Introduction
Subarachnoid hemorrhage (SAH) is a common
cerebrovascular accident which is associated with high morbidity.
SAH can be divided into spontaneous and traumatic SAH (1). The annual incidence of spontaneous SAH
is >10.5/100,000 individuals per year and, since the majority of
spontaneous SAH cases are induced by aneurysm rupture, the annual
incidence of aneurysm rupture-induced spontaneous SAH is
6–35.5/100,000 individuals per year (2). In recent years it has been demonstrated
that the incidence of SAH is increasing (3). The American Heart Association
demonstrated that the total mortality of SAH has reached 25%
(4,5). Spontaneous SAH often results in
numerous complications, of which cerebral vasospasm (CVS) is the
most severe (6). CVS occurs due to
the continuous contraction of one or several areas of smooth muscle
in the intracranial vasculature, or vascular injury induced by
sanguis stimulation, which leads to changes in the luminal
morphological and intracranial vascular stenosis or spasm, whose
incidence is 30–90% (7). According
to clinical presentation, CVS may be divided into two types, early
brain injury (EBI) and delayed cerebral vasospasm (DCVS) (8). EBI predominantly occurs in patients
several minutes to half an hour post-SAH, as the blood which
directly and mechanically stimulates the intracranial vasculature
flows into cerebrospinal fluid (CSF) following SAH (8). DCVS predominantly occurs at 4–15 days
post-SAH. DCVS often reaches vasospasm fastigium after 7–10 days,
ultimately leading to severe ischemia of the local brain tissue,
d(elayed ischemic injury or cerebral infarction, which are the
predominant causes of morbidity and mortality in patients with DCVS
(9). Patients with DCVS require
long-term care and active nursing, therefore they are an increased
burden on their family and limited medical resources, and suffer
from decreased quality of life. Eventually, this may impede the
development of social economic construction (10).
The occurrence of DCVS is associated with multiple
factors and procedures. Previous studies have demonstrated that
numerous factors influence the occurrence mechanism of DCVS, such
as hemolysate, inflammatory response and endothelial dysfunction,
which are associated with the occurrence of DCVS following SAH
(11–13). The stabilization of endothelial cells
has a key role in the normal physiological function of the human
body. Vascular endothelial cells are cable of generating various
vasoactive substances, including nitrogen oxide and prostacylin,
which can dilate blood vessels, and endothelin (ET), arachidonic
acid and cyclooxygenase, which can constrict vasculature (14). A previous study has investigated the
dysfunction of vascular endothelin cells (15).
Caveolin is a type of integral membrane protein with
a molecular weight of 17–24 kD, and is located on the inner surface
of the alveolar cell membranes (16). Three members of the caveolin family,
caveolin-1, caveolin-2 and caveolin-3, have previously been cloned
(17). Although the structure and
function of the caveolin gene family is highly conserved in
mammals, the cellular distribution is diverse. Caveolin-1 is highly
expressed in adipocytes, endotheliocytes and fibrocytes (18,19).
Caveolin can form complexes by combining with various signal
molecules in order to regulate the activated state of numerous key
signaling molecules and control transmembrane signal transduction
(20). Furthermore, caveolin
participates in vital cell activities in numerous cells, including
endocytosis, the transportation of cholesterol, cytomembrane
assembly and signal transduction (21–24).
Caveolin-1, which has a molecular weight of 21–24
kD, is associated with cholesterol homeostasis, molecular
transportation and transmembrane signal transduction. In
particular, a previous study has demonstrated that caveolin-1 has a
role in signal transduction, the pathological and biological
processes (25) of cell
differentiation, proliferation, tumorigenesis, myopathy,
angiopathy, neurodegenerative disorders and senility. Caveolin is a
repository of calcium ions and is key for its transportation.
Furthermore, previous studies have demonstrated that endothelial
nitric oxide synthase (eNOS), which is a key signaling molecule
associated with the precise regulation and control of calcium ions,
is assembled around caveolin (26).
Jasmin et al (26) have
previously utilized caveolin knockout mice models in order to
investigate the function of caveolin in cerebral ischemic injury.
In addition, Jasmin et al (26) demonstrated that caveolin-1 gene
knockout resulted in an increase in the cerebral infarct volume, as
compared with the wild type, and the speed of endotheliocyte
proliferation of the wild type mice with cerebral ischemia markedly
increased, as compared with the caveolin-1 gene knockout mice.
Furthermore, the eNOS levels of wild type mice with cerebral
ischemia increased, whereas no notable alterations were detected in
the caveolin-1 gene knockout mice. Therefore, these results
demonstrated that cerebral ischemia induced an increase in the
number of endotheliocytes and the expression of genes associated
with angiogenesis was impaired by cerebral ischemia in mice with
caveolin-1 knockout; therefore, the number of apoptotic cells
increases. Using a mouse model of cerebral ischemia induced by
arterial occlusion, Shen et al (27) investigated the function of eNOS in
the regulation of caveolin-1. The results demonstrated that NO
regulates the expression of caveolin-1, and reduced caveolin-1
expression is associated with the generation of NO in the ischemic
brain. These previous studies demonstrated that caveolin-1 may
serve a crucial function in the pathogenesis of cerebral ischemia
and participates in the regulation of physiological mechanisms
following cerebral ischemia (26,27).
Furthermore, it has previously been demonstrated that caveolin-1 is
associated with various types of vascular disease, including
atherosclerosis and hypertension, indicating that caveolin-1 may be
associated with the differentiation of vascular endothelial cells
(15). As endothelial cells have
abundant cell membrane alveoli and caveolin-1, the authors of the
present study hypothesize that caveolin-1 may be correlated with
DCVS following SAH. To the best of our knowledge, the present study
is the first to investigate whether caveolin-1 is associated with
the development of SAH-induced DCVS.
Using a mouse model of SAH, generated by
suboccipital pool double hemorrhage injection, the present study
investigated the pathogenesis of DCVS by observing alterations in
the levels of caveolin-1 in the basilar arteries of mice with DCVS
following SAH. The results of the present study may provide novel
theories for the future treatment of patients with DCVS following
SAH.
Materials and methods
Experimental animals
A total of 150 clean-grade male Sprague-Dawley mice,
weighing 300±10 g and aged 8–10 weeks, were purchased from Shanghai
Slyke Experimental Animals, Co., Ltd. (SCXK 2007-0005; Shanghai,
China). Ethical approval was obtained from the Ethical Committee of
the First Affiliated Hospital of Wenzhou Medical College (Wenzhou,
China).
Apparatus
P627 microscale pipettor; Thermostat (4304057; both
Shanghai Medical Instrument Factory, Shanghai, China); Electronic
analytical balance (ESJ182-4; Hangzhou Medical Equipment Factory,
Hangzhou, China); Optical microscope (P7220; Thermo Fisher
Scientiifc, Inc., Waltham, MA, USA), Fluorescence microscope
(AF6000; Leica Microsystems GmbH, Wetzlar, Germany); Operating
microscope (HR900A/2-11; Germany); Mice stereotaxic apparatus
(YH51500D; Wuhan, China); Dural refrigerator (BCD-575WDBI; Haier
Group, Qingdao, China); Micrograph system (Leica Microsystems
GmbH); Thermostat water bath (W1933; Shanghai Cairrao Apparatus
Company, Shanghai, China); T-Gradient polymerase chain reaction
(PCR) amplifier (Biometra GmbH, Göttingen, Germany); Sigma 1–14
centrifuge (Sigma Laborzentrifugen GmbH, Osterode am Harz,
Germany); and concentrator table (TP22; Wuhan, China).
