Introduction
COPD is a chronic inflammatory disease characterized
by airflow limitation, with a high morbidity and mortality.
According to previously reported statistics, COPD ranks fourth in
the cause of mortality worldwide, and its rank is expected to move
up to third in 2030 (1). The
prevalence rate of COPD among people over age 40 is 8.2% in China
(2). COPD has already become an
important global public health problem. Therefore, further study
and understanding of COPD are of great significance.
Epidemiological studies have shown that smoking is
the most important pathogenic factor of human COPD (3). Long-term exposure to tobacco smoke can
cause the infiltration and recruitment of lung inflammatory cells,
which can then release various cytokines and finally cause the
formation of COPD through a variety of mechanisms (4). Among COPD patients, 80–90% are smokers
or ex-smokers (5,6). Studies showed that ~15–20% of smokers
eventually develop COPD (7), and a
survey in China indicated that the prevalence of COPD among smokers
was 2–4 times of that among non-smokers, and was directly
associated with the duration and quantity of smoking, i.e., a
longer duration and a greater amount of smoking results in a higher
prevalence of COPD (8).
Adiponectin is an adipokine secreted by adipocytes.
Previously, a study identified that airway epithelial cells also
secrete adiponectin, which through autocrine and paracrine
mechanisms regulates chronic inflammation in COPD (9). Tomoda et al (10) used ELISA to detect the serum level of
adiponectin in male patients with COPD and found that adiponectin
levels in the serum of patients with COPD was significantly higher
compared with that of a normal control, suggesting that COPD may be
closely associated with adiponectin. Chan et al (11) studied the association between
adiponectin, interleukin (IL)-6, IL-8 and C-reactive protein with
COPD, and found that adiponectin, IL-6 and C-reactive protein were
significantly elevated in patients with COPD. Miller et al
(12) used an adiponectin null mouse
model to study the association between adiponectin and emphysema
and observed an increase in diseased animals. Daniele et al
(13) used ELISA technique and
demonstrated that in patients with COPD, adiponectin levels were
significantly upregulated, suggesting the involvement of
adiponectin in the occurrence and development of COPD.
Previous have shown that adiponectin serves an
anti-inflammatory role in the occurrence and development of COPD.
The study of Tilg and Moschen (14)
indicated that in patients with COPD, the anti-inflammatory effect
of adiponectin was primarily achieved through inhibiting the
activity of macrophages and the release of pro-inflammatory
cytokines, such as IL-6 and tumor necrosis factor (TNF)-α, from
macrophages. In addition, adiponectin was shown to reduce the
proliferation of lymphocytes, reduce lymphocyte reactivity, and
induce the production of anti-inflammatory cytokines, such as IL-10
and IL-1 receptor antagonist, in monocytes and macrophages
(14). Wert (15) injected exogenous adiponectin into
wild-type and adiponectin-deficient mice and induced the production
of TNF-α in macrophages by lipopolysaccharide stimulation; it was
observed that the quantity of TNF-α generation reduced in
adiponectin-null mice compared with the control, suggesting the
anti-inflammatory effect of adiponectin.
A previous demonstrated that adiponectin has a
pro-inflammatory function in COPD. Miller et al (9) found that adiponectin can bind to the
inducible adiponectin receptor 1 on airway epithelial cells and
macrophages, promote the production of IL-8, TNF-α and other
inflammatory mediators, and recruit the aggregation of various
inflammatory cells (shch as macrophages and neutrophils) by
chemotaxis. The synthesis and secretion of IL-8 further promotes
the production of adiponectin and induces the synthesis of
adiponectin receptor 1 on airway epithelial cells and macrophages,
resulting in a cascade effect via autocrine and paracrine
mechanisms and thereby exerting an anti-inflammatory effect. Thus,
adiponectin serves dual anti- and pro-inflammatory roles in the
occurrence and development of COPD; however, further investigation
is required in order to determine which role exerts the leading
effect. In the present study, cigarette smoke extract (CSE)
stimulates human bronchial epithelial cells to simulate the
occurrence of COPD, and the mechanism of adiponectin in this
process is researched.
Materials and methods
Cell culture
Human bronchial epithelial cells (16HBECs) were
purchased from ScienCell Research Laboratories, Inc. (Carlsbad, CA,
USA), and cultured in Minumum Essential Medium (MEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) at 37°C
in a 5% CO2 incubator.
