Introduction
Nasopharyngeal carcinoma (NPC) is a squamous cell
carcinoma that develops in the epithelial lining of the
nasopharynx. The disease occurs among individuals in Southern China
and North Africa, as well as Inuits from Alaska and Bidayuhs from
East Malaysia (1,2). Patients with locoregionally advanced
stages are treated by concurrent chemotherapy and radiotherapy with
or without adjuvant chemotherapy. Unfortunately, conventional
cancer therapies are often associated with toxicities. Alternative
approaches that selectively kill tumor cells whilst having weaker
or negligible effects on healthy tissues are required (3).
Cinnamomum burmannii Blume (Lauraceae)
is a tree-like shrub that is native to Southeast Asia and Indonesia
(4). The aromatic bark is used for
medicine and making spices for the flavor industry (5). Trans-cinnamaldehyde (TCA) has
been identified as one of the bioactive compounds in C.
burmannii(6). Numerous reports
on the anti-cancer activities of Cinnamomum have been
published (7–9). Additionally, studies have
demonstrated that TCA inhibits cell proliferation and induces cell
apoptosis (10–13). Despite the extensive research on
the effect of TCA on a variety of cell lines, studies concerning
its anti-proliferative effect on NPC cells are limited. Therefore,
the present study addresses the anti-neoplastic potential of the
methanol extract of the C. burmannii stem and its
constituent, TCA, on HK1 and C666-1 NPC cell lines. We also
explored the potential of combining TCA with conventional cytotoxic
chemotherapy for more successful elimination of NPC cells.
Materials and methods
Chemicals and reagents
Curcumin and TCA were purchased from Sigma-Aldrich
(St. Louis, MO, USA). All other solvents, reagents and chemicals
used were of analytical or high-performance liquid chromatography
(HPLC) grade from Sigma-Aldrich or Merck (Darmstadt, Germany).
Plant material and extraction
The stem bark of C. burmannii was collected
from Agroforestry Garden, Sumatra, Indonesia and was authenticated
by Mr Onrizal S. Hut, Forestry Department, Universitas Sumatera
Utara, Indonesia. A voucher specimen (BO.0032998) was stored for
future reference. The stem bark was air-dried and ground into a
powder. The powdered plant material (250 g) was extracted with
methanol (2.5 liters) using Soxhlet apparatus. The extract was
concentrated to dryness under reduced pressure and temperature
(40°C). The yield (w/w) of the extract from the dry stem bark was
9.8%. The extract was stored in a desiccator at room temperature
until analysis.
Gas chromatography-mass spectrometry
(GC-MS) of the extract
GC-MS was performed using the Agilent 7000 GC-MS/MS
Triple Quadrupole mass spectrometer (Agilent Technologies, Inc.,
Santa Clara, CA, USA) operated under the full scan mode. Stock
solution (1 mg/ml) of the authentic TCA standard was prepared in
methanol. Calibration standards were prepared from the stock
solution at a concentration range of 1 to 100 μg/ml. One
milligram portions of methanol extract were weighed and dissolved
in 1 ml methanol. The solution was diluted accordingly to fit into
the calibration range prior to analysis. The GC was operated at an
initial temperature of 73°C, then increased by 50°C/min until a
final temperature of 280°C was reached. It was held at this
temperature for 1 min. The column used was a DB-5 column (length,
30.0 m; diameter, 250 μm) with helium as the carrier gas.
The injection volume was 2 μl, operated in split mode with a
ratio of 1:10.
Cell culture
The NPC cell line, HK1 (14), was maintained in the exponential
growth phase in RPMI-1640 medium (Gibco Life Technologies,
Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal
calf serum (FCS; Gibco Life Technologies), 10 U/ml penicillin
(Gibco Life Technologies) and 10 μg/ml streptomycin (Gibco
Life Technologies) at 37°C in a 5% CO2 humidified
atmosphere. C666-1 (15), also an
NPC cell line, was maintained similarly; however, the FCS
concentration was increased to 15%. The immortalized human skin
keratinocyte, HaCaT, represented normal cells. HaCaT (16) was purchased from CLS Cell Lines
Service (Eppelheim, Germany) and was maintained in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco Life Technologies)
supplemented with 10% FCS. Tests for detection of mycoplasma using
an e-Myco™ Mycoplasma polymerase chain reaction (PCR) Detection kit
(Intron Biotechnology, Seoul, Korea) were conducted routinely and
contamination-free cells were used throughout this study.
Cell viability assay
The effect of the methanol extract and TCA was
determined using the CellTiter 96® AQueous One Solution
Cell Proliferation (MTS) assay (Promega Corporation, Madison, WI,
USA). Cells (5,000–15,000 cells/well) were seeded into 96-well
microtiter plates in 100 μl culture medium and then
incubated. Old medium from the exponentially growing cells was
replaced with medium containing the desired concentrations of test
substances for 24 h at 37°C in a 5% CO2 atmosphere. The
concentrations used for the study were 100–450 μg/ml
methanol extract or 2–16 μg/ml TCA. The methanol extract was
dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) with a final
concentration of 0.5%. Vehicle control cultures received DMSO
alone. Cells treated with cisplatin served as the positive control.
Absorbance at 490 nm was read using the EnVision multilabel plate
reader (Perkin-Elmer, Waltham, MA, USA) and the non-specific
absorbance measured at 630 nm was subtracted. Wells containing the
appropriate culture medium without cells served as the blank
measurement. Cell viability was estimated as the percentage of
population growth compared with control cells (arbitrarily assigned
as 100% viable). The IC50 values, defined as the
concentration that inhibited the cell growth by 50% relative to
that of control cells, was graphically obtained from the
dose-response curves. All experiments were performed in triplicate.
The results were expressed as the mean ± standard deviation
(SD).
xCELLigence cell proliferation assay
Cells (5,000–15,000 cells/well) were seeded into
E-Plate 16 devices (ACEA Biosciences Inc., San Diego, CA, USA)
containing 100 μl culture medium. Upon reaching the
logarithmic phase, the culture medium was aspirated and replaced
with culture medium containing either 100 μg/ml methanol
extract or 4–8 μg/ml TCA. The methanol extract was dissolved
in DMSO, to a final concentration not exceeding 0.5%. Vehicle
control cultures received DMSO alone. Cells treated with cisplatin
served as the positive control. Dynamic monitoring of the growth
inhibition pattern was determined by the impedance-based
xCELLigence system (Roche Applied Science, Mannheim, Germany) for
24 h at 37°C in a humidified atmosphere of 5% CO2. The
cell index, automatically calculated from the change in electrical
impedance as the living cells interacted with electrodes in the
E-Plate wells, correlated with the number of cells, viability
and/or cytotoxicity over time.