Reagents
Chloral hydrate (302-17-0; Wuhan Boster Biological
Technology, Ltd., Wuhan, China); paraformaldehyde (Z-P193265;
Sinopharm Chemical Reagent Co., Ltd., Shanghai, China); rabbit
polyclonal caveolin-1 primary antibodies (ab2910; Abcam, Cambridge,
MA, USA); protein inhibitors (85-00-4975-03; Beijing Aolilaiyin
Technology Co., Ltd., Beijing, China); peroxidase-AffiniPure goat
anti-rabbit (111-035-003) and goat anti-mouse (115-035-003; both
Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA)
immunoglobulin (Ig)G (H+L) fluorescent secondary antibodies; and
goat anti-human IgG secondary antibodies (ZF-0308; Shanghai Boyun
Chemical Reagent Co., Ltd., Shanghai, China). GAPDH (TA-08;
Zhongshan Jinqiao, Beijing, China); enhanced chemiluminescence
(ECL) kit (sc-2048; Cell Signaling Technology, Inc., Danvers, MA,
USA). TRIzol kit (15596–018); caveolin-1 mRNA primer design; TaqMan
DNA polymerase (4405495), diethylpyrocarbonate-treated water and
4′,6-diamidino-2-phenylindole (62247) (all Invitrogen; Thermo
Fisher Scientific, Inc.). DNase I kit (D8071-25; Solarbio Science
& Technology Co., Ltd., Beijing, China). Reverse
transcription-quantitative PCR Master Mix (QPK-201; Toyobo Co.,
Ltd., Osaka, Japan). Finland reagent; hematoxylin and eosin
(00-8011 and E-18); anti-fluorescence quencher (P0126);
bicinchoninic acid assay (BCA) kit (P0011); ammonium persulfate
(ST005); 30% acrylamide; 10% sodium dodecyl sulfate (SDS); 1 M
Tris-HCl (pH 6.8); 1.5 M Tris-HCl (pH 8.8);
tetramethylethylenediamine (ST066); phenylmethylsulfonyl fluoride
(ST506); horseradish without IgG secondary antibodies marked
(P0203); polyvinylidene difluoride (PVDF) membrane (A0793); and
Tween-20 (ST825; all Biyuntian Nantong, Jiangsu, China).
Primary reagent compounds
i) 10% Chloral hydrate: 10 g Chloral hydrate powder
was supplemented with 100 ml distilled water, incubated in a 55°C
water bath unit and discontinuously stirred until the solution was
clear. Solution was maintained in the dark. ii) 4%
Paraformaldehyde: 40 g Paraformaldehyde was supplemented with 100
ml phosphate-buffered saline (PBS; 0.1 mol/l), discontinuously
stirred until the solution was clear and the pH value was
subsequently adjusted to 7.4 prior to preservation at 4°C. iii) 5X
Tris-buffered saline (TBS): 20.375 g Tris-HCl; 22.5 g sodium
chloride, double-distilled water; and 1X TBS: 100 ml TBS (5X) + 400
ml double-distilled water. iv) TBS with Tween-20 (TBST): 500 µl
Tween-20 + 500 ml TBS (1X). v) 10% SDS-polyacrylamide gel
electrophoresis (SDS-PAGE): 15.1 g Tris; 72 g glycine, 5 g SDS, 500
ml double-distilled water; 1% SDS-PAGE: 100 ml (10X) + 900 ml
double-distilled water. vi) 10X Transfer buffer: 15 g Tris, 72 g
glycine, 5 g SDS, 500 ml double-distilled water; and 1X transfer
buffer: 100 ml (10X) + 200 ml carbinol + 700 ml double-distilled
water. vii) 5% sealing milk: 5 g skim milk powder + 100 ml TBST
liquid. viii) ECL solution: ECL solution A + ECL solution B (mixed
1:1).
Experimental groups
Mice were allocated into three groups: Blank group
(n=14), the saline group (n=56) and the operational group (n=80).
The normal saline and operational groups were further divided into
four subgroups: Days 3, 5, 7 and 14 post-SAH. The mice in the
operational group were injected autologous arterial blood into
subarachnoid space. The saline group was injected equivalent saline
into subarachnoid space. The blank group did not receive any
treatment.
Murine model of SAH-induced DCVS
In the present study a model of SAH was created
using the method of double hemorrhage of cisterna magna, as
previously described (28). Mice
were intraperitoneally anesthetized using 10% chloral hydrate (3
ml/100 g) and fixed in the prone position. Following disinfection
of the preserved skin using 75% ethanol, the fascia muscle was
incised along the middle wire, using an operational microscope.
Mice were subsequently placed in the supine position and the skin
of the inguinal region was disinfected prior to the incision of the
skin to expose the femoral artery, which was subsequently separated
and fixed. Following these procedures, 0.01 ml heparin (25 U) was
injected into the artery using a hollow 1-ml needle and mice were
fixed into the prone position with their heads bent forward to
extract 0.10 ml/100 g autologus arterial blood by weight.
Subsequently, autologus arterial blood was slowly injected into the
cisternas magna (speed, 0.1 ml/min) from the foramen occipital
magnum using a needle inserted at 1 mm. A ‘breakthrough’ sensation
was detected when the needle was inserted into the atlantooccipital
membrane and the puncture was immediately sealed with a gelatin
sponge. After hemostasis, the muscular layer and skin were sutured
layer-by-layer. Subsequently, mice were placed in a dorsal elevated
position for 30 min, and placed in a cage. After 48 h, blood was
harvested from the femoral artery and injected for the second time.
The method remained the same. A total of 24 h after the second
injection was regarded as day 1 post-SAH, and so on.
Grading of neurological impairment in
the SAH group
Neurological impairment was assessed according to
grading system proposed by Bederson (29). Grading was conducted on days 3, 5 and
14 post-SAH, according to the condition of the tail, particularly
its bend and whirl, and resistance of its bilateral forelegs, and
the condition of the whirl. Scoring was classified as follows: 0,
Normal; 1, mild neurological impairment; 2, moderate neurological
impairment; and 3, severe neurological impairment.
Specimen collection
New specimen collection
Following establishment of the mouse model of DCVS,
mice were anesthetized using 3 ml/100 g chloral hydrate (10%) on
days 3, 5 and 14 post-SAH, according to the sub-groupings.
Following this, the respective mouse brains were harvested, placed
on an iced plate in order to separate basilar artery and rapidly
preserved in liquid nitrogen.
Preparation of paraffin specimen
The abdominal skin of the mice was cut off, the
ensisternum was lifted using a vessel clamp, and the ribs were cut
along the two sides of sternum using scissors in order to expose
the heart, ventriculus sinister, and right auricular. A 24-gauge
transfusion needle was inserted into the ventriculus sinister and,
simultaneously, using the hemostatic forceps to clamp the
transfusion needles and the left ventricular wall, normal saline
was quickly infused. Immediately, scissors were used to cut the
right auricular in order to release the blood until the effluent
was clear. Following this, 300 ml paraformaldehyde (4%), which had
been prepared previously, was injected quickly at first then slowly
until the heads and necks of the mice stiffened. Upon completion of
the injection, the mice were immediately decapitated and the scalps
were simultaneously removed. The skulls were subsequently removed
and the brainstems, which contains basal arteries, were
respectively harvested. All the mice brainstems were fixed in 4%
paraformaldehyde and, following 24 h, were paraffin embedded. The
basal artery brainstem specimens were trimmed to 10×5×4 mm and
placed in the embedding boxes. Specimens were processed according
to the following protocol: Overnight incubation with 75% ethyl
alcohol; once with 85% ethyl alcohol for 2 h; twice for hour with
95% ethyl alcohol; twice for 45 min with absolute ethyl alcohol to
dehydrate; twice for 45 min with xylene to make the cells
transparent; wax I overnight; wax II for 2 h; and the wax was
subsequently injected into the specimen. After the wax had cooled
down, the specimen was sliced using the microtome, with the slicing
knife parallel with the transverse section to ensure with each
section was 4 µm-thick. Each specimen was subsequently cut into 10
continuous pieces and subjected to HE staining and
immunofluorescence analysis, respectively.
HE staining
Staining with HE was performed in order to measure
the interfacial inner diameter of the basilar artery and the
thickness of the vessel wall of the basilar artery. Five section
pieces were selected from each specimen, were dried for 60 min at
65°C and stained as follows: Deparaffinization with xylene twice
for 5 min; hydration using an alcohol gradient: 100% Ethyl alcohol
twice for 5 min, 95% ethyl alcohol once for 5 min, 90% ethyl
alcohol once for 5 min, 70% ethyl alcohol once for 5 min, and
washing three times with distilled water for 5 min; staining with
hematoxylin for 10 min, incubation at 37°C for 3 min in warm water
to ensure the sections are stained blue; washing with running water
for 10 min; followed by counterstaining with eosin for 5 min; 95%
ethyl alcohol twice for 5 min, xylene twice for 10 min, and
subsequent mounting using neutral gum. The interfacial inner
diameter of basilar artery was calculated using the Image Pro Plus
5.1 image analyzing system (Media Cybernetics, Inc., Rockville, MD,
USA) and the thickness of the vessel wall was measured and the mean
values were calculated. Following dewaxing, the sections were
treated with 0.3% H2O2 in methanol for 10
min, washed with distilled water supplemented with 0.01 M citrate
solution (pH 6.0) and heated in an oven for 10 min. Following
cooling to 25°C, the sections were washed three times with PBS
prior to immunohistochemical analysis.
