
formation assay was performed for 7 or 12 days. The cells were
fixed with 10% formalin (F8775, Sigma-Aldrich; Merck KGaA) for 10
min and stained with 0.1% crystal violet (V5265, Sigma-Aldrich) at
room temperature for 1 h. The residual dye was washed off with
water and the plate was air-dried.
Statistical analysis
All quantitative data are presented as the mean ± SD
of at least three independent experiments. A 2-group comparison was
conducted using the 2-sided, 2-sample Student's t-test. A
simultaneous comparison of >2 groups was conducted using one-way
ANOVA followed by Tukey's post hoc tests. The SPSS statistical
package (version 12; SPSS Inc.) was used for the analyses. Values
of P<0.05 were considered to indicate statistically significant
differences.
Results
EGFR transactivation is required for
the Wnt3A-induced the Warburg effect, as well as cell
proliferation, colony formation and cancer cell migration
Wnt3A was used to stimulate A431, SCC12, and SCC13
human epidermal carcinoma cells, U251, LN229 and LN18 human GBM
cells, and MDA-MB-231 human breast carcinoma cells, in order to
determine whether the Wnt-induced EGFR transactivation has effects
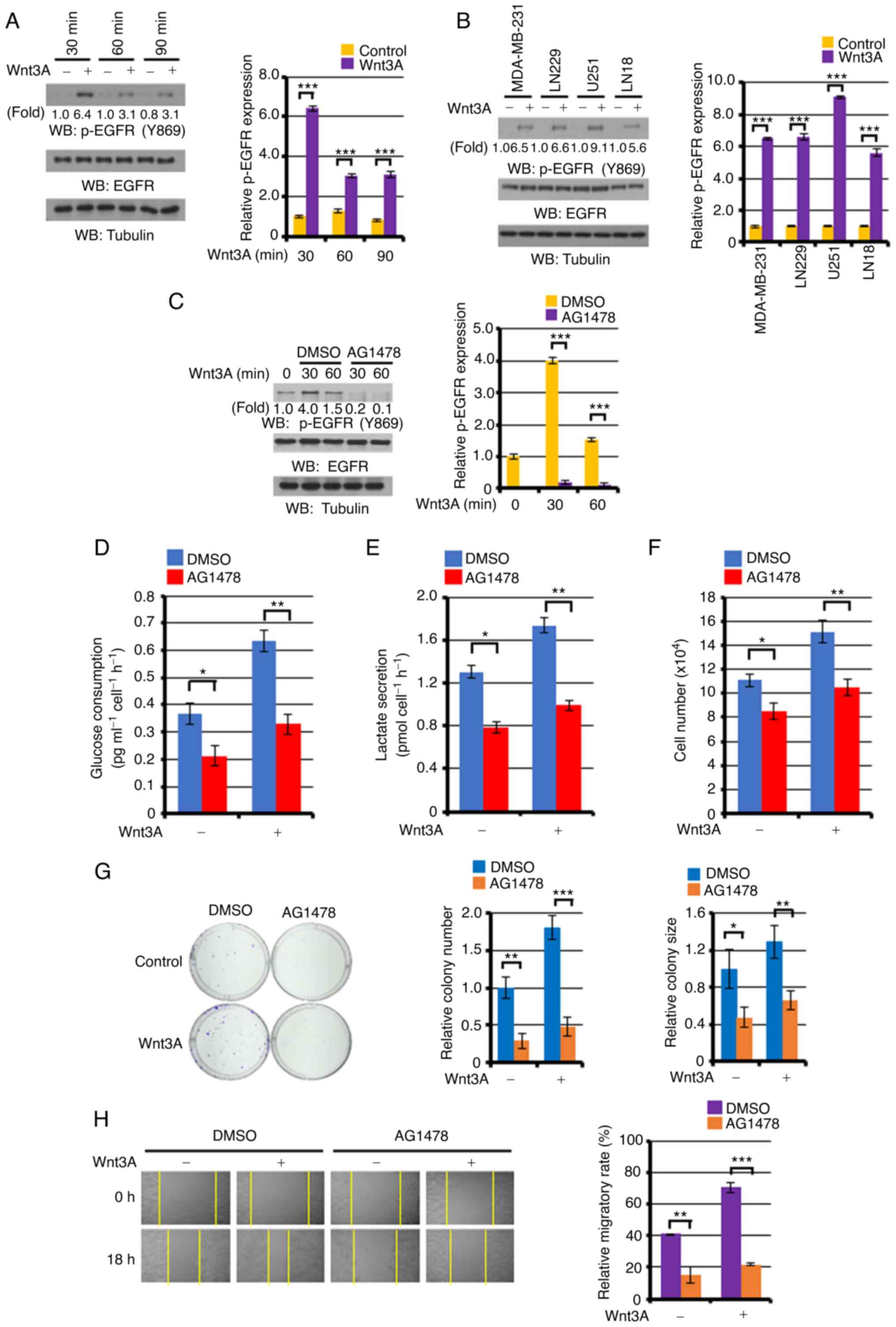
on tumor development or not. As presented in Fig. 1A, Wnt3A stimulation significantly
induced EGFR phosphorylation levels at 30, 60 and 90 min, as
compared with the unstimulated control A431 cell EGFR levels.
