Introduction
Cutaneous squamous cell carcinoma (SCC) originates
in the epidermis or adnexal keratinocytes and has the highest
incidence rate, next to basal cell carcinoma (BCC), of any
non-melanocyte cell carcinoma (1).
Due to its metastasis, the degree of malignancy and the risk of
mortality for individuals with SCC is significantly higher than for
those with BCC (2,3). A number of studies have indicated
that ultraviolet (UV) light, ionizing radiation and chemical
carcinogens play important roles in the development of SCC
(1,4,5);
however, its pathogenesis remains unknown. Currently, the main
therapy for SCC is surgical excision (4,5).
However, a carcinoma of a large size and the subsequently large
excision in the affected area may severely affect the quality of
life of the patient.
Metformin has been used in a wide range of
applications for more than half a century. It is the first-line
oral hypoglycemic agent for the treatment of type 2 diabetes and is
recommended by the American Diabetes Association and the European
Association for the Study of Diabetes (6). Previous studies have demonstrated
that metformin is able to reduce the incidence rate of a variety of
tumors in patients with diabetes (7–12).
In in vitro studies, metformin has been found to reduce the
volume of prostate tumors in nude mice (13) and inhibit the growth and
proliferation of breast (14),
prostate (13), cervical (15) and ovarian (16) cancer cells. However, the antitumor
mechanism of metformin remains unclear. Akt, a serine/threonine
protein kinase, has a vital function in multiple cellular processes
including the regulation of cell growth, proliferation and
apoptosis (17). PI3K is the most
important upstream molecule to activate Akt (17). Previous studies have shown that the
increased activities of PI3K and Akt play an important role in the
development of numerous types of tumors (18).
In the present study, the A431 human squamous cell
carcinoma cell line was cultured to investigate the effect of
metformin on the proliferation of A431 cells and the underlying
molecular mechanisms.
Materials and methods
Cells and reagents
SCC cell line A431 was purchased from the Cell
Culture Center of the Chinese Academy of Sciences (Shanghai,
China). Rabbit anti-human polyclonal antibodies against Akt
(sc-8312), phosphorylated (p)-Akt (Ser 473; sc-135651) and PI3K
(sc-423) were purchased from Santa Cruz Biotechnology (Dallas, CA,
USA). The polyclonal rabbit anti-human antibody against β-actin was
purchased from Sigma-Aldrich (SAB2100037; St. Louis, MO, USA). The
secondary polyclonal goat anti-rabbit antibody labeled with
horseradish peroxidase (HRP) was purchased from EarthOx LLC
(E030120-01; San Francisco, CA, USA). Metformin hydrochloride with
98.8% purity was purchased from Shouguang Fukang Pharmaceutical
Co., Ltd. (Shouguang, China) and was diluted with
phosphate-buffered saline (PBS) into 1 M solution for storage.
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum and
0.25% trypsin digestion solution were purchased from Gibco (Grand
Island, NY, USA). TRIzol® reagent was purchased from
Invitrogen Life Technologies (Carlsbad, CA, USA). 2X Power Taq
polymerase chain reaction (PCR) MasterMix and the Bioteke Super
reverse transcription (RT)-kit were purchased from BioTeke
Corporation (Beijing, China). RIPA lysis buffer and
phenylmethanesulfonylfluoride (PMSF) were purchased from Beyotime
Institute of Biotechnology (Jiangsu, China). The Cell Counting
kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies,
Inc. (Kunamoto, Japan). The polyvinylidene fluoride (PVDF) membrane
was purchased from the Pall Corporation (Port Washington, NY, USA).
The study was approved by the Ethics Committee of Shandong
University (Jinan, China).
Cell culture and metformin treatment
A431 cells were cultured in DMEM containing 10%
fetal bovine serum at 37°C in a 5% CO2 incubator.
Following three stable passages, the A431 cells were seeded onto
6-well plates with a density of 5×104 cells/well. After
24 h of inoculation, the culture medium was replaced with
serum-free medium and different concentrations of metformin (0, 15,
30, 45 and 60 mM) were added for 12, 24 and 36 h. The control group
received the corresponding volume of PBS.
Cell morphology observation
The morphology of the A431 cells with the treatment
of 45 mM metformin for 24 h was observed under an inverted
microscope (Olympus CKX31; Olympus Corporation, Tokyo, Japan).
These values were selected as they produced the most marked
morphological results.