CSE preparation
CSE was prepared as described by Su et al
(16) with modifications. Briefly, a
stick of Da Qian Men cigarette (containing tar 0.013 g, nicotine
0.001 g and CO 0.014 g; Taiyuan Tobacco Company, Taiyuan, China)
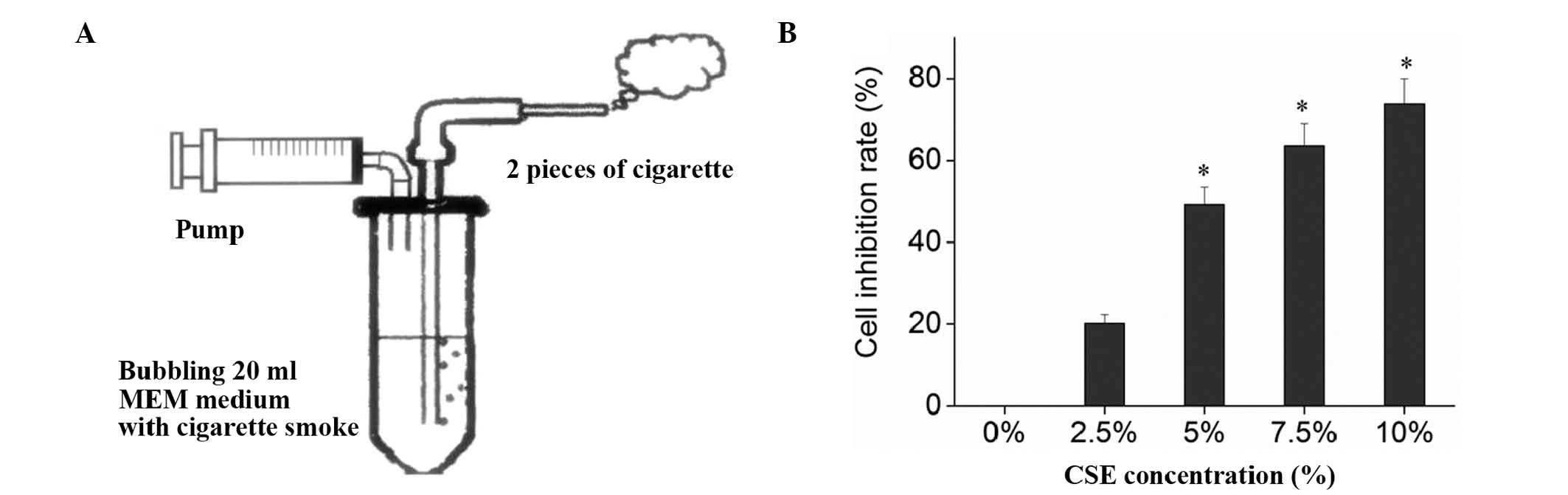
was connected to a syringe driving instrument (Fig. 1A). The syringe was filled with 20 ml
serum-free MEM, and the cigarette was lit. To simulate human
smoking, the sucking lasted 2 sec, and the pause was 58 sec, to
fully absorb the smoke. Each cigarette took ~7–8 min, and two
cigarettes were tested continuously. The 20 ml serum-free MEM were
then prepared into CSE stock solution after being adjusted to pH
6.8–7.2 with l N NaOH (Amresco, Inc., Farmingham, MA, USA) and
sterilized by filtering through a 0.22 µm filter membrane (Merck
Millipore, Shanghai, China). The CSE stock solution was then
diluted with serum-free MEM into 0 (no CSE intervention group),
2.5, 5, 7.5 and 10% culture media, which were then used in the
experiment within 30 min.
MTT assay
16HBECs in the exponential growth were digested,
resuspended, adjusted into 5×104 cells/ml, and seeded
into 96-well plates at 200 µl/well in triplicate. After the cells
were grown into 70–80% confluence 24 h later and washed twice with
D-Hank's solution (Gibco; Thermo Fisher Scientific, Inc.), each
well was added 200 µl of serum-free MEM containing 0, 2.5, 5.0, 7.5
and 10.0% diluted CSE stock solution. After another 24 h of
culture, the CSE medium in each well was replaced with fresh medium
containing 20 µl MTT (Amresco, Inc.) and 180 µl MEM, and the cells
were cultured for another 4 h. The MTT medium was removed, and the
plate was added 150 µl DMSO/well and shaken for 10 min. The
absorbance was then measured on an automatic microplate reader
(Bio-Rad 680; Bio-Rad Laboratories, Inc. Hercules, CA, USA) at 490
nm wavelength (A490). The inhibition rates were calculated based on
the A490 values of the control (without CSE) and the blank (without
CSE and MTT) wells. The CSE concentration with a cell inhibition
rate of 50% was defined as CSE IC50 (inhibitory
concentration of 50%). Inhibition rate = (control well -
experimental well) / (control well - blank well) × 100%.
Detection of TNF-α, IL-8 and
4-hydroxy-nonenal (HNE) with ELISA
The cells inoculated onto 6-well plate at
1×105 cells/ml were randomly divided into the following
8 groups: i) CSE group, 2 ml MEM/well containing 100 µl of CSE (5%
CSE); ii) negative control group, equal volume of D-Hank's
solution/well; iii) high molecular weight (HMW) groups, first
incubated in medium containing 5, 10 and 20 µg/ml HMW (Gibco;
Thermo Fisher Scientific, Inc.) for 2 h at 37°C, and then cultured
in 5% CSE medium; iv) globular domain (gAd) groups, firstly
incubated in medium containing 5, 10 and 20 µg/ml gAd (Gibco;
Thermo Fisher Scientific, Inc.) for 2 h at 37°C, and then cultured
in 5% CSE medium. The above groups were cultured for 24 h, and the
cells and supernatants were harvested and used for the subsequent
experiments. The experiments were repeated three times, and the
supernatants were collected to detect the contents of TNF-α (cat.
no. EHC103a; Human TNF-α ELISA kit; Neobioscience, Shenzhen,
China), IL-8 (cat. no. EHC008; Human IL-8 ELISA kit;
Neobioscience), and 4-HNE (cat. no. E01H0203; Human 4-HNE ELISA
kit; BlueGene Biotech Co., Ltd., Shanghai, China) proteins by
ELISA.
