Introduction
Atherosclerosis is a chronic, progressive systemic
disease affecting arteries, particularly those in the brain, heart,
and lower extremities. Arteriosclerosis obliterans (ASO) of the
lower extremities is one of the most significant health-related
issues worldwide and may have devastating consequences, including
intermittent claudication, resting pain and gangrene (1–3). Notable
progress has been made in endovascular surgical techniques to treat
ASO; however, restenosis following angioplasty remains a primary
limiting factor for the long-term success of artery reconstruction
(4). Vascular smooth muscle cells
(VSMCs) comprise the main component of blood vessel walls, and
excessive proliferation and migration of VSMCs is a critical event
in the pathogenesis of atherosclerosis and vascular stenosis
(5,6). It is unclear whether the molecular
mechanism underlying the proliferation and migration of human
arterial smooth muscle cells (HASMCs) is associated with ASO.
MicroRNAs (miRNAs or miRs) have previously been
shown to serve important roles in cardiovascular systems (7–9). miRNAs
comprise a class of non-coding, short (18–22 nucleotides),
endogenous RNAs that negatively regulate their targets through
binding of the 3′-untransslated region (3′-UTR) at a
post-translational level. Aberrant miRNA expression serves a key
role in the development of ASO (10). There have been notable breakthroughs
in studies of gene expression regulation in recent decades, and so
miRNAs have been investigated for their functions in VSMC
differentiation, proliferation, migration and apoptosis (11,12). A
previous study by the present authors demonstrated that miR-21
promoted HASMC proliferation and migration, and inhibited apoptosis
in ASO of the lower extremities (11). Another study revealed that miR-663
regulated the human VSMC phenotypic switch and vascular neointimal
formation by targeting JunB/myosin light chain 9 expression, which
suggests that targeting miR-663 or its targets may provide a
promising approach for the treatment of proliferative vascular
diseases (12).
Although miR-31 is involved in various biological
processes, including tumor development (13–16),
angiogenesis (17,18) and inflammation (19), the role of miR-31 in ASO,
particularly the regulatory mechanism, remains elusive. The present
study aimed to elucidate the potential roles of miR-31 in HASMCs
within ASO processes, and to identify the underlying molecular
mechanism.
Materials and methods
Source of artery specimens
ASO artery specimens were obtained from 6 patients
with ASO (4 males and 2 females) with an average age of 65.3 years
who had undergone major amputations, and normal artery specimens
were acquired from 6 healthy organ donors (3 males and 3 females)
with an average age of 55.1 years. Specimens were obtained between
June 2013 and December 2014. ASO artery specimens were used for
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) analyses, in situ hybridization (ISH),
immunofluorescence, and 4′,6-diamidino-2-phenylindole (DAPI) and
haematoxylin and eosin (H&E) staining. Artery specimens were
harvested in a sterile state and some specimens were snap-frozen in
liquid nitrogen immediately after arteriectomy and stored at −80°C
for RNA extraction, while other specimens were fixed with
paraformaldehyde (4%) at 4°C overnight and embedded in paraffin for
further analysis. The present study was approved by the research
ethics committee of the First Affiliated Hospital, Sun Yat-Sen
University (Guangzhou, China) and prior informed consent was
provided by the patients.
RT-qPCR analysis
The separation of three layers of artery samples was
performed using a light microscope with anatomical lens
(magnification, ×5), micro scissors and micro forceps (both
Shanghai Medical Instruments (Group) Ltd., Corp. Surgical
Instruments Factory, Shanghai, China). The normal tissues or
untreated cells were used as control. Total RNA was extracted from
arterial specimens or cells using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc., USA), according to the
manufacturer's protocol. RNA was reverse transcribed using a miRNA
RT kit (Takara Biotechnology Co., Ltd., Dalian, China) at 37°C for
60 min, then 85°C for 5 sec. The cDNA was used template with the
SYBR primeScript miRNA Real-Time PCR kit (Takara Biotechnology Co.,
Ltd.) for qPCR analysis, according to the manufacturer's protocol
with the following primers: miR-31 5′-CTACGTTCTGGCATAGCTGAAA-3′, U6
5′-ACGCAAATTCGTGAAGCGTT-3′. The volume of the total PCR reaction
was 25 µl and the contained the following: ddH2O (10.5 µl), miRNA
Primer Mix (1 µl), cDNA (1 µl), 2X SYBR® Premix Ex Taq™
II (12.5 µl). PCR was performed using a Roche Lightcycler 480
Real-Time PCR System (Roche Diagnostics, Basel, Switzerland) under
the following conditions: 95°C for 10 sec, followed by 40 cycles at
95°C for 5 sec and 60°C for 20 sec. U6 was used as a reference
gene. Relative expression was analyzed using the 2−∆∆Cq
method (20). All experiments were
independent and performed at least in triplicate.
