Introduction
Acute lymphoblastic leukemia (ALL) is the most
common hematologic malignant disorder of children. It is divided
into two subtypes: B-cell acute lymphoblastic leukemia (B-ALL) and
T-cell acute lymphoblastic leukemia (T-ALL) (1). Among adults, B-ALL constitutes 75% of
cases, with T-ALL constituting the other 25% (1,2). In
childhood ALL, T-ALL accounts for 15% of pediatric ALL cases
(3). In Taiwan ~200 new cases of
ALL are diagnosed annually. These patients develop comorbidities
including hypothyroidism, obesity, metabolic syndrome and
subclinical cardiac dysfunction (4). In recent years, several new drugs
such as resveratrol, everolimus and selumetinib have been used to
treat ALL, and compared with previously used drugs, they cause
higher complete remission and lower drug toxicity (5–8).
However, due to drug resistance and relapse, the prognosis of
patients with T-ALL is still not satisfactory. Therefore,
developing a new strategy to treat recurrent ALL and decrease the
side effects has become an important issue.
Niclosamide has been a traditional oral
anti-helminthic drug for about half a century, with FDA-approval
for treating parasitic infections worldwide. According to previous
reports, niclosamide is relatively safe and non-toxic to mammals
(oral LD50 of >5 g/kg in rats) (9,10). A
previous study reported that an abnormally low ratio of
Bcl-2-associated X protein (Bax)/B-cell lymphoma 2 (Bcl-2), and a
spontaneous loss of caspase-3 processing are positively correlated
with the relapse rate of childhood ALL (11). Therefore, the metabolic pathway
that regulates apoptosis may be a useful target to prevent the
relapse of ALL. It was also reported that niclosamide regulates
multiple signaling pathways, including nuclear factor (NF)-κB,
Wnt/β-catenin, mechanistic target of rapamycin complex 1 (mTORC1),
signal transducer and activator of transcription 3 (STAT3) and
Notch, thereby achieving its anticancer actions (12). Niclosamide, through activation of
these apoptotic pathways, reduces the expression of Ki-67 cells and
p-STAT3Try705 in melanoma. Furthermore, it can inhibit
metastasis in lung cancer (13).
In osteosarcoma, niclosamide was found to inhibit osteosarcoma cell
proliferation through cell apoptosis and inhibition of cell cycle
progression. In the mouse xenograft tumor model of human
osteosarcoma cells, niclosamide inhibited tumor growth (14). On the other hand, autophagy is an
evolutionary process in cells. This process involves the transport
of damaged organelles, misfolded proteins and other macromolecular
substances to lysosome for degradation. In cancer, cells are
self-phagocytic. Reduced autophagy inhibits degradation of damaged
organelles and increases oxidative stress. These changes lead to
cancer development (15,16). Previous studies have shown that
glucocorticoids are useful in treating B-ALL by inducing cellular
autophagy and causing cell death (17,18).
In the ALL Jurkat cell model, Timosaponin AIII was found to induce
cell apoptosis and autophagy for its antitumor action (19). Autophagy is therefore widely used
in treating ALL. Niclosamide inhibits the growth of colorectal
cancer both in vitro and in vivo by inhibiting Wnt
signaling through internalizing Wnt receptors and degrading
Dishevelled 2 and β-catenin proteins of the Wnt signaling pathway.
Niclosamide-induced inhibition of Wnt signaling was found to be
regulated by autophagosome (20).
Apoptosis and autophagy hence play important roles in regulating
the development of leukemia. As demonstrated above, niclosamide
displays anticancer activity in different cancers. However, few
reports have been published concerning the effects of niclosamide
on T-ALL development. We hypothesized here that niclosamide has
anticancer effects on T-ALL. Niclosamide likely acts to ameliorate
T-ALL in different ways, such as autophagy and apoptosis. To this
end, the anticancer effects of niclosamide on T-ALL were
investigated in the present study.
Materials and methods
Cells and drug treatments
Human T-ALL cell lines Jurkat and CCRF-CEM (obtained
from the American Type Culture Collection; ATCC), were kept at 37°C
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% heat-inactivated fetal bovine serum (FBS;
HyClone; GE Healthcare Life Sciences), 1 mM sodium pyruvate
(HyClone; GE Healthcare Life Sciences), 100 U/ml penicillin, and
100 µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.), and maintained in a humidified atmosphere containing 95% air
and 5% CO2. Cells were treated with niclosamide (ACROS
Organics™) at different doses and time courses.
Cell viability assay
The MTT assay was performed to assess the effects of
niclosamide on the viability of T-ALL cells. Thiazolyl blue
tetrazolium bromide (Sigma-Aldrich; Merck KGaA) was prepared from a
stock concentration of 5 mg/ml in PBS, and then diluted with
RPMI-1640 medium to a working concentration of 0.5 mg/ml. To
evaluate the dose-dependent effects of niclosamide, Jurkat and
CCRF-CEM cells were seeded at a density of 1×105
cells/well in a 96-well cell culture plate. They were treated at
37°C for 24 h with either the vehicle control (DMSO) or different
doses of niclosamide (0.125, 0.25, 0.5, 1.0, 2.0 and 4.0 µM). To
evaluate the time-dependent effects of niclosamide, Jurkat cells
were treated with vehicle control (DMSO) or niclosamide (2 µM) for
24, 48, and 72 h. CCRF-CEM cells were also treated with vehicle
control (DMSO) or niclosamide (1 µM) for 24, 48 and 72 h. After
incubation, thiazolyl blue tetrazolium bromide (0.5 mg/ml) was then
added, and cells were incubated for 2 h in dark. Formazan crystals
were dissolved in DMSO, and the absorbance was measured at 570 nm
using an ELISA reader (BioTek Synergy HT). Each experiment was
conducted three times.
Flow cytometry of Ki-67 staining
To assess the cell proliferation, Jurkat and
CCRF-CEM cells were seeded at a density of 2×106
cells/well in a 24-well cell culture plate, while Jurkat cells were
treated with either vehicle control (DMSO) or niclosamide (1.0, 2.0
and 4.0 µM); and CCRF-CEM cells were treated with either vehicle
control (DMSO) or niclosamide (0.5, 1.0 and 2.0 µM) at 37°C for 24
h. After niclosamide treatment, the cells were harvested and fixed
with 70% ethanol and stored at −20°C. Then, the cells were washed
and stained with Alexa Fluor® 488 anti-human Ki-67
antibody (cat. no. 151204, 1:200 dilution; BioLegend®)
in the dark for 30 min. The mean fluorescence intensity (MFI) of
Ki-67 expression was recorded using a FACSCalibur flow cytometer
using CellQuest software (version 5.2; BD Biosciences).
Apoptotic assays
Jurkat and CCRF-CEM cells were seeded at a density
of 5×105 cells/well in a 24-well cell culture plate;
Jurkat cells were treated with either the vehicle control (DMSO) or
niclosamide (2 µM) while CCRF-CEM cells were treated with vehicle
control (DMSO) or niclosamide (1 µM) at 37°C for 24 h. After
incubation, the cells were washed twice with PBS, and then labeled
with Annexin V-fluorescein isothiocyanate and propidium iodide (PI)
according to the manufacturer's instructions (Elabscience).
Fluorescence intensities were measured with flow cytometry. Each
experiment was performed in duplicate, for at least three times
independently.