Cell proliferation assay
A431 cells were inoculated into 96-well plates with
a density of 5×104 cells/well at 37°C in a 5%
CO2 incubator for 24 h. Metformin was added to the wells
with final concentrations of 0, 15, 30, 45 and 60 mM for an
incubation period of 12, 24 and 36 h. A total of 10 μl CCK-8
solution was added to each well and the optical density (OD) values
were measured at 450 nm using a quantitative automatic microplate
reader (model no. 2010; Anthos Labtec Co., Ltd., Salzburg,
Austria). The rate of cell growth inhibition (%) was calculated as
follows: (OD control - OD metformin)/OD control ×100.
Total RNA extraction and RT-PCR
analysis
Total RNA was extracted according to the
manufacturer’s instructions of the TRIzol® reagent and
RT was carried out using the RT-kit for the total RNA. For each
sample, 1 μg total RNA, random primers and M-MuLV reverse
transcriptase enzyme were added to the 20 μl RT reaction system.
Complementary DNA (cDNA) was synthesized under the following
conditions: 42°C for 50 min and then 70°C for 10 min. The primers
were provided by the Sangon Biotech Co., Ltd. (Shanghai, China),
and are shown in Table I. PCR was
performed in a 50-μl reaction system including 5 μl cDNA, 2.5 μl
upstream and downstream primer, 25 μl 2X PCR mix and 15 μl
triple-distilled water. The PCR cycler used was the Icycling 96
Gradient PCR Instrument (BioTeke Corporation, Beijing, China). The
amplification conditions of the PCR were as follows: 94°C for 5
min, followed by 33 cycles at 94°C for 30 sec, 55°C for 30 sec,
72°C for 30 sec, and finally 72°C for 10 min. The amplification
products of PCR were electrophoresed in a 2% agarose gel stained
with ethidium bromide. The optical density of each sample band was
captured using a UV gel imaging system (Tanon-2500R; Tanon Science
& Technology Co., Ltd, Shanghai, China) and analyzed using
ImageJ 1.47v software (National Institutes of Health, Bethesda, MD,
USA). The OD ratio of the target band to GAPDH was defined as the
relative mRNA expression of the target band.
 | Table IPrimer sequences for reverse
transcription-polymerase chain reaction (RT-PCR). |
Table I
Primer sequences for reverse
transcription-polymerase chain reaction (RT-PCR).
| Target genes | Primer sequence | Product size
(bp) |
|---|
| PI3K | Forward:
5′-ACTTTGCGACAAGACTGC-3′
Reverse: 5′-GCCCTATCCTCCGATTAC -3′ | 337 |
| Akt | Forward:
5′-ACAGCAAAGCAGGAGTATAAGA-3′
Reverse: 5′-CCAAACGAAACCAAGTCAA-3′ | 336 |
| GAPDH | Forward:
5′-AAGTACTCCGTGTGGATCGG-3′
Reverse: 5′-ATGCTATCACCTCCCCTGTG-3′ | 360 |
Western blot analysis
RIPA protein lysis buffer containing 1 mM PMSF was
used to lyse cells on ice and the protein concentration was
determined using a bicinchoninic acid (BCA) assay kit (Beyotime
Institute of Biotechnology, Shanghai, China). A total of 50 μg
protein from every sample was loaded onto a 10% SDS-PAGE gel for
constant voltage electrophoresis and transferred to a PVDF membrane
under a constant 1-mA/cm2 current. The nonspecific
binding sites on the PVDF membranes were blocked by Tris-buffered
saline and Tween 20 (TBST) buffer containing 5% skimmed milk for 1
h. The PVDF membranes were subsequently incubated with primary
antibodies (dilution, 1:1,000) at 4°C overnight followed by
incubation with the corresponding secondary antibody labeled with
HRP (dilution, 1:10,000) at room temperature for 2 h. The membranes
were developed on film using enhanced chemiluminescence (ECL) and
scanned using an electrophoresis gel imaging analysis system
(Tanon-5000R; Tanon Science & Technology Co., Ltd). β-actin was
used as the internal control. The relative absorbance ratios of
target protein to β-actin were defined as the relative values of
target protein.
