1. Introduction
Evolution can be viewed as the accidental result of
random mutations, in both gene-coding and non-coding regions of the
DNA, leading to the amazing diversity of life forms currently
observed on earth. Still, it is the genetic, epigenetic,
epitranscriptomic and socio-environmental events that act upon the
developmental mechanisms, that guide evolution and dictate the
survival of a single living unit and a species' reproductive
success across and along generations. Genetic alterations that can
‘switch off’ genes or enhance their expression, or even relegate
them to non-coding segments of DNA, are merely part of the
equation. Genetic and environmental challenges are determining
evolution in terms of the survival of the fittest. Natural
selection guides the evolutionary process by favoring genes for
their ability to propagate their information through
generations.
Evolutionary and developmental biology, namely
evo-devo, refers to the mechanisms through which developmental
processes determine and modulate variations, and dictate the
evolutionary outcomes throughout the course of history. The main
focus of evo-devo, as previously described by Hendrikse et
al, is the capacity of developmental systems for adaptive
evolution through these variations, or else evolvability (1). Robustness, that is the ability of a
system to be resistant to change, is a basic concept of
evolvability. Robust organisms have the ability to accumulate
genetic variations with no effects on their phenotype; however,
this cryptic evolutionary potential of their genomes can be
released in a new environmental and genetic background, enhancing
the probability of the organisms to adapt (2).
Cancer can be viewed as a robust, evolvable system,
and focusing on the evolutionary origins of cancer can be used as
an alternative approach (3). The
genes that are responsible for the processes of proliferation
inhibition, cell death, division of labor, resource allocation and
extracellular environment maintenance, are the genes that sustain
the viability of complex multicellular organisms (4). They are also the genes that
malfunction in cancer (5). A wide
number of oncogenes and tumor suppressors have been discovered over
the past 50 years, and almost all cancers are driven by genetic
alterations in these genes. Hence, the genetic basis of cancer has
an ancient past. According to the atavistic theory, cancer is the
result of the accumulation of mutations that reprogram the cell
into adopting a primitive phenotype, by reactivating an ancient
behavior characterized by highly conserved survival (6). This phenotype is characterized by the
upregulation of ancestral genes with unicellular evolutionary
origins, disrupting the genetic regulatory network that rules upon
complex multicellular organisms and leading to uncontrolled
proliferation in adverse environmental conditions (7-9).
As such, cancer events can be considered as a
drawback or a side-effect of evolution. However, under the scope of
evo-devo as an interplay of yin and yang, it can be argued that
cancer events can be viewed as an integral component of evolution
(10). At a (cancer) cell-level,
this phenotype depicts a ‘drive for survival’ with its evolutionary
roots in the early transition stages from unicellularity to
multicellularity that lead to a diversity burst, whereas at the
(host) organism-level, a cancer event outlines the ‘unfitness’ of
the host for natural selection. The trading between two
evolutionary states, an ancestral one characterized by
stochasticity and plasticity/flexibility with a high evolutionary
rate and a recent, synchronous equilibrium with a refined
regulatory machinery (however threatened by a variety of
stressors), by activating and deactivating ‘ancient’ genes may be
the gear which sets in motion the forces that formulate life by
survival selection and thus, shaping evo-devo.
In the following sections, focus is paid on
elucidating the role of ancestral genes involved in signaling,
metabolism and transcription pathways as stake holders of evo-devo
under the prism of reduced survival and disease through ‘cancer’.
Collectively, a list of inhouse curated genes that are involved in
signaling, metabolism and transcription in cancer, by harvesting
the NCBI Gene Database (11) with
the respective keywords and filtering out the noise, is provided in
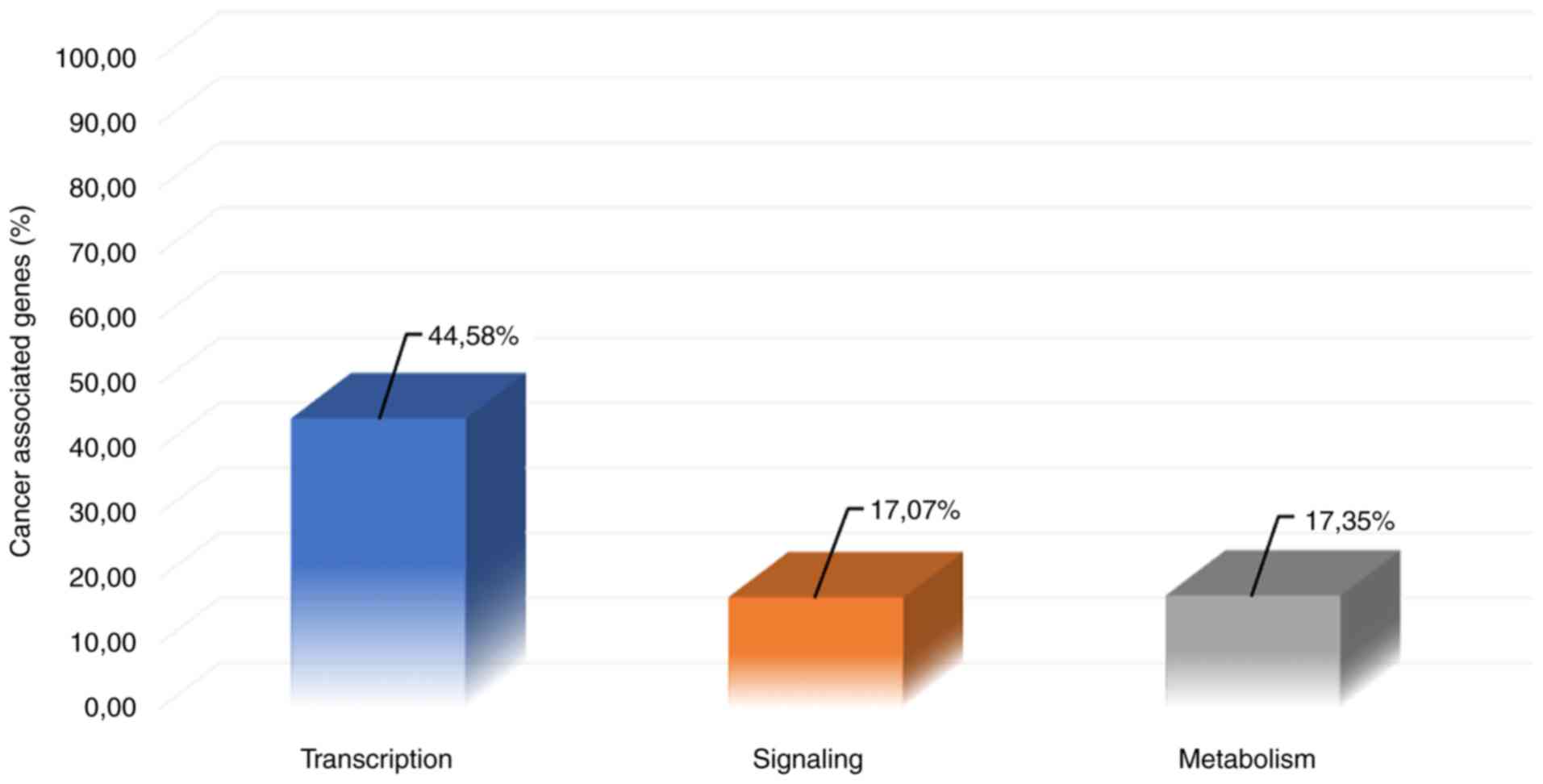
Table SI. Notably, as shown in
Fig. 1, 44.58% of
cancer-associated genes are involved in transcription pathways,
whereas the co-occurrence of signaling- and metabolism-associated
genes in cancer is 17.07 and 17.35%, respectively.