Cytosolic and mitochondrial protein
isolation
Cytosolic and mitochondrial protein fractions were
extracted from the vehicle-treated or niclosamide-treated Jurkat or
CCRF-CEM cells using the Mitochondria/Cytosol Fractionation Kit
(cat. no. K256-100, BioVision Inc.). Briefly, 1×107
cells were harvested, washed and centrifuged at 600 × g for 5 min
at 4°C. Next, the cells were resuspended in cytosol extraction
buffer mix, incubated on ice for 10 min, and homogenized on ice
using the XL-2020 Sonicator Ultrasonic Processor (Heat Systems
Inc.) The homogenate was centrifuged at 700 × g for 10 min at 4°C.
The supernatant was collected and centrifuged at 10,000 × g for 30
min at 4°C. That supernatant was used as the cytosolic fraction.
The pellet was resuspended in mitochondrial extraction buffer mix,
vortexed for 10 sec, and used as the mitochondrial fraction.
Western blot analysis
Autophagy- and apoptosis-related proteins were
determined using western blot analysis. Jurkat and CCRF-CEM cells
were seeded at a density of 1.5×106 cells/well in a
12-well cell culture plate. Jurkat cells were treated with either
vehicle control (DMSO) or niclosamide (1.0, 2.0 and 4.0 µM), and
CCRF-CEM cells were treated with either vehicle control (DMSO) or
niclosamide (0.5, 1.0 and 2.0 µM) at 37°C for 24 h. After
niclosamide treatment, the cells were harvested and protein
expression levels were evaluated with western blotting using
appropriate antibodies. Antibodies for apoptosis protein markers
(Bcl-2, cleaved caspase-3, caspase-3 and cytochrome c) and
autophagy protein markers (LC3B, p62 and ATG5) were used in
experiments. In brief, cells were first washed with PBS before
collection. Cells were lysed in RIPA buffer (Biomed, Taiwan), and
centrifuged at 13,000 × g for 10 min at 4°C. The supernatant was
used for protein quantification with a Coomassie protein assay
reagent (Thermo Fisher Scientific, Inc.). Equal amounts (15
µg/lane) of protein were loaded to each well and separated by
SDS-PAGE. Samples were finally transferred to PVDF membrane.
Immunoblotting was conducted using the following antibodies: Bcl-2
(cat. no. 15071; Cell Signaling Technology; 1:1,000 dilution),
cleaved caspase-3 (cat. no. 9664; Cell Signaling Technology;
1:1,000 dilution), caspase-3 (cat. no. E-AB-30756; Elabscience;
1:1,000 dilution), LC3B (cat. no. 83506; Cell Signaling Technology;
1:1,000 dilution), p62 (cat. no. 5114; Cell Signaling Technology;
1:1,000 dilution), ATG5 (cat. no. E-AB-22149; Elabscience; 1:1,000
dilution), cytochrome c (cat. no. GTX108585; GeneTex;
1:1,500 dilution) and β-actin (cat. no. E-AB-20034; Elabscience;
1:2,000 dilution), followed by incubation with secondary anti-mouse
IgG, HRP-linked antibody (cat. no. 7076P2; Cell Signaling
Technology; 1:3,000 dilution) or goat anti-rabbit IgG antibody
(cat. no. A0545; Sigma-Aldrich; Merck KGaA; 1:3,000 dilution).
Labeled proteins were detected using the ECL Detection Kit
(Millipore) by the Alliance Q9 (UVITEC, UK). Normalization was
performed with the β-actin antibody.
T-ALL xenograft murine model
Female NOD/SCID mice, 8 weeks of age and body weight
18 to 22 g, were obtained from the National Laboratory Animal
Center. They were housed under specific pathogen-free conditions
with at a 12:12-h dark/light cycle with food and water ad
libitum, at the Animal Center of Kaohsiung Veterans General
Hospital. The study protocol was approved by the Animal Care and
Use Committee of Kaohsiung Veterans General Hospital
(2020–2021-A031). CCRF-CEM cells (1×106 cells/100
µl/mouse) suspended in a 1:1 mixture of BD Matrigel™ (basement
membrane matrix, growth factor reduced, phenol red-free; BD
Biosciences, cat. no. 356231) and RPMI-1640 medium were injected
subcutaneously into the flank region of NOD/SCID mice. Tumor sizes
were measured using a digital caliper three times a week until
sacrifice. Mice were randomly assigned to different experimental
groups (n=5/group) as follows: vehicle control group, 20 mg/kg of
niclosamide treatment group, 5 mg/kg of niclosamide treatment group
and 1 mg/kg of dexamethasone (DEX) treatment group. Drugs were
delivered through intraperitoneal injections, three times a week,
starting on day 7 after inducing the xenograft mouse model.
Twenty-six days after tumor cell implantation, all mice were
anesthetized by sevoflurane (3%) (Baxter Healthcare Corp.), then
they were received CO2 (flow rate: 30%/min, for 7 min)
for euthanasia. Tumor tissues were collected for hematoxylin and
eosin (H&E) staining and immunohistochemistry staining.
Hematoxylin and eosin (H&E)
staining
Tumor tissues were removed from the xenograft mice
and fixed in 10% formalin for 24 h at room temperature. Tissues
were then embedded in paraffin and sectioned for staining with
hematoxylin and eosin (H&E). Microscopic images were captured
with a light microscope (Olympus BX53).
Immunohistochemistry staining (IHC
staining)
Tumor tissues were first removed from the xenograft
mice. Tumor specimens were fixed in 10% phosphate-buffered formalin
and then dissected and embedded in paraffin. Paraffin sections (5
µm thick) were incubated with 0.3% hydrogen peroxide for 15 min,
blocked for 1 h at room temperature, and incubated with cleaved
caspase-3 (cat. no. 9664; Cell Signaling Technology) and LC3B (cat.
no. 83506; Cell Signaling Technology) overnight at 4°C. After
washing with DPBS, sections were processed with the
DakoEnVision®+ Dual Link System-HRP (DAB+) kit, and
immediately stained with DAB (Dako) as required.
Statistical analyses
Data are expressed as mean ± standard error of mean.
Statistical analyses were performed using one-way ANOVA followed by
Tukey's test for post-hoc comparisons. Results were analyzed using
the software GraphPad Prism (version 6; GraphPad Software, Inc.).
P<0.05 was indicative of a statistically significant
difference.
Results
Niclosamide suppresses the viability
of T-ALL cells dose- and time-dependently
To investigate the effects of niclosamide on T-ALL
cell viability, Jurkat and CCRF-CEM human T leukemia cells were
initially treated with different doses of niclosamide or vehicle
control for 24 h. The cytotoxic activity of niclosamide on T-ALL
cells was measured with the MTT assays. Results showed that
niclosamide (0.5 to 4 µM) exhibited significant cytotoxicity on
Jurkat and CCRF-CEM cells and the toxic effects were dose-dependent
(P<0.05; Fig. 1A and B). In
addition, the effects of viability of niclosamide were also
investigated on Jurkat and CCRF-CEM cells at progressively longer
times (24, 48 and 72 h). The results showed that niclosamide at 2
µM significantly reduced the viable numbers of Jurkat cells, and
similarly at 1 µM in CCRF-CEM cells, both in a time-dependent
manner, when compared with the vehicle control (P<0.05; Fig. 1C and D). To further investigate the
cell proliferation, Ki-67 staining was performed. The results of
the Ki-67 analysis showed that niclosamide significantly reduced
cell proliferation in Jurkat and CCRF-CEM cells at all doses tested
(P<0.05; Fig. 1E and F). Taken
together, the results demonstrated that niclosamide had suppressed
the growth of T-ALL cells in a dose- and time-dependent manner.