Quantitative real time PCR
Cells were grown to near confluence, then were
either treated with 4–8 μg/ml TCA or left untreated for 24
h. Total RNA was extracted with TRIzol reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA). DNase I (Promega Corporation)
treatment was performed according to the manufacturer’s
instructions. cDNA was synthesized from 2 μg total RNA using
the High Capacity cDNA Reverse Transcription kit (Applied
Biosystems, Foster City, CA, USA), then amplified using Power
SYBR-Green Master Mix (Applied Biosystems) in the Applied
Biosystems 7500 Fast Real Time PCR system and analyzed with 7500
software v.2.0.4. The primers for Ki67 were as follows: Ki67
forward: 5′-ATGGAAGCGTCACGGGAA-3′ and reverse:
5′-TCTTCCGCAGGTTCAATTCTT-3′. β-actin (ACTB) and peptidylprolyl
isomerase A (PPIA) were amplified as the internal controls using
the following primers: ACTB forward: 5′-TCATCACCATTGGCAATGAG-3′ and
reverse: 5′-CACTGTGTTGGCGTACAGGT-3′; PPIA forward:
5′-GGCCAGGCTCGTGCCGTTTT-3′ and reverse:
5′-TGCTGTCTTTGGGACCTTGTCTGC-3′.
Western blotting
Cells were grown to near confluence, then the old
media was aspirated and replaced with media with or without 4–8
μg/ml TCA. The cells were then re-incubated. Cells were
lysed in 1X RIPA lysis buffer and then boiled for 10 min. The
quantity of protein in the cell lysate was determined by protein
assay (Bio-Rad, Hercules, CA, USA). The protein (10 μg) was
resolved in NuPAGE® Novex Bis-Tris Mini Gels (Invitrogen
Life Technologies) and electro-transferred to polyvinylidene
fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The
membranes were blocked with 5% skimmed milk and incubated overnight
with primary antibodies diluted in 5% skimmed milk. Primary
antibodies (Cell Signalling Technology, Inc., Danvers, MA, USA)
used were monoclonal mouse anti-glyceraldehyde 3-phosphate
dehydrogenase (GAPDH), monoclonal mouse anti-proliferating cell
nuclear antigen (PCNA) and rabbit anti-phospho-histone H2AX
(Ser139). The secondary antibody reaction was performed with
anti-mouse or anti-rabbit horse-radish peroxidase-conjugated IgG
(Promega Corporation). Western Lightning® Plus enhanced
chemiluminescence (ECL) substrate (Perkin-Elmer) and
autoradiography were used for visualization of protein expression.
Densitometric analysis of X-ray films was performed on an Alpha
Imager System (ProteinSimple, Santa Clara, CA, USA) using Alpha
View software. The band intensities were normalized with respect to
the GAPDH product.
Morphological observation
Cells were seeded at a density of 5,000–15,000
cells/well into 96-well microtiter plates and were left to grow
exponentially at 37°C in a 5% CO2 atmosphere. Then, the
cells were treated with 4–8 μg/ml TCA or 20–30 μg/ml
cisplatin. After 24 h incubation, morphological changes were
observed under a Leica DM IRB (Leica Microsystems, Wetzlar,
Germany) inverted microscope.
Caspase-3/7 activity assay
Cells were seeded and treated as described above.
After the appropriate exposure times, caspase-3/7 activation was
measured using the ApoTox-Glo™ Triplex assay (Promega Corporation)
kit according to the manufacturer’s instructions. The luminescence
signal was read using the EnVision multilabel plate reader
(Perkin-Elmer).
Apoptosis analysis assay
HK1 cells were seeded in 10-cm culture dishes to
reach confluence overnight. Following this, the cells were treated
with 8 μg/ml TCA, 30 μg/ml cisplatin or left
untreated (as the control). All culture dishes were re-incubated
for a further 24 h. Apoptosis was determined with a FACSCalibur
flow cytometer (BD Biosciences, San Jose, CA, USA), using the
fluorescein isothiocyanate (FITC) Annexin V apoptosis detection kit
(BD Pharmingen, San Diego, CA, USA), according to the
manufacturer’s instructions.
Combined drug analysis
For combined drug analysis, a non-fixed ratio
combination of TCA and cisplatin was evaluated. Drug dilutions and
combinations were made in RPMI-1640 medium immediately before use.
Following drug addition, the 96-well microtiter plates were
incubated for 24 h and the MTS assay was performed to determine
cell viability. Drug interaction was determined by the
combination-index (CI) and isobologram methods (17). Dose-response curves, dose-effect
analysis and CI for the combination treatment group were generated
using CalcuSyn 2 software (Biosoft, Cambridge, UK). CI >1
implies antagonism, CI =1 is additive and CI <1 implies
synergism.
Nitric oxide radical inhibition
assay
The scavenging effect of the extracts on nitric
oxide radicals was measured according to the method of Rao
(18). Curcumin was used as a
positive reference compound. Measurements were performed six times.
The percentage inhibition of nitric oxide radical generation was
calculated. The inhibitory concentration at which 50% of the nitric
oxide radical was scavenged by test samples (IC50) was
determined from the graph of inhibition percentage against sample
concentration.
Statistical analysis
Statistical analysis was performed using SPSS 12.0
statistical software for Windows (SPSS Inc., Chicago, IL, USA).
Differences between mean values were evaluated with the Student’s
t-test or one-way analysis of variance (ANOVA) and Tukey’s or
Dunnett’s T3 post hoc analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
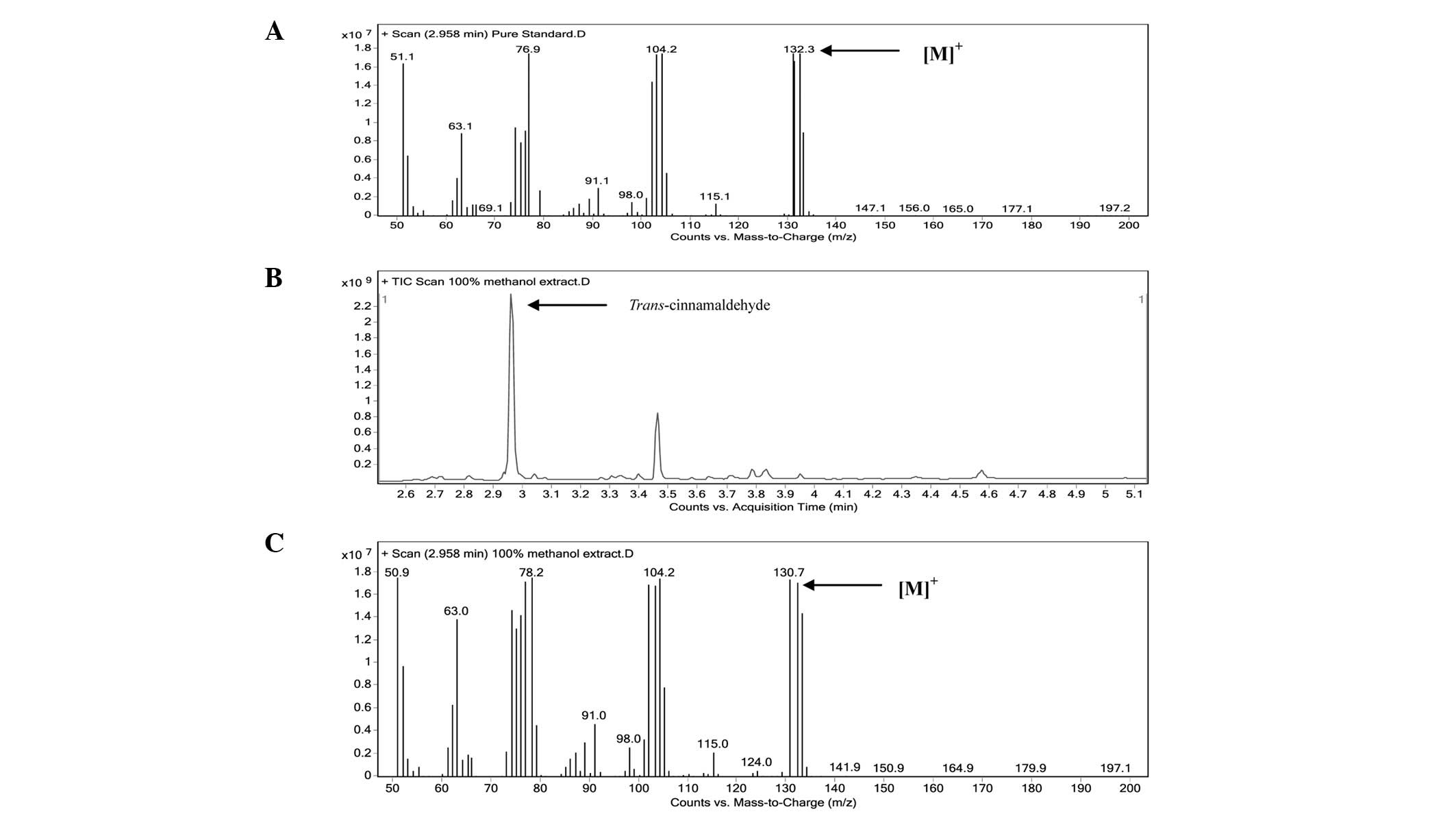
GC-MS analysis
The marker (TCA) was identified in the extracts by
comparing the retention time and mass spectrum with that of the
authentic standard. The mass spectrum of the authentic TCA standard
is shown in Fig. 1A. The total ion
chromatogram (TIC) of the methanol extract and mass spectrum of the
peak-matching TCA are shown in Fig. 1B
and C, respectively. TCA was the major peak in the extract. The
TCA component of the extracts presented the [M]+ ion at
132 m/z, which corresponded to 132.3 m/z observed in the mass
spectrum of the authentic TCA standard, confirming a similar
identity. The content of TCA in the methanol extract was
standardized to 13.61% w/w by an external standardization
method.