Similar results were also observed in other types of cancer cells:
MDA-MB-231, LN229, U251 and LN18 (Fig.
1B), and SCC12 and SCC13 (Fig.
S1). Pre-treatment with the specific EGFR tyrosine kinase
inhibitor, AG1478, following the successful blocking of
Wnt3A-induced EGFR phosphorylation (Fig.
1C), markedly abrogated both basal and Wnt3A-enhanced glucose
consumption (Fig. 1D), lactate
production (Fig. 1E), proliferation
(Fig. 1F), colony-forming ability
(Fig. 1G) and migration (Fig. 1H) in the A431 cells. These results
indicated that Wnt signaling successfully may transactivate EGFR in
cancer cells, which is required for the Wnt-induced the Warburg
effect, as well as proliferation, colony formation and cancer cell
migration.
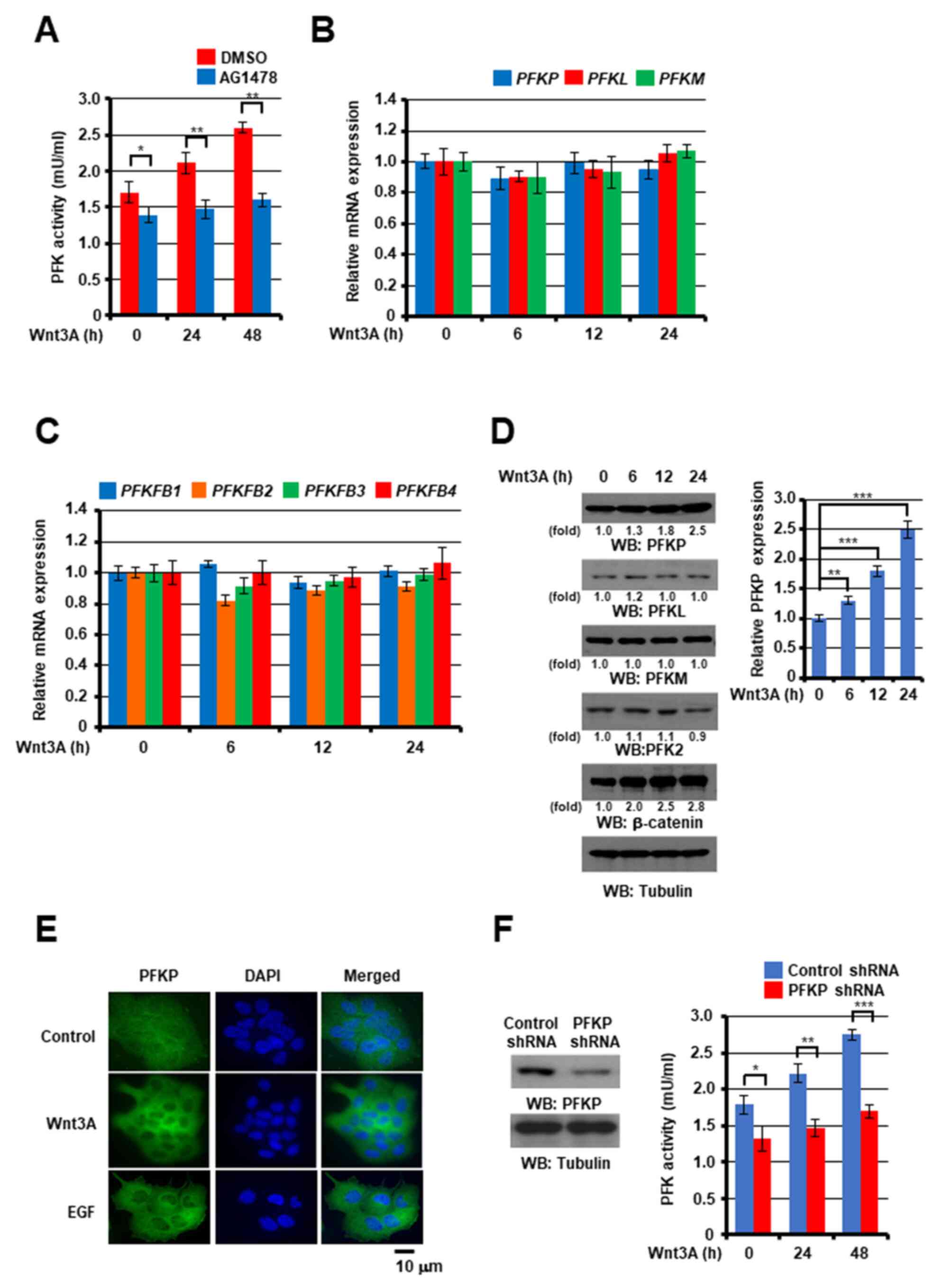
Wnt3A induces PFK enzyme activity and
PFKP expression
PFK is critical for glycolysis (33), and its activity and expression is
important for tumor development (37,39,40). Thus,
the effect of Wnt signaling on PFK enzyme activity was examined in
the present study. As shown in Fig.