Statistical analyses
Quantitative data are expressed as the mean ±
standard deviation. Statistical analyses were performed using the
SPSS statistical software package, version 20.0 (IBM SPSS, Armonk,
NY, USA). The Student’s t-test was used to compare the difference
between two groups and P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of metformin on A431 cell
morphology
Following treatment with 45 mM metformin for 24 h,
poor growth of the A431 cells with increasing karyopyknosis was
observed. There were scattered areas with no cell growth and the
number of floating cells was increased significantly in the
treatment group compared with that in the untreated group. The A431
cells that were not subjected to metformin treatment were
distributed evenly and were of a similar size (Fig. 1).
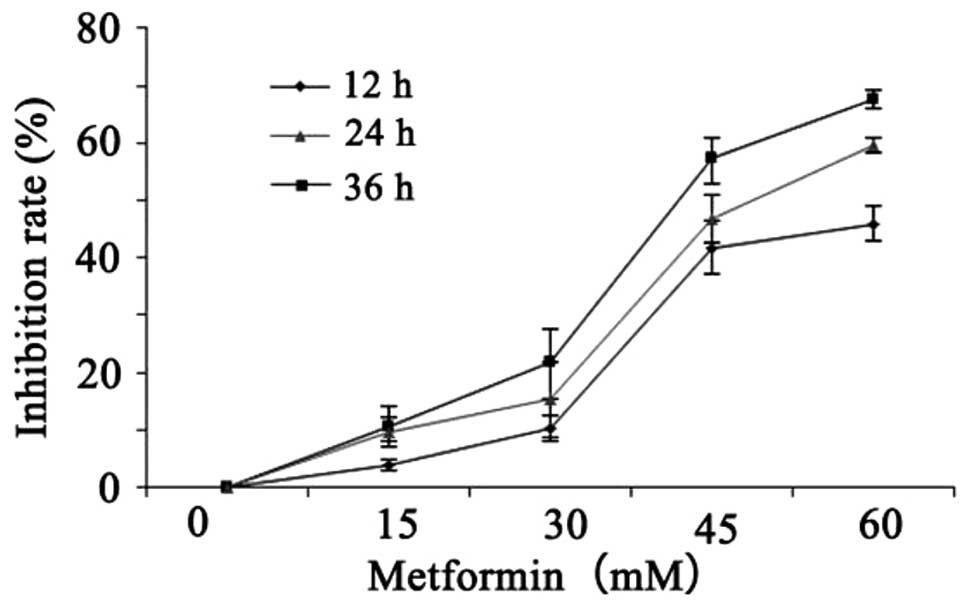
Inhibitory effect of metformin on the
proliferation of A431 cells
A431 cells were seeded onto 96-well plates.
Following treatment with metformin at different concentrations (0,
15, 30, 45 and 60 mM) for 12, 24 and 36 h, cell activity was
detected by the CCK-8 method. With the increase in treatment
concentration and duration, the inhibitory effect of metformin on
A431 cell proliferation gradually increased (Table II and Fig. 2). These results demonstrated that
metformin inhibited the proliferation of A431 cells in a time- and
dose-dependent manner.
| Table IIInhibition of A431 cell proliferation
by metformin treatment with different concentrations and
durations. |
Table II