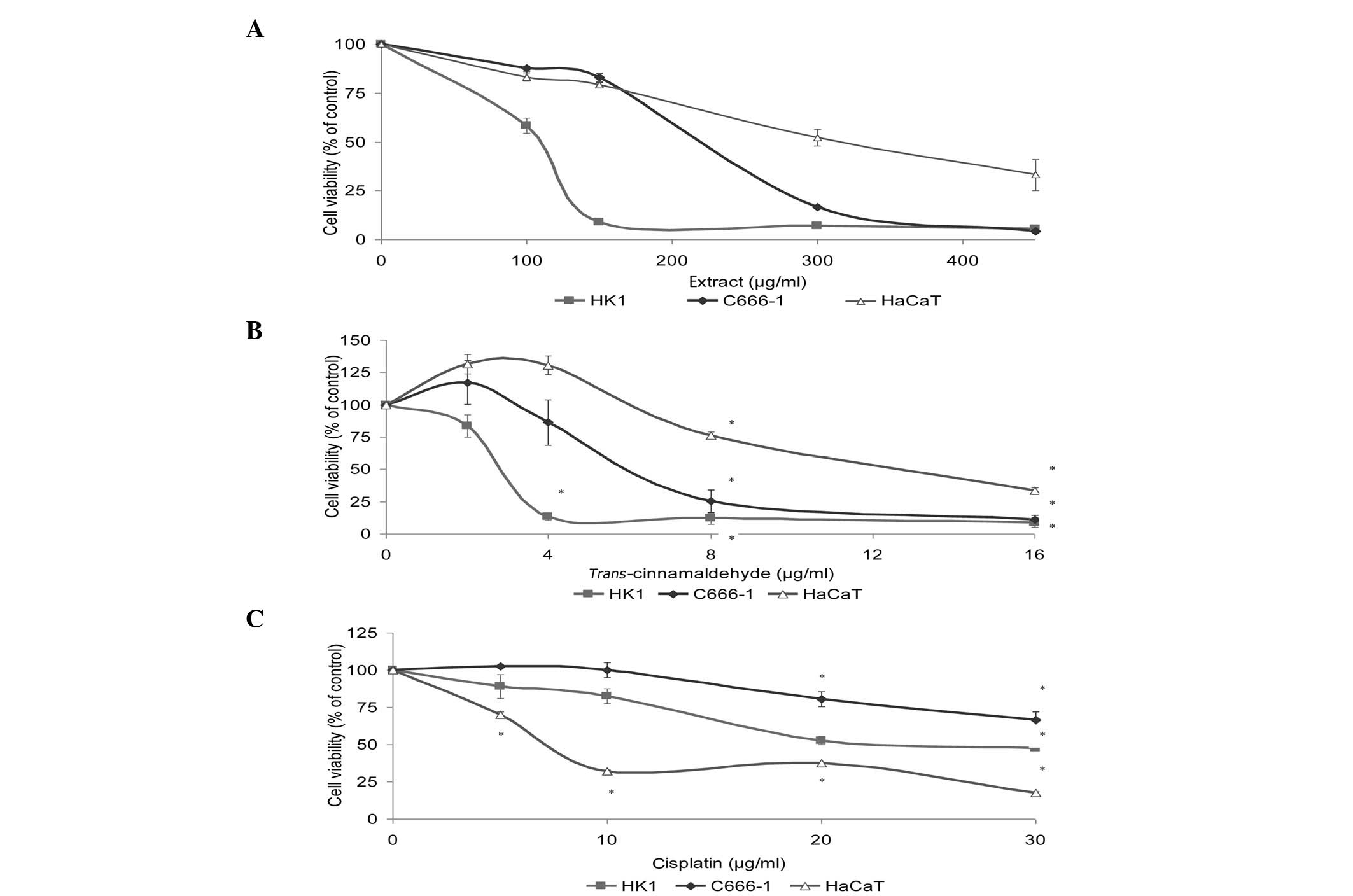
Determination of viable cells
We assessed the effects of the standardized methanol
extract and TCA on the HK1 and C666-1 NPC cell lines. NPC cell
growth was inhibited (Fig. 2A and
B) in a concentration-dependent manner. TCA at 16 μg/ml
caused almost 90% inhibition of cell growth (Fig. 2B). To determine whether normal cell
growth was also inhibited, we investigated cell viability in the
immortalized human skin keratinocyte, HaCaT. The order of cells
affected by the activity of test substances was HK1 > C666-1
> HaCaT. Cisplatin (positive control drug) demonstrated a much
higher toxic effect in normal cells, affecting HaCaT to a greater
extent than the NPC cell lines (Fig.