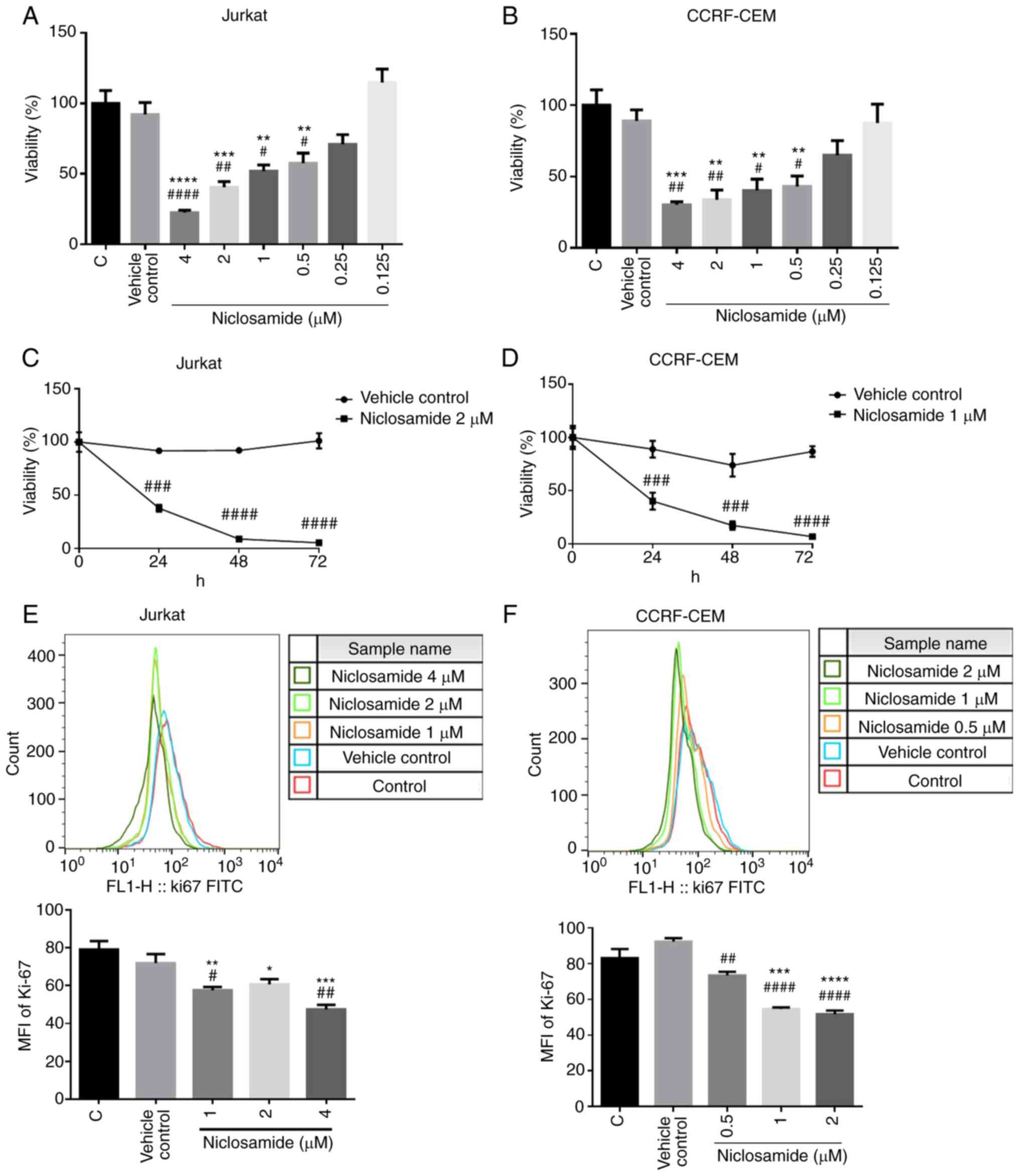 | Figure 1.Niclosamide effectively inhibits the
viability of T-ALL cells in a dose- and time-dependent manner.
Human T-ALL cells were treated with niclosamide for 24 h. Cell
viability and proliferation was measured by MTT assays and flow
cytometry of Ki-67 staining, respectively. MTT assays showed that
niclosamide inhibited the proliferation of (A) Jurkat and (B)
CCRF-CEM cells dose-dependently. (C) Jurkat and (D) CCRF-CEM cells
were treated with niclosamide for 24, 48 and 72 h. MTT assays
showed that niclosamide inhibited the proliferation of Jurkat and
CCRF-CEM cells time-dependently. MFI of Ki-67 were examined by flow
cytometry after incubation of (E) Jurkat and (F) CCRF-CEM cells
treated with different doses of niclosamide for 24 h. Niclosamide
treatment effectively inhibited the proliferation of T-ALL cells
dose-dependently. The results are expressed as mean ± SEM.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, vs. the
control group; #P<0.05, ##P<0.01,
###P<0.001, ####P<0.0001, vs. the
vehicle control group. C, control; vehicle control, DMSO; MFI, mean
fluorescence intensity; T-ALL, T-cell acute lymphoblastic
leukemia. |
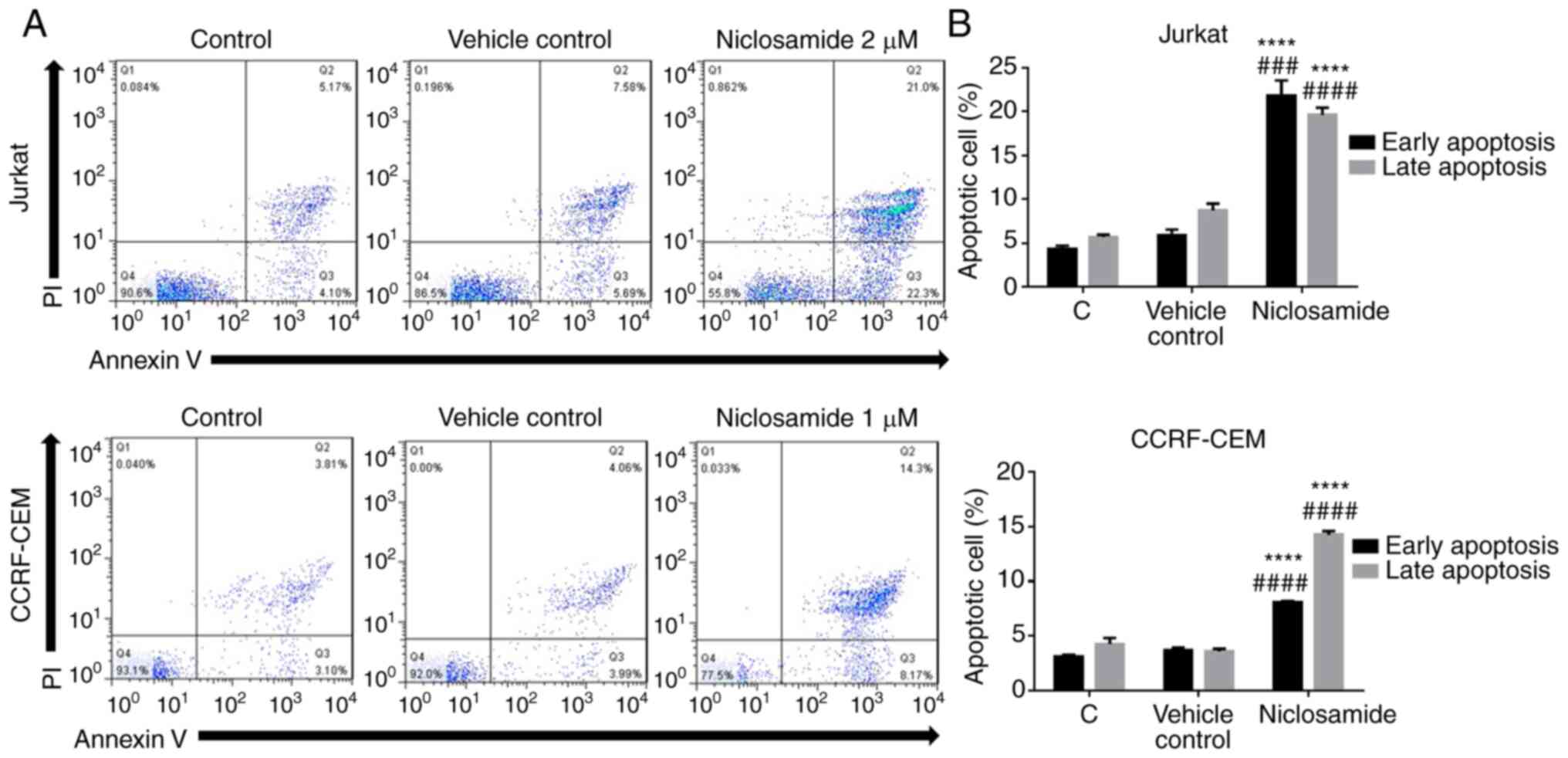
Niclosamide promotes the apoptosis of
T-ALL cells
