Introduction
Lower back pain (LBP) is one of the most common
musculoskeletal disorders, and ~40% of LBP involves degeneration of
the intervertebral discs (IVDs) (1). IVDs are composed of two distinct
components: The inner gel-like core nucleus pulposus (NP) and the
outer firm annulus fibrosus (AF). Relying upon a delicate balance
between matrix synthesis and degradation, the extracellular matrix
(ECM), including collagen and proteoglycans, undergoes a process of
remodeling in normal IVDs. However, in degenerative IVDs the net
increase of matrix-degrading proteinase activity disrupts the
normal balance and leads to the breakdown of ECM (2).
Inflammatory cytokines, such as tumor necrosis
factor-α (TNF-α) and interleukin-1β (IL-1β), are highly expressed
in degenerative IVDs and contribute to a degenerative IVD phenotype
by inhibiting the production of ECM (3,4).
While not directly degrading the IVD, TNF-α and IL-1β act
indirectly by promoting the production of degradative enzymes, such
as matrix metalloproteinase (MMP) and a disintegrin and
metalloproteinase with thrombospondin motifs (ADAMTS) (5–7).
Autophagy is involved in the control of cell death
(8). Macroautophagy (hereafter
referred to as autophagy) is a vacuolar lysosomal degradation
pathway for organelles and cytoplasmic macromolecules (9). It occurs during tissue and organ
formation and has a critical role in the pathogenesis of
degenerative diseases, such as osteoarthritis and Alzheimer's
disease (10,11). In IVDs, autophagy is also present
and associated with the increased pathological process of IVD
degeneration in rats. Furthermore, autophagy of AF cells may be
secondary to endoplasmic reticulum stress (12,13). In addition, Shen et al
(14) reported that the autophagy
of rat AF cells was induced by serum deprivation in vitro
and that IL-1β upregulated serum deprivation-induced autophagy in a
dose-dependent manner. Ma et al (15) revealed that compression activated
autophagy in NP cells and that compression-induced autophagy was
closely associated with intracellular reactive oxygen species
production.
In inflammatory conditions the inhibition of
autophagy increased the expression of OA-like genes, such as
MMP13 and ADAMTS5, while the induction of autophagy
suppressed these genes (16,17). Regardless, the effect of autophagy
on the catabolic effect of inflammatory cytokines in NP cells
remains unclear. Additionally, TNF-α and IL-1β activated the
autophagy of chondrocyte cells and murine fibrosarcoma L929 cells
(16,18). Nuclear factor κB (NF-κB) and
mitogen-activated protein kinase (MAPK) signaling pathways,
including extracellular signal-regulated kinases (ERK), c-Jun
N-terminal kinase (JNK) and p38 MAPK signaling pathways, are
involved in the autophagy process. However, the molecular mechanism
appears to be cell-type dependent. Certain studies have identified
those signaling pathways as a potent negative regulator of
autophagy, while others have shown them to be a potent positive
regulator (18–25). Thus far, the impact and molecular
mechanisms of cytokines on the autophagy of NP cells have remained
elusive.
The overall objective of the present study was to
demonstrate the impact of autophagy in catabolic factors regulation
by cytokines and the effect and mechanism of cytokines, TNF-α and
IL-1β, on autophagy in NP cells.
Materials and methods
Reagents
3-Methyladenine (3-MA; autophagy inhibitor),
rapamycin (autophagy activator), SM7368 (NF-κB inhibitor), PD98059
(ERK inhibitor), SP600125 (JNK inhibitor) and SB203580 (p38 MAPK
inhibitor) were purchased from Calbiochem (Danvers, MA, USA). TNF-α
and IL-1β were obtained from Peprotech, Inc. (Rocky Hill, NJ, USA).
Beclin-1 (#3495), LC3 (#12741), GAPDH (#2118) antibody and rabbit
immunoglobulin G (IgG) conjugated with horseradish peroxidase were
obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA),
while the MMP3 (#ab52915) and COX2 (#ab179800) antibodies were
purchased from Abcam (Cambridge, UK). NF-κB reporter construct,
psPAX2, pMD2.G, pRL-TK and pLKO.1 plasmids were kindly provided by
Dr D Xiao (Nanfang Medical University, Guangzhou, China) (26). IKKβ shRNA (TRCN0000018917)
was purchased from Dharmacon, Inc. (Lafayette, CO, USA), and the
knockdown sequence was ATGTTCAAGATATGAACCAGC.