2C). TCA was quite potent against NPC cells with
IC50 values lower than that of cisplatin, demonstrating
far higher efficacy than cisplatin against the NPC cells (Table I).
 | Table IIC50 values for the
anti-proliferative activity of cinnamon methanol extract,
trans-cinnamaldehyde and cisplatin against cancer and normal
cells. |
Table I
IC50 values for the
anti-proliferative activity of cinnamon methanol extract,
trans-cinnamaldehyde and cisplatin against cancer and normal
cells.
| IC50
value (μg/ml)
|
|---|
| Cell line | Extract |
Trans-cinnamaldehyde | Cisplatin |
|---|
| HK1 | 108.32±3.43 | 2.94±0.17 | 26.89±0.26 |
| C666-1 | 224.32±3.17 | 6.30±0.74 | >60 |
| HaCaT | 320.29±30.41 | 12.92±0.40 | 7.65±0.16 |
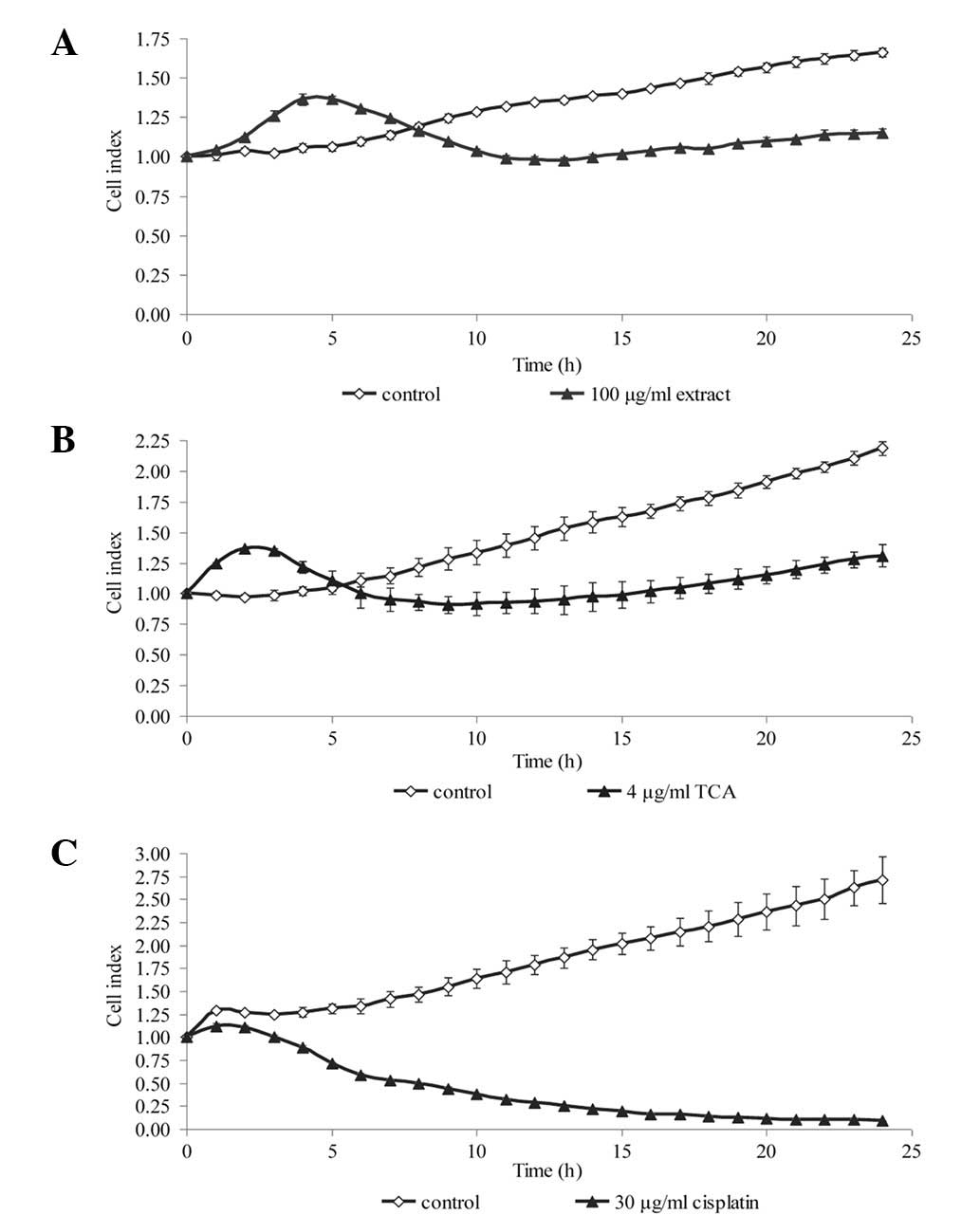
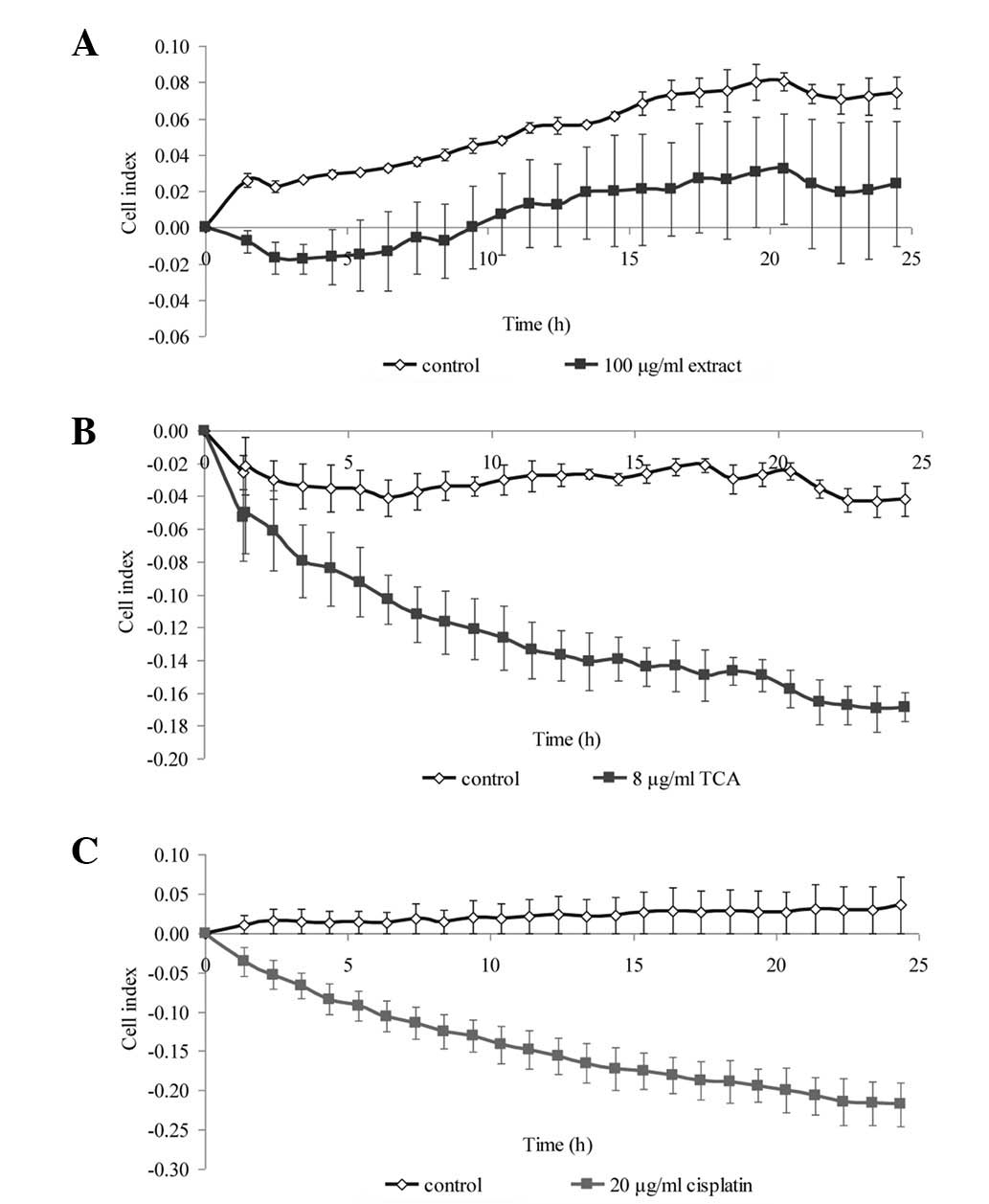
Dynamic monitoring of cell
proliferation
To confirm the results from the viability assay,
real-time cell proliferation was monitored using the xCELLigence
assay, whereby the cell index generated represents growth over
time. We selected one concentration from the MTS cell viability
assay that produced a significant reduction in cell viability.