To further characterize the cell death induced by
niclosamide in T-ALL cells, Annexin V-FITC/PI double staining flow
cytometry was conducted. Jurkat and CCRF-CEM cells were treated
either with vehicle control (DMSO) or niclosamide for 24 h. Annexin
V/PI staining demonstrated that niclosamide at 2 µM significantly
increased the early and late apoptosis in Jurkat cells, and
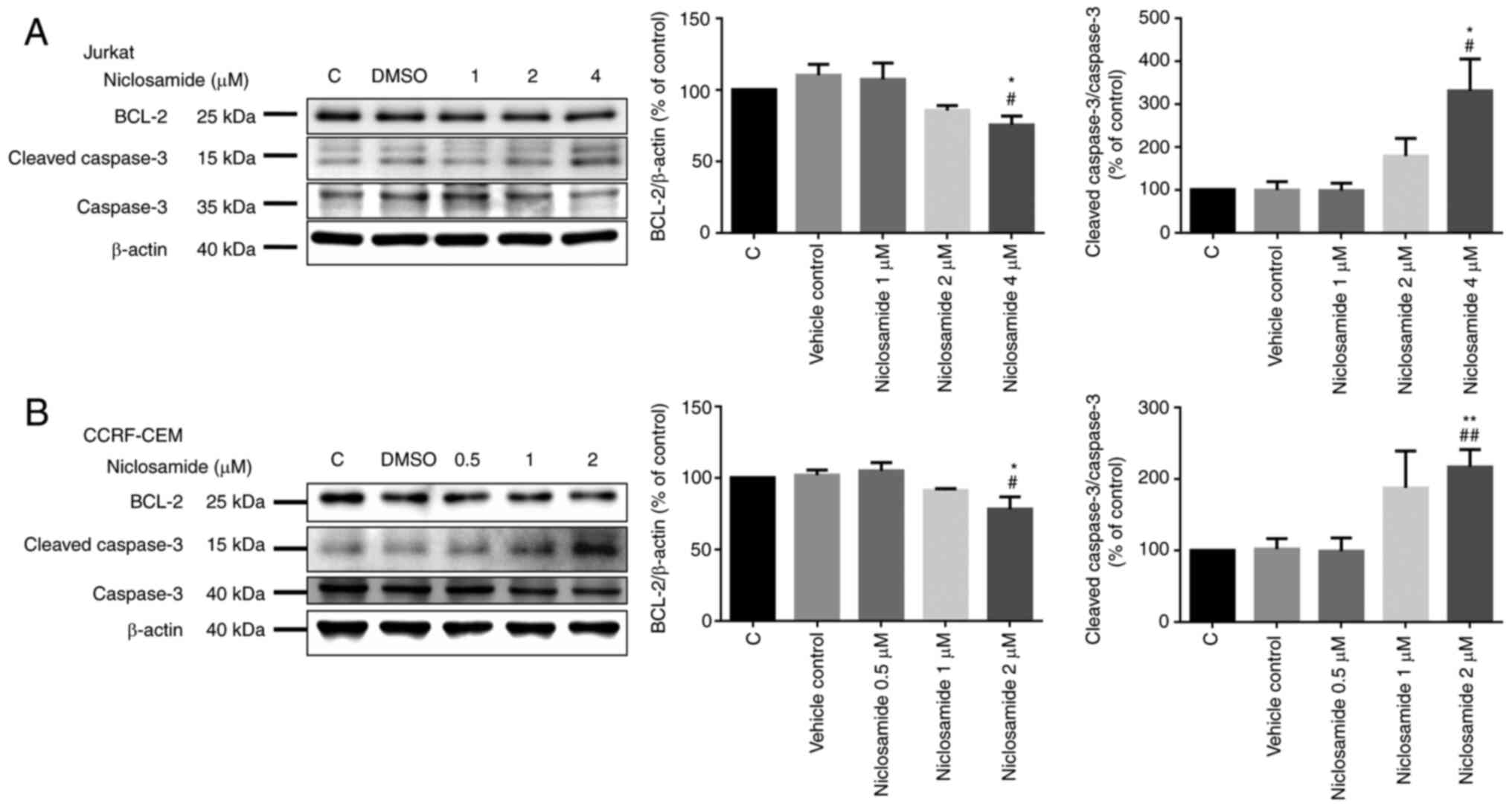
similarly at 1 µM in CCRF-CEM cells (P<0.05; Fig. 2). Moreover, apoptosis-related
proteins were also assessed. The Jurkat and CCRF-CEM cells treated
with different doses of niclosamide for 24 h were analyzed with
western blot analysis. The results showed that the level of Bcl-2
decreased significantly, and the level of cleaved caspase-3
increased significantly in a dose-dependent manner after
niclosamide treatment for 24 h in both Jurkat and CCRF-CEM cells
(P<0.05; Fig. 3). According to
our findings, there was no significant difference in cytochrome
c expression in the cytosolic/mitochondrial ratio, which
revealed that niclosamide affects cell apoptosis by
non-mitochondrial signaling pathways for the activation of caspases
(Fig. S1). Collectively, these
findings demonstrated that niclosamide promoted the apoptosis of
T-ALL cells in a dose-dependent manner.