ISH, immunofluorescence, and DAPI and
H&E staining
ISH was performed using a 5′-Digoxigenin (DIG)- and
3′-DIG-labeled miRCURY LNA Detection Probe (Exiqon A/S, Vedbaek,
Denmark) to human miR-31 (5′-AGCTATGCCAGCATCTTGCCT-3′) in
4-µm-thick sections of tissues. The tissues were fixed with 4%
paraformaldehyde at 4°C overnight, and embedded in paraffin then
stored at room temperature for further analysis. Briefly, the
sections were deparaffinized and rehydrated in graded dilutions of
ethanol (100, 95, 75 and 60%, digested with 40 µg/ml proteinase K
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 10 min,
rinsed in 0.2% glycine/PBS for 5 min, fixed with 4%
paraformaldehyde at room temperature for 10 min, and acetylated for
10 min. Sections were subsequently prehybridized in a hybridization
buffer [50X Denhardts's solution, 60% formamide, 300 µg/ml transfer
RNA, 20X Saline Sodium Citrate (SSC) Buffer and 1 M dithiothreitol
(Invitrogen; Thermo Fisher Scientific, Inc.)] at 49°C for 30 min
and then hybridized with the miR-31 probe (1:500) at 49.5°C
overnight. Sections were subsequently washed with 2X SSC for 5 min
at 49.5°C and then blocked for 1 h at room temperature using 2%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
incubated with anti-DIG antibodies (1:200; cat. no. 11093274910;
Roche Diagnostics) at 37°C for 2.5 h. Sections were incubated with
tyramide signal amplification buffer (1:50; Focofish, Inc.,
Guangzhou, China) at room temperature for 20 min in the dark,
washed with PBS and covered with a cover slip. Immunofluorescence
was performed to determine SM-α-actin localization using
anti-SM-α-actin primary antibodies (1:500; cat. no. ab5694; Abcam,
Cambridge, MA, USA) at 37°C for 2 h (11) and Alexa Fluor™ 488-conjugated
secondary antibodies (1:1,000; cat. no. A12379; Thermo Fisher
Scientific, Inc.) at room temperature for 30 min.
DAPI staining was used as a nuclear counterstain in
fluorescence microscope. Following the fluorescence staining of
SM-α-actin, DAPI staining was performed using a SlowFade™ Diamond
Antifade Mountant with DAPI reagent (cat. no. 36968; Invitrogen;
Thermo Fisher Scientific, Inc.) in of tissue sections at room
temperature overnight in the dark. Images of the sections were
captured by an inverted fluorescence microscope (magnification,
×100; Axio Observer Z1, Carl Zeiss AG, Oberkochen, Germany) and
analyzed by calculating the integrated optical density (IOD) value
using Image Pro Plus software (version 6.0, Media Cybernetics,
Inc., Rockville, MD, USA).
H&E staining was used to demonstrate arterial
structures. The tissue sections were deparaffinized and rehydrated
in graded dilutions of ethanol (100, 100, 95 and 75%) for 5 min
each. The slides were the incubated with hematoxylin solution for
20 sec, then washed the slides with H2O for 1 min. The
slides were then stained with 1% eosin solution for 3 min and
dehydrated with graded dilutions of ethanol (75, 95, 100 and 100%)
for 1 min each. Alcohol was extracted with two changes of xylene
and covered with a cover slip. All of the above steps were
performed at room temperature. Images of H&E sections were
observed by a light microscope (magnification, ×100).
Western blotting
The HASMCs harvested from the femoral arterial walls
of healthy organ donors were lysed at 0°C for 15 min in a lysis
buffer (cat. no. 9806S; Cell Signaling Technology, Inc., Danvers,
MA, USA) supplemented with a protease inhibitor cocktail (cat. no.
04693132001; Roche Diagnostics) and centrifuged in 12,500 × g at
4°C for 15 min. The proteins (20 µg/lane) were separated by 10%
SDS-PAGE and transferred onto a polyvinylidene difluoride membrane
(EMD Millipore, Billerica, MA, USA). The membranes were blocked
with 5% non-fat milk in TBS at 37°C for 2 h, incubated with a
rabbit anti-mitofusin-2 (MFN2) monoclonal antibody (1:1,000; cat.
no. ab50843; Abcam) at 4°C overnight and washed with TBST at room
temperature, three times for 10 min. The signals were detected
using Luminol reagent (Pierce; Thermo Fisher Scientific, Inc.),
imaged using a GE ImageQuant Las 4000 mini phosphorimager (GE
Healthcare Life Sciences, Chalfont, UK) with ImageJ (version 1.48;
National Institutes of Health, Bethesda, MD, USA) and presented as
the density ratio vs. GAPDH (1:1,000; cat. no. 2118S; Cell
Signaling Technology, Inc.). Densitometric analyses were taken as
an average of three experiments.
Cell culture
The primary HASMCs were obtained from the femoral
arterial walls of amputation from patients who suffered serious
trauma, and the cells were prepared by the explant method,
previously established in our lab (11). HASMCs from healthy donors were used
in all subsequent experiments and the ASO cell model was
established by platelet-derived growth factor-BB (PDGF-BB)
treatment. The HASMCs were identified via staining with
anti-SM-α-actin antibodies at 37°C for 2 h (1:1,000; cat. no.
ab5694; Abcam), and passages from 4 to 9 were utilized in the
present study. The cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS at 37°C in a humidified atmosphere
containing 5% CO2, then the cells were added to a cell
cryopreservation solution (10% dimethylsulphoxide and 90% FBS) and
stored in liquid nitrogen.
HASMC transfection
Cells (2–3×103 cells/well) were seeded
into wells and transfection was performed using Lipo RNA iMax
(Invitrogen; Thermo Fisher Scientific, Inc.) 24 h later. For miR-31
function analysis, miR-31 mimics (50 nmol/l), miR-31 inhibitor (100
nmol/l) and negative control oligos (50 nmol/l; all Guangzhou
RiboBio Co., Ltd., Guangzhou, China) were transfected into the
cells, following the manufacturer's protocol.
Measurement of HASMC
proliferation
Cell Counting Kit-8 (CCK-8) and EdU assays were
performed to measure cell proliferation. HASMCs were seeded into
96-well plates (2–3×103 cells/well). Cells were
incubated at 37°C in serum-free DMEM with or without 20 ng/ml
PDGF-BB (R&D Systems, Inc., Minneapolis, MN, USA) for 24 h
following transfection. For the CCK-8 (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) assay, 10 µl CCK-8 solution
was added to each well, and the absorbance was measured at 450 nm
following 3 h of incubation at 37°C.