Immunofluorescence analysis
Five sections were taken from each specimen and the
section was dried for 60 min at 65°C; deparaffinized with xylene
twice for 5 min; hydrated using an alcohol gradient (100% ethyl
alcohol twice for 5 min, 95% ethyl alcohol once for 5 min, 90%
ethyl alcohol once for 5 min, 70% ethyl alcohol once for 5 min, and
washed three times with PBS water for 5 min). Antigen retrieval was
performed using sodium citrate, the sections were washed three
times with PBS for 5 min, immersed in 3% H2O2
to wipe off the peroxidase, sealed using serum, incubated overnight
with rabbit polyclonal caveolin-1 primary antibodies antibody
(1:1,000). Following this, the sections were washed three times
with PBS water for 5 min and incubated for 1.5 h with secondary
fluorescence antibody (1:1,000) in the dark room. Following washing
three times with PBS water for 5 min,
4′,6-diamidino-2-phenylindole, and fluorescence agent quenching
mounting, the sections were observed under a fluorescence
microscope.
Western blot analysis
Extraction and measurement of protein
Protein extraction was performed as follows, 50 mg
cryopreserved basilar artery was placed in the grinding rod and
supplemented with 500 µl organization lysate and 5 µl proteinase
inhibitor prior to full grinding on ice for 30 min The liquid was
collected, placed in 1.5-ml non-enzyme tubes, centrifuged for 5 min
at 12,500 × g, prior to supplementing the dissolution liquid with
150 µl protein preserving liquid, which can be adjusted by the
precipitation capacity. After repeatedly blowing and beating the
sediment solution, the dissolution was placed in a 0.5-ml
centrifuge tube and 20–25 µl was preserved at 80°C. Protein
concentration was measured using a BCA kit, according to the
manufacturer's protocol, where the solubility of the protein was
measured using a microplate reader.
Western blot analysis
Proteins were separated by 10% SDS-PAGE, which was
selected according to the molecular weight of the protein to be
measured, caveolin-1. Following albuminous degeneration (100°C for
5 min), 20 µl protein was loaded into the respective wells using a
microscale pipette, and an equal quantity of buffer solution was
subsequently loaded in order to avoid edge effects. Electrophoresis
was run in 1X liquid at 35 mA for 90 min, until the leading edge of
the dye had migrated to the bottom of the gel. Proteins were
transferred to a PVDF membrane and were rinsed using
double-distilled water for 5 min. The membrane was transferred at
350 mA for 60 min in the 1X transferring membrane liquid. In order
to seal the PVDF membrane, it was removed, washed with deionized
water and incubated with 5% skim milk for 2 h on the concentrator
table. Subsequently, the membrane was washed three times with TBST
for 10 min and incubated with caveolin-1 and GAPDH primary
antibodies (1:1,000) overnight at 4°C. Following this, the membrane
was again washed three times with TBST for 10 min and incubated
with goat anti-rabbit secondary antibodies (1:2,500), for 2 h at
25°C on the concentrator table. The membrane was washed three times
with TBST for 10 min prior to development and exposure. According
to the ECL protocol, ECL solutions A and B were mixed (ratio, 1:1),
and incubated with the PVDF membrane in the dark room to develop
for 3–5 min prior to exposure. Data were analyzed using ImageJ
protein grayscale software (National Institutes of Health,
Bethesda, MA, USA).
PCR
Extraction, concentration determination and
reverse transcription of RNA
RNA was extracted by grinding 50–100 mg tissue into
powder in liquid nitrogen and subsequently injecting 1 ml TRIzol
prior to further grinding for 15 min. Following this, the
homogenate was transferred into an Eppendorf (EP) tube and left to
dissociate for 5 min at room temperature prior to centrifugation
12,000 × g for 10 min at 4°C. Using a new EP tube, the supernatant
was collected and supplemented with chloroform at the ratio of
0.2:1 ml TRIzol. The EP tube was shaken for 15 sec by hand prior to
incubation for 3 min at room temperature and subsequent
centrifugation at 12,000 × g for 15 min at 4°C. The supernatant was
transferred to a new EP tube, supplemented with isopropanol at the
ratio of 0.5 ml to 1 ml TRIzol, incubated for 10 min at room
temperature and subsequently centrifuged at 12,000 × g for 10 min
at 4°C. After discarding the supernatant, the pellet was washed
with 75% ethyl alcohol at the ratio of ≤1 ml ethyl alcohol to 1 ml
TRIzol. Following shaking, the supernatant was discarded and the
sedimentary RNA was left to dry naturally at room temperature. Once
dry, the pellet was suspended in 20–40 µl
diethylpyrocarbonate-treated non-enzyme water and stored at
−70°C.
RNA concentration was determined by suspending 2 µl
RNA in 98 µl non-RNase water and centrifuging at 12,000 × g for 10
min at 4°C, prior to measuring the optical density (OD) 260/280
value using a spectrophotometer (1OD=40 µg RNA). When the OD
260/280 value of the RNA sample was detected as 1.8–2.0, the purity
was regarded as high. In general, the normal concentration value
was determined to be 1,000 ng/ml, although when the concentration
was deemed too high, it was attenuated by diluting the sample with
an appropriate volume of non-RNase water and the measurement was
repeated. Reverse transcription was performed using TaqMan DNA
polymerase by initially placing RNA at 65°C for 51 min, and
immediately transferring the sample to ice to cool. Subsequently,
the reverse transcription reaction was performed for 15 min,
followed by enzyme inactivation for 5 min. Upon completion, the
subsequent cDNA underwent DNase treatment and was stored at
−20°C.
RT-qPCR analysis
PCR reaction mixtures contained: 5 µl SYBR Green
Realtime PCR Master Mix-Plus; 1 µl Plus Solution; 1 µl upstream and
downstream primers (10 µmol/l), respectively; and 1 µl template
supplemented with sterile deionized water to a final reaction
volume of 10 µl. Reaction conditions were as follows: 94°C for 3
min, then 94°C for 5 sec, 60°C for 20 sec, all for 40 cycles. Mouse
actin was used as the reference gene, against which the expression
levels of the target genes were normalized to calculate the
standard curves for the real-time quantification of the caveolin-1
target genes. The following primers were used: Caveolin-1, upstream
5′-GACCCCAAGCATCTCAACGA-3′ and downstream
5′-GCCATAGGGATGCCGAA-GA-3′; and actin internal reference, upstream
5′-AGAGGGAAATCGTGCGTGAC-3′ and downstream
5′-AGAGGTCTTTACGGATGT-CAACG-3′. In order to increase the
credibility of the results, each specimen was repeated in 6 tubes,
3 of which were target genes, and the remainder were internal
reference. RT-qPCR analysis of the target gene and internal
reference was completed in triplicate in order to determine the
mean Cq value. Relative mRNA expression levels of caveolin-1 were
calculated by dividing the mean Cq of actin by the mean Cq of
caveolin-1, according to the 2−∆∆Cq method.