2A, Wnt3A stimulation markedly induced PFK enzyme activity,
which was significantly inhibited by treatment with AG1478 EGFR
inhibitor, suggesting that Wnt signaling induced PFK activity
through EGFR transactivation. Subsequently, the effect of Wnt
signaling on PFK expression was determined. PFK includes PFK1 and
PFK2 isoforms (33). Additionally,
PFK1 includes PFKP, PFKM and PFKL isoforms. Furthermore, PFK2 [also
known as 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
(PFKFB)], a bi-functional enzyme encoded by four different genes
(PFKFB1, PFKFB2, PFKFB3 and PFKFB4) with both a
kinase and a phosphatase domain, is responsible for the production
and degradation of fructose-2,6-bisphosphate (41). The aforementioned enzymes and their
corresponding isoforms have been originally detected in different
tissues: PFKFB1 in the liver and skeletal muscle, PFKFB2 in the
heart tissue, PFKFB3 in the brain and PFKFB4 in the testis
(42). The analyses of the isoform
expression profiles using RT-qPCR and western blot analysis
revealed that Wnt3A treatment did not markedly alter the PFK1
(Fig. 2B) and PFK2 (Fig. 2C) isoform mRNA expression levels and
in A431 cells, whereas the PFKP protein expression levels, but not
those of PFKL, PFKM and PFK2, were upregulated in the A431 cells
(Fig. 2D), and the LN18, MDA-MB-231,
SCC12 and SCC13 cells (Fig. S2) in
response to Wnt3A stimulation. Consistent with these findings,
immunofluorescence analyses with anti-PFKP antibody revealed that
Wnt3A treatment enhanced PFKP expression in A431 cells (Fig. 2E). Taken together, it was observed
that Wnt signaling increased PFK activity and PFKP expression. In
order to determine the role of PFKP expression in Wnt-induced PFK
activation, PFKP expression was depleted with the use of shRNA. It
was then demonstrated that a reduction in PFKP expression
significantly decreased the Wnt3A-induced total PFK enzyme activity
in A431 cells (Fig. 2F), suggesting a
crucial role of PFKP expression in Wnt signaling-induced PFK