2. Signaling in cancer
Cells are able to respond and adapt to their
environment through signal transduction. Signaling pathways
coordinate intra- and intercell communication, as well as the
communication between cells and the extracellular matrix. Genetic
and epigenetic alterations often lead to the disruption of
signaling networks and enable cancer cells to escape regular
control mechanisms (12). These
alterations are involved in cancer progression, such as cell growth
and proliferation, angiogenesis and inflammation. Oncogenic
mutations can cause the hyperactivation of the signaling pathways
and the overexpression of the affected genes or produce mutated
proteins with deficient activity, and deletions or other mutations
can inactivate tumor suppressors that normally act as negative
regulators in signaling (13).
Even though the mutational profile of tumors is highly diverse,
there are a small number of mutations with a causative role in
oncogenesis, termed ‘drivers’, that lead the cancer cell to a
fitness advantage (14,15). ‘Passengers’ on the other hand are
estimated to account for ~97% of mutations in cancer and are mostly
considered random non-functional mutations, although there are
indications of a certain fitness cost for the tumor upon elevated
passengers load (16,17). Driver mutations have been shown to
affect a limited number of signaling pathways that regulate three
core cellular processes: Cell fate, cell survival and genome
maintenance, rendering the focus towards a consensus of mutated
driver pathways rather than individual driver genes (18-20).
A representative example of driver mutation is p53
tumor suppressor protein, that is a crucial component in the
regulation of cell cycle in multicellular organisms (21). The evolutionary history of the p53
pathway can be traced back to the beginning of multicellularity and
is activated upon cellular stress signals (22,23).
The previous evolutionary study by Belyi et al traced the
origins of the p53 family genes back to the unicellular
choanoflagellates and the early metazoan sea anemone, where the
ancestral gene was found to be related to the p63/p73-like gene,
and upon gene duplication in the early vertebrate lineage, the
resulting product was closely associated with that of the p53 gene
(24). The p53 protein is a
transcription factor that binds to specific DNA sequences and acts
as an anticancer promoting agent, and is therefore also known as
the ‘genome guardian’ (25). It
activates DNA-repair proteins, induces growth arrest and initiates
apoptosis. p53 is encoded by the TP53 gene, which is the most
frequently mutated gene in human cancers. In almost half of human
cancer types, p53 is inactivated by mutation, leading to a severely
reduced tumor suppressive activity (26).
Another example of genetic alterations deregulating
the signal transduction and promoting tumor progression is the
phosphoinositide 3-kinase (PI3K)/Akt signaling pathway. The
PI3K/Akt pathway is evolutionary conserved and regulates
metabolism, proliferation, cell survival, growth, motility and
apoptosis in response to extracellular signals (27). Both PI3K and Akt enzymes have a
wide phylogenetic distribution and have been identified in
unicellular organisms and have later evolved through complex
duplication patterns (28,29). Activating mutations in PI3K, Akt
and PIK3R1 have been described in cancer and result in persistent
amplification of the PI3K/Akt pathway and aberrant cell
proliferation. Inactivating mutations and deletions of phosphatase
and tensin homolog deleted on chromosome 10 (PTEN) and inositol
polyphosphate-4-phosphatase, type II (INPP4B) tumor suppressors
lead to unrestrained downstream signaling (30). Mutations in TSC1 and TSC2 tumor
suppressors hyperactivate signaling by mammalian target of
rapamycin (mTOR)C1, a crucial target of the PI3K/Akt pathway
(31,32).
The Notch signaling pathway is involved in cell
development and differentiation, and has also been found to be
aberrantly activated in a number of different solid tumors through
mutations (33). The Notch pathway
is based on cell-cell contacts for signal transduction and is
highly involved in tumor metastasis (34,35).
Of note, both gain- and loss-of-function mutations in NOTCH
isoforms are described, reverting the ability of the Notch
signaling pathway from an oncogene to a tumor-suppressor in a
highly context-dependent manner (36,37).
A gene that is present in unicellular organisms and
associated with cancer in multicellular organisms is the
helicase-associated endonuclease for fork-structured DNA (Hef). Hef
is a protein found in Archaea and is required for the processing of
blocked replication forks. The vertebrate Hef ortholog appears to
participate in the Fanconi anemia-related (FA) tumor suppressor
pathway, while the archaebacterial Hef processes stalls replication
forks in order for them to be repaired by homologous recombination
(38). Vertebrate Hef seems to
also play a role in DNA repair mechanisms, where it interacts
directly with DNA structures that are DNA replication intermediates
and may contribute to resolving DNA crosslinks through a complex
association with the FA complementation group C gene (38).