Statistical analysis
Experimental data were statistically analyzed using
SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA). Data were
expressed as the mean ± standard deviation. Inter-group and
intragroup comparisons were performed using Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Establishment of the murine model
During the observation period, 10 mice succumbed to
SAH-induced DCVS in the day 3 (n=3), 5 (n=5) and 7 (n=2) groups. No
mortality was observed in the blank control and normal saline
groups. In the experimental group, blood clots were observed around
the basilar artery of the respective brainstems during the
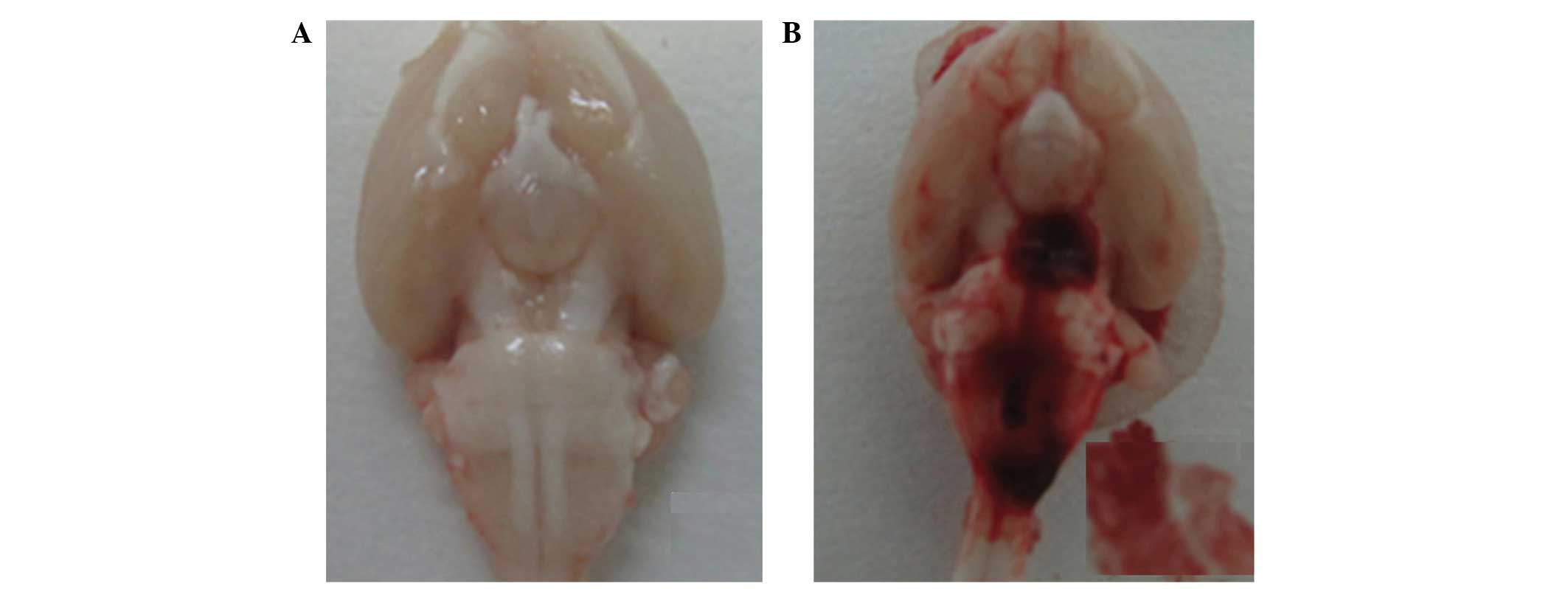
craniotomy (Fig. 1A and B).
Furthermore, all mice in the SAH group had Bederson neurological
severity scores ≥1, which indicated the success of the model
(Table I). No neurological
impairment was detected in the blank and normal saline groups.
 | Table I.Bederson grading of neurological
impairment in the mouse model of subarachnoid hemorrhage (SAH)
group (n=20). |
Table I.
Bederson grading of neurological
impairment in the mouse model of subarachnoid hemorrhage (SAH)
group (n=20).
|
| Bederson grade of
neurological impairment |
|
|---|
|
|
|
|
|---|
| Duration of
SAH | 0 | 1 | 2 | 3 | Mortality (%) |
|---|
| 3 days | 0 | 9 | 8 | 0 | 15 |
| 5 days | 0 | 3 | 6 | 6 | 25 |
| 7 days | 0 | 3 | 11 | 4 | 10 |
| 14 days | 0 | 14 | 6 | 0 | 0 |
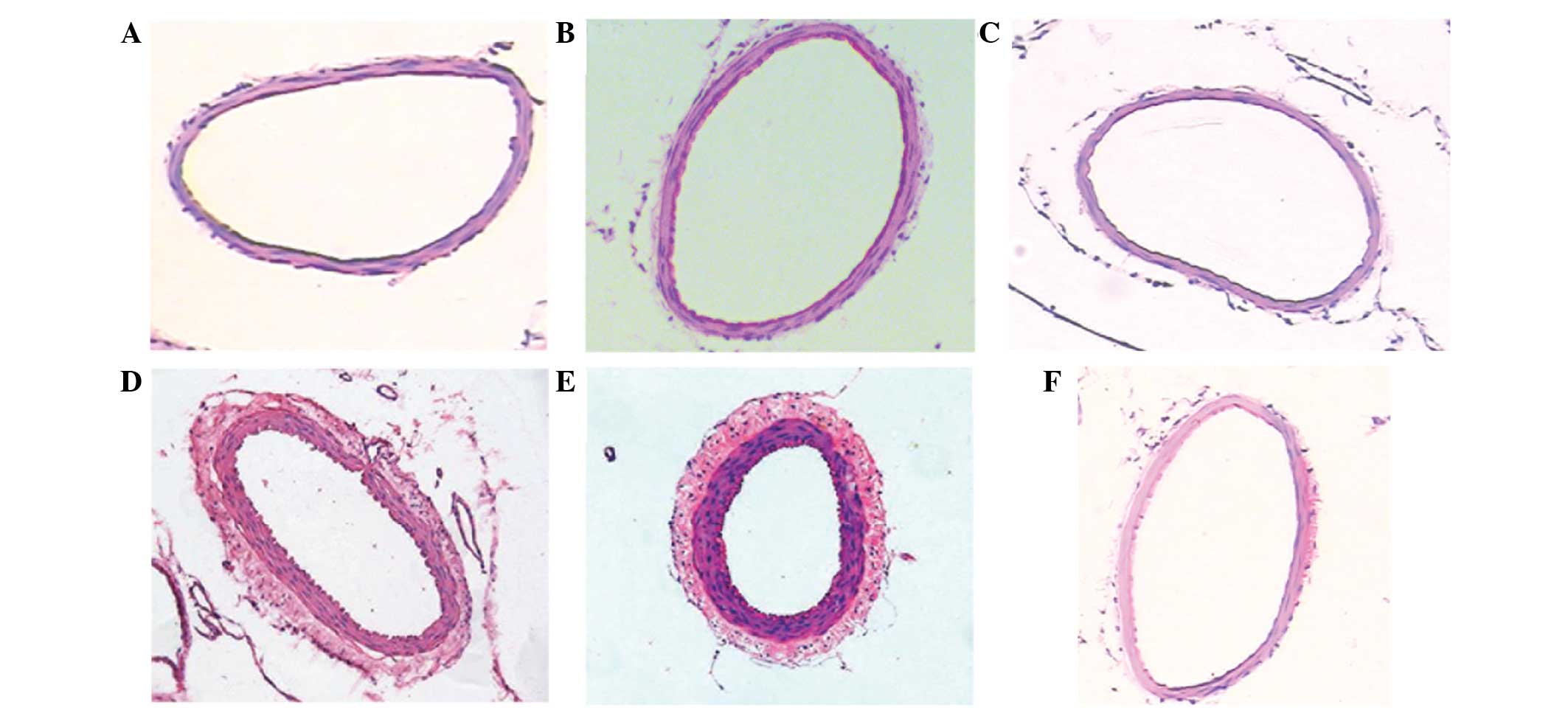
As detected by light microscopy, HE staining of the
basilar artery demonstrated that the inner membranes of the basilar
arteries appeared smooth in the blank and normal saline groups and
the inner elasticity and integrity were maintained. In the SAH
groups, the inner perimeter of the basilar artery was reduced, the
thickness of the arterial wall of the basilar artery increased
under the light microscope and its inner membrane appeared to be
buckling, as compared with the blank and normal saline groups
(Fig. 2).
SAH significantly reduces the internal
diameter of the basilar artery
The internal diameter of the basilar artery was
measured in all the groups at various time points (Table II). Across all durations of SAH, the
internal diameter of the basilar artery was significantly reduced
in the SAH group at the same time point, as compared with the
normal saline and blank groups (P<0.01). The t-values for the
SAH group, and the normal saline and blank groups were 9.72 and
10.13, respectively (data not shown). No significant differences
were detected between the normal saline and blank groups (t-value,
3.49; P>0.05).
| Table II.Internal diameter (µm) of the basilar
artery in the blank, normal saline and subarachnoid hemorrhage
(SAH) groups. |
Table II.