Within 24 h of treatment, the growth kinetics of the NPC cells was
noticeably decreased compared with that of the untreated control
(Fig. 3 and 4). The growth kinetics of the cells
treated with cisplatin is shown as a positive control.
Since TCA, rather than the methanol extract,
exhibited greater efficacy at inhibiting growth, all further
experiments conducted concerned TCA only. We further verified the
ability of TCA to inhibit growth by demonstrating that it decreased
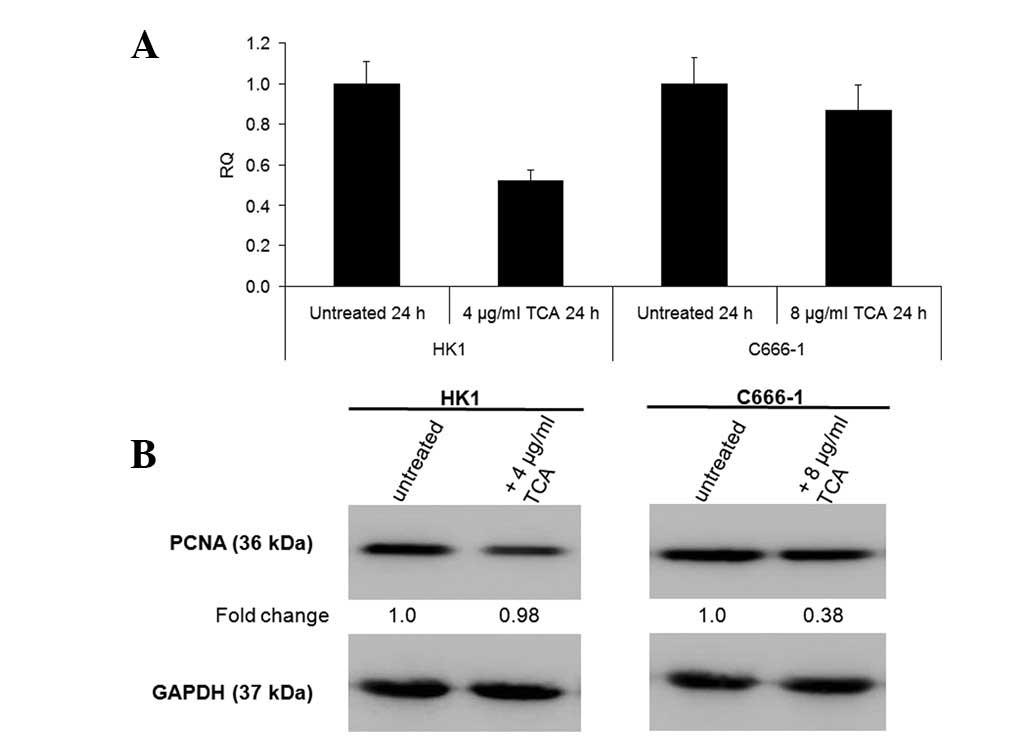
the mRNA levels of Ki67 proliferation antigen and protein levels of
PCNA (Fig. 5).
Effects of TCA on morphological
changes
We examined the morphologic characteristics
associated with growth inhibition (Fig. 6). HK1 are adherent cells. It was
evident that treatment with the methanol extract and TCA within 24
h caused characteristic features of apoptosis, including loss of
adherence and roundness in shape. In the C666-1 cell line, control
cells were flattened and adherent with few rounded cells. Treated
cells detached from the plates to become floating aggregates. These
morphological changes were in agreement with the decreased cell
index determined in the xCELLigence assay (Fig. 3 and 4). Plasma membrane blebbing and debris,
possibly apoptotic bodies, in the culture medium were also
apparent. The morphological changes associated with cisplatin
treatment were used for comparison. Cisplatin is used in cancer
therapy due to its apoptosis-inducing activity.
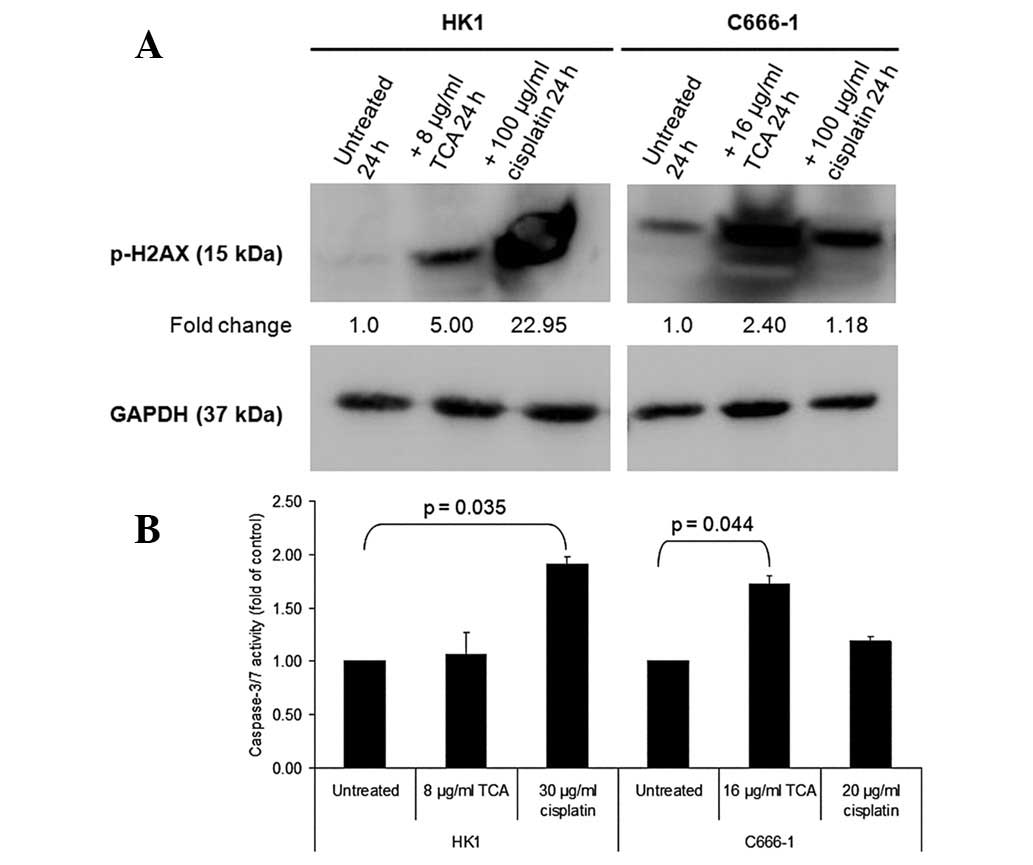
TCA induces phosphorylation of histone
H2AX and activation of caspase-3/7
To elucidate the mechanism of apoptosis, we analyzed
the phosphorylation of histone H2AX and changes in caspase-3/7
activity. Clear differences between TCA-treated NPC cells and
control cultures were observed with regard to the phosphorylation
of histone H2AX, a marker of DNA damage (Fig. 7A). In addition, cisplatin, a drug
that crosslinks DNA and interferes with DNA replication, markedly
increased H2AX phosphorylation. Fig.