Niclosamide induces the autophagy of
T-ALL cells
To investigate the role of autophagy induced by
niclosamide, Jurkat and CCRF-CEM cells were treated for 24 h with
either vehicle control (DMSO) or niclosamide at different doses. We
found that the level of Microtubule Associated Protein 1 Light
Chain 3β (LC3B) was increased significantly and the level of p62
was decreased significantly in a dose-dependent manner after
niclosamide treatment in both Jurkat and CCRF-CEM cells. But we
found no change in the level of Autophagy Related 5 (ATG5)
(Fig. 4). Taken together, the
results demonstrated that niclosamide activated autophagy in T-ALL
cells in a dose-dependent manner.
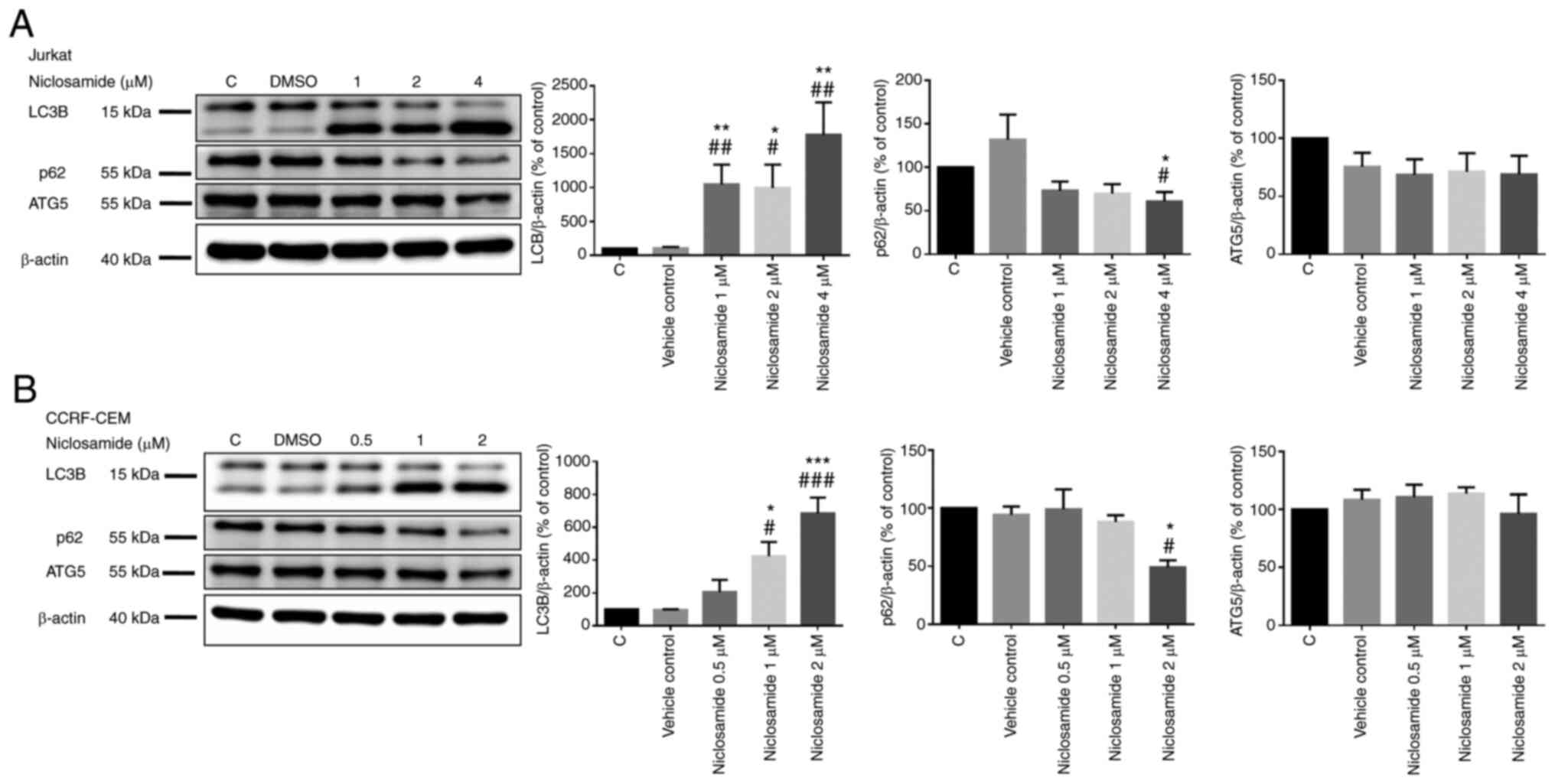 | Figure 4.Niclosamide induces T-ALL cell
autophagy in a dose-dependent manner. The cells were treated with
different doses of niclosamide for 24 h, and the expression of
LC3B, p62 and ATG5 were measured by western blot analysis in (A)
Jurkat and (B) CCRF-CEM cells. β-actin protein was used in these
experiments as the loading control. The results are expressed as
mean ± SEM. *P<0.05, **P<0.01, ***P<0.001, vs. the control
group; #P<0.05, ##P<0.01,
###P<0.001, vs. the vehicle control group. C,
control; vehicle control, DMSO; T-ALL, T-cell acute lymphoblastic
leukemia. |
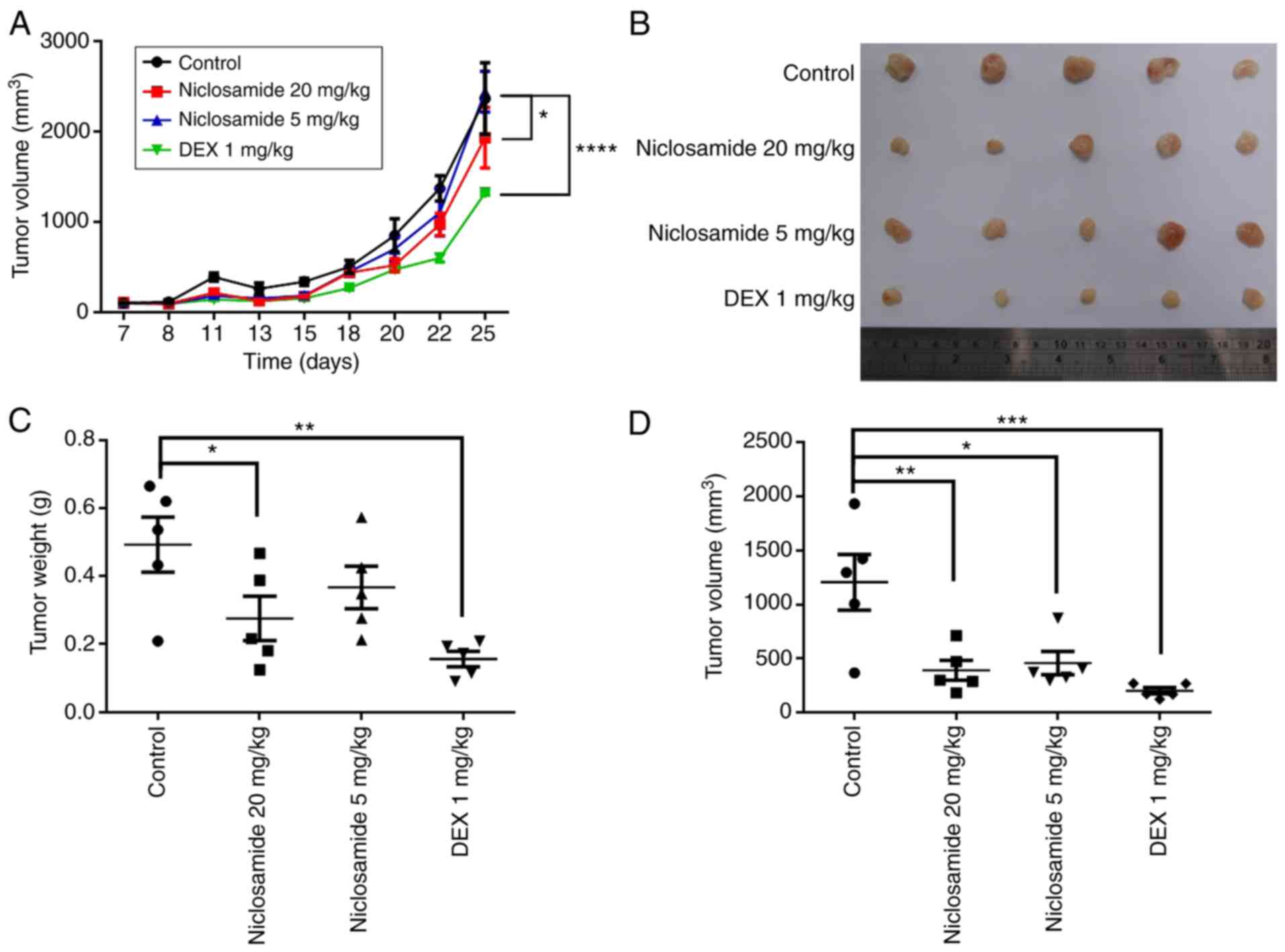
Niclosamide suppresses CCRF-CEM
leukemia cell growth in an in vivo xenograft mouse model
To confirm the in vitro results, the
antitumor effects of niclosamide in vivo were investigated
by measuring its effects in the CCRF-CEM xenograft murine model.