Internal diameter (µm) of the basilar
artery in the blank, normal saline and subarachnoid hemorrhage
(SAH) groups.
| Duration of
SAH | Blank group | Normal saline
group | SAH group |
|---|
| 3 days | 1,002±0.24 | 1,001±0.45 | 701±8.34 |
| 5 days | 1,003±0.56 | 1,003±0.32 | 686±6.23 |
| 7 days | 1,003±0.29 | 1,002±0.26 | 638±5.68 |
| 14 days | 1,001±0.31 | 1,002±0.67 | 698±4.65 |
SAH significantly increases the wall
thickness of the basilar artery
The wall thickness of the basilar artery was
measured in all the groups at various time points (Table III). Across all durations of SAH,
the wall thickness of the basilar artery was significantly
increased in the SAH group at the same time point, as compared with
the normal saline and blank groups. The t-values for the SAH group,
and the normal saline and blank groups were 8.63 and 8.19,
respectively (P<0.01; data not shown). No significant
differences were detected between the normal saline and blank
groups (t-value, 2.18; P>0.05).
| Table III.Wall thickness (µm) of basilar artery
in the blank, normal saline and subarachnoid hemorrhage (SAH)
groups. |
Table III.
Wall thickness (µm) of basilar artery
in the blank, normal saline and subarachnoid hemorrhage (SAH)
groups.
| Duration of
SAH | Blank group | Normal saline
group | SAH group |
|---|
| 3 days | 9.80±0.34 | 9.26±0.13 | 22.68±0.36 |
| 5 days | 9.47±0.29 | 9.43±0.20 | 36.74±0.52 |
| 7 days | 9.58±0.27 | 9.39±0.32 | 64.86±0.31 |
| 14 days | 9.24±0.12 | 9.35±0.19 | 32.16±0.45 |
SAH significantly reduces the
expression of caveolin-1 in the basilar artery, as detected by
fluorescence immunoassay
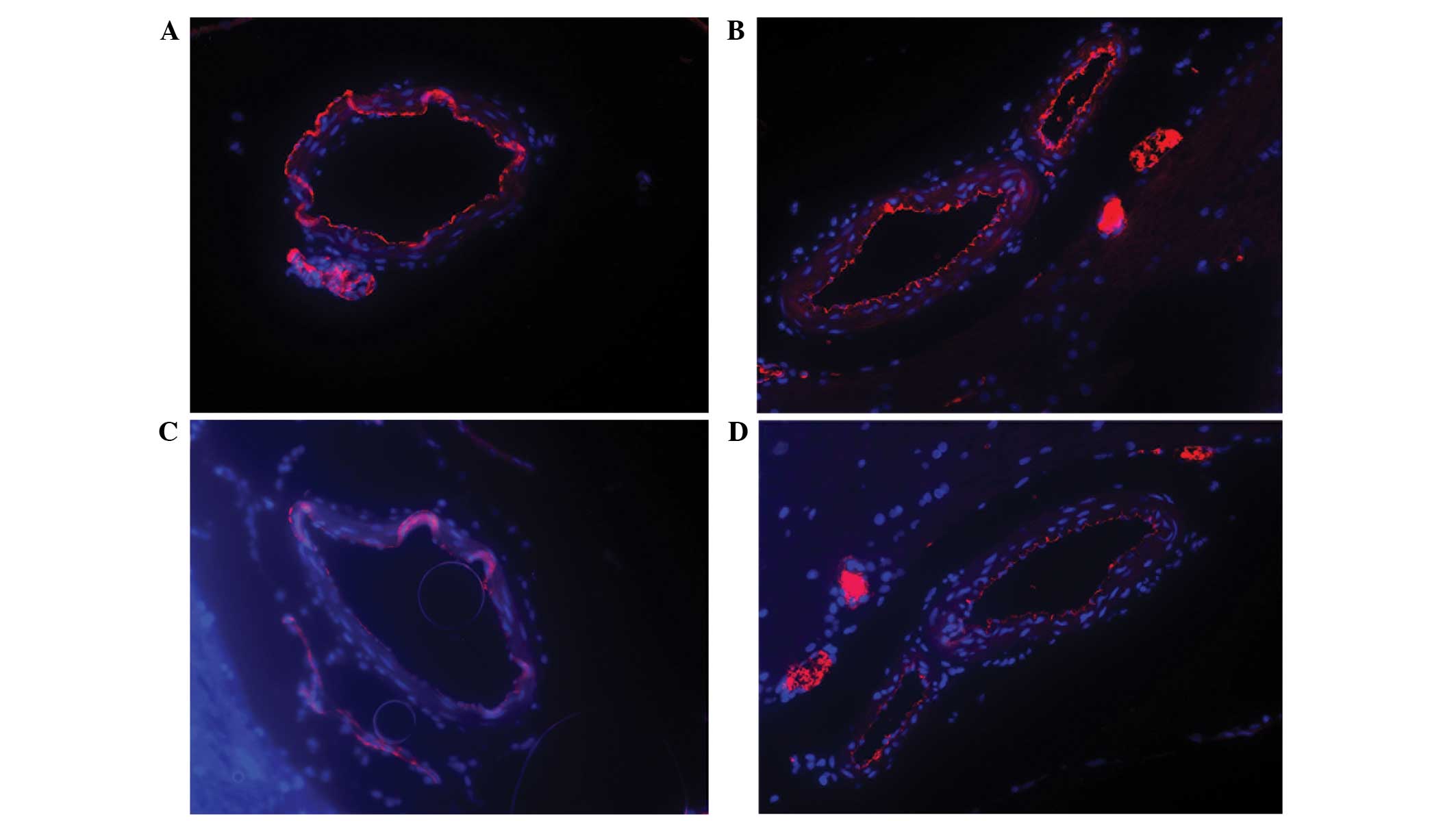
Immunofluorescence analysis of caveolin-1 expression
in the basilar artery was performed on all groups (Fig. 3). The results demonstrated that
caveolin-1 expression levels in the endothelial cells of the
basilar artery were markedly reduced in the experimental group
subjected to SAH-induced DCVS for 5 days, as compared with the
blank and normal saline groups. Although caveolin-1 expression
notably improved by day 7 of SAH, expression levels remained
reduced, as compared with the blank and normal saline groups. No
significant differences were detected between the normal saline and
blank groups.