Reactive oxygen species (ROS)
determination
After 24 h of culture, the 6-well plate was removed
from the incubator, and the cells were washed three times with
preheated sterile D-Hank's buffer. The cells in each group were
then added 1.5 ml medium containing 10 µM DCFH-DA (NKKCBio,
Nanjing, China) and incubated for 40 min at 37°C in a 5%
CO2 incubator. After incubation, the cells were washed
three times with D-Hank's solution to remove the extracellular
fluorescent substances and reduce the background. The cells were
then collected and analyzed by flow cytometry (Beckman Coulter,
Inc., Brea, CA, USA) and detected by Cytomics FC flow cytometry
(Beckman Coulter, Inc.) within 1 h.
Apoptosis
The cells were harvested by digestion with EDTA-free
trypsin (Gibco; Thermo Fisher Scientific, Inc.) and washed twice
with D-Hank's solution (by centrifugation at 200 × g for 5
min at room temperature). The cells (1–5×105) were then
resuspended in 500 µl binding buffer, mixed with 5 µl annexin
V-FITC (KenGen Biotech Co., Ltd., Nanjing, China) and 5 µl
propidium iodide sequentially, incubated at room temperature in the
dark for 5–15 min, and detected by flow cytometry (Beckman Coulter,
Inc., Brea, CA, USA) within 1 h.
Statistical analysis
All data were presented as the mean ± standard
deviation and statistically analyzed using SPSS version 17.0
software (SPSS, Inc., Chicago, IL, USA). The comparison among
groups was performed using one-way analysis of variance, and the
pairwise comparison between groups was conducted using Fisher's
least significant difference t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Inhibitory effect of CSE on
16HBECs
Bronchial epithelial cells are the first barrier to
protect the airway tissue from the invasion of harmful substances,
with a wide range of physiological functions. Bronchial epithelial
cells are the major and first target cells to suffer from injuries
in acute and chronic airway diseases. Upon activation by the
stimuli of external harmful substances, they promote the expression
of inflammatory cytokines and actively participate in the process
of airway inflammation via secretion of inflammatory mediators,
cytokines and growth factors (9).
Therefore, the present study used 16HBECs as a model to investigate
the effects of CSE and adiponectin. MTT assay indicated that CSE
significantly inhibited the proliferation of 16HBECs (F=1808.88,
P<0.01) in a dose-dependent manner, and the IC50 of
CSE on 16HBECs was ~5% (Fig.
1B).
Adiponectin reverses the
CSE-upregulated expression of TNF-α and IL-8 in 16HBECs
TNF-α and IL-8 serve important roles in the chronic
airway inflammation of COPD. A previous study demonstrated that the
levels of IL-8 and TNF-α are significantly elevated in patients
with COPD compared with the normal control, and are significantly
higher in the acute exacerbation phase compared with in the stable
phase (17). They are important
inflammatory markers in evaluating the condition and prognosis of
COPD (18). ELISA detection of the
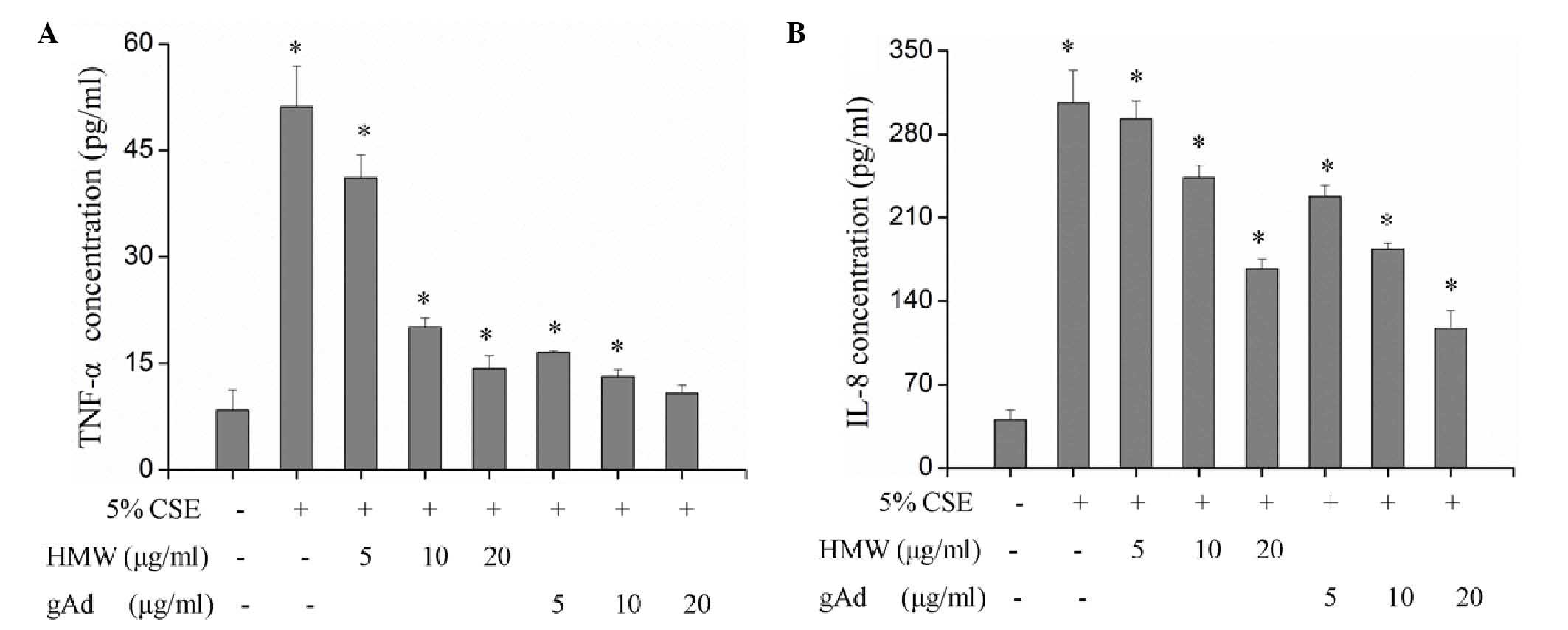
secretion of TNF-α and IL-8 from 16HBECs treated with 5% CSE
indicated significantly increased expression of TNF-α and IL-8
(Table I; P<0.01), suggesting
that CSE may promote the expression of TNF-α and IL-8 and thereby
cause injury to 16HBECs. In the serum, adiponectin proteins exist
in the forms of full length structure (triploid, hexaploid and HMW)
and gAd (14). The present study
explored the specific functions of different forms of adiponectin.