For the EdU (Guangzhou RiboBio Co., Ltd.) assay, the
cells were incubated at 37°C in EdU solution (50 nmol/l) for 2 h,
and fixed at room temperature in 4% formaldehyde for 30 min. The
cells were subsequently exposed to 1,000 µl Cell-Light™ EdU
Apollo®488 In Vitro Imaging kit (100T) (938 µl
dH2O, 50 µl reaction buffer, 10 µl catalyst solution, 3
µl fluorescent dye solution, 9 mg buffer additive) for 30 min,
followed by nuclear staining at room temperature with Hoechst
33,342 for 30 min (all Guangzhou RiboBio Co., Ltd.). An inverted
fluorescence microscope was used to capture and analyze the images
(magnification, ×100; Axio Observer Z1).
Measurement of HASMC migration
Transwell and wound closure assays were used to
assess cell migration. For the Transwell assay (Corning
Incorporated, Corning, NY, USA), cells were resuspended following
transfection and added to the upper chambers with 200 µl serum-free
DMEM (5×105 cells/ml), and the lower chamber was filled
with 500 µl serum-free DMEM with or without PDGF-BB (20 ng/ml).
After 16 h, the migrated cells on the lower face of the chamber
membrane were fixed with 4% formaldehyde and stained with 0.1%
crystal violet.
For the wound closure assay, the HASMCs were seeded
into 12-well plates following transfection (10,000 cells/well), and
a straight scratch wound was created using a sterilized 200 µl
disposable pipette tip in each well. The scratch wounds were
visualized every hour at 37°C using a live cell imaging system
(magnification, ×100; Axio Observer Z1). Image Pro Plus software
(version 6.0) was used to measure the widths of the scratch
wounds.
Luciferase reporter assay
The MFN2 mRNA 3′-UTR, containing putative or mutated
binding sites for miR-31, were amplified by PCR using the
aforementioned protocol and cloned into the pmiR-RB-Report vector
(Guangzhou RiboBio Co., Ltd.). The HEK 293T cells (American Type
Culture Collection, Manassas, VA, USA) were co-transfected with
miR-31 mimics (50 nmol/l) or negative control oligos (50 nmol/l)
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Luciferase values were determined 48 h later
using the Dual-Luciferase Reporter Assay System kit (Promega
Corporation, Madison, WI, USA), according to the manufacturer's
protocol.
Statistical analysis
The data are presented as the mean ± standard
deviation. Student's t-test and one-way analysis of variance, with
multiple comparisons using the Newman-Keuls test, were used for the
statistical analysis. The data analysis was performed with SPSS
17.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-31 is upregulated in ASO arteries
of the lower extremities
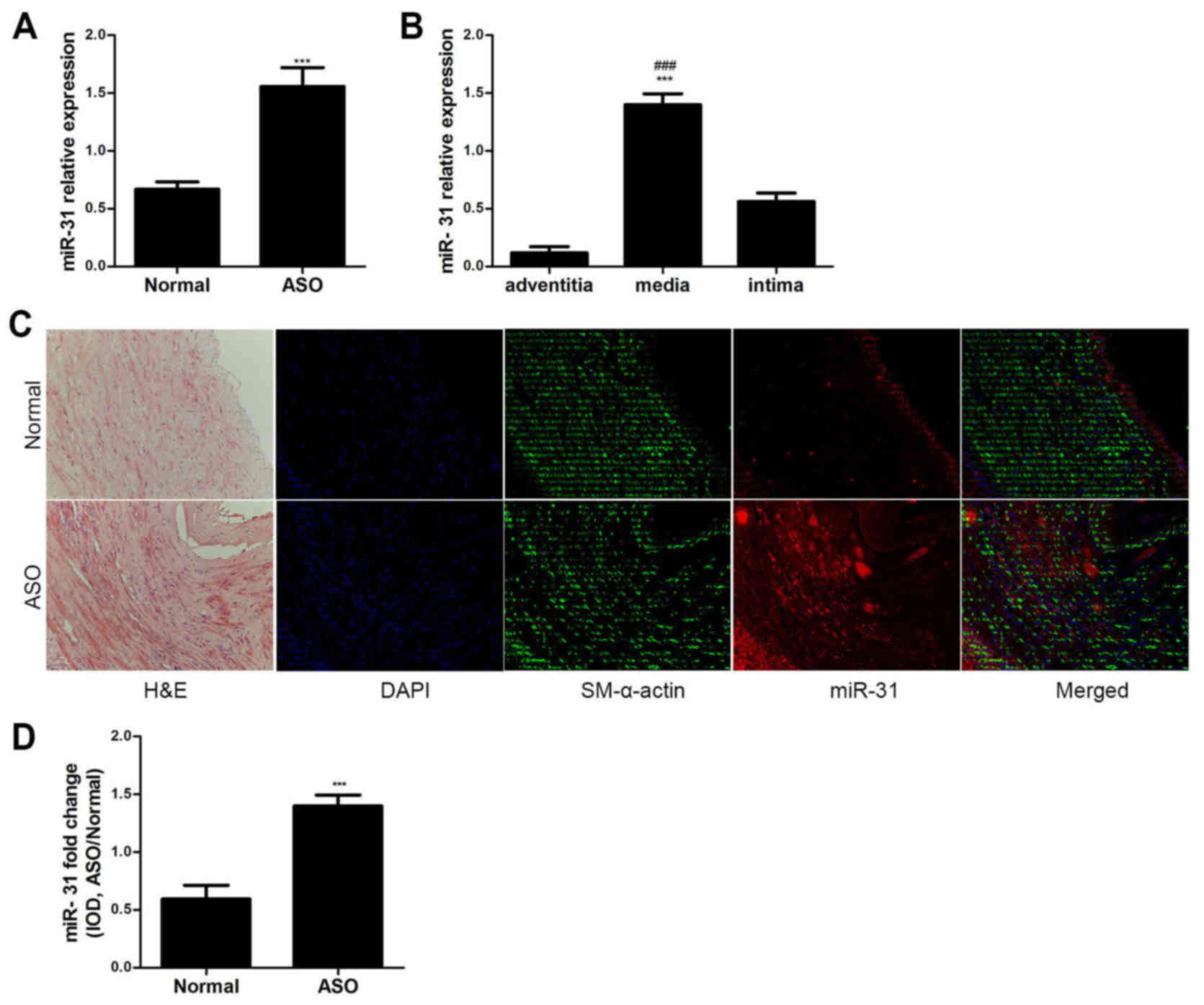
The miR-31 expression levels were analyzed in ASO
and normal arterial walls from the lower extremities via RT-qPCR.