7B shows that treatment with ≥8 μg/ml TCA activated
caspase-3/7. A considerable increase in caspase-3/7 activation was
noted in C666-1 cells.
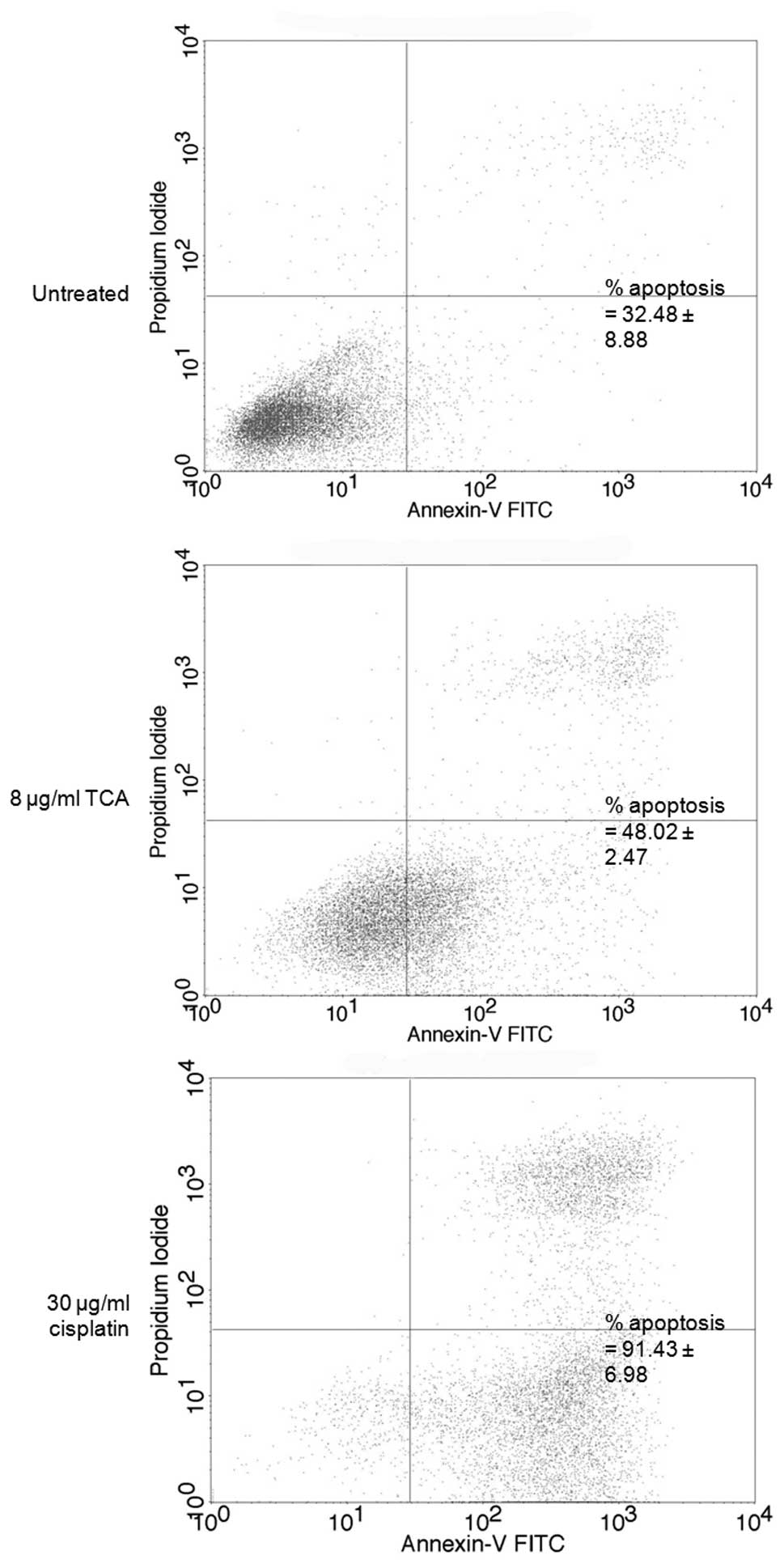
TCA contributes to apoptosis
To confirm the phenomenon of apoptosis, we performed
an apoptosis assay by flow cytometry. Approximately 16% of cells
underwent apoptosis following correction of background apoptosis,
compared with the control (Fig.
8). Cisplatin, used for NPC chemotherapy, was used for clear
comparison of apoptosis induction.