Treatment of 16HBECs with different concentrations of HMW and gAd
found that both forms inhibited the expression of TNF-α and IL-8 in
a dose-dependent manner and significantly blocked the CSE-induced
upregulation of TNF-α and IL-8 (Fig.
2; Table II). However, the
inhibitory effect of gAd was more evident; while 5 µg/ml gAd
significantly inhibited the expression of TNF-α and IL-8, a
significant effect of HMW was only observed when its concentration
reached 10 µg/mL (Fig. 2). At the
same concentration, gAd displayed a stronger inhibitory effect on
the expression of TNF-α and IL-8 compared with HMW (Fig. 2).
 | Table I.Injury of 5% CSE on 16HBECs (±
standard deviation). |
Table I.
Injury of 5% CSE on 16HBECs (±
standard deviation).
| Group | TNF-α (pg/ml) | IL-8 (pg/ml) | 4-HNE (µg/ml) | ROS (mean
fluorescence intensity) |
|---|
| Control |
8.43±2.85 | 40.19±8.27 | 0.16±0.13 | 17.33±3.06 |
| 5% CSE | 51.12±5.75 | 306.43±27.09 | 2.21±0.24 | 346.52±13.71 |
| T-value | −11.52 | −16.28 | −12.93 | −40.59 |
| P-value | <0.01 | <0.01 | <0.01 | <0.01 |
| Table II.Correlation of HMW, gAd and TNF-α,
IL-8, 4-HNE, ROS. |
Table II.
Correlation of HMW, gAd and TNF-α,
IL-8, 4-HNE, ROS.
| Group |
| TNF-α | IL-8 | 4-HNE | ROS |
|---|
| HMW | R-value | −0.919 | −0.958 | −0.930 | −0.931 |
|
| P-value | <0.01 | <0.01 | <0.01 | <0.01 |
| gAd | R-value | −0.755 | −0.959 | −0.933 | −0.965 |
|
| P-value | <0.01 | <0.01 | <0.01 | <0.01 |
Adiponectin blocks the upregulating
effect of CSE on 4-HNE and ROS levels in 16HBECs
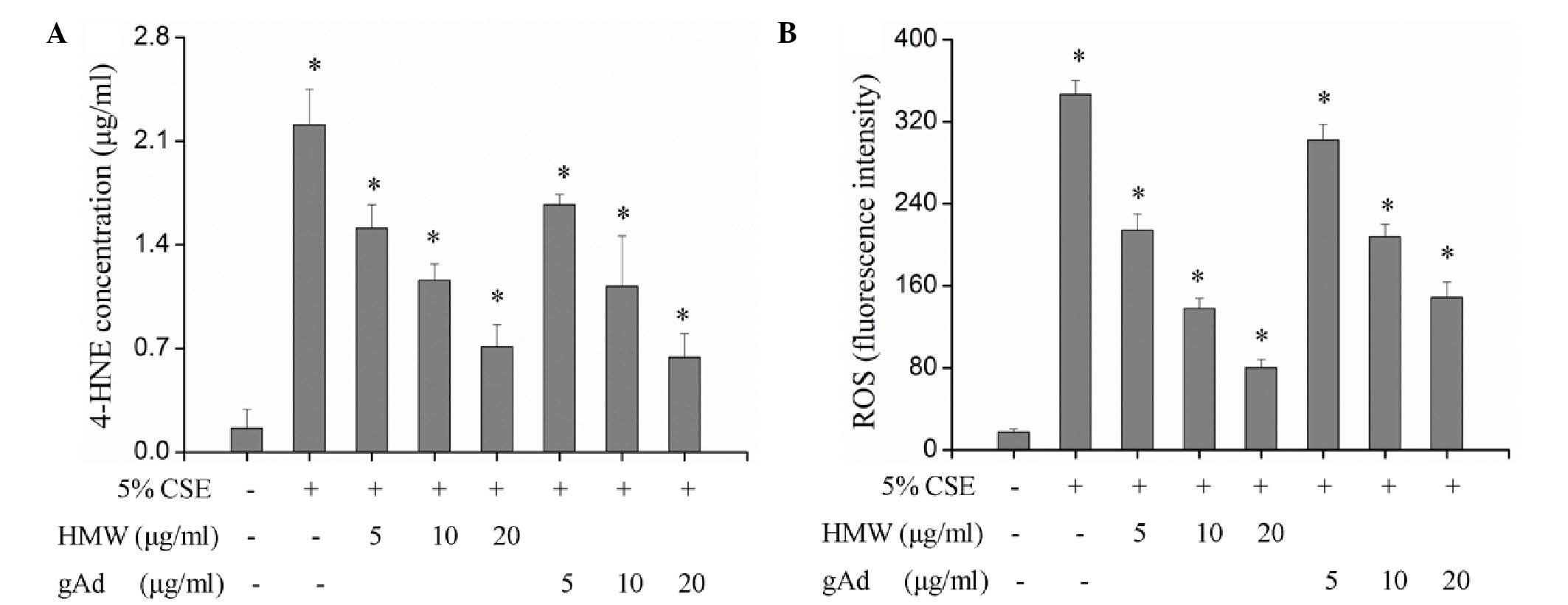
Tobacco smoke can induce inflammatory cells to
infiltrate into the lung tissue and produce ROS (19). An excessive production of ROS can
cause peroxidation of cellular proteins, lipids, DNA and
carbohydrates. As one of the most stable representative terminal
products of lipid peroxidation under pulmonary oxidative stress,
4-HNE is more stable than ROS and can diffuse to and exert effects
in distant tissues. The detection of 4-HNE and ROS indicated that
both HMW and gAd inhibited the production of 4-HNE and ROS in a
dose-dependent manner and significantly blocked the upregulation of
4-HNE and ROS by CSE (P<0.01; Fig.