The 70-kDa family of heat shock proteins (HSP70) is
regarded as one of the most conserved groups of proteins in
evolution. The majority of organisms include multiple members of
the protein, while some, such as archaebacteria, include at least
one (39). Hsp70 proteins are
molecular chaperones that participate in a diverse group of
processes, including protein folding and remodeling as they act
virtually at all stages of protein life, from synthesis to
degradation, and thus are essential in protein homeostasis
(40). Cancer cells rely heavily
on the mechanisms of HSP70s regulation for survival. The majority
of human tumors, as an example, overexpress HSP70 family members,
and this overexpression can be used as a biomarker for a poor
prognosis (39). This
overexpression in the case of mammary carcinoma may be largely due
to the proliferation of misfolded proteins and overexpressed
oncoproteins that trigger the transcription of HSP genes. A prime
example is the ability of Tp53 to suppress the HSP70 promoter, and
the loss of this protein, a common event in cancer, may lead to an
increase in the expression of said chaperone proteins (41).
3. Metabolism in cancer
Metabolic reprogramming is a hallmark of cancer. The
activation of cancer genes and the progression of a cancer event
alter the metabolic process of the cell, in order for it can meet
the high demands of cancer cells in energy and nutrient resources.
The majority of malignant cells switch to aerobic glycolysis as a
preferred metabolic pathway, a phenomenon also known as the Warburg
effect, producing a high amount of secreted lactate (42,43).
The acquired glycolytic behavior of cancer cells holds for the
sustainability of their bioenergetics, biosynthesis and redox
demands. Even though oxidative phosphorylation is a much more
efficient source of ATP compared to glycolysis, it has been shown
that the high glycolytic rates in cancer cells are favored due to
the increased levels of precursor metabolites for anabolic pathways
(44). In addition, glycolysis
enables cancer cells to deal with unfavorable conditions, such as
hypoxia and a low nutrient supply, and to adapt to their highly
heterogeneous microenvironment, thus maintaining an evolutionary
advantage (45).
The activation of specific oncogenes and the loss of
tumor suppressors, along with the upregulation of the PI3K pathway
is controlling the metabolic switch in cancer (46,47).
The activation of Myc, Ras and Akt, and the inactivation of p53
have been shown to upregulate glycolytic enzymes and glucose
transporters and stimulate glycolysis (48,49).
Additionally, to maintain this glycolytic phenotype, cancer cells
upregulate a number of plasma membrane transporters, such as
monocarboxylate transporters (MCTs), that mediate the proton-linked
transport of metabolic monocarboxylic acid (50). Specifically, MCT1 and MCT4 isoforms
are highly involved in maintaining the metabolic phenotype of
cancer cells by facilitating the transport of lactate across the
plasma membrane and regulating the intracellular pH by
co-transporting a proton (51).
MCT4 has the lowest affinity for lactate among MCTs and is the main
isoform that mediates lactic acid efflux from glycolytic cells,
including white skeletal muscle fibers, astrocytes, immune cells,
chondrocytes and hypoxic cells (52). MCT4 expression is upregulated under
hypoxic conditions and oxidative stress by hypoxia-inducible factor
1α (HIF-1α), favoring lactate extrusion from the cell MCTs and
preventing intracellular acidification, thus promoting a number of
carcinogenic processes (53-55).
MCT4 is also recognized for its role in metastasis and is shown to
be upregulated in the tumor stroma by oncogenes Ras and nuclear
factor (NF)-κB (56). The
increased expression of MCT4 and its ancillary protein, CD147, is
also associated with a poor prognosis in a number of types of
cancer (57). c-Myc has been shown
to increase the expression of MCT1, as in with pyruvate
dehydrogenase kinase-1 (PDK-1) that phosphorylates the pyruvate
dehydrogenase enzyme and lactate dehydrogenase A (LDH-A), an enzyme
that catalyzes the conversion of lactate to pyruvate (58,59).
As such, MCTs are considered as promising therapeutic targets for
disrupting the glycolytic cascade of cancer cells.
One of the key enzymes found in all organisms and a
key component in the metabolic reprogramming of cancer, is
glutamine synthetase (GS). GSs is responsible for nitrogen
metabolism, where it participates in the biochemical reaction of
ammonia assimilation and in glutamine biosynthesis. It has been
demonstrated that GS expression is induced by the Myc oncogene,
resulting in increased glutamine anabolism that is associated with
increased cell proliferation, survival and transplant tumor growth.
All of the above lead to the conclusion that GS expression plays an
important role in Myc-driven carcinogenesis (60).
Three types of GS have been reported from previous
studies. The two most basic types are GSI and GSII. GSI has been
identified in prokaryotic organisms, whereas GSII has been
identified mainly in eukaryotes, but also in some prokaryotes
belonging to the families Rhizobiaceae, Frankiaceae and
Streptomycetaceae (61). Finally,
the third type of GS is GSIII, which has been observed in the
anaerobes Bacteroides fragilis (62) and Butyrivibrio fibrisolvens
(63). These types differ in both
their primary and tertiary structure. GSI consists of 12 identical
subunits, which are structured in 2 layers, each consisting of 6
subunits. At the active site, the synthetase contains a pair of
Mn++ ions and forms 2 antisense π-structures, one in the
carboxyl terminal end of a subunit and the other in the N-terminal
end of the adjacent subunit. GSII and GSIII contain fewer subunits
than GSI, with GSII consisting of 8 subunits and GSIII of 6. It is
still worth noting that a fourth type of synthetase has been
identified in Rhizobium leguminosarum and is reported as
glnT, which has more common elements with prokaryotic GSI (64).