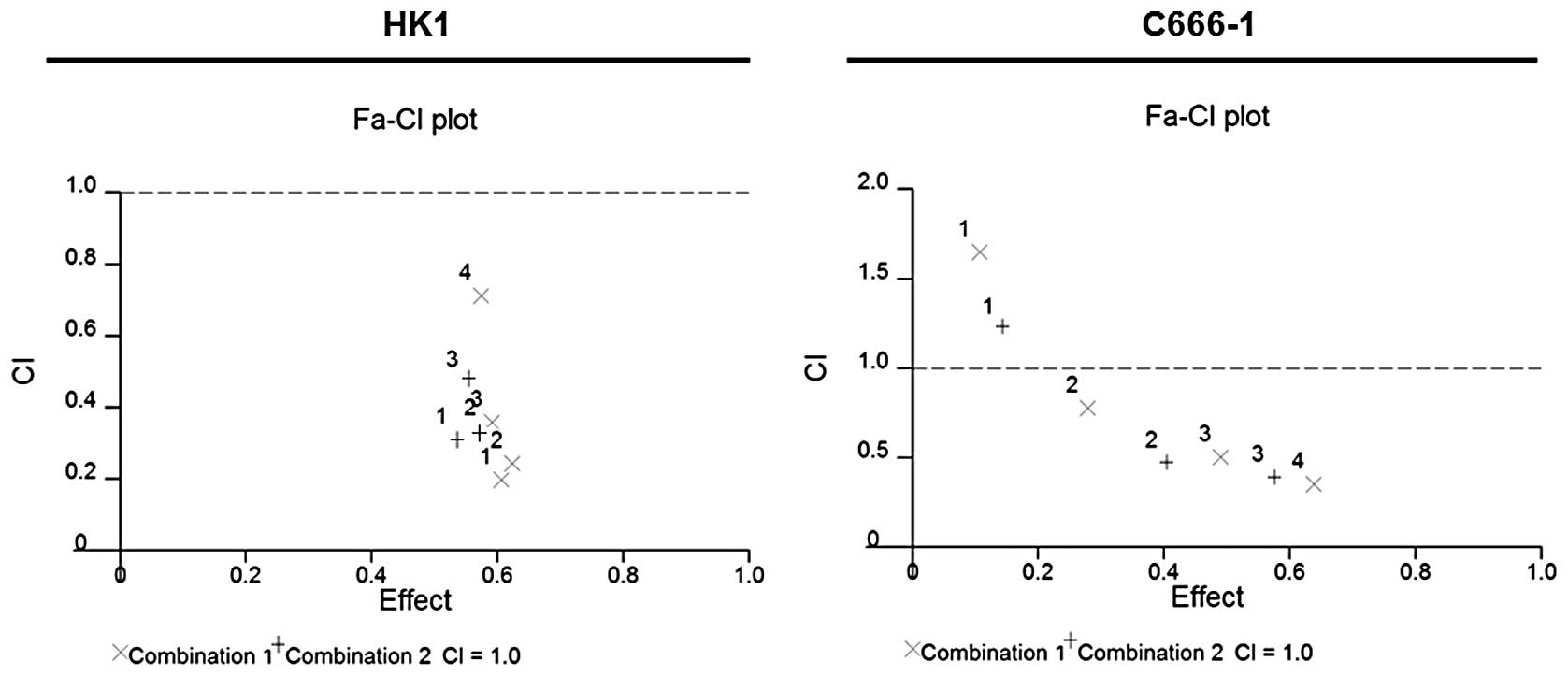
TCA exhibits synergistic effects in
combination with cisplatin
Using CalcuSyn software, we determined the CI to
ascertain synergism (CI <1), antagonism (CI >1) or additive
effect (CI =1). The CI values are presented in Table II. The CI method (17) revealed that the simultaneous
combination of TCA and cisplatin produces synergistic
anti-proliferative effects (Fig.
9).
| Table IIDescription of CI values for each
fraction of cells affected and the corresponding DRI. |
Table II
Description of CI values for each
fraction of cells affected and the corresponding DRI.
| NPC cell line | Non-fixed ratio
combination | Fa | CI | Description | DRIa
|
|---|
| TCA | Cisplatin |
|---|
| HK1 | 1 | 0.606 | 0.199 | Strong
synergism | 15.824 | 7.365 |
| 0.624 | 0.245 | Strong
synergism | 16.630 | 5.417 |
| 0.592 | 0.358 | Synergism | 15.232 | 3.415 |
| 0.574 | 0.713 | Moderate
synergism | 14.512 | 1.552 |
| 2 | 0.536 | 0.311 | Synergism | 8.748 | 5.086 |
| 0.581 | 0.334 | Synergism | 9.858 | 4.294 |
| 0.553 | 0.469 | Synergism | 9.149 | 2.779 |
| C666-1 | 1 | 0.107 | 1.652 | Antagonism | 13.318 | 0.634 |
| 0.279 | 0.779 | Moderate
synergism | 21.684 | 1.365 |
| 0.490 | 0.504 | Synergism | 31.649 | 2.118 |
| 0.639 | 0.355 | Synergism | 40.803 | 3.023 |
| 2 | 0.145 | 1.218 | Moderate
antagonism | 5.129 | 0.977 |
| 0.405 | 0.472 | Synergism | 9.141 | 2.758 |
| 0.574 | 0.392 | Synergism | 12.143 | 3.228 |
Nitric oxide inhibition activity
The nitric oxide-scavenging activity of the methanol
extract and TCA were examined using sodium nitroprusside as a
nitric oxide donor in vitro. The samples demonstrated
concentration-dependent inhibitory activity against the nitric
oxide radical. The IC50 values for curcumin (positive
control), methanol extract and TCA were 32.45±1.56, 30.67±1.12 and
24.74±1.25 μg/ml, respectively.
Discussion
There is a requirement to ensure the quality control
of herbal medicinal plant products by using modern techniques and
applying suitable standards, since diverse medicinal extracts from
the same plant material may vary with respect to their bioactive
contents and consequently their therapeutic effects. Therefore, the
objective of the GC-MS in this study was to standardize the
methanol extract of C. burmannii stem bark using TCA as a
marker, before proceeding with the anti-proliferative studies. TCA
was selected as a marker for this study since it has been
identified as one of the bioactive compounds in the
Cinnamomum species (6). The
GC-MS method was suitable for separating and quantifying TCA from
the methanol extract.
The present study demonstrated that the methanol
extract of C. burmannii stem bark and TCA impair NPC cell
proliferation. The anti-proliferative activity of TCA in the NPC
cells was greater than that of the methanol extract, suggesting
that TCA was responsible for the cytotoxicity exhibited by the
methanol extract (Table I). There
was a moderate degree of selectivity against NPC cell lines
compared with human skin keratinocytes, with the anti-proliferative
activity of TCA two- to six-fold higher in NPC cells (Table I). Apoptosis is characterized by
morphological changes, including cell shrinkage, membrane blebbing,
DNA fragmentation, activation of caspases and cell breakdown into
apoptotic bodies (19). H2AX, a
histone H2A variant becomes phosphorylated at serine 139 to form
γH2AX upon DNA double-strand breakage (20). A number of drugs and cytotoxic
agents are associated with the formation of γH2AX (21). H2AX phosphorylation is considered
to be the earliest known marker of DNA damage (22) and its detection provides a more
sensitive and efficient measure of DNA damage than other techniques
(20). In the current study, we
detected that phosphorylated H2AX (Ser 139) was much more
upregulated in the TCA-treated groups than in the untreated control
(Fig. 7A). Caspases are proteases
involved in the execution of apoptotic cell death (23). The activation of caspase-3/7
demonstrated in the NPC cells in our study (Fig. 7B) suggests a caspase-dependent
mechanism for TCA. Collectively with the morphologic observations
(Fig. 6) and flow cytometry
apoptosis assay (Fig. 8), we infer
that TCA induces apoptosis in NPC cells.
The concomitant treatment of cultured NPC cells with
cisplatin and the flavonoid, quercetin, has been shown to enhance
cytotoxic effects (24). In our
study, we successfully demonstrated that TCA, a phenolic acid, is
able to enhance the anti-proliferative effect of cisplatin in
cultured NPC cells (Fig. 9 and
Table II). In nature, TCA and its
derivatives serve as important intermediates for the biosynthesis
of flavonoids.
The nitric oxide-scavenging activities of the
extracts may be attributed to the phenolic contents in the
extracts, including TCA. TCA and its derivatives have been reported
to inhibit nitric oxide production(25). Phenolic compounds are known to
suppress nitric oxide production and also scavenge nitric oxide in
an acellular system using sodium nitroprusside under physiological
conditions at a micromolar range (26). The Environmental Protection Agency
(EPA) has not classified nitrogen oxides for potential
carcinogenicity. Nevertheless, it has been reported that exposure
to high levels of nitric oxide may result in membrane damage, since
nitric oxide reacts with oxygen to form nitrogen dioxide, which in
biological systems initiates the auto-oxidation of fatty acids in
lipid membranes (27). The nitric
oxide-scavenging activity of the extracts indicates that infusions
of C. burmannii may serve as therapeutic agents for
scavenging free radicals and thereby inhibit the pathological
conditions caused by excessive generation of free radicals and
their oxidation products.