3, Table II). Although there
was no significant difference between the inhibitory effects of HMW
and gAd on 4-NHE production, the suppression of ROS by HMW was more
effective compared with gAd. The inhibitory effects of 5 and 10
µg/ml HMW were equivalent to those of 10 and 20 µg/ml gAd,
respectively (Fig. 3).
Effects of CSE and adiponectin on the
apoptosis of 16HBECs
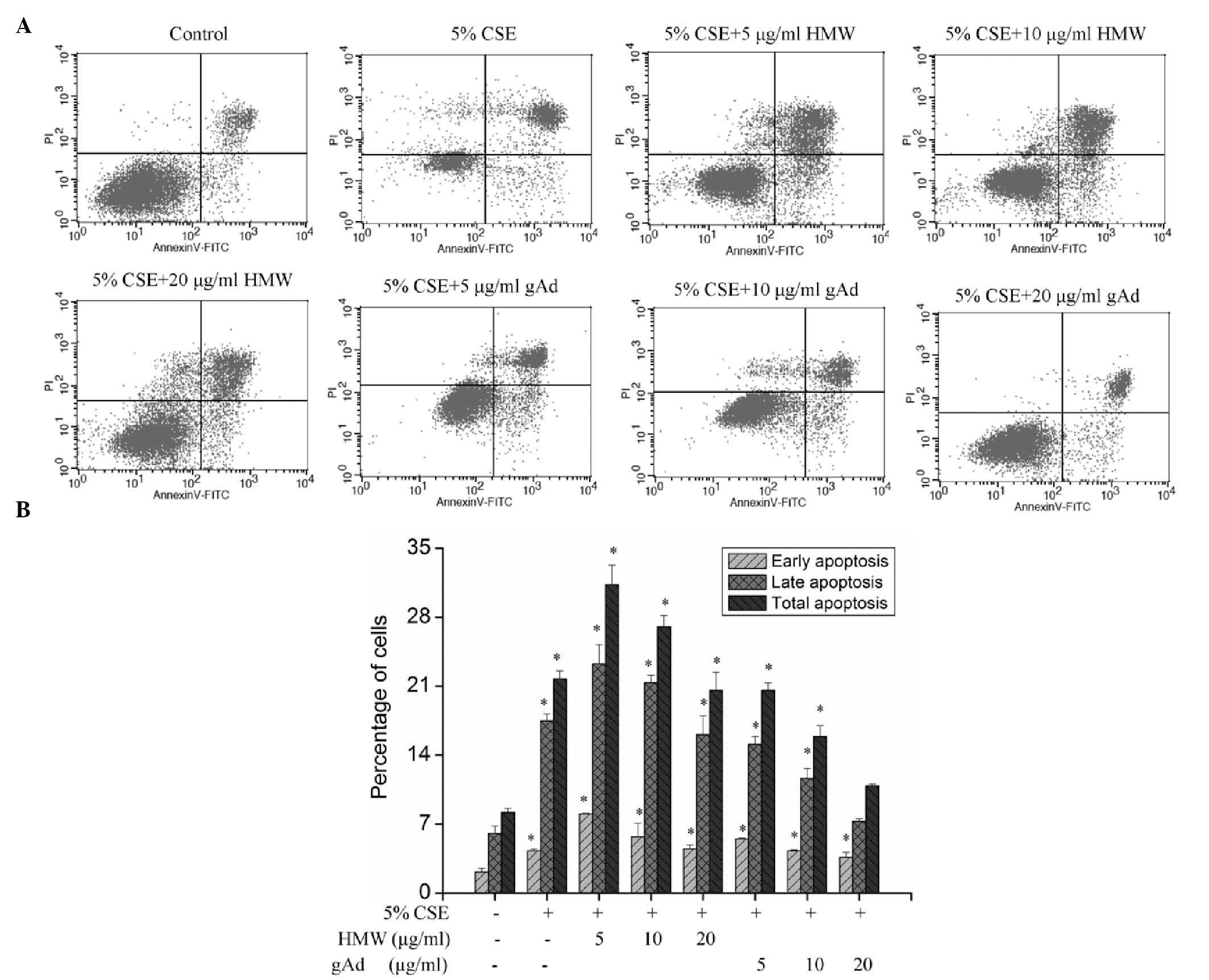
CSE treatment significantly increased the apoptosis
of 16HBEC (P<0.01; Fig. 4),
suggesting that CSE can promote apoptosis in these cells.