CCRF-CEM cells were subcutaneously injected into NOD/SCID mice for
26 days and treated with either 5 or 20 mg/kg of niclosamide, or 1
mg/kg of dexamethasone three times a week. The results showed that
mice treated with niclosamide (20 mg/kg) and dexamethasone had
significantly attenuated tumor growth compared with the controls
(P<0.05; Fig. 5A). At the end
of the study, tumors were isolated from the mice and weighed. The
overall weight and volume of the tumors in the niclosamide-treated
group and dexamethasone-treated group were significantly lower than
those of the controls (P<0.05; Fig.
5B-D). After 26 days, the mean tumor weight in the
niclosamide-treated group (20 mg/kg) was 0.2756±0.06513 g, and in
the dexamethasone-treated group was 0.1562±0.02282 g, both were
significantly lower than the controls (0.4934±0.08122 g)
(P<0.05; Fig. 5C). The mean
tumor volume in the niclosamide-treated group (20 mg/kg) was
392.8±92.49 mm3, in the niclosamide-treated group (5
mg/kg) was 458.4±106.2 mm3, and in the
dexamethasone-treated group was 202.9±29.08 mm3, all
statistically significantly smaller than the controls (1,206±257.0
mm3) (P<0.05; Fig.
5D). The survival rates of all the mouse groups were 100%
(Fig. S2). Therefore, the in
vivo results showed that niclosamide inhibited CCRF-CEM
leukemia cell tumor growth.
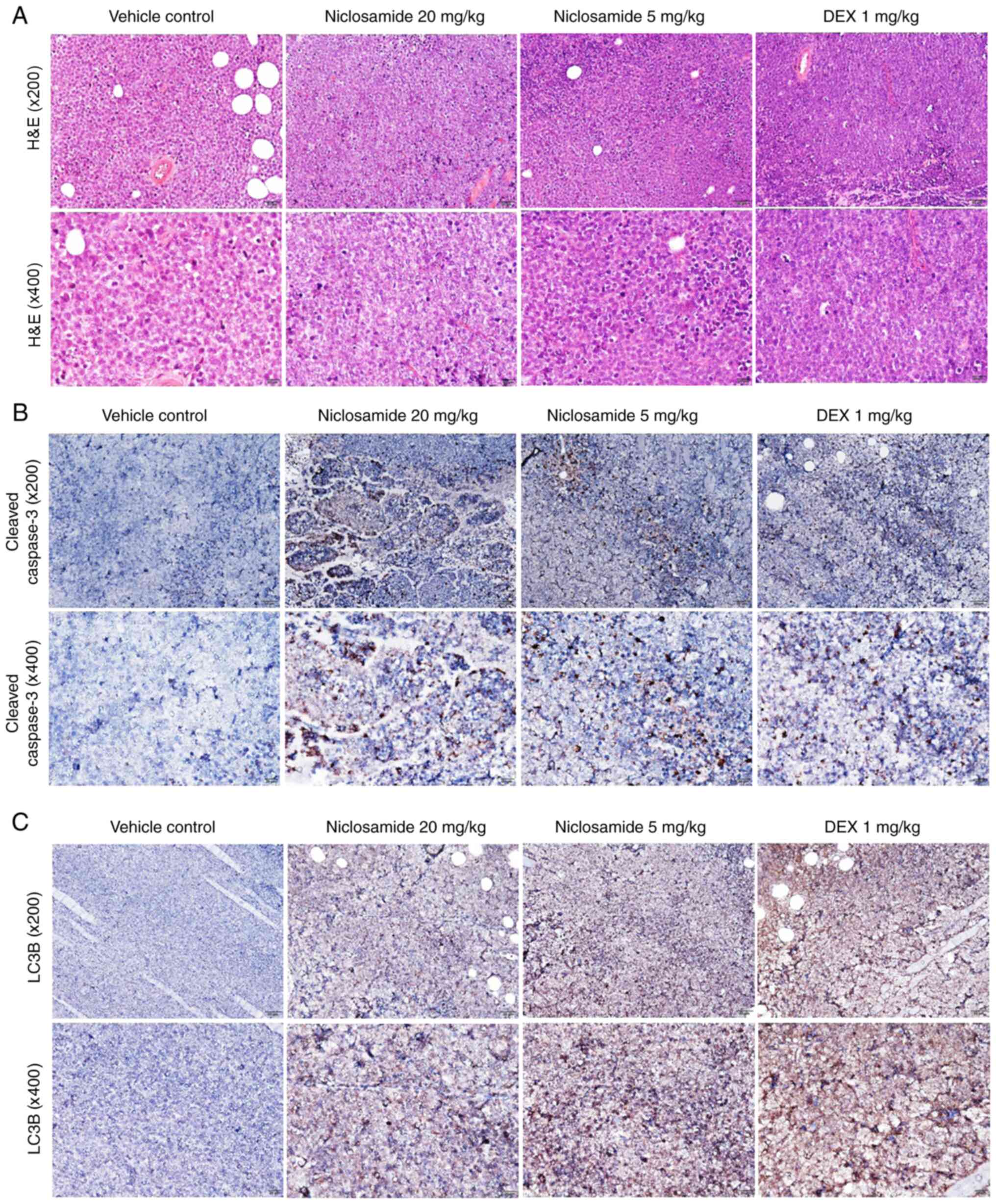
Niclosamide activates apoptosis and
autophagy proteins in the CCRF-CEM leukemia cell xenograft mouse
model
To investigate the effects of niclosamide on
apoptosis- and autophagy-related proteins, tumors from the CCRF-CEM
leukemia cells xenograft mouse model were collected for H&E
staining and immunohistochemical analysis. H&E staining showed
that tumor masses contained leukemia cells, with a discohesive
pattern of medium to large atypical lymphoid cells showing brisk
apoptosis and mitotic activities (Fig.