Inhibition of A431 cell proliferation
by metformin treatment with different concentrations and
durations.
| Time | Untreated (%) | 15 mM (%) | 30 mM (%) | 45 mM (%) | 60 mM(%) |
|---|
| 12 h | 0 | 0.0388±0.0087 | 0.1030±0.0225 | 0.4181±0.0470 | 0.4593±0.0297 |
| 24 h | 0 | 0.1051±0.0349 | 0.2192±0.0561 | 0.5723±0.0375 | 0.6748±0.0179 |
| 36 h | 0 | 0.0964±0.0262 | 0.1527±0.0655 | 0.4678±0.0428 | 0.5962±0.0137 |
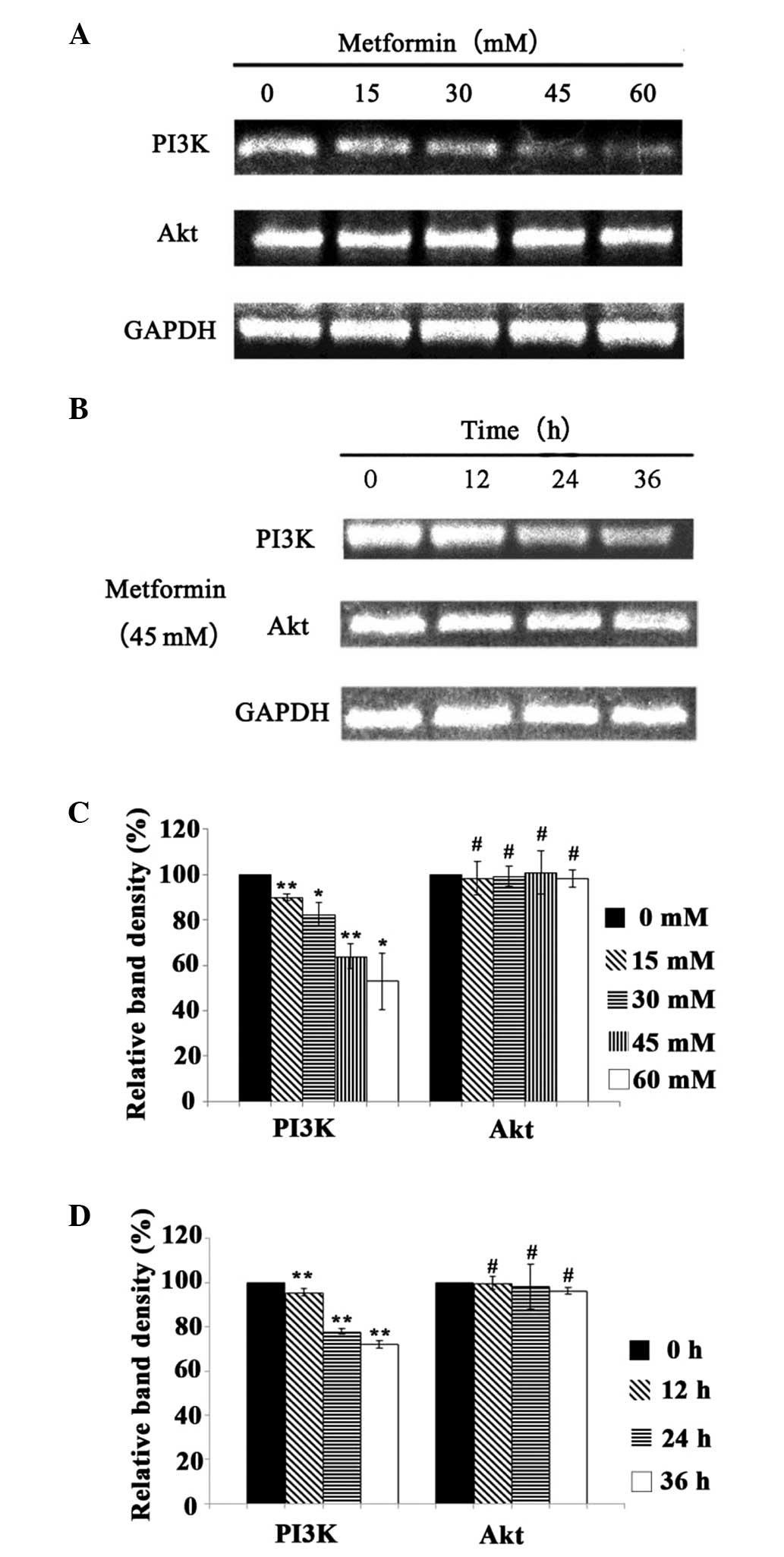
Effect of metformin on the mRNA
expression levels of PI3K and Akt
Following treatment with metformin at different
concentrations (0, 15, 30, 45 and 60 mM) for 24 h, the mRNA
expression levels of PI3K and Akt were detected by RT-PCR (Fig. 3). In addition, following treatment
with 45 mM metformin for 12, 24 and 36 h, the mRNA expression
levels of PI3K and Akt were also detected by RT-PCR (Fig. 3). The mRNA expression level of PI3K
decreased with the increase in the concentration and duration of
metformin treatment (P<0.05), which indicated that metformin
inhibited the mRNA expression of PI3K in a time- and
concentration-dependent manner. Following treatment with metformin
at different concentrations, the mRNA expression of Akt showed no
significant difference compared with that of the control group
(P>0.05). Furthermore, following treatment with 45 mM metformin
for different durations, the mRNA expression levels of Akt also
demonstrated no significant difference from that in the control
(P>0.05). Thus, metformin did not have a significant effect on
the gene expression of Akt.