miR-31 expression was significantly upregulated in the ASO arteries
compared with the normal arteries (P<0.001; Fig. 1A). The expression level of miR-31 in
the media was significantly higher compared with the adventitia and
intima in ASO arteries (P<0.001; Fig.
1B). Furthermore, the ISH and immunofluorescence results
demonstrated that miR-31 was primarily located in the artery media
and neointima of ASO (Fig. 1C). The
colocalization of miR-31 and SM-α-actin indicates that miR-31 is
primarily expressed in ASO HASMCs. The IOD value also indicated
that miR-31 expression was significantly increased in the ASO
arteries compared with the normal arteries (P<0.001; Fig. 1D). These results suggest that miR-31
is upregulated in ASO HASMCs.
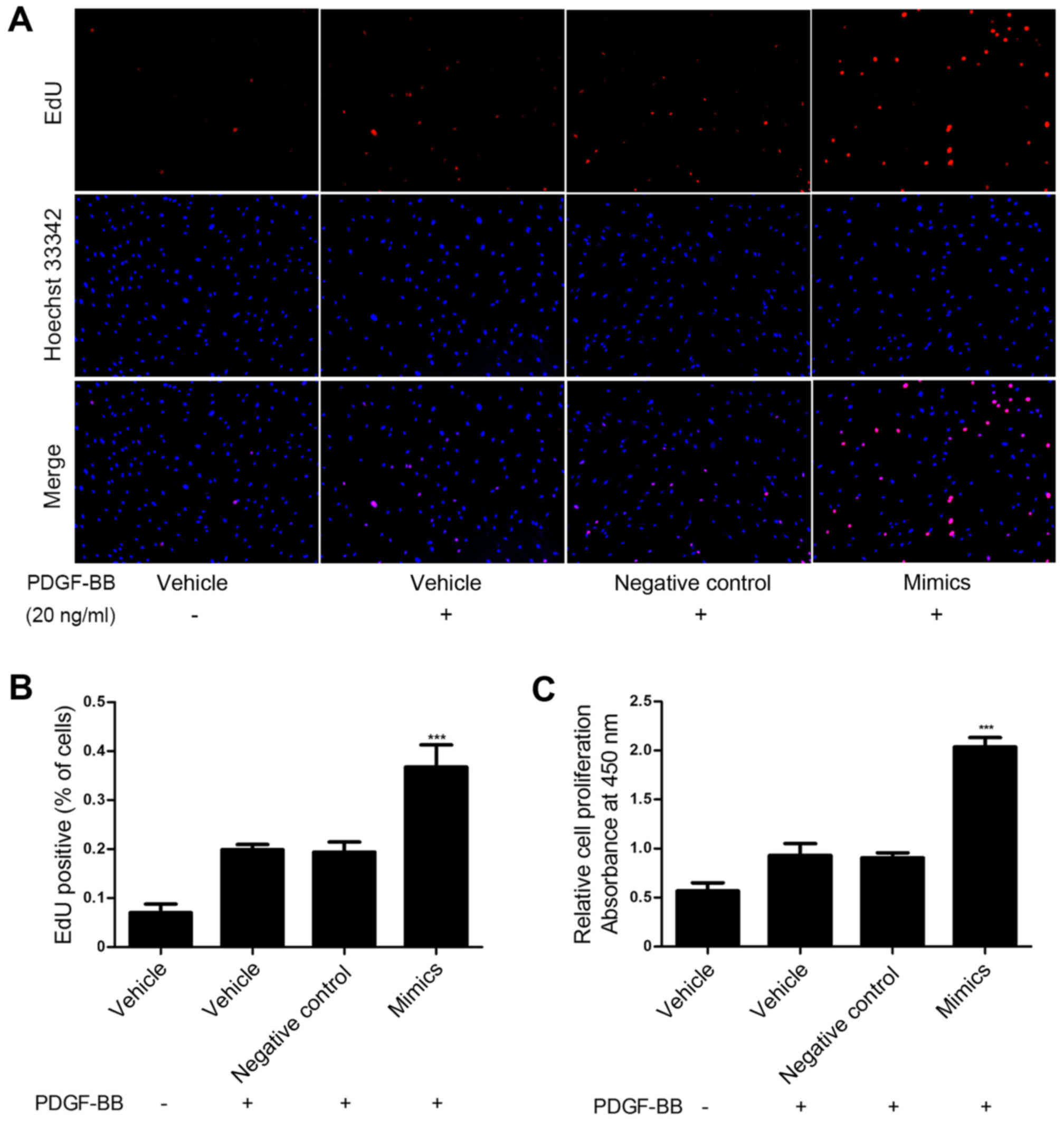
Mir-31 promotes proliferation in
PDGF-BB-induced HASMCs
To investigate the role of miR-31 in HASMC
proliferation induced by PDGF-BB, miR-31 mimics and negative
control oligos were transfected into HASMCs and proliferation was
assessed via an EdU and a CCK-8 assay. These assays demonstrated
that upregulation of miR-31 significantly promoted HASMC
proliferation compared with the control group (P<0.001; Fig. 2). These results suggest that miR-31
overexpression promotes HASMC proliferation in vitro.