activation.
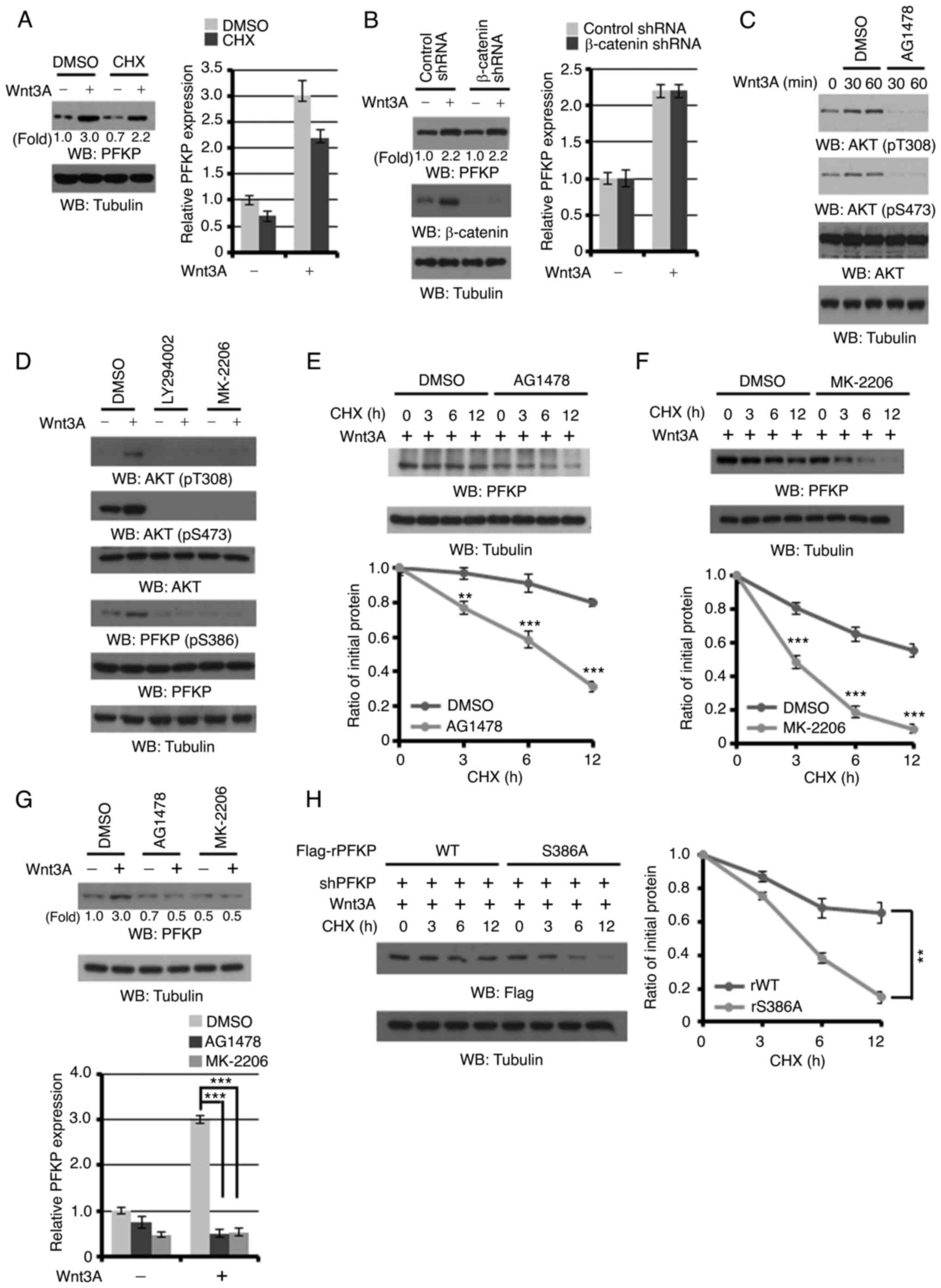
Wnt3A induces PFKP expression through
EGFR/PI3K/AKT activation-induced PFKP S386 phosphorylation
The A431 cells were pre-treated with CHX to block
protein synthesis, in order to determine whether PFKP protein
expression is regulated by Wnt signaling. CHX treatment exerted a
limited effect on Wnt3A-induced PFKP expression (Fig. 3A). These results suggest that Wnt
signaling enhances PFKP expression primarily by enhancing PFKP
stability. Of note, β-catenin depletion in A431 cells revealed that
a reduction in β-catenin expression did not affect Wnt3A-induced
PFKP protein expression (Fig. 3B),
suggesting that Wnt signaling induces PFKP expression in the
canonical Wnt-independent manner in cancer cells.
It has been previously reported by the authors that
AKT phosphorylates PFKP at S386, which results in the inhibition of
TRIM21 E3 ligase binding to PFKP and the subsequent TRIM21-mediated
polyubiquitylation and degradation of PFKP, ultimately resulting in
an increased PFKP expression (37).
Wnt3A stimulation successfully induced AKT phosphorylation, which
was blocked by treatment with AG1478 EGFR inhibitor (Fig. 3C). In addition, Wnt3A induced PFKP
S386 phosphorylation in A431 cells (Fig.
3D; first and second lanes). As expected, treatment of the A431
cells with the PI3K inhibitor, LY294002, or the AKT inhibitor,
MK-2206, successfully blocked Wnt3A-induced AKT T308/S473 and PFKP
S386 phosphorylation (Fig. 3D).
Furthermore, the Wnt3A-induced half-life of endogenous PFKP
(Fig. 3E and F) and PFKP expression
(Fig. 3G) were markedly decreased
following treatment with the AG1478 EGFR inhibitor or MK-2206 AKT
inhibitor, respectively. In line with this finding, the
Wnt3A-induced half-life of the PFKP S386A mutant was much shorter
than that of its wild-type (WT) counterpart (Fig. 3H). These results indicated that
EGFR/PI3K/AKT activation-induced PFKP S386 phosphorylation is
required for Wnt-enhanced PFKP protein expression.