6A). Immunohistochemical analyses showed that CCRF-CEM leukemia
cell xenograft mice treated with niclosamide (5 and 20 mg/kg) and
DEX (1 mg/kg) exhibited markedly increased expressions of both
cleaved caspase-3 and LC3B compared with the vehicle controls
(Fig. 6B and C).
Discussion
Our present study is the first to report that
niclosamide effectively inhibits the growth of T-cell acute
lymphoblastic leukemia (T-ALL). Specifically, we analyzed the
effects of niclosamide treatment on apoptosis and autophagy in
T-ALL cells and in the xenograft mouse model. The results showed
that niclosamide inhibited the viability of T-ALL cells in a dose-
and time-dependent manner. Furthermore, we demonstrated that
niclosamide effectively inhibited the viability of T-ALL cells and
tumor growth through activating apoptosis and autophagy.
Niclosamide is an anti-helminthic agent approved by
the FDA. Previous studies have reported that niclosamide exhibits
anticancer activity by regulating the apoptotic pathways. Our
present results of niclosamide, regarding its suppression of ALL
cell viability via inducing apoptotic cell death, agree with
earlier reports that niclosamide induces apoptosis in various
cancer types, such as chronic myeloid leukemia (21), breast cancer (22) and adrenocortical carcinoma
(23). In the present study,
niclosamide exhibited anticancer effects by upregulation of cleaved
caspase-3 and downregulation of Bcl-2. According to our findings,
niclosamide did not affect expression of cytochrome c
(Fig. S1). The amount of
cytochrome c expression fluctuates with different times
(24). Therefore, our findings
showed that niclosamide affects cell apoptosis by non-mitochondrial
signaling pathways for activation of caspases.
In tumorigenesis, the role of autophagy as a tumor
suppressor or tumor promoter remains controversial. During
autophagy, p62 binds to LC3 to promote the degradation of
ubiquitinated cell organisms (25). High levels of p62 in human acute
myeloid leukemia are associated with a poor prognosis (26). Induction of autophagy by rapamycin
has effects of anti-leukemia in ALL (27). Therefore, all of these findings
indicate that autophagy has tumor-suppressive effects. In contrast,
other studies have reported that autophagy has tumor-promoter
action. Autophagy driven by genes or by pharmacological inhibition
was found to induce leukemia cell death (28,29).
What is the role of autophagy in niclosamide-treated T-ALL cells?
In the present study, niclosamide treatment increased LC3B
expression and decreased p62 expression, leading to T-ALL cell
death. Our results are in line with previous studies. Increased
autophagy could cause leukemia cell death. Niclosamide was found to
induce apoptosis and autophagic cell death by regulating
mitochondria dynamics (30).
Induction of apoptosis and autophagy both are known to cause T-ALL
cell death. For example, tamoxifen treatment was found to induce
apoptosis and autophagy in Jurkat cells (31). Previous studies have indicated that
Bcl-2 family members regulate autophagy. Bcl-2 family members
inhibit autophagy regulator beclin 1, which limits the formation of
autophagosome and inhibits autophagy (32,33).
In human HL60 cells, downregulation of Bcl-2 was found to induce
autophagy without impairing the mitochondrial functions (34). Therefore, this evidence indicates
that Bcl-2 exerts an inhibitory effect on autophagy. This evidence
is consistent with our findings. Niclosamide inhibited the
expression of Bcl-2 and induced autophagy by regulating the
formation of autophagosomes. Therefore, the present study showed
that apoptosis and autophagy are both activated by niclosamide,
leading to the death of T-ALL cells, both in vitro and in
vivo.
Most current leukemia studies have adopted the
xenograft mouse model by subcutaneously injecting tumor cells,
followed by measuring the volume and weight of the subcutaneous
tumors. Other xenograft mouse models use intravenous injection of
tumor cells. The number of tumor cells in the body after drug
treatment are evaluated by IVIS imaging to determine effects of
treatment. In the present study, we used the T-ALL xenograft mouse
model by subcutaneously injecting leukemia cells to evaluate the
effect of niclosamide treatment. Our study showed that niclosamide
effectively inhibited tumor growth in the T-ALL xenograft mice.
Pathological analyses showed that niclosamide treatment caused an
increase in pro-apoptosis marker cleaved caspase-3 and autophagy
marker LC3B. Most studies, including our present one, with
subcutaneous tumor cell injection in leukemia xenograft mice all
adopted volume and weight measurements of tumors to evaluate tumor
growth (35,36).
There are some limitations to the present study.
First, we observed that niclosamide activated apoptosis and
autophagy in T-ALL cells both in vitro and in vivo,
but there was no direct evidence to confirm that the inhibition of
autophagy affected apoptosis. To verify the relationship between
autophagy and apoptosis, one may need to use siRNA. Second, the
present study did not use clinical samples for further
verification. There are patients with a type of relapsed/refractory
T-ALL that are difficult to treat. In the future, other T-ALL
patients should be further investigated to ascertain whether
niclosamide can be their effective treatment.
In conclusion, the effects of niclosamide treatment
were assessed on T-ALL cells and in a T-ALL xenograft mouse model.
The results showed that niclosamide has the potential for
suppressing T-ALL cell viability in vitro, and in
suppressing tumor growth in T-ALL xenograft mice in vivo,
both by activating the apoptosis and autophagy pathways. Our
findings provide new insight into the application of niclosamide
for T-ALL treatment.
Supplementary Material
Supporting Data
Acknowledgements
We thank Dr Wen-Long Cho (Institute of Biomedical
Sciences, MacKay Medical College, Sanzhi, New Taipei City, Taiwan,
R.O.C.) for his suggestion for our experimental design. We thank Dr
Ren-Ching Wang (Department of Pathology and Laboratory Medicine,
Taichung Veterans General Hospital, Taichung, Taiwan, R.O.C.) for
providing assistance in the H&E staining analysis.
Funding
The present work was supported by grants from Taichung Veterans
General Hospital (TCVGH-NCHU1097611, TCVGH-NCHU1107609 and
TCVGH-1106503C) and Ministry of Science and Technology (MOST
110-2314-B-075A-002).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
FLH, SJY and CLL designed the experiments. FLH, SJY
and CLL performed the experiments and the statistical analyses for
the molecular biology experimental results. ECL, LYL and PWS
acquired and analyzed the data. FLH, SJY and CLL drafted the
initial manuscript. FLH and CLL confirmed the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The study protocol for the mouse xenograft
experiment was approved by the Animal Care and Use Committee of
Kaohsiung Veterans General Hospital (2020–2021-A031).
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
References
|
1
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Terwilliger T and Abdul-Hay M: Acute
lymphoblastic leukemia: A comprehensive review and 2017 update.
Blood Cancer J. 7:e5772017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Karrman K and Johansson B: Pediatric
T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer.
56:89–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Corella Aznar EG, Ayerza Casas A, Carboné
Bañeres A, Calvo Escribano MÁC, Labarta Aizpún JI and Samper
Villagrasa P: Quality of life and chronic health conditions in
childhood acute leukaemia survivors. Med Clin (Barc). 152:167–173.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh SK, Banerjee S, Acosta EP, Lillard
JW and Singh R: Resveratrol induces cell cycle arrest and apoptosis
with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and
p27KIP1 pathway. Oncotarget. 8:17216–17228. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cai Y, Xia Q, Su Q, Luo R, Sun Y, Shi Y
and Jiang W: mTOR inhibitor RAD001 (everolimus) induces apoptotic,
not autophagic cell death, in human nasopharyngeal carcinoma cells.