Intervention with different concentrations of HMW found that HMW
further aggravated apoptosis; however, with the increase in HMW
concentration, its apoptosis-promoting effect declined (Fig. 4). Use of different concentrations of
gAd for intervention showed that gAd dose-dependently inhibited
CSE-induced apoptosis (Fig. 4).
These results indicate that different forms of adiponectin may have
different activities, and that gAd has an anti-apoptotic
effect.
Discussion
COPD is a common disease threatening public health
worldwide. With a trend of increasing incidence, it has currently
become the fourth biggest cause mortality worldwide (20). Present therapies cannot block the
disease progression of COPD patients, which severely affects the
treatment efficacy and prognosis of COPD (21). Therefore, further study on the
pathogenesis and risk factors of COPD may provide a better
theoretical basis for the prevention and treatment of COPD.
Previous studies have shown that cytokines serve
important roles in the pathogenesis of COPD; among them,
adiponectin has drawn increasing attention (17,19).
Adiponectin serves an important role in the body's metabolic
processes; it has multiple biological functions, such as regulating
lipid metabolism, improving insulin resistance, protecting the
cardiovascular system, regulating bone formation, suppressing
inflammation and tumor growth (22).
In addition, a previous study demonstrated that adiponectin is
associated with the pathophysiology of airway inflammation
(23). Sull et al (24) studied 2,500 healthy male Koreans and
found that smoking reduced human serum adiponectin levels, and that
such reduction was significantly associated with the quantity of
cigarettes smoked. Kirdar et al (25) showed that the serum levels of
adiponectin, erythrocyte sedimentation rate and C-reactive protein
in the acute exacerbation phase of COPD were significantly higher
compared with those in the stable phase, and hence believed that
adiponectin may be a marker of systemic inflammation in COPD.
A previous study demonstrated that adiponectin
serves a proinflammatory role in the pathogenesis of COPD (9). However, further investigation indicated
that adiponectin exerted anti-inflammatory effects in COPD. Summer
et al (26) studied a mouse
model and confirmed that adiponectin was present in the lungs and
exerted an anti-inflammatory effect via reducing the release of
TNF-α and matrix metalloproteinase 12 through inhibiting the
activation of alveolar macrophages; adiponectin deficiency or
insufficiency changed the type of pulmonary emphysema in mice.
Nakanishi et al (27)
reported that adiponectin deficiency led to the occurrence of COPD
in mice, accompanied by systemic inflammation and extrapulmonary
effects; they also found that hypoadiponectinemia is associated
with the loss of function of endothelial cells and may therefore
serve a key role in the progression of COPD and concomitant
complications. Yoon et al (28) reported a seven year cohort study
which indicated that an increase of serum adiponectin levels
reduced the level of mortality of patients with COPD from
cardiovascular diseases. Therefore, the function of adiponectin
needs to be further verified in future investigations.
The present study on 16HBEC cells revealed that both
HMW and gAd dose-dependently inhibit the expression of TNF-α and
IL-8 as well as the generation of 4-HNE and ROS; they also
significantly block the upregulating effect of CSE on the above
factors. TNF-α is an important inflammatory and immune-regulatory
factor in the pathogenesis of COPD. A previous study demonstrated
that TNF-α serves a key role in tobacco smoke-induced chronic
airway inflammation and connective tissue injury (29). The primary biological function of
IL-8 is chemotactic effect, which is the major cause of the
increase of pulmonary neutrophils in patients with COPD. Meanwhile,
IL-8 is an important indicator used to assess the severity of
chronic airway inflammation (30).
Adiponectin can antagonize the upregulating effect of CSE on TNF-α
and IL-8, indicating that adiponectin has an anti-inflammatory
effect. In addition, oxidative stress serves a key role in the
development and progression of COPD. Oxidative stress generates an
excessive quantity of ROS, which may cause pathophysiological
changes through various pathways (31). Lipid peroxidation products from ROS,
such as 4-HNE, may serve as signal molecules to activate nuclear
factor-κB, causing pulmonary and systemic inflammation in patients
with COPD; these inflammatory responses generate more
oxidative-stress products, forming a vicious cycle (32).
A previous study showed that the expression of 4-HNE
significantly increased in mouse bronchial epithelial cells and
type II alveolar epithelial cells upon exposure to tobacco smoke
(33), suggesting that 4-HNE may be
involved in the injuries to bronchial mucosa caused by smoking. The
inhibitory effect of adiponectin on 4-HNE and ROS production in the
present study demonstrates its anti-inflammatory function. However,
it was observed that gAd exerted a stronger inhibitory effect on
the expression of TNF-α and IL-8 compared with HMW, while HMW more
significantly inhibited the generation of ROS compared with gAd,
suggesting that different forms of adiponectin may have different
activities.