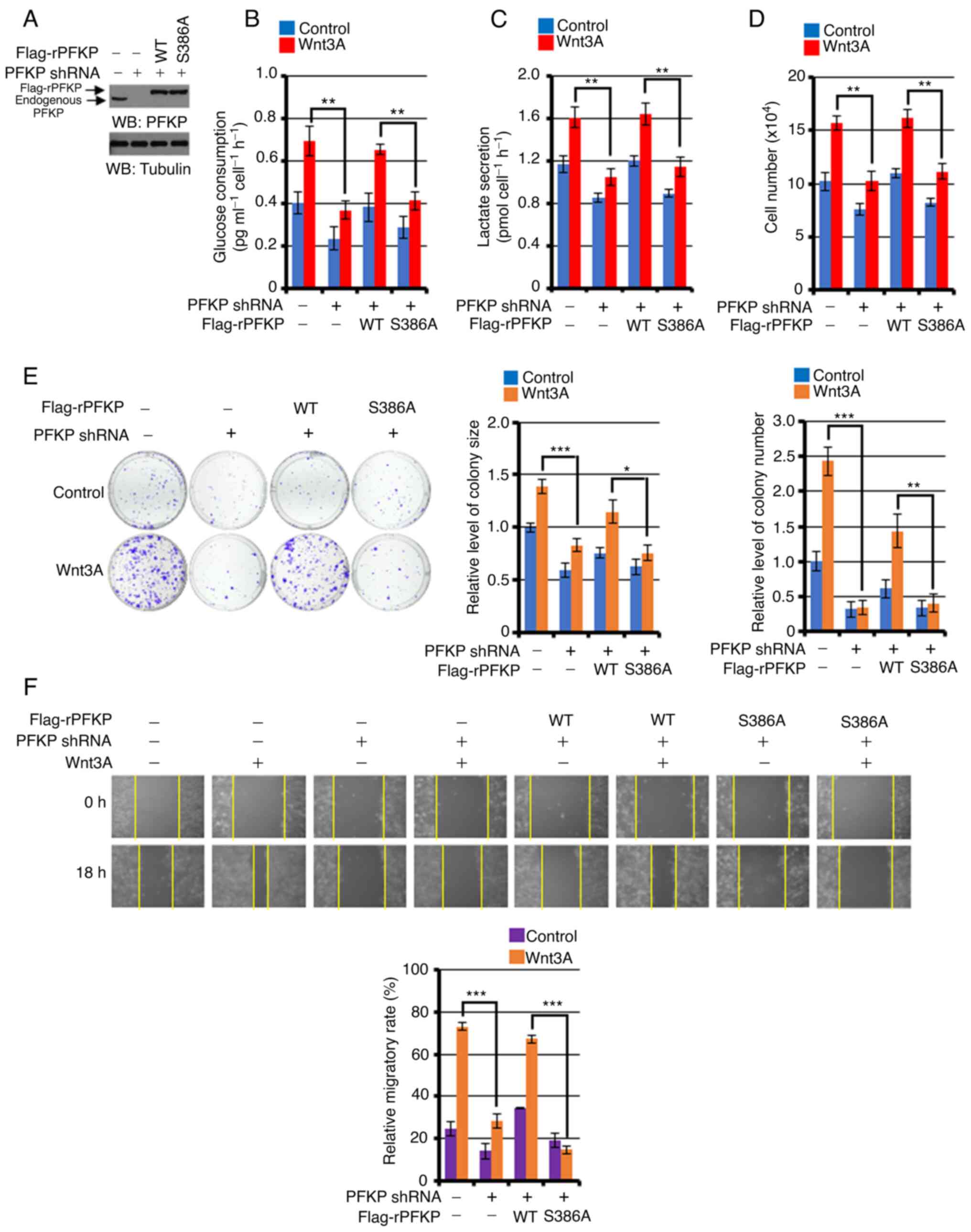
PFKP S386 phosphorylation is required
for the Wnt3A-induced the Warburg effect, proliferation, colony
formation and cancer cell migration
To investigate the role of PFKP S386 phosphorylation
in the Wnt signaling-induced Warburg effect, cell proliferation,
colony formation and migration, endogenous PFKP was depleted in
A431 cells and the expression of RNAi-resistant (r) WT Flag-rPFKP
or Flag-rPFKP S386A was reconstituted in these cells. The depletion
of PFKP (Fig. 4A) markedly impaired
Wnt3A-induced glucose consumption (Fig.
4B), lactate production (Fig.
4C), proliferation (Fig. 4D),
colony formation ability (Fig. 4E)
and cell migration (Fig. 4F); this
inhibition was reversed by the expression of WT rPFKP, but not by
the expression of rPFKP S386A mutant in A431 cells (Fig. 4A-F). Of note, Wnt3A stimulation or
PFKP depletion did not affect LDHA enzyme expression and activity
in A431 cells (Fig. S3). These
results suggested that PFKP S386 phosphorylation may be important
for the Wnt-induced the Warburg effect, as well as for
proliferation, colony formation and cancer cell migration.
Discussion
Metabolic alterations have been observed in cancer
cells, of which the Warburg effect is considered a common feature
and critical for tumor development. Metabolic changes in cancer
cells are regulated by microenvironmental and genetic factors. Wnt
has been reported to be one of the environmental factors (10,11), and
the abnormal activation of Wnt signaling has been observed in a
number of human tumor types, which is considered important for
tumor development (7–9). However, how Wnt signaling regulates
metabolic alterations to promote tumor development remains unknown.