According to the results of previous evolutionary
analyses in which the genes encoding GS were aligned in different
organisms, it was found that the genes of GSI and GSII existed
1,700 million years prior to the divergence of prokaryotes and
eukaryotes (60). In the study by
Shatters and Kahn, it was denoted that the common ancestor of the
GSII genes in Rhizobiaceae and the host plant was older than the
plant itself (65). Moreover, it
is surprising that the genes encoding the rice and pea chloroplast
GSII enzymes are more closely related than the corresponding genes
of the same species. Finally, it is estimated that mitochondrial
GSII is 1,050 million years old (60).
4. Transcription in cancer
Dysregulation in the gene expression program is also
a signature in cancer. Transcriptional regulation and gene
expression are controlled by a vast number of transcription
enzymes, transcription factors, co-factors and chromatin regulators
that are interacting in a highly coordinated manner and
perturbations on the transcriptional mechanism controls are evident
in all tumors (66). Gene
alterations that result in the activation of oncogenes, the
inactivation of tumor suppressors and the upregulation of protein
kinases can promote transcription and subsequently drive cell
proliferation.
RNA polymerases are highly conserved in evolution,
and their subunits exhibit a common structural framework, while
being operated by closely related molecular mechanisms (67). In their study, Werner and Grohmann
(67) denoted that the last
universal common ancestor of bacteria, Archaea, and Eukarya carried
an RNA polymerase very similar to the simplest form of contemporary
RNAPs found in bacteria, while Shin et al indicated that
archaeal RNAPs share more properties with their eukaryotic homologs
(68). RNA polymerase I (Pol I) is
the most highly engaged enzyme in the general transcriptional
machinery, accounting for >60% of the overall cell
transcriptional activity (69).
Cancer cells have a higher biosynthetic demand, exhibiting
upregulated ribosome biogenesis, known as ‘nucleolar hypertrophy’,
to support cell growth and uncontrolled cell proliferation.
Ribosome production is strictly dependent on the Pol I
transcription machinery in the nucleolus; thus, Pol I activity is
further increased in proliferating cells (70). A number of oncogenic factors
driving accelerated Pol I transcription have been reported over the
years. The oncogenic activity of Myc has been shown to stimulate
Pol I transcription and enhance ribosomal biogenesis (71,72).
Activated mTOR induces Pol I transcription and ribosome synthesis
by activating upstream binding factor (UBF) and transcription
initiation factor 1A (TIF1A) (73). The inactivation of PTEN tumor
suppressor results in the constitutive activation of the oncogenic
PI3K/AKT pathway and tumorigenesis (74). A number of tumor cells harbor
mutations that affect both pRb and p53 tumor suppressors, that
normally suppress Pol I transcription and inhibit cellular rRNA
synthesis, having an added impact on Pol I activity (75).
Cancer cells also exhibit an upregulated activity of
RNA polymerase II (Pol II) to produce a high number of transcripts,
including oncogenes and anti-apoptotic factors, supporting rapid
growth and resistance to apoptosis (76). The control of Pol II is highly
regulated by transcriptional factors and non-coding RNAs (ncRNAs)
and is critical for the cell homeostasis. Genetic variations, such
as mutations in transcription factors that control Pol II can
result in a disruption of the pause release and elongation process.
Increased levels of c-Myc cause transcriptional amplification by
accumulating at promoters regions and producing high levels of
transcripts, thus inducing tumorigenesis (77). Gene fusion events have been shown
to alter the transcription elongation, as in the case of the
chromatin regulator MLL in leukemias (78). Long non-coding RNAs have also been
implicated for their role in cancer progression, functioning as
transcriptional regulators. The lncRNA ANRIL induces the
transcriptional repression of members of the INK4A/ARF/INK4B locus,
which encode tumor suppressors whose deactivation is associated
with various types of cancer (79).
5. Computational methods for pharmacological
targeting in cancer
Developing efficient therapeutic methods for
anticancer drug design remains a challenge, even though success
stories have been reported over the past years. Cancer is a
heterogeneous disease and a deeper understanding of the underlying
molecular mechanisms that drive its initiation, progression and
metastasis is crucial for providing effective treatment and
improved diagnostics. Through the advances of bioinformatics and
omics technologies, a wide variety of computational methods can be
applied for pharmacological targeting in cancer.
Structure and ligand-based drug design are principal
methodologies for drug discovery and lead optimization.
Ligand-based approaches use the information of known active and/or
inactive molecules to generate SAR models, whereas structure-based
approaches use the structural information of the protein target to
discover lead molecules as potent inhibitors (80). Homology modelling techniques are
applied when an experimentally determined structure of a protein is
not available. Molecular docking and molecular dynamics are
extensively used to simulate the conformational state of a protein
target and protein-ligand interactions to estimate the binding
affinity. Virtual high-throughput screening through docking of
small molecule libraries have been successfully applied for the
identification of novel inhibitors with anticancer properties, as
in the case of targeting Cdc25A phosphatases and protein kinase CK2
(81,82). The physicochemical properties of
small molecules with potent activity can be also analyzed through
statistical methods and the applications of artificial intelligence
(AI), such as machine learning, in the pursuit of a selective
inhibitor. A key factor for the success of these strategies is to
target specific biomolecules involved in functional molecular and
biological traits that distinguish cancer cells from normal cells,
known as the hallmarks of cancer.
Omics data are accumulating rapidly and can be
assessed through statistical analysis and computational methods to
uncover potential targets for efficacious therapeutics in cancer.
Systems biology approaches, including computational modeling,
network analysis, gene signature analysis, functional genomics,
protein-protein interactions and high-throughput screening, are
efficient tools for advancing the prediction of efficient
therapeutics in complex diseases, such as cancer (83). In network analysis, computational
models of signaling networks are designed and used to predict
systems properties that can indicate and prioritize protein targets
for cancer therapy (84).
Computational approaches have been used for the prediction of
functional impact of mutations and discriminate driver from
passenger mutations, based on evolutionary conservation, protein
structure modifications and observed recurrence in existing cancer
datasets (85). Chen et al
illustrated the oncogenic signatures in the tyrosine kinase family
through an evolutionary analysis concluding that gain-of-function
mutations are causing reverse evolution on the oncogenes supporting
the cancer atavistic model (86).