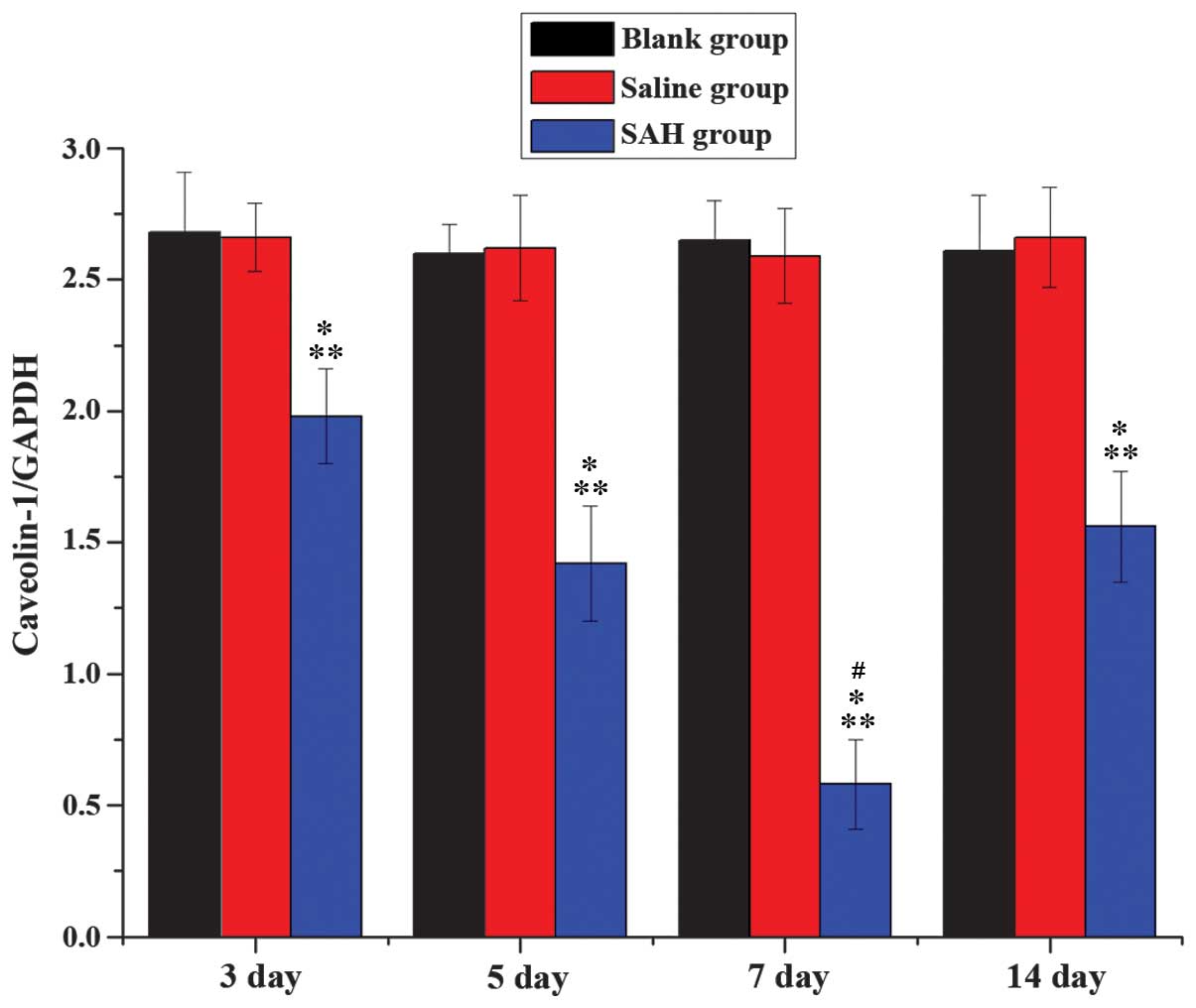
Western blot analysis for the
detection of caveolin-1 protein expression in the basilar
artery
The t-values of SAH group, and the normal saline and
blank groups were 21.72 and 53.66, respectively, at each time point
(P<0.01; data not shown). No significant differences were
detected between the normal saline and blank groups (t-value, 2.18;
P>0.05). Caveolin-1 expression levels were significantly reduced
in the SAH groups at all time points, as compared with the normal
saline and blank groups (Figs. 4 and
5). When the SAH groups were
compared, the reduction in caveolin-1 expression levels detected on
the day 7 was significantly lower than the other SAH group time
points.
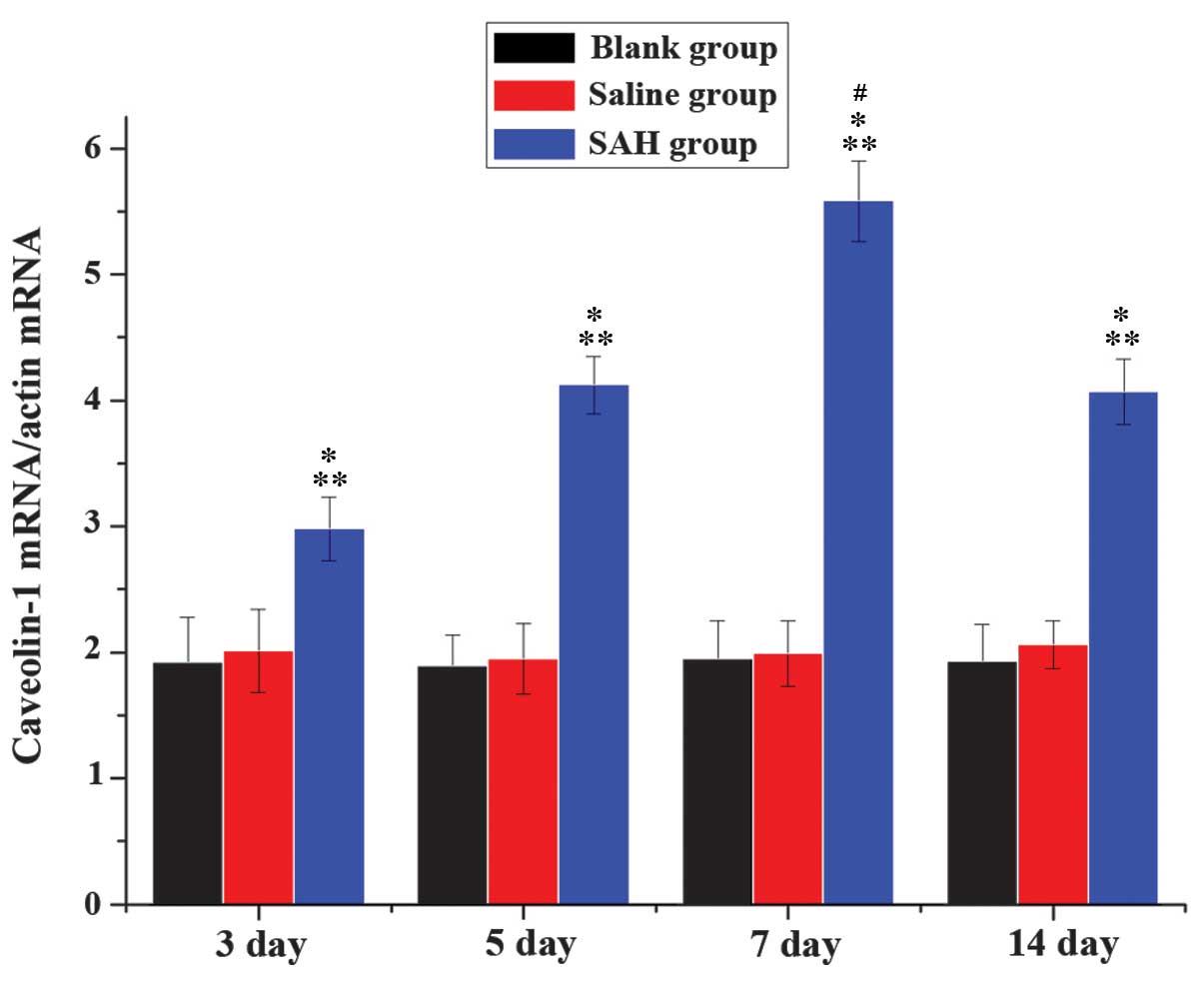
RT-qPCR analysis of caveolin-1 mRNA
expression
RT-qPCR analysis was performed to compare the
relative mRNA expression levels of caveolin-1 in the normal saline,
blank and SAH groups at various time points (Fig. 6). The results demonstrated that
caveolin-1 mRNA expression levels were significantly increased
across all time points in the SAH group, peaking on day 7 of SAH
(P<0.05). No significant differences were detected between the
blank and normal saline groups.
Discussion
SAH is a common cerebrovascular emergency. A total
of 30% patients with arterial aneurysm SAH will experience DCVS,
which is a common complication of SAH that remains the predominant
cause of patient morbidity and mortality (28). Cerebral angiospasm typically occurs
on days 3–5 following SAH, peaks on days 7–10 and ceases after 2–3
weeks. Cerebral angiospasm causes delayed ischemic brain damage
(30). Although a previous study has
investigated the pathogenesis of DCVS in patients following SAH,
the mechanisms underlying the pathogenesis of DCVS are yet to be
elucidated (31).
DCVS is the most severe complication of SAH and is
the predominant factor affecting the prognosis of patients post-SAH
(32). Therefore, it is crucial that
the underlying mechanisms of post-SAH DCVS are investigated and
potential clinical treatment options are elucidated. However, a
simple practicable animal model of post-SAH DCVS is required in
order to further study the pathogenesis and clinical treatment of
DCVS. The establishment of a good DCVS animal model is the key
finding of the present study. Previous experimental animal models
have been created to study DCVS via methods such as, puncturing the
murine middle cerebral artery to induce SAH (33), mice pulvinar next blood injection and
the recognized method of murine cisterna magna double hemorrhage
injection which was used to establish the present mouse model of
SAH (34). As a good DCVS animal
model involves the production of a blood clot circling around
basicranial blood vessel (34,35),
mice occipital bone double hemorrhage injection was adopted in the
present study to form the most effective animal model possible.