In the present study, it was demonstrated that Wnt signaling
directly regulated glycolysis through β-catenin
signaling-independent PFKP upregulation. Mechanistically, Wnt3A
signaling transactivated EGFR, leading to PI3K/AKT activation and
culminating in increased PFKP stability through PFKP S386
phosphorylation. Wnt3A-induced PFKP S386 phosphorylation increased
PFKP expression and PFK activity and promoted the Warburg effect,
as well as proliferation, colony formation and cancer cell
migration (Fig. 5). The current
findings uncovered a novel mechanism, to the best of our knowledge,
through which Wnt signaling directly regulates glycolysis in a PFKP
S386 phosphorylation-dependent manner to promote tumor
development.
EGFR overexpression has been detected in various
human tumors, including epidermoid and breast carcinomas and GBM
tumors and has been correlated with poor clinical prognosis
(37,43,44). It
has been reported that G protein-coupled receptors (GPCRs)
stimulation could transactivate EGFR, in which metalloproteases
(MMPs), such as a disintegrin and metalloproteases (ADAMs), matrix
metalloproteinase-2 (MMP2) and matrix metalloproteinase-9 (MMP9),
mediate EGFR ligand cleavage, leading to the activation of
downstream signals (45–48). Frizzled family members have been
functionalized as canonical GPCRs (49,50) that
contain an N-terminal extra-cellular cysteine-rich domain to bind
with glycoproteins of the Wnt family (51). Upon Wnt/Frizzled binding, diverse
signaling pathways are activated through the phosphoprotein
Dishevelled (22). Schlange et
al (52) and Civenni et al
(53) have reported that Wnt
signaling induced the activation of EGFR through Dishevelled, Src,
and the MMP-dependent release of soluble EGFR ligands, leading to
the activation of ERK, inducing proliferation. Consistent with
these reports, in the present study, Wnt3A stimulation induced the
phosphorylation of EGFR in several cancer types. In addition,
Wnt3A-induced EGFR phosphorylation was blocked by Src inhibition
(data not shown). Since EGFR is amplified in most tumor cells, in
addition to the canonical pathway Wnt signaling can activate more
diverse signals, in order to promote tumor development through EGFR
transactivation.
PFK1 is a rate-limiting enzyme in glycolysis
(33). It has been reported both by
the authors and others that PFKP is the prominent PFK1 isoform in
GBM, ascites tumor cells, and breast carcinoma (37,54–56). In a
previous study by the authors, it was revealed that PFKP was
overexpressed in human GBM cell lines, primary GBM cells and GBM
specimens (37). Furthermore, AKT
activation enhanced the phosphorylation of PFKP at S386, resulting
in the suppression of TRIM21 E3 ligase-mediated polyubiquitylation
and the proteasomal degradation of PFKP. PFKP S386 phosphorylation
enhanced PFKP expression and promoted aerobic glycolysis and brain
tumor growth (37). In the present
study, it was demonstrated that Wnt signaling induced PFKP S386
phosphorylation in an EGFR/PI3K/AKT activation-dependent manner,
resulting in PFKP expression upregulation and PFK enzyme activity
increase. PFKP S386 phosphorylation was important for the
Wnt-induced Warburg effect, proliferation, colony formation and
cancer cell migration.
In conclusion, the present study was demonstrated
for the first time, to the best of our knowledge, that Wnt
signaling directly regulated glycolysis-associated enzyme
expression through the non-canonical Wnt pathway, in order to
promote tumor development. The findings of the present study may
provide insight, concerning the critical role of PFKP in
fundamental biological processes for Wnt-induced tumor
development.
Supplementary Material
Supporting Data
Acknowledgements
LN18 and LN229 GBM cells were kindly provided by Dr
Hyunggee Kim (Korea University, Seoul, Korea). U251 GBM cells were
kindly provided by Dr Kyu Heo (Dongnam Institute of Radiological
and Medical Sciences, Busan, Korea). The human epidermal squamous
carcinoma cell lines, SCC12 and SCC13, were kindly provided by Dr
Tae-Jin Yoon (Gyeongsang National University and Hospital, Jinju,
Korea).
Funding
The present study was supported by the Dong-A
University research fund.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SMJ, JSL, SHP and J-HL designed and performed
experiments and analyzed data. SMJ and J-HL wrote the manuscript.
SMJ, JSL, and SHP confirmed the authenticity of all the raw data.