In conclusion, compared with conventional drugs,
including cisplatin, the lower toxicity of TCA in normal cells,
whilst maintaining or improving the anti-proliferative effect in
NPC cells, may contribute to its potential use and benefit as a
herbal medicinal product. TCA not only inhibits cell proliferation
but also induces characteristic features of apoptosis.
Additionally, TCA synergized with cisplatin to produce
anti-proliferative effects in NPC cells. C. burmannii stem
bark extracts and TCA are able to trap and scavenge free radicals,
which may be a mechanism involved in their therapeutic effects.
These results merit further investigation into the modulation of
anti-proliferative action and/or induction of apoptosis.
Acknowledgements
We wish to thank the Director General
of Health (Malaysia) for permission to publish this article and the
Director of the Institute for Medical Research for her support.
This study was financially supported by research grants from
Universiti Sains Malaysia (304/PFARMASI/6311040) and the Ministry
of Health, Malaysia (NMRR-12-996-13910). HK1 was kindly provided by
George Tsao (Department of Anatomy, University of Hong Kong, Hong
Kong, China). C666-1 was a gift from Kwok-Wai Lo (Department of
Anatomical and Cellular Pathology, Chinese University of Hong Kong,
Hong Kong, China).
References
|
1.
|
Wei WI and Sham JS: Nasopharyngeal
carcinoma. Lancet. 365:2041–2054. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Tao Q and Chan AT: Nasopharyngeal
carcinoma: molecular pathogenesis and therapeutic developments.
Expert Rev Mol Med. 9:1–24. 2007.PubMed/NCBI
|
|
3.
|
van Leeuwen IMM and Laín S:
Pharmacological manipulation of the cell cycle and metabolism to
protect normal tissues against conventional anticancer drugs.
Oncotarget. 2:274–276. 2011.PubMed/NCBI
|
|
4.
|
Cao H, Graves D and Anderson R: Cinnamon
extract regulates glucose transporter and insulin-signaling gene
expression in mouse adipocytes. Phytomed. 17:1027–1032. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bandar E: Pharmaceutical applications and
phytochemical profile of Cinnamomum burmannii. Pharmacog
Rev. 6:125–131. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lv G, Huang W, Yang F, Li J and Li S:
Pressurized liquid extraction and GC-MS analysis for simulateneous
determination of seven components in Cinnamomum cassia and
effect of sample preparation. J Sep Sci. 33:2341–2348. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Gomez-Flores R, Hernández-Martínez H,
Tamez-Guerra P, et al: Antitumor and immunomodulating potential of
Coriandrum sativum, Piper nigrum and Cinnamomum
zeylanicum. J Nat Prod. 3:54–63. 2010.
|
|
8.
|
Koppikar S, Choudhari A, Suryavanshi S,
Kurnari S, Chattopadhyay S and Kaul-Ghanekar R: Aqueous cinnamon
extract (ACE-c) from the bark of Cinnamomum cassia causes
apoptosis in human cervical cancer cell line (SiHa) through loss of
mitochondrial membrane potential. BMC Cancer. 10:2102010.PubMed/NCBI
|
|
9.
|
Kwon HK, Hwang JS, So JS, et al: Cinnamon
extract induces tumor cell death through inhibition of NFκB and
AP1. BMC Cancer. 10:3922010.PubMed/NCBI
|
|
10.
|
Wu S, Ng L and Lin C:
Cinnamaldehyde-induced apoptosis in human PLC/PRF/5 cells through
activation of the proapoptotic Bcl-2 family proteins and MAPK
pathway. Life Sciences. 77:938–951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Cabello CM, Bair WB III, Lamore SD, et al:
The cinnamon-derived Michael acceptor cinnamic aldehyde impairs
melanoma cell proliferation, invasiveness and tumor growth. Free
Radic Biol Med. 46:220–231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Zhang JH, Liu LQ, He YI, Kong WJ and Huang
SA: Cytotoxic effect of trans-cinnamaldehyde on human leukemia K562
cells. Acta Pharmacologica Sinica. 31:861–866. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Ng LT and Wu SJ: Antiproliferative
activity of Cinnamomum cassia constituents and effects of
pifithrin-alpha on their apoptotic signaling pathways in Hep G2
cells. Evid Based Complement Alternat Med. 2011:4921482011.
|
|
14.
|
Huang DP, Ho JH, Poon YF, et al:
Establishment of a cell line (NPC/HK1) from a differentiated
squamous carcinoma of the nasopharynx. Int J Cancer. 26:127–132.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Cheung ST, Huang DP, Hui AB, et al:
Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring
Epstein-Barr virus. Int J Cancer. 83:121–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Boukamp P, Petrussevska RT, Breitkreutz D,
Hornung J, Markham A and Fusenig NE: Normal keratinization in a
spontaneously immortalized aneuploid human keratinocyte cell line.
J Cell Biol. 106:761–771. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Rao M: Nitric oxide scavenging by
curcuminoids. J Pharm Pharmacol. 49:105–107. 1997. View Article : Google Scholar
|
|
19.
|
Unlu M, Ergene E, Unlu G, Zeytinoglu H and
Vural N: Composition, antimicrobial activity and in vitro
cytotoxicity of essential oil from Cinnamomum zeylanicum
Blume (Lauraceae). Food Chem Toxicol. 48:3274–3280. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Dickey J, Redon C, Nakamura A, Baird B,
Sedelnikova O and Bonner W: H2AX: functional roles and potential
applications. Chromosomes. 118:683–692. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kuo L and Yang L: Gamma-H2AX - a novel
biomarker for DNA double-strand breaks. In Vivo. 22:305–309.
2008.PubMed/NCBI
|
|
22.
|
Ayoub N, Jeyasekharan A, Bernal J and
Venkitaraman A: Paving the way for H2AX phosphorylation: chromatin
changes in the DNA damage response. Cell Cycle. 8:1494–1500. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chung W, Seung H, Jae W, et al:
2-Hydroxycinnamaldehyde inhibits SW620 colon cancer cell growth
through AP-1 inactivation. J Pharmacol Sci. 104:19–28. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Daker M, Ahmad M and Khoo ASB:
Quercetin-induced inhibition and synergistic activity with
cisplatin - a chemotherapeutic strategy for nasopharyngeal
carcinoma cells. Cancer Cell Int. 12:342012. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Seung H, Sun Y, Dong J, et al: Inhibitory
effect of 2-hydroxycinnamaldehyde on nitric oxide production
through inhibition of NFκB activation in RAW 264.7 cells. Biochem
Pharmacol. 69:791–799. 2005.
|
|
26.
|
Jagetia S, Balgia M and Babu K: Evaluation
of nitric oxide scavenging activity of certain herbal formulation
in vitro. Phytother Res. 18:561–565. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Korkmaz B, Buharalioglu K, Sahan-Firat S,
Cuez T, Demiryurek T and Tunctan B: Activation of
MEK1/ERK1/2/iNOS/sGC/PKG pathway associated with peroxynitrite
formation contributes to hypotension and vascular hyporeactivity in
endotoxemic rats. Nitric Oxide. 24:160–172. 2011. View Article : Google Scholar : PubMed/NCBI
|