Isolation, culture and treatment of NP
cells
Consistent with the Institutional Review Board
guidelines of Sun Yat-sen University (Guangzhou, China), human NP
tissue samples of Pfirrmann grades 1–2 (27) were obtained from two female
thoracolumbar fracture patients undergoing spinal fusion. Informed
consent for sample collection was obtained from each patient. All
the Sprague-Dawley rats were obtained from the Laboratory Animal
Center of Sun Yat-sen University. Experimental procedures were
approved by the Animal Care and Use Committee of Sun Yat-sen
University.
NP cells were isolated as described by Ye et
al (28). For isolation of
rat NP cells, following euthanization by an overdose of
pentobarbital (100 mg/kg body weight), the lumbar IVDs of
Sprague-Dawley rats, aged 2 months, were collected. Subsequently,
NP tissues were separated from AF tissues under the microscope.
Later, the NP tissues from the same rats were cut into small
pieces, digested with 0.2% pronase medium (Sigma, St. Louis, MO,
USA) for 1 h and subsequently cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco-BRL, Gaithersburg, MD, USA) with 10%
fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and
100 U/ml streptomycin) at 37°C in a 5% CO2 incubator.
The medium was refreshed every 3 days. Subsequent to reaching 80%
confluence, the NP cells were treated with TNF-α or IL-1β and at
corresponding time-points the cell RNA or protein extraction was
performed. The inhibitor or activator was added 1 h before TNF-α or
IL-1β.
Immunofluorescence microscopy
Rat NP cells were plated in 96-well plates
(6×103 cells/well). After the treatment with TNF-α and
IL-1β for 24 h, NP cells were fixed with 4% paraformaldehyde,
permeabilized with 1% Triton X-100 for 10 min and blocked with
phosphate-buffered saline (PBS) containing 5% FBS serum for 1 h at
room temperature. The cells were subsequently incubated with
antibodies against LC3-II antibody (1:200; Cell Signaling
Technology, Inc.) at 4°C overnight. The following day, NP cells
were washed with PBS and were incubated with Alexa Fluor
488-conjugated anti-rabbit (Invitrogen Life Technologies, Carlsbad,
CA, USA) secondary antibody at a dilution of 1:100 for 1 h and 50
µM propidium iodide for 15 min at room temperature. The
images were captured with a fluorescent microscope.
Transfections and dual-luciferase
reporter assay
Rat NP cells were seeded in 48-well plates
(4×104 cells/well) with 2% Opti-MEM. The following day,
250 ng of NF-κB reporter construct and 250 ng pRL-TK plasmids were
premixed with the transfection reagent, Lipofectamine 2000
(Invitrogen Life Technologies) and were co-transfected cells. At 48
h after transfection, the cells were treated with TNF-α (50 ng/ml)
or IL-1β (10 ng/ml) for 24 h, and subsequently the cells were
harvested. Firefly and Renilla luciferase activities were
measured by a dual-luciferase reporter assay (Promega Corporation,
Madison, WI, USA). All the luciferase assays were performed in
triplicate and every experiment was repeated ≥3 times.
IKKβ knockdown
As described previously (28), HEK 293T human embryonic kidney
cells at a density of 3×106 cells/10-cm plate were
seeded in DMEM with 10% heat-inactivated FBS. Approximately 24 h
later, cells were transfected with 9 µg of shRNA control
sequence or IKK shRNA plasmids, along with 6 µg
psPAX2 and 3 µg pMD2.G. The transfection medium was replaced
with DMEM with 10% heat-inactivated FBS 16 h later. At 48 and 60 h
after transfection, the plasmid medium containing lentiviral
particles was harvested from HEK 293T cells. Subsequently, a virus
solution replaced the medium in the plate-seeded human NP cells at
a density of 0.5×106 cells/10-cm plate, along with 6
mg/ml polybrene. Five days later, cells were harvested for protein
extraction.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted with TRIzol reagent
(Invitrogen Life Technologies) following the manufacturer's
instructions. Single-stranded cDNA templates were prepared from
2,000 ng total RNA using SuperScript III Reverse Transcriptase
(Invitrogen Life Technologies). Template cDNA and gene-specific
primers were added to Fast SYBR Green Master mix (Applied
Biosystems, Foster City, CA, USA) and mRNA expression was
quantified using the 7900HT Fast Real-Time PCR System (Applied
Biosystems). Hprt was used to normalize the expression. Each
sample was analyzed in duplicate. All the primers used were
synthesized by Shanghai Sangon Biological Engineering Technology
& Services Co., Ltd. (Shanghai, China).