According to the grading system of neurological impairment proposed
by Bederson (29), mice in the SAH
day 3, 5 and 7 experimental groups exhibited Bederson neurological
severity scores ≥1; whereas no neurological impairment was detected
in normal saline and normal control groups. The SAH groups
demonstrated varying degrees of neurological impairment,
particularly in the day 5 and 7 SAH groups, where neurological
impairment was graded as the most severe. This result is consistent
with the time of the appearance of DCVS symptoms following SAH.
Therefore, the results of the present study suggested that a mouse
model of DCVS following SAH was successfully established, thus
supporting the accuracy and objectivity of the experimental
data.
DCVS following SAH is a complex pathological
process, which is associated with vascular endothelial function
impairment, shrinkage and proliferation of vascular smooth muscle,
immune inflammatory reaction and gene regulation (36). It has previously been demonstrated
that cerebrovascular endothelial cell damage is the key
characteristic of cerebral artery pathology in patients following
SAH (37). Thus, it may be
associated with the pathogenic mechanism of DCVS. Endothelial cells
dilate vessels by generating NO, whereas they constrict vessels via
ET. Under normal pathology, there is a dynamic balance between the
two to maintain homeostasis via the dilation and constriction of
vessels (38). Disorder of this
balance following SAH is a key factor in the pathogenesis of DCVS.
In recent years, studies in this field have focused on NO, which is
generated by eNOS and L-arginine, and is a key factor for the
development of DCVS (39,40). At the same time, the organism also
generates a kind of inductive NOS. The generation of NO in
vivo is controlled by a negative-feedback mechanism that is
dependent on NO content, the feedback affection of NOS and
methylate of L-arginine. NO adjusts vasodilatation via the cyclic
guanosine monophosphate (cGMP) signaling pathway (41). When the endothelial cells secrete NO,
NO acts on smooth muscle cells, activating guanylate cyclase in the
inner cytoplasm to generate cGMP, which facilitates the opening
calcium channels on the endoplasmic reticulum membrane (42). Calcium ions subsequently flow into
the endoplasmic reticulum, inducing the dilation of smooth muscle.
As a result, oxygen and hemoglobin consumption increases and the
generation of endogenous NO decreases, which in turn decreases the
bioavailability of NO on endothelial cells, leading to
vasoconstriction and stenosis of the lumen (43). ET-1 is released from the vestibular
nucleus in endothelial cells and is activated by receptor binding
to generate diacyl glycerol (DAG), leading to vasoconstriction via
DAG-protein kinase C (PKC) (44).
Caveolin-1 is localized between endothelial cells and vascular
smooth muscle cells and is a key constitutive protein for the
formation of caveolae. The oligomerization domain and caveolin
scaffolding domain (CSD) are key to the structure and function of
this area. On endothelial and vascular smooth muscle cells,
caveolin-1 prevents PKC from activating CSD its by preventing
interaction and subsequent enzymatic activity (45). Therefore, this can results in a
reduction in the concentration of the ET compound and subsequent
vasodilatation. Furthermore, caveolin-1 is capable of attenuating
the excitation of downstream protein molecules to reduce the
release of eNOS, which weakens the negative feedback NO release by
combining CSD microcell and eNOS (46). This increases the release of NO,
leading to vasodilatation (2). Shen
et al (28) demonstrated that
the loss of caveolin-1 is associated with the generation of NO in
the brain.
The present study demonstrated that the inner tube
of the basilar artery significantly narrowed on day 3 following
SAH, peaking on day 7, and subsequently attenuating on day 14, as
compared with day 7. All the results were obtained via observation
of inner diameter and thickness of the basilar artery. The results
of the present study also indicated that the smooth muscle layer
became thicker on day 5 post-SAH, once again peaking on day 7 and
attenuating by day 14. Caveolin-1 expression in the basilar artery
was detected using immunofluorescence and western blot analyses;
the results of which demonstrated that caveolin-1 protein
expression significantly reduced by day 5 following SAH
(P<0.05), as compared with the normal saline and blank groups.
Caveolin-1 mRNA expression levels were detected using RT-qPCR. The
results demonstrated that the mRNA expression level of caveolin-1
significantly increased following SAH, and peaked on day 7, as
compared with the blank and normal saline groups (P<0.01). These
results were consistent with previous findings (47) and demonstrated similar time-related
development of symptoms in patients with SAH-induced DCVS.
Therefore, the results of the present study suggested that
caveolin-1 may be crucially involved in the development of DCVS
following SAH.
In conclusion, the method of cisterna magna double
hemorrhage injection may be used to successfully establish a model
of DCVS following SAH. Furthermore, the downregulation of
caveolin-1 expression detected in the basilar artery of mice with
DCVS following SAH suggested that caveolin-1 may be associated with
the development of DCVS following SAH. Therefore, intervening and
regulating caveolin-1 and its associated mechanisms may be a
potential target for the treatment of DCVS. However, the underlying
molecular mechanisms are yet to be elucidated, therefore, further
studies are required in the future.
References
|
1
|
Wencel K, Dyaczyńska-Herman A and Wencel
T: Plasma clotting and fibrinolysis parameters in patients with
spontaneous and posttraumatic SAH. Clin Neuro Neurosurg. 99(Suppl
1): 70. 1997. View Article : Google Scholar
|
|
2
|
Arreche ND, Sarati LI, Martinez CR, Fellet
AL and Balaszczuk AM: Contribution of caveolin-1 to ventricular
nitric oxide in age-related adaptation to hypovolemic state. Regul
Pept. 179:43–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Agyemang C, van Oeffelen AA, Norredam M,
et al: Ethnic disparities in ischemic stroke, intracerebral
hemorrhage, and subarachnoid hemorrhage incidence in The
Netherlands. Stroke. 45:3236–3242. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Laskowitz DT and Kolls BJ: Neuroprotection
in subarachnoid hemorrhage. Stroke. 41(Suppl 10): S79–S84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Connolly ES Jr, Rabinstein AA, Carhuapoma
JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech
AM, Ogilvy CS, et al: Guidelines for the management of aneurysmal
subarachnoid hemorrhage: A guideline for healthcare professionals
from the American heart association/American stroke association.
Stroke. 43:1711–1737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pluta RM: Introduction to problems of
postsubarachnoid hemorrhage delayed cerebral vasospasmAnimal Models
of Acute Neurological Injuries II. Humana Press; New York, NY: pp.
459–464. 2012, View Article : Google Scholar
|
|
7
|
Anderson GB, Ashforth R, Steinke DE, et
al: CT angiography for the detection of cerebral vasospasm in
patients with acute subarachnoid hemorrhage. Am J Neuroradiol.
21:1011–1015. 2000.PubMed/NCBI
|
|
8
|
Specogna AV: Subarachnoid hemorrhage
diagnosis. JAMA. 311:2012014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang W and Zhou Z: Research advances on
pathogenesy of cerebral vasospasm after suharachnoid hemorrhage.
Zhong Guo Nao Xue Guan Bing Za Zhi. 7:215–219. 2010.(In
Chinese).
|
|
10
|
Roos YB, Dijkgraaf MG, Albrecht KW, Beenen
LF, Groen RJ, de Haan RJ and Vermeulen M: Direct costs of modern
treatment of aneurysmal subarachnoid hemorrhage in the first year
after diagnosis. Stroke. 33:1595–1599. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pluta RM: Delayed cerebral vasospasm and
nitric oxide: Review, new hypothesis, and proposed treatment.
Pharmacol Ther. 05:23–56. 2005. View Article : Google Scholar
|
|
12
|
Suzuki H, Kanamaru K, Tsunoda H, Inada H,
Kuroki M, Sun H, Waga S and Tanaka T: Heme oxygenase-1 gene
induction as an intrinsic regulation against delayed cerebral
vasospasm in rats. J Clin Invest. 104:59–66. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Varsos VG, Liszczak TM, Han DH, Kistler
JP, Vielma J, Black PM, Heros RC and Zervas NT: Delayed cerebral
vasospasm is not reversible by aminophylline, nifedipine, or
papaverine in a ‘two-hemorrhage’ canine model. J Neurosurg.