An interesting computational analysis in gene expression data was
previously presented by Trigos et al (87), where 7 solid tumors were
investigated with regards to their corresponding gene ages using
phylostratigraphy. The results indicated that a common feature in
tumors was a trend for the preferential expression of more ancient
genes. The authors reported that these cellular processes assigned
to a unicellularity origin were more active in tumors. Additional
research demonstrates the effectiveness of evolutionary network
analysis to identify prognostic cancer modules (88). These results provide a completely
new perspective in identifying suitable pharmacological targets
based on the evolutionary age of their encoding genes.
6. Conclusions
Cancer is a complex disease and its underlying
mechanisms have yet to be fully elucidated. However, critical nodes
can be identified under the scope of its evolutionary origins. The
emergence of long-living multicellular animals demands the
evolution of mechanisms that operate on various levels (including
on individual cells, tissue organization, and the whole body) in
order to maintain an appropriate number of cells within a specific
tissue and limit cancer growth (89).
Another aspect of the evolutionary link of cancer to
unicellularity concerns lateral gene transfer between bacteria and
eukaryotes, particularly when symbiotic relationships are present
(90). Given that human somatic
cells are in a state of coexistence with various kinds of bacteria,
the integration of bacterial genetic material is hypothesized to
disrupt tumor suppressor genes or proto-oncogenes, acting as a
mutagen, or the possibility for integrated bacterial gene to be
transcribed by the mechanisms of the recipient human cell, leading
to the production of a polypeptide with unpredictable repercussions
on the cell (91). In a 2017
study, the tissues of patients suffering from esophageal cancer
were analyzed, affirming the presence of the bacterium
Fusobacterium nucleatum. Through microarray analysis, it was
found that the presence of the bacterium affected cellular pathways
in the cancer tissues, namely the cytokine-to-cytokine receptor
interaction. These findings suggested that the presence of the
bacterium in the cells induced the activation of chemokines, such
as CCL20, contributing to tumorigenesis (92). Other researchers have begun to
elucidate the effects of bacteria-human lateral gene transfer on
the development of various cancer types. Bacterial DNA integrations
have been found in human mitochondrial genome and more
specifically, in samples of acute myeloid leukemia, identifying
bacterial integration in genes known to be upregulated in stomach
adenocarcinoma, an integration that appeared to take place in the
5'-UTR and 3'-UTR of those proto-oncogenes (93).
The regulatory network of living systems has been
finely tuned through evolution and genetic and environmental
perturbations can compromise the viability equilibrium of the cell
and ancestral genes that are dysregulated can be encountered in all
the critical cell signaling pathways. Upon tumorigenesis,
uncontrolled proliferation and metastasis, a number of properties
of multicellular organisms are dysregulated or lost (94). Genes originating from unicellular
ancestors are either specifically activated or required for
maintenance of cancer phenotype (95). It is argued herein that these
ancestral cancer genes represent an integral part of evolution, by
disrupting the acquired balance of the multicellular organisms and
driving disease through cancer as a means for change and
evolvability.
Cancer itself may be considered as an evolutionary
system, in which cancer cells acquire mutations that allow them to
survive, compete for space and resources, evade the immune system,
and even cooperate in order to disperse and colonize new organs
(96). Several factors, from
radiation to chemicals to aging, can promote the evolution of
cancer by increasing mutation frequency and promoting the selection
of adaptive mutations. In direct correspondence with animal
evolution, cancer cells respond to environmental adversities by
selecting the clone which is most fit for survival. Therefore, it
appears that cancer and tumor-suppressive mechanisms are engaged in
an evolutionary arms race with each other (10). Considering this, the evolutionary
aspect of cancer may help to predict the response of cancers to
drugs and therapy, and lay the foundation for optimal treatment
with immunotherapy, drugs, or chemotherapy.
Supplementary Material
Table SI. List of all
cancer-associated genes and their co-occurrence in transcription,
signaling and metabolism pathways.
Acknowledgements
Not applicable.
Funding
DV would like to acknowledge funding from: i)
Microsoft Azure for Genomics Research Grant (CRM:0740983); ii)
FrailSafe Project (H2020-PHC-21-2015-690140) ‘Sensing and
predictive treatment of frailty and associated co-morbidities using
advanced personalized models and advanced interventions’, co-funded
by the European Commission under the Horizon 2020 research and
innovation program; iii) Amazon Web Services Cloud for Genomics
Research Grant (309211522729); iv) AdjustEBOVGP-Dx
(RIA2018EF-2081): Biochemical Adjustments of native EBOV
Glycoprotein in Patient Sample to Unmask target Epitopes for Rapid
Diagnostic Testing. A European and Developing Countries Clinical
Trials Partnership (EDCTP2) under the Horizon 2020 ‘Research and
Innovation Actions’ DESCA. EE would like to acknowledge funding by
the project ‘INSPIRED-The National Research Infrastructures on
Integrated Structural Biology, Drug Screening Efforts and Drug
Target Functional Characterization’ (Grant MIS 5002550) and by the
project: ‘OPENSCREEN-GR An Open-Access Research Infrastructure of
Chemical Biology and Target-Based Screening Technologies for Human
and Animal Health, Agriculture and the Environment’ (Grant MIS
5002691), which are implemented under the Action ‘Reinforcement of
the Research and Innovation Infrastructure’, funded by the
Operational Programme ‘Competitiveness, Entrepreneurship and
Innovation’ (NSRF 2014-2020) and co-financed by Greece and the
European Union (European Regional Development Fund). EP would like
to acknowledge funding by the State Scholarships Foundation
(IKY)-European Union (European Social Fund-ESF) and Greek national
funds through the action entitled ‘Strengthening Human Resources
Research Potential via Doctorate Research’ in the framework of the
Operational Program ‘Human Resources Development Program, Education
and Lifelong Learning’ of the National Strategic Reference
Framework (NSRF) 2014-2020.