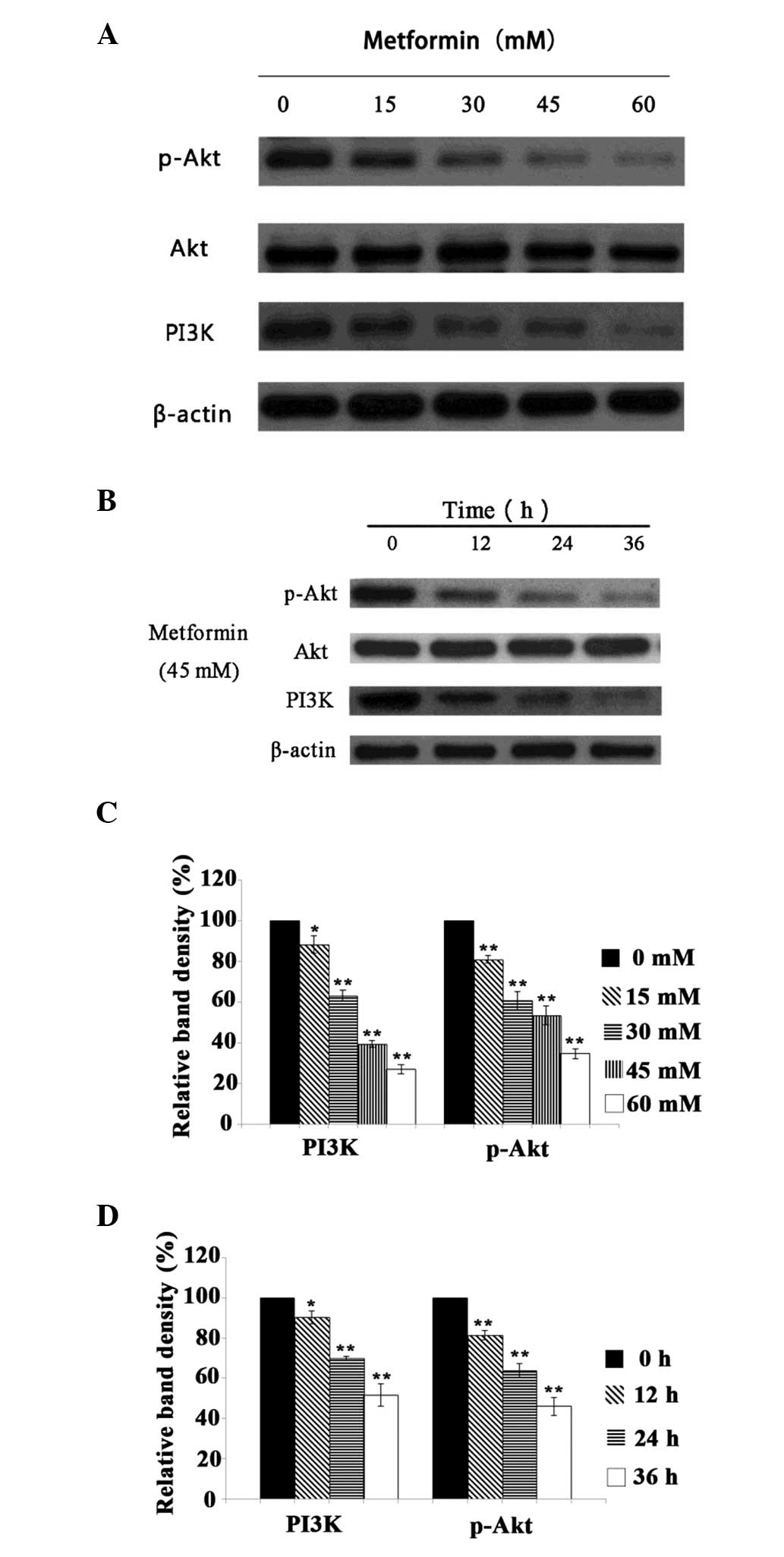
Effect of metformin on the protein
expression levels of PI3K and p-Akt
It is accepted that Akt plays a key role in the
regulation of the growth and proliferation of tumor cells (17). The inhibition of Akt activity may
suppress cancer cell growth and promote the apoptosis of these
cells. The present study observed that treatment with metformin
decreased the protein expression levels of PI3K and p-Akt in A431
cells in a time- and concentration-dependent manner (P<0.05;
Fig. 4). These results indicate
that the mechanism by which metformin inhibits the proliferation of
A431 cells may involve modulation of the PI3K/Akt signaling
pathway.