All authors have read and approved the final manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Acebron SP, Karaulanov E, Berger BS, Huang
YL and Niehrs C: Mitotic wnt signaling promotes protein
stabilization and regulates cell size. Mol Cell. 54:663–674. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Atlasi Y, Noori R, Gaspar C, Franken P,
Sacchetti A, Rafati H, Mahmoudi T, Decraene C, Calin GA, Merrill BJ
and Fodde R: Wnt signaling regulates the lineage differentiation
potential of mouse embryonic stem cells through Tcf3
down-regulation. PLoS Genet. 9:e10034242013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esen E, Chen J, Karner CM, Okunade AL,
Patterson BW and Long F: WNT-LRP5 signaling induces Warburg effect
through mTORC2 activation during osteoblast differentiation. Cell
Metab. 17:745–755. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Angers S and Moon RT: Proximal events in
Wnt signal transduction. Nat Rev Mol Cell Biol. 10:468–477. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clevers H, Loh KM and Nusse R: Stem cell
signaling. An integral program for tissue renewal and regeneration:
Wnt signaling and stem cell control. Science. 346:12480122014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lang CMR, Chan CK, Veltri A and Lien WH:
Wnt signaling pathways in keratinocyte carcinomas. Cancers (Basel).
11:12162019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee Y, Lee JK, Ahn SH, Lee J and Nam DH:
WNT signaling in glioblastoma and therapeutic opportunities. Lab
Invest. 96:137–150. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Milovanovic T, Planutis K, Nguyen A, Marsh
JL, Lin F, Hope C and Holcombe RF: Expression of Wnt genes and
frizzled 1 and 2 receptors in normal breast epithelium and
infiltrating breast carcinoma. Int J Oncol. 25:1337–1342.
2004.PubMed/NCBI
|
|
11
|
Benhaj K, Akcali KC and Ozturk M:
Redundant expression of canonical Wnt ligands in human breast
cancer cell lines. Oncol Rep. 15:701–707. 2006.PubMed/NCBI
|
|
12
|
Zheng W, Yao M, Fang M, Pan L, Wang L,
Yang J, Dong Z and Yao D: Oncogenic Wnt3a: A candidate specific
marker and novel molecular target for hepatocellular carcinoma. J
Cancer. 10:5862–5873. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaur N, Chettiar S, Rathod S, Rath P,
Muzumdar D, Shaikh ML and Shiras A: Wnt3a mediated activation of
Wnt/β-catenin signaling promotes tumor progression in glioblastoma.
Mol Cell Neurosci. 54:44–57. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jing Q, Li G, Chen X, Liu C, Lu S, Zheng
H, Ma H, Qin Y, Zhang D, Zhang S, et al: Wnt3a promotes
radioresistance via autophagy in squamous cell carcinoma of the
head and neck. J Cell Mol Med. 23:4711–4722. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He S, Lu Y, Liu X, Huang X, Keller ET,
Qian CN and Zhang J: Wnt3a: Functions and implications in cancer.
Chin J Cancer. 34:554–562. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Verras M, Brown J, Li X, Nusse R and Sun
Z: Wnt3a growth factor induces androgen receptor-mediated
transcription and enhances cell growth in human prostate cancer
cells. Cancer Res. 64:8860–8866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grumolato L, Liu G, Mong P, Mudbhary R,
Biswas R, Arroyave R, Vijayakumar S, Economides AN and Aaronson SA:
Canonical and noncanonical Wnts use a common mechanism to activate
completely unrelated coreceptors. Genes Dev. 24:2517–2530. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Katoh M: Canonical and non-canonical WNT
signaling in cancer stem cells and their niches: Cellular
heterogeneity, omics reprogramming, targeted therapy and tumor
plasticity (Review). Int J Oncol. 51:1357–1369. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Polakis P: The many ways of Wnt in cancer.
Curr Opin Genet Dev. 17:45–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reya T and Clevers H: Wnt signalling in
stem cells and cancer. Nature. 434:843–850. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Duchartre Y, Kim YM and Kahn M: The Wnt
signaling pathway in cancer. Crit Rev Oncol Hematol. 99:141–149.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
24
|
Corda G and Sala A: Non-canonical WNT/PCP
signalling in cancer: Fzd6 takes centre stage. Oncogenesis.
6:e3642017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kurayoshi M, Oue N, Yamamoto H, Kishida M,
Inoue A, Asahara T, Yasui W and Kikuchi A: Expression of Wnt-5a is
correlated with aggressiveness of gastric cancer by stimulating
cell migration and invasion. Cancer Res. 66:10439–10448. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weeraratna AT, Jiang Y, Hostetter G,
Rosenblatt K, Duray P, Bittner M and Trent JM: Wnt5a signaling
directly affects cell motility and invasion of metastatic melanoma.
Cancer Cell. 1:279–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pate KT, Stringari C, Sprowl-Tanio S, Wang
K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell
MA, et al: Wnt signaling directs a metabolic program of glycolysis
and angiogenesis in colon cancer. EMBO J. 33:1454–1473. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G,
Han SI, Park HG and Kang HS: Wnt/Snail signaling regulates
cytochrome C oxidase and glucose metabolism. Cancer Res.