58:11–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Manabe K, Ito H, Matsuda H, et al:
Hyperpolarization induced by vasoactive substances in intact
guinea-pig endocardial endothelial cells. J Physiol. 484:25–40.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rabinstein AA, Lanzino G and Wijdicks EF:
Multidisciplinary management and emerging therapeutic strategies in
aneurysmal subarachnoid haemorrhage. Lancet Neurol. 9:504–519.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rothberg KG, Heuser JE, Donzell WC, Ying
YS, Glenney JR and Anderson RG: Caveolin, a protein component of
caveolae membrane coats. Cell. 68:673–82. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang H and Zou W: Caveolin-1 and Breast
Cancer. Chinese Zhong Guo Sheng Wu Hua Xue Yu Fen Zi Sheng Wu Xue
Bao. 23:20–26. 2007.(In Chinese).
|
|
18
|
Sampson MJ, Lovell RS and Craigen WJ: The
murine voltage-dependent anion channel gene family. Conserved
structure and function. J Biol Chem. 272:18966–18973. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hurlstone AF, Reid G, Reeves JR, Fraser J,
Strathdee G, Rahilly M, Parkinson EK and Black DM: Analysis of the
CAVEOLIN-1 gene at human chromosome 7q31.1 in primary tumours and
tumour-derived cell lines. Oncogene. 18:1881–1890. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Prinetti A, Prioni S, Loberto N, Aureli M,
Chigorno V and Sonnino S: Regulation of tumor phenotypes by
caveolin-1 and sphingolipid-controlled membrane signaling
complexes. Biochim Biophys Acta. 1780:585–596. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Botos E, Klumperman J, Oorschot V, Igyártó
B, Magyar A, Oláh M and Kiss AL: Caveolin-1 is transported to
multi-vesicular bodies after albumin-induced endocytosis of
caveolae in HepG2 cells. J Cell Mol Med. 12:1632–1639. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arakawa R, Abedohmae S, Asai M, Ito JI and
Yokoyama S: Involvement of caveolin-1 in cholesterol enrichment of
high density lipoprotein during its assembly by apolipoprotein and
THP-1 cells. J Lipid Res. 41:1952–1962. 2001.
|
|
23
|
Yu Q, Chen X, Fang X, Chen Q and Hu C:
Caveolin-1 aggravates cigarette smoke extract-induced MUC5AC
secretion in human airway epithelial cells. Int J Mol Med.
35:1435–1442. 2015.PubMed/NCBI
|
|
24
|
Smart EJ, Graf GA, McNiven MA, Sessa WC,
Engelman JA, Scherer PE, Okamoto T and Lisanti MP: Caveolins,
liquid-ordered domains, and signal transduction. Mol Cell Biol.
19:7289–7304. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song L, Liu J, Zou W and An LJ: Role of
Caveolin-1 in cell senescence and senescent dieases. Zhong Guo
Sheng Wu Hua Xue Yu Fen Zi Sheng Wu Xue Bao. 08:712–720. 2011.(In
Chinese).
|
|
26
|
Jasmin JF, Malhotra S, Singh Dhallu M,
Mercier I, Rosenbaum DM and Lisanti MP: Caveolin-1 deficiency
increases cerebral ischemic injury. Circ Res. 100:721–729. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen J, Ma S, Chan P, Lee W, Fung PC,
Cheung RT, Tong Y and Liu KJ: Nitric and oxide down-regulates
caveolin-1 expression in rat brains during focal cerebral ischemia
and reperfusion injury. J Neurochem. 96:1078–1089. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JY, Huang DL, Keep R and Sagher O:
Characterization of an improved double hemorrhage rat model for the
study of delayed cerebral vasospasm. J Neurosci Methods.
168:358–366. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bederson JB, Pitts LH, Tsuji M, Nishimura
MC, Davis RL and Bartkowski H: Rat middle cerebral artery
occlusion: Evaluation of the model and development of a neurologic
examination. Stroke. 17:472–476. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vellimana AK, Milner E, Azad TD, Harries
MD, Zhou ML, Gidday JM, Han BH and Zipfel GJ: Endothelial nitric
oxide synthase mediates endogenous protection against subarachnoid
hemorrhage-induced cerebral vasospasm. Stroke. 42:776–782. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kassell NF, Sasaki T, Colohan AR and Nazar
G: Cerebral vasospasm following aneurysmal subarachnoid hemorrhage.
Stroke. 1985.16:562–572. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jia L and Sun BL: Establishment of an
animal model of cerebral vasospasm following subarachnoid
hemorrhage. J Clinical Rehabilitative Tissue Engineering Research.
13:8147–8150. 2009.
|
|
33
|
Sugawara T, Ayer R, Jadhav V and Zhang JH:
A new grading system evaluating bleeding scale in filament
perforation subarachnoid hemorrhage rat model. J Neurosci Methods.
167:327–334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marbacher S, Fandino J and Kitchen ND:
Standard intracranial in vivo animal models of delayed cerebral
vasospasm. Br J Neurosurg. 24:415–434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Güresir E, Raabe A, Jaiimsin A, Dias S,
Raab P, Seifert V and Vatter H: Histological evidence of delayed
ischemic brain tissue damage in the rat double-hemorrhage model. J
Neurol Sci. 293:18–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yanamoto H, Kataoka H, Nakajo Y and Iihara
K: The role of the host defense system in the development of
cerebral vasospasm: Analogies between atherosclerosis and
subarachnoid hemorrhage. Eur Neurol. 68:329–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chaichana KL, Pradilla G, Huang J and
Tamargo RJ: Role of inflammation (leukocyte-endothelial cell
interactions) in vasospasm after subarachnoid hemorrhage. World
Neurosurg. 73:22–41. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Anuntasethakul T, Srikiatkhachorn A,
Maneesri S, Patumraj S and Kasantikul V: Ultrastructural changes in
endothelial cells of cerebral microvessels after exposure to nitric
oxide donor. Neuropathol. 19:259–266. 1999. View Article : Google Scholar
|
|
39
|
Pluta RM, Andre D, George G, Gladwin MT
and Oldfield EH: Nitrite infusions to prevent delayed cerebral
vasospasm in a primate model of subarachnoid hemorrhage. JAMA.
293:1477–1484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kevil CG and Patel RP: Preserving vessel
function during ischemic disease: New possibilities of inorganic
nitrite therapy. Expert Rev Cardiovasc Ther. 6:1175–1179. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Duncan C, Dougall H, Johnston P, Green S,
Brogan R, Leifert C, Smith L, Golden M and Benjamin N: Chemical
generation of nitric oxide in the mouth from the enterosalivary
circulation of dietary nitrate. Nat Med. 1:546–551. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dora KA, Doyle MP and Duling BR: Elevation
of intracellular calcium in smooth muscle causes endothelial cell
generation of NO in arterioles. PNAS. 94:6529–6534. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Corpas FJ, Barroso JB, Carreras A, Quirós
M, León AM, Romero-Puertas MC, Esteban FJ, Valderrama R, Palma JM,
Sandalio LM, et al: Cellular and subcellular localization of
endogenous nitric oxide in young and senescent pea plants. Plant
Physiol. 136:2722–2733. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Russell FD and Molenaar P: The human heart
endothelin system: ET-1 synthesis, storage, release and effect.
Trends Pharmacol Sci. 21:353–359. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thyberg J: Caveolin-1 and caveolae act as
regulators of mitogenic signaling in vascular smooth muscle cells.
Arterioscler Thrombo Vasc Biol. 23:1481–1483. 2003. View Article : Google Scholar
|
|
46
|
Penumathsa SV, Koneru S, Samuel SM, Maulik
G, Bagchi D, Yet SF, Menon VP and Maulik N: Strategic targets to
induce neovascularization by resveratrol in hypercholesterolemic
rat myocardium: role of caveolin-1, eNOS, HO-1 and VEGF. Free Radic
Biol Med. 45:1027–1034. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang X, Zhou Z, Liu J, Feng B, Zhang J
and Huang W: Relationship between caveolin-1 and proliferation of
basilar artery smooth muscle in rabbit model of subarachnoid
hemorrhage. Journal of Third Military Medical University.
33:830–834. 2011.
|