Discussion
Metformin, the first-line drug for the treatment of
type 2 diabetes, can reduce hepatic glucose output and increase
glucose uptake by activating the AMP-activated protein kinase
(AMPK) signaling pathway (19).
Decensi et al performed a meta-analysis on 11 studies and
found that compared with other antidiabetic drugs, metformin led to
a 31% reduction in the total relative risk (RR) of cancer (RR,
0.69; 95% confidence interval, 0.61–0.79) (8). At present, studies investigating the
inhibition of tumor cell proliferation by metformin remain focused
on the AMPK signaling pathway (20).
Zakikhani et al reported that the inhibitory
effect of metformin on the proliferation of the MCF-7 human breast
cancer cell line decreased after the expression of the AMPK subunit
was inhibited by siRNA (14). Yung
et al (15) observed that
the growth of cervical cancer cells was inhibited by metformin in a
time- and dose-dependent manner. Similar results were obtained
following treatment with AMPK activators including
5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide (AICAR) and
A23817, which further confirmed that the activation of AMPK was
able to suppress cervical cancer cell growth. However, Ben Sahra
et al demonstrated that the inhibitory effect of metformin
on human prostate cancer cell proliferation did not decrease when
the two catalytic subunits of AMPK were suppressed by siRNA. This
difference in the response to AMPK suppression may have been due to
the different sources of the cancer cells used and the incomplete
inhibition of AMPK (13). Byekova
et al found that compared with the expression of p-AMPK in
normal keratinocytes and HaCaT cells, that in A431 cells was
significantly higher and metformin was able to increase the
expression of p-AMPK in a dose-dependent manner in HaCaT cells, but
not in A431 cells (21).
In addition to the classic AMPK signaling pathway,
the PI3K/Akt signaling pathway also plays an important role in the
regulation of cell proliferation by metformin. Zakikhani et
al reported that metformin inhibited the growth of breast
cancer cells through the activation of AMPK and the inhibition of
Akt to suppress certain regulatory molecules, including mTOR and S6
kinase (S6K) (14). Yung et
al demonstrated that the activation of AMPK induced by 25 mM
metformin caused the upregulation of p-AMPK expression and the
downregulation of p-Akt expression, and the phosphorylation of
FOXO3a and Forkhead Box M1 (FOXM1) in cervical cancer cell line
C33A. The study also revealed that following the inhibition of
FOXO3a by siRNA, the expression of FOXM1 was not significantly
altered by metformin, which indicated that metformin may have
suppressed cervical cancer cell growth through activation of the
AMPK and inhibition of the Akt/FOXO3a/FOXM1 signaling pathways
(15). Karnevi et al found
that the antiproliferative effect of metformin on pancreatic cancer
cells was associated with the activation of AMPK Thr172, which was
able to suppress the phosphorylation of Akt and eventually inhibit
cell proliferation (22).
The present study showed that metformin treatment
induced morphological changes and inhibited the proliferation of
A431 cells in a significant time- and dose-dependent manner. The
mRNA expression level of PI3K and protein expression levels of PI3K
and p-Akt were significantly decreased by metformin in a time- and
dose-dependent manner. The mRNA expression of Akt did not alter
with metformin treatment. Thus, it is speculated that metformin
significantly inhibits phosphorylation in the PI3K/Akt signaling
pathway, thereby suppressing A431 cell proliferation. The
activation of Akt regulates cell cycle progression through multiple
downstream pathways. The cyclin-dependent kinase inhibitors,
p27kip1 on T157 and p21Clip/WAF1 on T145, are phosphorylated by Akt
to prevent the localization of p27 and p21 to the nucleus, which
attenuates their inhibitory cell-cycle effects (17). The stability of c-jun, c-myc and
cyclins D and E, the important molecules in the G1 to S phase
transition of the cell cycle, can be enhanced by Akt through the
inhibition of GSK3, which leads to cell cycle arrest in the G1
phase (23,24). Akt activates eIF4E by inhibiting
4EBP1 through the mTORC1 pathway to enhance the mRNA expression
levels of cyclin D1 and c-Myc (25). The DNA damage effector kinase Chk1
on serine 280 can be directly phosphorylated by Akt, thereby
stimulating the movement of Chk1 into the cytoplasm which blocks
its function and promotes cell cycle progression from the G2 to the
M phase (26). However, the exact
molecular mechanism of the inhibition of PI3K/Akt pathway by
metformin and the resulting suppression of cell proliferation
remains unclear and requires clarification by further study.