Availability of data and materials
A list of Homo sapiens genes associated with
cancer, signaling, metabolism and transcription as downloaded by
the NCBI Gene Database (66) are
reported and their occurrence in cancer, signaling, metabolism and
transcription databases are colour coded. All data generated or
analyzed during this study are included in this published article
or are available from the corresponding author on reasonable
request.
Authors' contributions
DV, EP, AE, FB, GG, GPC, EE all equally contributed
to the writing, drafting, revising, editing, reviewing, and the
conception and design of the study. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hendrikse JL, Parsons TE and Hallgrímsson
B: Evolvability as the proper focus of evolutionary developmental
biology. Evol Dev. 9:393–401. 2007.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pavlicev M and Wagner GP: Coming to Grips
with evolvability. Evolution: Educ Outreach. 5:231–244. 2012.
|
|
3
|
Tian T, Olson S, Whitacre JM and Harding
A: The origins of cancer robustness and evolvability. Integr Biol
(Camb). 3:17–30. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Aktipis CA, Boddy AM, Jansen G, Hibner U,
Hochberg ME, Maley CC and Wilkinson GS: Cancer across the tree of
life: Cooperation and cheating in multicellularity. Philos Trans R
Soc Lond B Biol Sci. 370(pii: 20140219)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bussey KJ, Cisneros LH, Lineweaver CH and
Davies PCW: Ancestral gene regulatory networks drive cancer. Proc
Natl Acad Sci USA. 114:6160–6162. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chu XY, Jiang LH, Zhou XH, Cui ZJ and
Zhang HY: Evolutionary origins of cancer driver genes and
implications for cancer prognosis. Genes (Basel). 8(pii:
E182)2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen H, Lin F, Xing K and He X: The
reverse evolution from multicellularity to unicellularity during
carcinogenesis. Nat Comm. 6(6367)2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Trigos AS, Pearson RB, Papenfuss AT and
Goode DL: Somatic mutations in early metazoan genes disrupt
regulatory links between unicellular and multicellular genes in
cancer. Elife. 8(pii: e40947)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Casás-Selves M and Degregori J: How cancer
shapes evolution, and how evolution shapes cancer. Evolution (N Y).
4:624–634. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pruitt KD, Tatusova T and Maglott DR: NCBI
reference sequence (RefSeq): A curated nonredundant sequence
database of genomes, transcripts and proteins. Nucleic Acids Res.
33 (Database Issue):D501–D504. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sever R and Brugge JS: Signal transduction
in cancer. Cold Spring Harb Perspect Med. 5(a006098)2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lodish H, Berk A, Zipursky SL, Matsudaira
P, Baltimore D and Darnell J: Oncogenic mutations affecting cell
proliferation. In: Molecular Cell Biology. 4th edition. W. H.
Freeman, New York, NY, 2000.
|
|
14
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Miller MA: Driver mutations take the wheel
in invasive yet nonmalignant disease. Sci Transl Med. 9(pii:
eaan8194)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
McFarland CD, Yaglom JA, Wojtkowiak JW,
Scott JG, Morse DL, Sherman MY and Mirny LA: The damaging effect of
passenger mutations on cancer progression. Cancer Res.
77:4763–4772. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
McFarland CD, Korolev KS, Kryukov GV,
Sunyaev SR and Mirny LA: Impact of deleterious passenger mutations
on cancer progression. Proc Natl Acad Sci USA. 110:2910–2915.
2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen J, Sun M and Shen B: Deciphering
oncogenic drivers: From single genes to integrated pathways. Brief
Bioinform. 16:413–1428. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang J and Zhang S: Discovery of cancer
common and specific driver gene sets. Nucleic Acids Res.
45(e86)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lu WJ, Amatruda JF and Abrams JM: p53
ancestry: Gazing through an evolutionary lens. Nat Rev Cancer.
9:758–762. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Jegga AG, Inga A, Menendez D, Aronow BJ
and Resnick MA: Functional evolution of the p53 regulatory network
through its target response elements. Proc Natl Acad Sci USA.
105:944–949. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Belyi VA, Ak P, Markert E, Wang H, Hu W,
Puzio-Kuter A and Levine AJ: The origins and evolution of the p53
family of genes. Cold Spring Harb Perspect Biol.
2(a001198)2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Levine AJ and Oren M: The first 30 years
of p53: Growing ever more complex. Nat Rev Cancer. 9:749–758.
2009.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Joerger AC and Fersht AR: The p53 pathway:
Origins, inactivation in cancer, and emerging therapeutic
approaches. Annu Rev Biochem. 85:375–404. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Philippon H, Brochier-Armanet C and
Perrière G: Evolutionary history of phosphatidylinositol-3-kinases:
Ancestral origin in eukaryotes and complex duplication patterns.
BMC Evol Biol. 15(226)2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kriplani N, Hermida MA, Brown ER and
Leslie NR: Class I PI 3-kinases: Function and evolution. Adv Biol
Regul. 59:53–64. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bertucci MC and Mitchell CA:
Phosphoinositide 3-kinase and INPP4B in human breast cancer. Ann N
Y Acad Sci. 1280:1–5. 2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: Effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Park S, Chapuis N, Tamburini J, Bardet V,
Cornillet-Lefebvre P, Willems L, Green A, Mayeux P, Lacombe C and
Bouscary D: Role of the PI3K/AKT and mTOR signaling pathways in
acute myeloid leukemia. Haematologica. 95:819–828. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Polychronidou E, Vlachakis D, Vlamos P,
Baumann M and Kossida S: Notch signaling and ageing. Adv Exp Med
Biol. 822:25–36. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li L, Tang P, Li S, Qin X, Yang H, Wu C
and Liu Y: Notch signaling pathway networks in cancer metastasis: A
new target for cancer therapy. Med Oncol. 34(180)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kwon OJ, Zhang L, Wang J, Su Q, Feng Q,
Zhang XH, Mani SA, Paulter R, Creighton CJ, Ittmann MM and Xin L:
Notch promotes tumor metastasis in a prostate-specific Pten-null
mouse model. J Clin Invest. 126:2626–2641. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Weng AP, Ferrando AA, Lee W, Morris JP IV,
Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster
JC: Activating mutations of NOTCH1 in human T cell acute
lymphoblastic leukemia. Science. 306:269–271. 2004.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lobry C, Oh P, Mansour MR, Look AT and
Aifantis I: Notch signaling: Switching an oncogene to a tumor
suppressor. Blood. 123:2451–2459. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Mosedale G, Niedzwiedz W, Alpi A, Perrina
F, Pereira-Leal JB, Johnson M, Langevin F, Pace P and Patel KJ: The
vertebrate Hef ortholog is a component of the Fanconi anemia
tumor-suppressor pathway. Nat Struct Mol Biol. 12:763–771.