Int J Mol Med. 31:904–912. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ciombor KK and Bekaii-Saab T: Selumetinib
for the treatment of cancer. Expert Opin Investig Drugs.
24:111–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang FL, Liao EC, Li CL, Yen CY and Yu
SJ: Pathogenesis of pediatric B-cell acute lymphoblastic leukemia:
Molecular pathways and disease treatments (Review). Oncol Lett.
20:448–454. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Al-Hadiya BM: Niclosamide: Comprehensive
profile. Profiles Drug Subst Excip Relat Methodol. 32:67–96. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Merschjohann K and Steverding D: In vitro
trypanocidal activity of the anti-helminthic drug niclosamide. Exp
Parasitol. 118:637–640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hogarth LA and Hall AG: Increased BAX
expression is associated with an increased risk of relapse in
childhood acute lymphocytic leukemia. Blood. 93:2671–2678. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen W, Mook RA Jr, Premont RT and Wang J:
Niclosamide: Beyond an antihelminthic drug. Cell Signal. 41:89–96.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu Y, Zuo W, Chen L, Bian S, Jing J, Gan
C, Wu X, Liu H, Su X, Hu W, et al: Repurposing of the
anti-helminthic drug niclosamide to treat melanoma and pulmonary
metastasis via the STAT3 signaling pathway. Biochem Pharmacol.
169:1136102019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liao Z, Nan G, Yan Z, Zeng L, Deng Y, Ye
J, Zhang Z, Qiao M, Li R, Denduluri S, et al: The anthelmintic drug
niclosamide inhibits the proliferative activity of human
osteosarcoma cells by targeting multiple signal pathways. Curr
Cancer Drug Targets. 15:726–738. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mathew R, Kongara S, Beaudoin B, Karp CM,
Bray K, Degenhardt K, Chen G, Jin S and White E: Autophagy
suppresses tumor progression by limiting chromosomal instability.
Genes Dev. 21:1367–1381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Teuffel O, Kuster SP, Hunger SP, Conter V,
Hitzler J, Ethier MC, Shah PS, Beyene J and Sung L: Dexamethasone
versus prednisone for induction therapy in childhood acute
lymphoblastic leukemia: A systematic review and meta-analysis.
Leukemia. 25:1232–1238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bhadri VA, Trahair TN and Lock RB:
Glucocorticoid resistance in paediatric acute lymphoblastic
leukaemia. J Paediatr Child Health. 48:634–640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang H, Dong R, Fan WW, Zheng XC, Li AM
and Wang WD: Timosaponin AIII induces autophagy of Tcell acute
lymphoblastic leukemia Jurkat cells via inhibition of the
PI3K/Akt/mTOR pathway. Oncol Rep. 41:2937–2944. 2019.PubMed/NCBI
|
|
20
|
Wang J, Ren XR, Piao H, Zhao S, Osada T,
Premont RT, Mook RA Jr, Morse MA, Lyerly HK and Chen W:
Niclosamide-induced Wnt signaling inhibition in colorectal cancer
is mediated by autophagy. Biochem J. 476:535–546. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jin B, Wang C, Shen Y and Pan J:
Anthelmintic niclosamide suppresses transcription of BCR-ABL fusion
oncogene via disabling Sp1 and induces apoptosis in
imatinib-resistant CML cells harboring T315I mutant. Cell Death
Dis. 9:682018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim
H, Yin T, Fan S and Shen Q: Activation of STAT3 and Bcl-2 and
reduction of reactive oxygen species (ROS) promote radioresistance
in breast cancer and overcome of radioresistance with niclosamide.
Oncogene. 37:5292–5304. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Satoh K, Zhang L, Zhang Y, Chelluri R,
Boufraqech M, Nilubol N, Patel D, Shen M and Kebebew E:
Identification of niclosamide as a novel anticancer agent for
adrenocortical carcinoma. Clin Cancer Res. 22:3458–3466. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Krause M and Durner J: Harpin inactivates
mitochondria in Arabidopsis suspension cells. Mol Plant Microbe
Interact. 17:131–139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu
HL, Yang C and Liu HF: p62 links the autophagy pathway and the
ubiqutin-proteasome system upon ubiquitinated protein degradation.
Cell Mol Biol Lett. 21:292016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nguyen TD, Shaid S, Vakhrusheva O,
Koschade SE, Klann K, Thölken M, Baker F, Zhang J, Oellerich T,
Sürün D, et al: Loss of the selective autophagy receptor p62
impairs murine myeloid leukemia progression and mitophagy. Blood.
133:168–179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gong Y, Wu J, Yang R, Zhang L and Ma Z:
Rapamycin-induced autophagy plays a pro-survival role by enhancing
up-regulation of intracellular ferritin expression in acute
lymphoblastic leukemia. Exp Oncol. 42:11–15. 2020.PubMed/NCBI
|
|
28
|
Rothe K, Lin H, Lin KB, Leung A, Wang HM,
Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM and Jiang X:
The core autophagy protein ATG4B is a potential biomarker and
therapeutic target in CML stem/progenitor cells. Blood.
123:3622–3634. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baquero P, Dawson A, Mukhopadhyay A, Kuntz
EM, Mitchell R, Olivares O, Ianniciello A, Scott MT, Dunn K,
Nicastri MC, et al: Targeting quiescent leukemic stem cells using
second generation autophagy inhibitors. Leukemia. 33:981–994. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park SJ, Shin JH, Kang H, Hwang JJ and Cho
DH: Niclosamide induces mitochondria fragmentation and promotes
both apoptotic and autophagic cell death. BMB Rep. 44:517–522.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Torres-López L, Maycotte P, Liñán-Rico A,
Liñán-Rico L, Donis-Maturano L, Delgado-Enciso I, Meza-Robles C,
Vásquez-Jiménez C, Hernández-Cruz A and Dobrovinskaya O: Tamoxifen
induces toxicity, causes autophagy, and partially reverses
dexamethasone resistance in Jurkat T cells. J Leukoc Biol.
105:983–998. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chong SJF, Marchi S, Petroni G, Kroemer G,
Galluzzi L and Pervaiz S: Noncanonical cell fate regulation by
Bcl-2 proteins. Trends Cell Biol. 30:537–555. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Saeki K, Yuo A, Okuma E, Yazaki Y, Susin
SA, Kroemer G and Takaku F: Bcl-2 down-regulation causes autophagy
in a caspase-independent manner in human leukemic HL60 cells. Cell
Death Differ. 7:1263–1269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ge C, Huang H, Huang F, Yang T, Zhang T,
Wu H, Zhou H, Chen Q, Shi Y, Sun Y, et al: Neurokinin-1 receptor is
an effective target for treating leukemia by inducing oxidative
stress through mitochondrial calcium overload. Proc Natl Acad Sci
USA. 116:19635–19645. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang L, Li M, Wang F, Zhen C, Luo M, Fang
X, Zhang H, Zhang J, Li Q and Fu L: Ceritinib enhances the efficacy
of substrate chemotherapeutic agent in human ABCB1-Overexpressing
leukemia cells in vitro, in vivo and ex-vivo. Cell Physiol Biochem.
46:2487–2499. 2018. View Article : Google Scholar : PubMed/NCBI
|