In the current study, analysis of apoptosis found
that both CSE and HNW promoted apoptosis in 16HBECs, but
interestingly, with an increase in concentration, the pro-apoptotic
effect of HMW weakened. In contrast, gAd exerted an anti-apoptotic
effect and dose-dependently inhibited the occurrence of CSE-induced
apoptosis. These data further demonstrate that different forms
adiponectin may have different mechanisms of action. Previous
investigations have shown that HMW serves an anti-inflammatory role
in monocyte-macrophages and vascular endothelial cells (34,35),
while gAd can induce macrophage apoptosis (36). However, it can be suggested that gAd
has a role in the resistance of vascular endothelial cells to
apoptosis and the protection of vascular endothelial cells, which
is consistent with the findings of the present study. For example,
a previous study demonstrated that gAd intervention of vascular
endothelial cells increased the level of endothelial type nitric
oxide (NO) synthase and thereby increased the production of NO,
exerting a protective effect on vascular endothelial cells and
serving an anti-atherosclerotic role (37). Zhao et al (38) reported that gAd significantly reduced
apoptosis in vascular endothelial cells induced by persistent or
fluctuating high glucose.
In conclusion, the present study explored the
effects of HMW and gAd, two different forms of adiponectin, on
CSE-induced apoptosis and injury in human bronchial epithelial
cells, and found that both HMW and gAd dose-dependently reversed
the upregulating effect of CSE on the levels of TNF-α, IL-8, 4-HNE
and ROS. In addition, gAd inhibited CSE-induced cell apoptosis.
These results suggest that adiponectin may form a novel therapeutic
target of COPD; however, in-depth studies with other lung
parenchyma cells and animal models are required.
Acknowledgements
The present study was supported by the Natural
Science Foundation of ShanXi Province (grant no. 2012011037-1) and
the Youth Science and Technology Research Foundation of ShanXi
Province (grant no. 2014021040-4).
References
|
1
|
Smolonska J, Wijmenga C, Postma DS and
Boezen HM: Meta-analyses on suspected chronic obstructive pulmonary
disease genes: A summary of 20 years' research. Am J Respir Crit
Care Med. 180:618–631. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhong N, Wang C, Yao W, Chen P, Kang J,
Huang S, Chen B, Wang C, Ni D, Zhou Y, et al: Prevalence of chronic
obstructive pulmonary disease in China: A large, population-based
survey. Am J Respir Crit Care Med. 176:753–760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Decramer M, Janssens W and Miravitlles M:
Chronic obstructive pulmonary disease. Lancet. 379:1341–1351. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muro S: Cigarette smoking is the most
important causal factor for developing chronic obstructive
pulmonary disease (COPD). Nihon Rinsho. 69:1735–1740. 2011.(In
Japanese). PubMed/NCBI
|
|
5
|
Løkke A, Lange P, Scharling H, Fabricius P
and Vestbo J: Developing COPD: A 25 year follow up study of the
general population. Thorax. 61:935–939. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bush A: COPD: A pediatric disease. COPD.
5:53–67. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Viegi G, Pistelli F, Sherrill DL, Maio S,
Baldacci S and Carrozzi L: Definition, epidemiology and natural
history of COPD. Eur Respir J. 30:993–1013. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou Y, Wang C, Yao W, Chen P, Kang J,
Huang S, Chen B, Wang C, Ni D, Wang X, et al: COPD in Chinese
nonsmokers. Eur Respir J. 33:509–518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller M, Cho JY, Pham A, Ramsdell J and
Broide DH: Adiponectin and functional adiponectin receptor 1 are
expressed by airway epithelial cells in chronic obstructive
pulmonary disease. J Immunol. 182:684–691. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tomoda K, Yoshikawa M, Itoh T, Tamak S,
Fukuoka A, Komeda K and Kimura H: Elevated circulating plasma
adiponectin in underweight patients with COPD. Chest. 132:135–140.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chan KH, Yeung SC, Yao TJ, Ip MS, Cheung
AH, Chan-Yeung MM and Mak JC: COPD Study Group of the Hong Kong
Thoracic Society: Elevated plasma adiponectin levels in patients
with chronic obstructive pulmonary disease. Int J Tuberc Lung Dis.
14:1193–1200. 2010.PubMed/NCBI
|
|
12
|
Miller M, Pham A, Cho JY, Rosenthal P and
Broide DH: Adiponectin-deficient mice are protected against
tobacco-induced inflammation and increased emphysema. Am J Physiol
Lung Cell Mol Physiol. 299:L834–L842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Daniele A, De Rosa A, Nigro E, Scudiero O,
Capasso M, Masullo M, de Laurentiis G, Oriani G, Sofia M and Bianco
A: Adiponectin oligomerization state and adiponectin receptors
airway expression in chronic obstructive pulmonary disease. Int J
Biochem Cell Biol. 44:563–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tilg H and Moschen AR: Adipocytokines:
Mediators linking adipose tissue, inflammation and immunity. Nat
Rev Immunol. 6:772–783. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wert SE: Does adiponectin play a role in
pulmonary emphysema? Am J Physiol Lung Cell Mol Physiol.
294:L1032–L1034. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Su Y, Han W, Giraldo C, De Li Y and Block
ER: Effect of cigarette smoke extract on nitric oxide synthase in
pulmonary artery endothelial cells. Am J Respir Cell Mol Biol.