2005.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Murphy ME: The HSP70 family and cancer.
Carcinogenesis. 34:1181–1188. 2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Rosenzweig R, Nillegoda NB, Mayer MP and
Bukau B: The Hsp70 chaperone network. Nat Rev Mol Cell Biol.
20:665–680. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Calderwood SK and Gong J: Molecular
chaperones in mammary cancer growth and breast tumor therapy. J
Cell Biochem. 113:1096–1103. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Lunt SY and Vander Heiden MG: Aerobic
glycolysis: Meeting the metabolic requirements of cell
proliferation. Annu Rev Cell Dev Biol. 27:441–464. 2011.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Alfarouk KO, Verduzco D, Rauch C,
Muddathir AK, Bashir Adil HH, Elhassan GO, Ibrahim ME, David Polo
Orozco J, Cardone RA, Reshkin SJ and Harguindey S: Glycolysis,
tumor metabolism, cancer growth and dissemination. A new pH-based
etiopathogenic perspective and therapeutic approach to an old
cancer question. Oncoscience. 1:777–802. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
DeBerardinis RJ and Chandel NS:
Fundamentals of cancer metabolism. Sci Adv.
2(e1600200)2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Dang CV: Links between metabolism and
cancer. Genes Dev. 26:877–890. 2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Phan LM, Yeung SC and Lee MH: Cancer
metabolic reprogramming: Importance, main features, and potentials
for precise targeted anti-cancer therapies. Cancer Biol Med.
11:1–19. 2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Halestrap AP: The monocarboxylate
transporter family-Structure and functional characterization. IUBMB
Life. 64:1–9. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Baltazar F, Pinheiro C, Morais-Santos F,
Azevedo-Silva J, Queirós O, Preto A and Casal M: Monocarboxylate
transporters as targets and mediators in cancer therapy response.
Histol Histopathol. 29:1511–1524. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Perez-Escuredo J, Van Hée VF, Sboarina M,
Falces J, Payen VL, Pellerin L and Sonveaux P: Monocarboxylate
transporters in the brain and in cancer. Biochim Biophys Acta.
1863:2481–2497. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Ullah MS, Davies AJ and Halestrap AP: The
plasma membrane lactate transporter MCT4, but not MCT1, is
up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J
Biol Chem. 281:9030–9037. 2006.PubMed/NCBI View Article : Google Scholar
|
|
54
|
San-Millan I and Brooks GA: Reexamining
cancer metabolism: Lactate production for carcinogenesis could be
the purpose and explanation of the Warburg Effect. Carcinogenesis.
38:119–133. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Payen VL, Mina E, Van Hée VF, Porporato PE
and Sonveaux P: Monocarboxylate transporters in cancer. Mol Metab.
33:48–66. 2020.
|
|
56
|
Martinez-Outschoorn UE, Curry JM, Ko YH,
Lin Z, Tuluc M, Cognetti D, Birbe RC, Pribitkin E, Bombonati A,
Pestell RG, et al: Oncogenes and inflammation rewire host energy
metabolism in the tumor microenvironment: RAS and NFκB target
stromal MCT4. Cell Cycle. 12:2580–2597. 2013.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Bovenzi CD, Hamilton J, Tassone P, Johnson
J, Cognetti DM, Luginbuhl A, Keane WM, Zhan T, Tuluc M, Bar-Ad V,
et al: Prognostic indications of elevated MCT4 and CD147 across
cancer types: A Meta-analysis. Biomed Res Int.
2015(242437)2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47.
2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wahlstrom T and Henriksson MA: Impact of
MYC in regulation of tumor cell metabolism. Biochim Biophys Acta.
1849:563–569. 2015.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Bott JA, Peng IC, Fan Y, Faubert B, Zhao
L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, et al: Oncogenic
Myc induces expression of glutamine synthetase through promoter
demethylation. Cell Metab. 22:1068–1077. 2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Kumada Y, Benson DR, Hillemann D, Hosted
TJ, Rochefort DA, Thompson CJ, Wohlleben W and Tateno Y: Evolution
of the glutamine synthetase gene, one of the oldest existing and
functioning genes. Proc Natl Acad Sci USA. 90:3009–3013.
1993.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Hill RT, Parker JR, Goodman HJ, Jones DT
and Woods DR: Molecular analysis of a nove glutamine synthetase of
the anaerobe Bacteroides fragilis. J Gen Microb.
135:3271–3279. 1989.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Goodman HJ and Woods DR: Cloning and
nucleotide sequence of the Butyrivibrio fibrisolvens gene
encoding a type III glutamine synthetase. J Gen Micro.
139:1487–1493. 1993.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Pesole G, Gissi C, Lanave C and Saccone C:
Glutamine synthetase gene evolution in bacteria. Mol Biol Evol.
12:189–197. 1995.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Shatters RG and Kahn JL: Glutamine
synthetase II in Rhizobium: Reexamination of the proposed
horizontal transfer of DNA from eukaryotes to prokaryotes. J Mol
Evol. 2:422–428. 1989.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Lee TI and Young RA: Transcriptional
regulation and its misregulation in disease. Cell. 152:1237–1251.
2013.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Werner F and Grohmann D: Evolution of
multi-subunit RNA polymerases in the three domains of life. Nat Rev
Microbiol. 9:85–98. 2011.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Shin DS, Pratt AJ and Tainer JA: Archaeal
genome guardians give insights into eukaryotic DNA replication and
damage response proteins. Archaea. 2014(206735)2014.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Warner JR: The economics of ribosome
biosynthesis in yeast. Trends Biochem Sci. 24:437–440.