72:3607–3617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mor I, Cheung EC and Vousden KH: Control
of glycolysis through regulation of PFK1: Old friends and recent
additions. Cold Spring Harb Symp Quant Biol. 76:211–216. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moreno-Sanchez R, Rodriguez-Enriquez S,
Marin-Hernandez A and Saavedra E: Energy metabolism in tumor cells.
FEBS J. 274:1393–1418. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dunaway GA and Kasten TP: Nature of the
subunits of the 6-phosphofructo-1-kinase isoenzymes from rat
tissues. Biochem J. 242:667–671. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kahn A, Meienhofer MC, Cottreau D,
Lagrange JL and Dreyfus JC: Phosphofructokinase (PFK) isozymes in
man. I. Studies of adult human tissues. Hum Genet. 48:93–108. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee JH, Liu R, Li J, Zhang C, Wang Y, Cai
Q, Qian X, Xia Y, Zheng Y, Piao Y, et al: Stabilization of
phosphofructokinase 1 platelet isoform by AKT promotes
tumorigenesis. Nat Commun. 8:9492017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee JH, Liu R, Li J, Wang Y, Tan L, Li XJ,
Qian X, Zhang C, Xia Y, Xu D, et al: EGFR-Phosphorylated platelet
isoform of phosphofructokinase 1 promotes PI3K activation. Mol
Cell. 70:197–210.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee JH, Shao F, Ling J, Lu S, Liu R, Du L,
Chung JW, Koh SS, Leem SH, Shao J, et al: Phosphofructokinase 1
platelet isoform promotes β-catenin transactivation for tumor
development. Front Oncol. 10:2112020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stine ZE and Dang CV: Stress eating and
tuning out: Cancer cells re-wire metabolism to counter stress. Crit
Rev Biochem Mol Biol. 48:609–619. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rider MH, Bertrand L, Vertommen D, Michels
PA, Rousseau GG and Hue L:
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: Head-to-head
with a bifunctional enzyme that controls glycolysis. Biochem J.
381((Pt 3)): 561–579. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Avraham R and Yarden Y: Feedback
regulation of EGFR signalling: Decision making by early and delayed
loops. Nat Rev Mol Cell Biol. 12:104–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Prenzel N, Zwick E, Daub H, Leserer M,
Abraham R, Wallasch C and Ullrich A: EGF receptor transactivation
by G-protein-coupled receptors requires metalloproteinase cleavage
of proHB-EGF. Nature. 402:884–888. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Blobel CP: ADAMs: Key components in EGFR
signalling and development. Nat Rev Mol Cell Biol. 6:32–43. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ohtsu H, Dempsey PJ and Eguchi S: ADAMs as
mediators of EGF receptor transactivation by G protein-coupled
receptors. Am J Physiol Cell Physiol. 291:C1–C10. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Roelle S, Grosse R, Aigner A, Krell HW,
Czubayko F and Gudermann T: Matrix metalloproteinases 2 and 9
mediate epidermal growth factor receptor transactivation by
gonadotropin-releasing hormone. J Biol Chem. 278:47307–47318. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Koval A and Katanaev VL: Wnt3a stimulation
elicits G-protein-coupled receptor properties of mammalian Frizzled
proteins. Biochem J. 433:435–440. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nichols AS, Floyd DH, Bruinsma SP,
Narzinski K and Baranski TJ: Frizzled receptors signal through G
proteins. Cell Signal. 25:1468–1475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang-Snyder J, Miller JR, Brown JD, Lai CJ
and Moon RT: A frizzled homolog functions in a vertebrate Wnt
signaling pathway. Curr Biol. 6:1302–1306. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schlange T, Matsuda Y, Lienhard S, Huber A
and Hynes NE: Autocrine WNT signaling contributes to breast cancer
cell proliferation via the canonical WNT pathway and EGFR
transactivation. Breast Cancer Res. 9:R632007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Civenni G, Holbro T and Hynes NE: Wnt1 and
Wnt5a induce cyclin D1 expression through ErbB1 transactivation in
HC11 mammary epithelial cells. EMBO Rep. 4:166–171. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sanchez-Martinez C and Aragon JJ: Analysis
of phosphofructokinase subunits and isozymes in ascites tumor cells
and its original tissue, murine mammary gland. FEBS Lett.
409:86–90. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Moon JS, Kim HE, Koh E, Park SH, Jin WJ,
Park BW, Park SW and Kim KS: Kruppel-like factor 4 (KLF4) activates
the transcription of the gene for the platelet isoform of
phosphofructokinase (PFKP) in breast cancer. J Biol Chem.
286:23808–23816. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang G, Xu Z, Wang C, Yao F, Li J, Chen C
and Sun S: Differential phosphofructokinase-1 isoenzyme patterns
associated with glycolytic efficiency in human breast cancer and
paracancer tissues. Oncol Lett. 6:1701–1706. 2013. View Article : Google Scholar : PubMed/NCBI
|