19:819–825. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Damiá Ade D, Gimeno JC, Ferrer MJ,
Fabregas ML, Folch PA and Paya JM: A study of the effect of
proinflammatory cytokines on the epithelial cells of smokers, with
or without COPD. Arch Bronconeumol. 47:447–453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garrod R, Marshall J, Barley E, Fredericks
S and Hagan G: The relationship between inflammatory markers and
disability in chronic obstructive pulmonary disease (COPD). Prim
Care Respir J. 16:236–240. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kode A, Yang SR and Rahman I: Differential
effects of cigarette smoke on oxidative stress and proinflammatory
cytokine release in primary human airway epithelial cells and in a
variety of transformed alveolar epithelial cells. Respir Res.
7:1322006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chorostowska-Wynimko J: The role of
inflammation in the pathogenesis of chronic obstructive pulmonary
disease. Pol Merkur Lekarski. 17:203–207. 2004.(In Polish).
PubMed/NCBI
|
|
21
|
Barnes PJ, Shapiro SD and Pauwels RA:
Chronic obstructive pulmonary disease: Molecular and cellular
mechanisms. Eur Respir J. 22:672–688. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Siasos G, Tousoulis D, Kollia C, Oikonomou
E, Siasou Z, Stefanadis C and Papavassiliou AG: Adiponectin and
cardiovascular disease: Mechanisms and new therapeutic approaches.
Curr Med Chem. 19:1193–1209. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bianco A, Mazzarella G, Tu'rchiarelli V,
Nigro E, Corbi G, Scudiero O, Sofia M and Daniele A: Adiponectin:
An attractive marker for metabolic disorders in Chronic Obstructive
Pulmonary Disease (COPD). Nutrients. 5:4115–4125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sull JW, Kim HJ, Yun JE, Park EJ, Kim G
and Jee SH: Serum adiponectin is associated with smoking status in
healthy Korean men. Endocr J. 56:73–78. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kirdar S, Serter M, Ceylan E, Sener AG,
Kavak T and Karadağ F: Adiponectin as a biomarker of systemic
inflammatory response in smoker patients with stable and
exacerbation phases of chronic obstructive pulmonary disease. Scand
J Clin Lab Invest. 69:219–224. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Summer R, Little FF, Ouchi N, Takemura Y,
Aprahamian T, Dwyer D, Fitzsimmons K, Suki B, Parameswaran H, Fine
A and Walsh K: Alveolar macrophage activation and an emphysema-like
phenotype in adiponectin-deficient mice. Am J Physiol Lung Cell Mol
Physiol. 294:L1035–L1042. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakanishi K, Takeda Y, Tetsumoto S,
Iwasaki T, Tsujino K, Kuhara H, Jin Y, Nagatomo I, Kida H, Goya S,
et al: Involvement of endothelial apoptosis underlying chronic
obstructive pulmonary disease-like phenotype in adiponectin-null
mice: Implications for therapy. Am J Respir Crit Care Med.
183:1164–1175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yoon HI, Li Y, Man SF, Tashkin D, Wise RA,
Connett JE, Anthonisen NA, Churg A, Wright JL and Sin DD: The
complex relationship of serum adiponectin to COPD outcomes COPD and
adiponectin. Chest. 142:893–899. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chung KF: Cytokines as targets in chronic
obstructive pulmonary disease. Curr Drug Targets. 7:675–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Celik H, Akpinar S, Karabulut H, Oktar P,
Dursun B, Erguden HC, Gunay S and Sipit T: Evaluation of IL-8 nasal
lavage levels and the effects of nasal involvement on disease
severity in patients with stable chronic obstructive pulmonary
disease. Inflammation. 38:616–622. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Antus B and Kardos Z: Oxidative stress in
COPD: Molecular background and clinical monitoring. Curr Med Chem.
22:627–650. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
MacNee W: Pulmonary and systemic
oxidant/antioxidant imbalance in chronic obstructive pulmonary
disease. Proc Am Thorac Soc. 2:50–60. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aoshiba K, Koinuma M, Yokohori N and Nagai
A: Immunohistochemical evaluation of oxidative stress in murine
lungs after cigarette smoke exposure. Inhal Toxicol. 15:1029–1038.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kobayashi H, Ouchi N, Kihara S, Walsh K,
Kumada M, Abe Y, Funahashi T and Matsuzawa Y: Selective suppression
of endothelial cell apoptosis by the high molecular weight form of
adiponectin. Circ Res. 94:e27–e31. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pang TT and Narendran P: The distribution
of adiponectin receptors on human peripheral blood mononuclear
cells. Ann N Y Acad Sci. 1150:143–145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Akifusa S, Kamio N, Shimazaki Y, Yamaguchi
N and Yamashita Y: Regulation of globular adiponectin-induced
apoptosis by reactive oxygen/nitrogen species in RAW264
macrophages. Free Radic Biol Med. 45:1326–1339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hattori Y, Suzuki M, Hattori S and Kasai
K: Globular adiponectin upregulates nitric oxide production in
vascular endothelial cells. Diabetologia. 46:1543–1549. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao HY, Zhao M, Yi TN and Zhang J:
Globular adiponectin protects human umbilical vein endothelial
cells against apoptosis through adiponectin receptor 1/adenosine
monophosphate-activated protein kinase pathway. Chin Med J (Engl).
124:2540–2547. 2011.PubMed/NCBI
|