1999.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Drygin D, Rice WG and Grummt I: The RNA
polymerase I transcription machinery: An emerging target for the
treatment of cancer. Annu Rev Pharmacol Toxicol. 50:131–156.
2010.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Arabi A, Wu S, Ridderstråle K, Bierhoff H,
Shiue C, Fatyol K, Fahlén S, Hydbring P, Söderberg O, Grummt I, et
al: c-Myc associates with ribosomal DNA and activates RNA
polymerase I transcription. Nat Cell Biol. 7:303–310.
2005.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Grandori C, Gomez-Roman N, Felton-Edkins
ZA, Ngouenet C, Galloway DA, Eisenman RN and White RJ: c-Myc binds
to human ribosomal DNA and stimulates transcription of rRNA genes
by RNA polymerase I. Nat Cell Biol. 7:311–318. 2005.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Mayer C and Grummt I: Ribosome biogenesis
and cell growth: mTOR coordinates transcription by all three
classes of nuclear RNA polymerases. Oncogene. 25:6384–6391.
2006.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhang C, Comai L and Johnson DL: PTEN
represses RNA Polymerase I transcription by disrupting the SL1
complex. Mol Cell Biol. 25:6899–6911. 2005.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Grummt I: Life on a planet of its own:
Regulation of RNA polymerase I transcription in the nucleolus.
Genes Dev. 17:1691–1702. 2003.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Luo Z, Lin C, Guest E, Garrett AS,
Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP and
Shilatifard A: The super elongation complex family of RNA
polymerase II elongation factors: Gene target specificity and
transcriptional output. Mol Cell Biol. 32:2608–2617.
2012.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Rahl PB, Lin CY, Seila AC, Flynn RA,
McCuine S, Burge CB, Sharp PA and Young RA: c-Myc regulates
transcriptional pause release. Cell. 141:432–445. 2010.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Smith E, Lin C and Shilatifard A: The
super elongation complex (SEC) and MLL in development and disease.
Genes Dev. 25:661–672. 2011.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Aguilo F, Zhou MM and Walsh MJ: Long
noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a
expression. Cancer Res. 71:5365–5369. 2011.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Sliwoski G, Kothiwale S, Meiler J and Lowe
EW Jr: Computational methods in drug discovery. Pharmacol Rev.
66:334–395. 2014.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Park H, Bahn YJ and Ryu SE:
Structure-based de novo design and biochemical evaluation of novel
Cdc25 phosphatase inhibitors. Bioorg Med Chem Lett. 19:4330–4334.
2009.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Vangrevelinghe E, Zimmermann K, Schoepfer
J, Portmann R, Fabbro D and Furet P: Discovery of a potent and
selective protein kinase CK2 inhibitor by high-throughput docking.
J Med Chem. 46:2656–2662. 2003.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Kreeger PK and Lauffenburger DA: Cancer
systems biology: A network modeling perspective. Carcinogenesis.
31:2–8. 2010.PubMed/NCBI View Article : Google Scholar
|
|
84
|
San Lucas FA, Fowler J, Chang K, Kopetz S,
Vilar E and Scheet P: Cancer in silico drug discovery: A systems
biology tool for identifying candidate drugs to target specific
molecular tumor subtypes. Mol Cancer Ther. 13:3230–3240.
2014.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Carter H, Chen S, Isik L, Tyekucheva S,
Velculescu VE, Kinzler KW, Vogelstein B and Karchin R:
Cancer-specific high-throughput annotation of somatic mutations:
Computational prediction of driver missense mutations. Cancer Res.
69:6660–6667. 2009.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Chen W, Li Y and Wang Z: Evolution of
oncogenic signatures of mutation hotspots in tyrosine kinases
supports the atavistic hypothesis of cancer. Sci Rep.
8(8256)2018.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Trigos AS, Pearson RB, Papenfuss AT and
Goode DL: Altered interactions between unicellular and
multicellular genes drive hallmarks of transformation in a diverse
range of solid tumors. Proc Natl Acad Sci USA. 114:6406–6411.
2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Zhou XH, Chu XY, Xue G, Xiong JH and Zhang
HY: Identifying cancer prognostic modules by module network
analysis. BMC Bioinformatics. 20(85)2019.PubMed/NCBI View Article : Google Scholar
|
|
89
|
DeGregori J: Evolved tumor suppression:
Why are we so good at not getting cancer? Cancer Res. 71:3739–3744.
2011.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Dunning Hotopp JC: Horizontal gene
transfer between bacteria and animals. Trends Genet. 27:157–163.
2011.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Robinson KM, Sieber KB and Dunning Hotopp
JC: A review of bacteria-animal lateral gene transfer may inform
our understanding of diseases like cancer. PLoS Genet.
9(e1003877)2013.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Baba Y, Iwatsuki M, Yoshida N, Watanabe M
and Baba H: Review of the gut microbiome and esophageal cancer:
Pathogenesis and potential clinical implications. Ann Gastroenterol
Surg. 1:99–104. 2017.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Riley DR, Sieber KB, Robinson KM, White
JR, Ganesan A, Nourbakhsh S and Dunning Hotopp JC: Bacteria-human
somatic cell lateral gene transfer is enriched in cancer samples.
PLoS Comput Biol. 9(e1003107)2013.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Cao Y: Tumorigenesis as a process of
gradual loss of original cell identity and gain of properties of
neural precursor/progenitor cells. Cell Biosci.
7(61)2017.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Trigos AS, Pearson RB, Papenfuss AT and
Goode DL: How the evolution of multicellularity set the stage for
cancer. Br J Cancer. 118:145–152. 2018.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Merlo LM, Pepper JW, Reid BJ and Maley CC:
Cancer as an evolutionary and ecological process. Nat Rev Cancer.
6:924–935. 2006.PubMed/NCBI View Article : Google Scholar
|