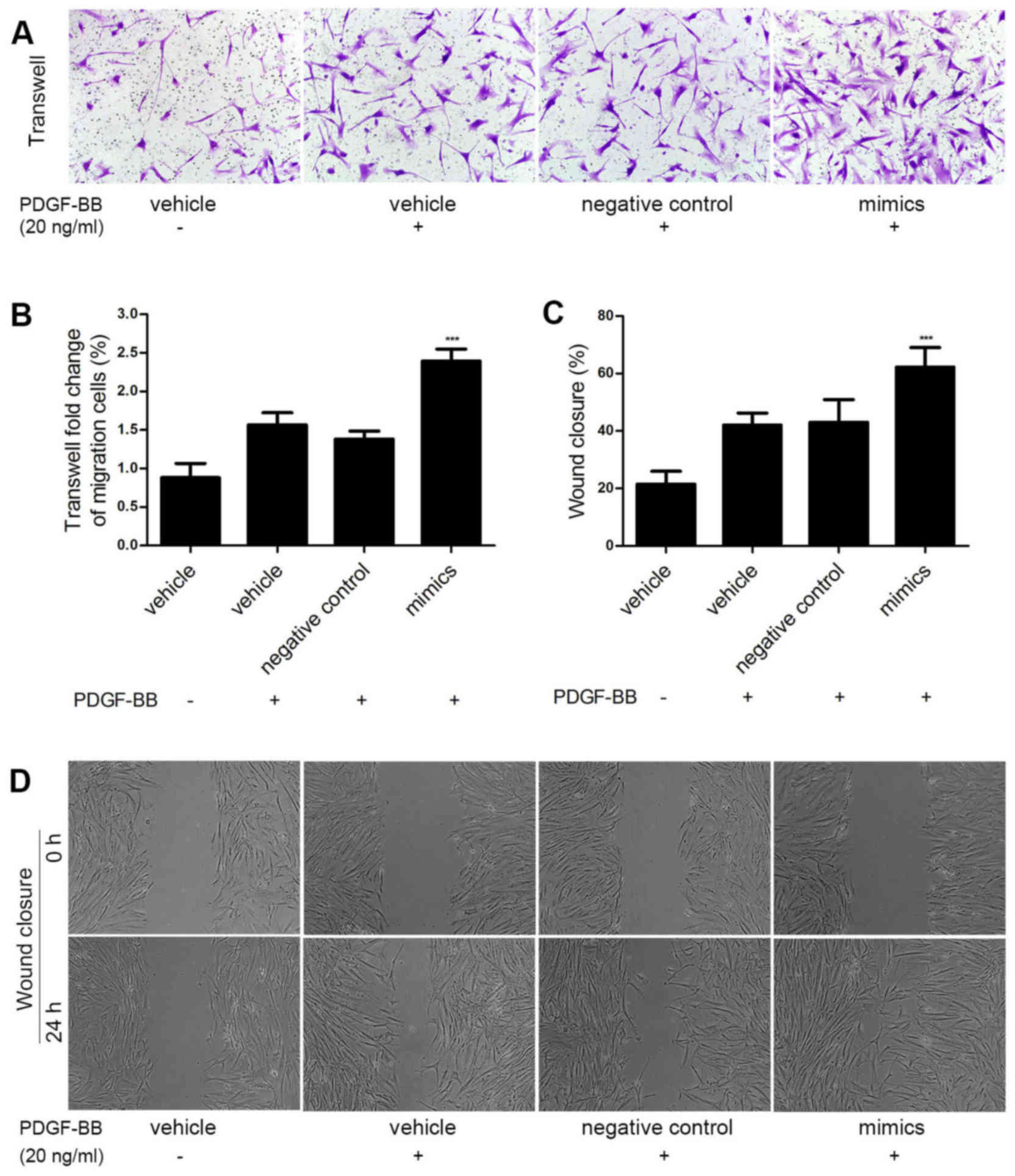
Mir-31 promotes migration in HASMCs
following PDGF-BB treatment
To explore whether miR-31 promoted HASMC migration
induced by PDGF-BB, miR-31 mimics and negative control oligos were
transfected into HASMCs to overexpress miR-31. Cell migration was
measured via Transwell assay. As shown in Fig. 3A and B, the miR-31 mimics
significantly enhanced PDGF BB-induced HASMC migration compared
with the control group (P<0.001). Similar results were obtained
in the wound closure assay (P<0.001; Fig. 3C and D). These results demonstrate
that miR-31 overexpression promotes HASMC migration in
vitro.