In summary, metformin inhibited the proliferation of
A431 cells in vitro in a time- and dose-dependent manner,
which was significantly associated with the phosphorylation level
of the PI3K/Akt signaling pathway. With increasing attention
devoted to the antitumor effect of metformin, the drug may lead to
improved treatment options for patients with SCC and become a
potential therapeutic treatment for SCC.
References
|
1
|
Samarasinghe V, Madan V and Lear JT:
Management of high-risk squamous cell carcinoma of the skin. Expert
Rev Anticancer Ther. 11:763–769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Macbeth AE, Grindlay DJ and Williams HC:
What’s new in skin cancer? An analysis of guidelines and systematic
reviews published in 2008–2009. Clin Exp Dermatol. 36:453–458.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van der Geer S, Siemerink M, Reijers HA,
Verhaegh ME, Ostertag JU, Neumann HA and Krekels GA: The incidence
of skin cancer in dermatology. Clin Exp Dermatol. 38:724–729.
2013.PubMed/NCBI
|
|
4
|
Nguyen TH and Ho DQ: Nonmelanoma skin
cancer. Curr Treat Options Oncol. 3:193–203. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Samarasinghe V and Madan V: Nonmelanoma
skin cancer. J Cutan Aesthet Surg. 5:3–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
American Diabetes Association. Standards
of medical care in diabetes - 2013. Diabetes Care. 36(Suppl 1):
S11–S66. 2013. View Article : Google Scholar
|
|
7
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta-analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar
|
|
9
|
Currie CJ, Poole CD, Jenkins-Jones S, Gale
EA, Johnson JA and Morgan CL: Mortality after incident cancer in
people with and without type 2 diabetes: Impact of metformin on
survival. Diabetes Care. 35:299–304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singh S, Singh H, Singh PP, Murad MH and
Limburg PJ: Antidiabetic medications and the risk of colorectal
cancer in patients with diabetes mellitus: A systematic review and
meta-analysis. Cancer Epidemiol Biomarkers Prev. 22:2258–2268.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gallagher EJ and LeRoith D: Diabetes,
antihyperglycemic medications and cancer risk: Smoke or fire. Curr
Opin Endocrinol Diabetes Obes. 20:485–494. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wright JL and Stanford JL: Metformin use
and prostate cancer in Caucasian men: Results from a
population-based case-control study. Cancer Causes Control.
20:1617–1622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yung MM, Chan DW, et al: Activation of
AMPK inhibits cervical cancer cell growth through AKT/FOXO3a/FOXM1
signaling cascade. BMC Cancer. 13:3272013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rattan R, Giri S, Hartmann LC and Shridhar
V: Metformin attenuates ovarian cancer cell growth in an AMP-kinase
dispensable manner. J Cell Mol Med. 15:166–178. 2011. View Article : Google Scholar
|
|
17
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou G, Myers R, Li Y, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bodmer M, Meier C, Krähenbühl S, Jick SS
and Meier CR: Long-term metformin use is associated with decreased
risk of breast cancer. Diabetes Care. 33:1304–1308. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Byekova YA, Herrmann JL, Xu J, Elmets CA
and Athar M: Liver kinase B1 (LKB1) in the pathogenesis of
UVB-induced murine basal cell carcinoma. Arch Biochem Biophys.
508:204–211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karnevi E, Said K, Andersson R and
Rosendahl AH: Metformin-mediated growth inhibition involves
suppression of the IGF-I receptor signalling pathway in human
pancreatic cancer cells. BMC Cancer. 13:2352013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei W, Jin J, Schlisio S, Harper JW and
Kaelin WG Jr: The v-Jun point mutation allows c-Jun to escape
GSK3-dependent recognition and destruction by the Fbw7 ubiquitin
ligase. Cancer Cell. 8:25–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Welcker M, Singer J, Loeb KR, Grim J,
Bloecher A, Gurien-West M, Clurman BE and Roberts JM: Multisite
phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol
Cell. 12:381–392. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mamane Y, Petroulakis E, Rong L, Yoshida
K, Ler LW and Sonenberg N: eIF4E - from translation to
transformation. Oncogene. 23:3172–3179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
King FW, Skeen J, Hay N and Shtivelman E:
Inhibition of Chk1 by activated PKB/Akt. Cell Cycle. 3:634–637.
2004. View Article : Google Scholar : PubMed/NCBI
|