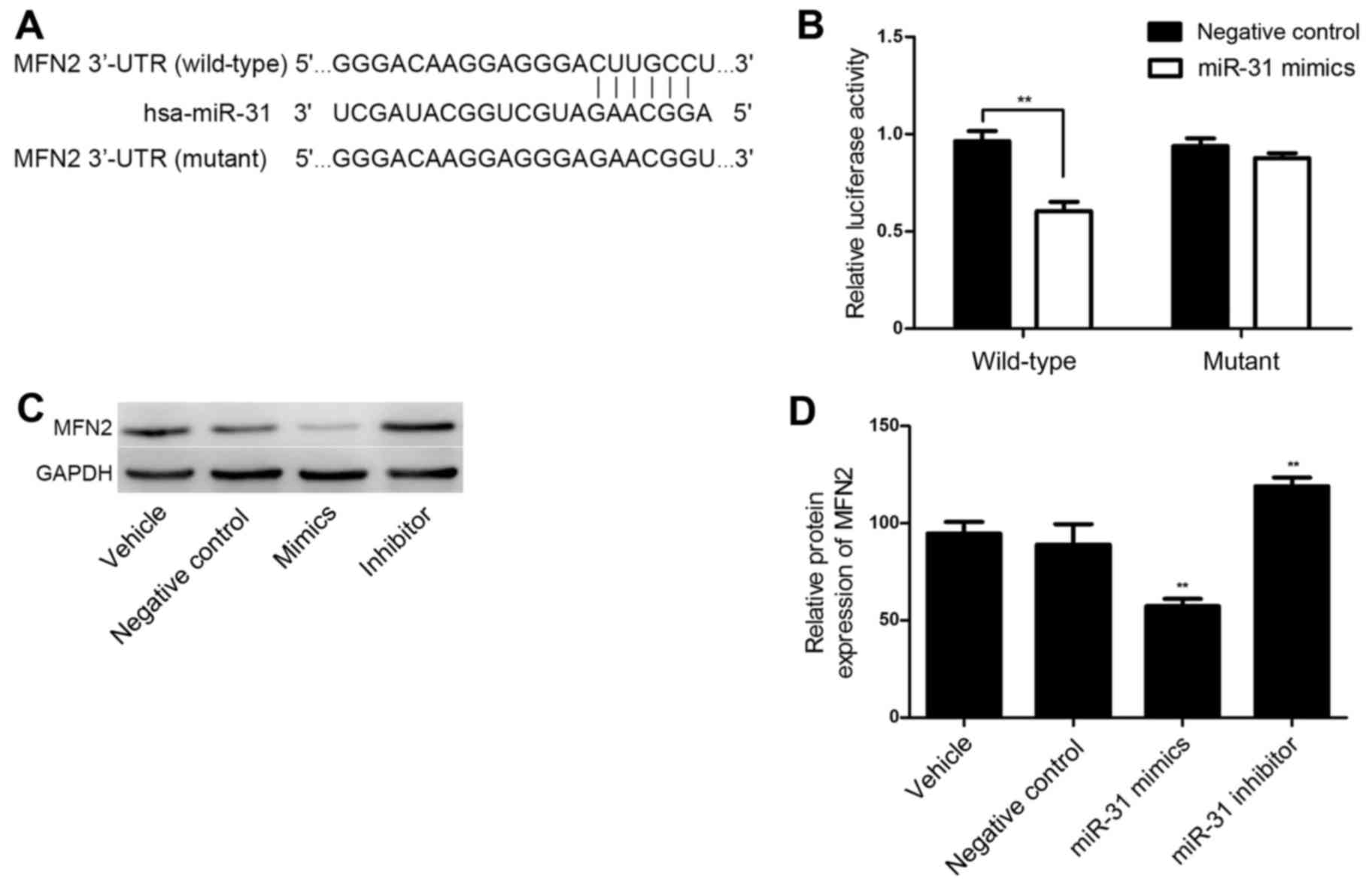
MiR-31 targets the 3′-UTR of MFN2 and
downregulates its expression
TargetScan online software was utilized for the
bioinformatics prediction and MFN2 was identified as a potential
target of miR-31. The putative seed sequences for miR-31 at the
3′-UTR of MFN2 were conserved (Fig.
4A). A dual-luciferase reporter assay was performed to verify
whether MFN2 was a direct target gene of miR-31. As shown in
Fig. 4B, in the wild-type MFN2 3′UTR
group, the miR-31 mimics significantly reduced Renilla/firefly
luciferase activity compared with the results in the negative
control group (P<0.01). However, in the mutant type MFN2 $3′UTR
group, there was no significant difference in Renilla/firefly
luciferase activity between the miR-31 mimics and the negative
control group. These results indicate that miR-31 directly binds to
the 3′-UTR of MFN2.
MFN2 expression is negatively
regulated by miR-31 at a transcriptional level in HASMCs
To further investigate whether MFN2 is a functional
target gene of miR-31 in HASMCs, HASMCs were transfected with
miR-31 negative control oligos, mimics and an inhibitor, and the
MFN2 expression levels were determined via western blotting. As
shown in Fig. 4C and D, the protein
level of MFN2 was significantly downregulated following miR-31
overexpression (P<0.01). However, MFN2 was significantly
upregulated in the HASMCs transfected with a miR-31 inhibitor
(P<0.01; Fig. 4C and D). These
observations demonstrate that miR-31 negatively regulates the
expression of MFN2 at a post-transcriptional level in HASMCs.
Discussion
It has been well established that miRNAs function as
key mediators in multiple physiological and pathological processes,
predominantly via modulating the expression of target genes at a
post-transcriptional level (21). In
the present study, miR-31 expression was markedly upregulated in
human ASO arterial walls compared with normal arterial walls.
Furthermore, an ASO cell model was established to investigate
whether miR-31 overexpression had an effect on HASMCs. To the best
of our knowledge, the present study demonstrated for the first time
that miR-31 promotes proliferation and migration in PDGF-BB-induced
HASMCs, most likely by regulating the expression of MFN2.
ASO of the lower extremities is one of the most
painful vascular diseases, with high morbidity and mortality; ASO
seriously affects survival and quality of life (22). It is well established that aberrant
proliferation and migration of VSMCs serves an important role in
arteriosclerosis and vascular stenosis (23). Despite great progress in the
understanding of VSMC biology (24),
the molecular mechanisms underlying the contribution of HASMCs to
the pathogenesis of ASO has yet to be elucidated. miRNAs have
previously been reported to serve important roles in cardiovascular
systems (7–9). miR-31 functions as an oncogene in the
majority of cancers, including colorectal cancer (13), lung cancer (14), and esophageal cancer (15); however, it has been demonstrated to
act as a tumor suppressor in glioblastoma and liver cancer
(16,25). In the present study, miR-31 was
significantly upregulated in ASO arterial walls compared with
normal arterial walls as determined by RT-qPCR and ISH. miR-31 was
located primarily in the media and neointima, which indicates that
HASMCs may be the main effector cells of miR-31 in the ASO
pathological process. The characteristics of miR-31 distribution in
he present study are consistent with the observations of a previous
study, which was performed in rat carotid arteries with neointimal
lesions (26).
The biological functions of miR-31 in the HASMC
physiological and pathological processes require further study. To
investigate the detailed roles of miR-31 in HASMCs in ASO, CCK-8
and EdU assays were performed to investigate cell proliferation,
wound closure and Transwell assays were used to detect cell
migration. In the present study, miR-31 was demonstrated to promote
proliferation and migration in PDGF-BB-induced HASMCs, which was
consistent with the observation of a previous study by Liu et
al (27), who reported that VSMC
proliferation in rats was significantly inhibited by miR-31
knockdown. Cell proliferation was increased by the overexpression
of Rattus norvegicus-miR-31, which demonstrates that miR-31
has a pro-proliferative effect on VSMCs in rats. Wang et al
(28) previously reported that
miR-31 was involved in the regulation of a human thoracic ASMC
phenotype switch by targeting the cellular repressor of
E1A-stimulated genes.
MFN2, also known as a hyperplasia suppressor gene,
has a crucial role in regulating cell proliferation, migration,
apoptosis and differentiation, which are involved in the
pathophysiological processes of severe cardiovascular diseases,
including atherosclerosis, restenosis after angioplasty,
hypertension and myocardial infarction (29–31). A
previous study demonstrated that MFN2 acted as an
anti-proliferation gene, by preventing VSMC proliferation in
cultured cells and a rat carotid artery balloon-injury model
(31). Another study revealed that
overexpression of MFN2 reduced VSMC proliferation and increased
apoptosis in human and experimental pulmonary arterial hypertension
model (32). In the present study, a
dual-luciferase reporter assay was applied to confirm that MFN2 was
a direct target gene of miR-31. In addition, via gain-of-function
and loss-of-function approaches, transfection with miR-31 mimics
reduced the protein expression of MFN2 in HASMCs; however,
transfection with an miR-31 inhibitor resulted in increased MFN2
expression. These previous observations and the results of the
present study suggest that miR-31 has a role in promoting
proliferation and migration in HASMCs, which is, at least in part,
dependent on directly targeting MFN2.
In conclusion, miR-31 was significantly increased in
arterial walls of patients with ASO. Furthermore, miR-31 promoted
the proliferation and migration of HASMCs, at least partially by
directly targeting the expression of MFN2. The results of the
present study may provide a novel insight into the mechanisms and
significant role of miR-31/MFN2 in the pathological processes of
ASO and offers a potential therapeutic target for the treatment of
ASO.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (nos. 81270378, 81070258 and
81370368), Guangdong Province Industry-Academia-Research Program
(no. 2011B090400117), Guangdong Department of Science &
Medicine Center grant (no. 2011A080300002) Guangdong Province
Medical Science and Technology Research Project (no. A2016424) and
Guangdong Science and Technology Plan Projects (no.
2017A020215124).
References
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al:
Heart disease and stroke statistics-2013 update: A report from the
American Heart Association. Circulation. 127:e6–e245. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohnishi H, Sawayama Y, Furusyo N, Maeda S,
Tokunaga S and Hayashi J: Risk factors for and the prevalence of
peripheral arterial disease and its relationship to carotid
atherosclerosis: The kyushu and okinawa population study (KOPS). J
Atheroscler Thromb. 17:751–758. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Diehm C, Allenberg JR, Pittrow D, Mahn M,
Tepohl G, Haberl RL, Darius H, Burghaus I and Trampisch HJ; German
Epidemiological Trial on Ankle Brachial Index Study Group, :
Mortality and vascular morbidity in older adults with asymptomatic
versus symptomatic peripheral artery disease. Circulation.
120:2053–2061. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Setacci C, Castelli P, Chiesa R, Grego F,
Simoni GA, Stella A, Galzerano G, Sirignano P, De Donato G and
Setacci F: Restenosis: A challenge for vascular surgeon. J
Cardiovasc Surg (Torino). 53:735–746. 2012.PubMed/NCBI
|
|
5
|
Hao H, Gabbiani G and Bochaton-Piallat ML:
Arterial smooth muscle cell heterogeneity: Implications for
atherosclerosis and restenosis development. Arterioscler Thromb
Vasc Biol. 23:1510–1520. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim J, Zhang L, Peppel K, Wu JH, Zidar DA,
Brian L, DeWire SM, Exum ST, Lefkowitz RJ and Freedman NJ:
Beta-arrestins regulate atherosclerosis and neointimal hyperplasia
by controlling smooth muscle cell proliferation and migration. Circ
Res. 103:70–79. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao Y, Samal E and Srivastava D: Serum
response factor regulates a muscle-specific microRNA that targets
Hand2 during cardiogenesis. Nature. 436:214–220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Condorelli G, Latronico MV and Cavarretta
E: microRNAs in cardiovascular diseases: Current knowledge and the
road ahead. J Am Coll Cardiol. 63:2177–2187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reilly S, Liu X, Carnicer R, Rajakumar T,
Sayeed R, Krasopoulos G, Verheule S, Fulga T, Schotten U and
Casadei B: Evaluation of the role of miR-31-dependent reduction in
dystrophin and nNOS on atrial-fibrillation-induced electrical
remodelling in man. Lancet. 385 Suppl 1:S822015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fish JE and Cybulsky MI: ApoE attenuates
atherosclerosis via miR-146a. Circ Res. 117:3–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang M, Li Wen, Chang GQ, Ye CS, Ou JS, Li
XX, Liu Y, Cheang TY, Huang XL and Wang SM: MicroRNA-21 regulates
vascular smooth muscle cell function via targeting tropomyosin 1 in
arteriosclerosis obliterans of lower extremities. Arterioscler
Thromb Vasc Biol. 31:2044–2053. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li P, Zhu N, Yi B, Wang N, Chen M, You X,
Zhao X, Solomides CC, Qin Y and Sun J: MicroRNA-663 regulates human
vascular smooth muscle cell phenotypic switch and vascular
neointimal formation. Circ Res. 113:1117–1127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu CW, Ng SC, Dong Y, Tian L, Ng SS, Leung
WW, Law WT, Yau TO, Chan FK, Sung JJ and Yu J: Identification of
microRNA-135b in stool as a potential noninvasive biomarker for
colorectal cancer and adenoma. Clin Cancer Res. 20:2994–3002. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu X, Sempere LF, Ouyang H, Memoli VA,
Andrew AS, Luo Y, Demidenko E, Korc M, Shi W and Preis M:
MicroRNA-31 functions as an oncogenic microRNA in mouse and human
lung cancer cells by repressing specific tumor suppressors. J Clin
Invest. 120:1298–1309. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang T, Wang Q, Zhao D, Cui Y, Cao B, Guo
L and Lu SH: The oncogenetic role of microRNA-31 as a potential
biomarker in oesophageal squamous cell carcinoma. Clin Sci (Lond).
121:437–447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim HS, Lee KS, Bae HJ, Eun JW, Shen Q,
Park SJ, Shin WC, Yang HD, Park M, Park WS, et al: MicroRNA-31
functions as a tumor suppressor by regulating cell cycle and
epithelial-mesenchymal transition regulatory proteins in liver
cancer. Oncotarget. 6:8089–8102. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu YH, Hu TF, Chen YC, Tsai YN, Tsai YH,
Cheng CC and Wang HW: The manipulation of miRNA-gene regulatory
networks by KSHV induces endothelial cell motility. Blood.
118:2896–2905. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shen J, Yang X, Xie B, Chen Y, Swaim M,
Hackett SF and Campochiaro PA: MicroRNAs regulate ocular
neovascularization. Mol Ther. 16:1208–1216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yan S, Xu Z, Lou F, Zhang L, Ke F, Bai J,
Liu Z, Liu J, Wang H and Zhu H: NF-κB-induced microRNA-31 promotes
epidermal hyperplasia by repressing protein phosphatase 6 in
psoriasis. Nat Commun. 6:76522015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Diehm C, Allenberg JR, Pittrow D, Mahn M,
Tepohl G, Haberl RL, Darius H, Burghaus I and Trampisch HJ; German
Epidemiological Trial on Ankle Brachial Index Study Group, :
Mortality and vascular morbidity in older adults with asymptomatic
versus symptomatic peripheral artery disease. Circulation.
120:2053–2061. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim J, Zhang L, Peppel K, Wu JH, Zidar DA,
Brian L, DeWire SM, Exum ST, Lefkowitz RJ and Freedman NJ:
Beta-arrestins regulate atherosclerosis and neointimal hyperplasia
by controlling smooth muscle cell proliferation and migration. Circ
Res. 103:70–79. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hao H, Gabbiani G and Bochaton-Piallat ML:
Arterial smooth muscle cell heterogeneity: Implications for
atherosclerosis and restenosis development. Arterioscler Thromb
Vasc Biol. 23:1510–1520. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rajbhandari R, McFarland BC, Patel A,
Gerigk M, Gray GK, Fehling SC, Bredel M, Berbari NF, Kim H, Marks
MP, et al: Loss of tumor suppressive microRNA-31 enhances
TRADD/NF-κB signaling in glioblastoma. Oncotarget. 6:17805–17816.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Steg PG, Bhatt DL, Wilson PW, D'Agostino R
Sr, Ohman EM, Röther J, Liau CS, Hirsch AT, Mas JL, Ikeda Y, et al
REACH Registry Investigators, : One-year cardiovascular event rates
in outpatients with atherothrombosis. JAMA. 297:1197–1206. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu X, Cheng Y, Chen X, Yang J, Xu L and
Zhang C: MicroRNA-31 regulated by the extracellular regulated
kinase is involved in vascular smooth muscle cell growth via large
tumor suppressor homolog 2. J Biol Chem. 286:42371–42380. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Yan CH, Li Y, Xu K, Tian XX, Peng
CF, Tao J, Sun MY and Han YL: MicroRNA-31 controls phenotypic
modulation of human vascular smooth muscle cells by regulating its
target gene cellular repressor of E1A-stimulated genes. Exp Cell
Res. 319:1165–1175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D,
Li P, Qiu X, Wen S, Xiao RP and Tang J: Dysregulation of HSG
triggers vascular proliferative disorders. Nat Cell Biol.
6:872–883. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma L, Liu Y, Geng C, Qi X and Jiang J:
Estrogen receptor β inhibits estradiol-induced proliferation and
migration of MCF-7 cells through regulation of mitofusin 2. Int J
Oncol. 42:1993–2000. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guo X, Chen KH, Guo Y, Liao H, Tang J and
Xiao RP: Mitofusin 2 triggers vascular smooth muscle cell apoptosis
via mitochondrial death pathway. Circ Res. 101:1113–1122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ryan JJ, Marsboom G, Fang YH, Toth PT,
Morrow E, Luo N, Piao L, Hong Z, Ericson K, Zhang HJ, et al:
PGC1α-mediated mitofusin-2 deficiency in female rats and humans
with pulmonary arterial hypertension. Am J Respir Crit Care Med.
187:865–878. 2013. View Article : Google Scholar : PubMed/NCBI
|