The primers were as follows: MMP2 sense,
GGTGGTGGTCACAGCTATTT and antisense, CCAGCCAGTCCGATTTGAT;
MMP3 sense, CAGGGAAAGTGACCCACATATT and antisense,
CGCCAAGTTTCAGAGGAAGA; MMP9 sense, CCCAACCTTTACCAGCTACTC and
antisense, GTCAGAACCGACCCTACAAAG; ADMATS4 sense,
GGAGATCGTGTTTCCAGAGAAG and antisense, CAAAGGCTGGTAATCGGTACA;
COX2 sense, TCAACCAGCAGTTCCAGTATC and antisense,
GTGTACTCCTGGTCTTCAATGT; and Hprt sense, GCTGACCTGCTGGATTACAT
and anti-sense, CCCGTTGACTGGTCATTACA.
Western blot analysis
Following the treatment, the plates with NP cells
were placed on ice immediately. The condition medium was collected
with corresponding collection tubes, and cells were washed with
ice-cold PBS twice and collected with scrapers. Following
centrifugation (10,000 × g), the cells were treated with lysis
buffer, including 1X Protease Inhibitor Cocktail (Roche Diagnostics
GmbH, Mannheim, Germany), NaCl (5 mM), NaF (200 µM),
Na3VO4 (200 µM) and dithio-threitol
(0.1 mM). Subsequently, total cell proteins (30 ng) qualified with
bicinchoninic acid reagent were resolved on 10–15%
SDS-polyacrylamide gels and transferred by electroblotting to PVDF
membranes (Bio-Rad, Hercules, Hercules, CA, USA). The membranes
were blocked with 5% non-fat dry milk in 50 mM Tris (pH 7.6), 150
mM NaCl and 0.1% Tween-20, and incubated overnight at 4°C with
anti-Beclin-1 (1:1,000), anti-LC3 (1:1,000), anti-COX2 (1:1,000),
anti-MMP3 (1:500), anti-IKKβ (1:1,000) and anti-GAPDH (1:3,000).
Finally, the membranes were incubated in anti-serum against rabbit
or mouse IgG conjugated with horseradish peroxidase (Cell Signaling
Technology, Inc.) (1:1,000–5,000) for 1 h and subsequently treated
with ECL Plus according to the manufacturer's instructions
(Amersham Pharmacia Biotech, Umeå, Sweden). Blot intensity was
determined by densitometric analysis using Kodak 1D 3.6 software
(Kodak, Rochester, NY, USA). Beclin-1, LC3-II, COX2, MMP3 and IKKβ
protein expression data were normalized to GAPDH expression, which
was the internal control.
Acridine orange staining for NP cell
autophagy
As a marker of autophagy, the volume of the cellular
acidic compartment was visualized by acridine orange staining;
acridine orange staining of rat NP cells was performed as described
by Paglin et al (29) and
Wang et al (30). Rat NP
cells (6×103 cells/well) were seeded in 96-well plates,
and at 50% confluence the cells were treated with TNF-α and SM7368
or SP600125. The cells were incubated with acridine orange (1
µg/ml) 24 h later at 37°C. After 30 min, the acridine orange
was removed and a confocal microscope was used to immediately
detect the autophagy; 488 nm excitation light, and 520–530 nm
(green) and 650 nm (red) emission light were used. The acidic
autophagic vacuoles exhibit red fluorescence, whereas the cytoplasm
and nucleus of the stained cells exhibit bright green
fluorescence.
Statistical analysis
All the experiments were performed in triplicate.
All the data are presented as the mean ± standard error.
Differences between the groups were analyzed by one-way analysis of
variance. P<0.05 were considered to indicate a statistically
significant difference.
Results
Autophagy decreases the catabolic effect
of IL-1β
Rapamycin and 3-MA are an autophagy activator and
inhibitor, respectively. To investigate the impact of autophagy on
the catabolic effect, rapamycin (1 µM) or 3-MA (5 mM) was
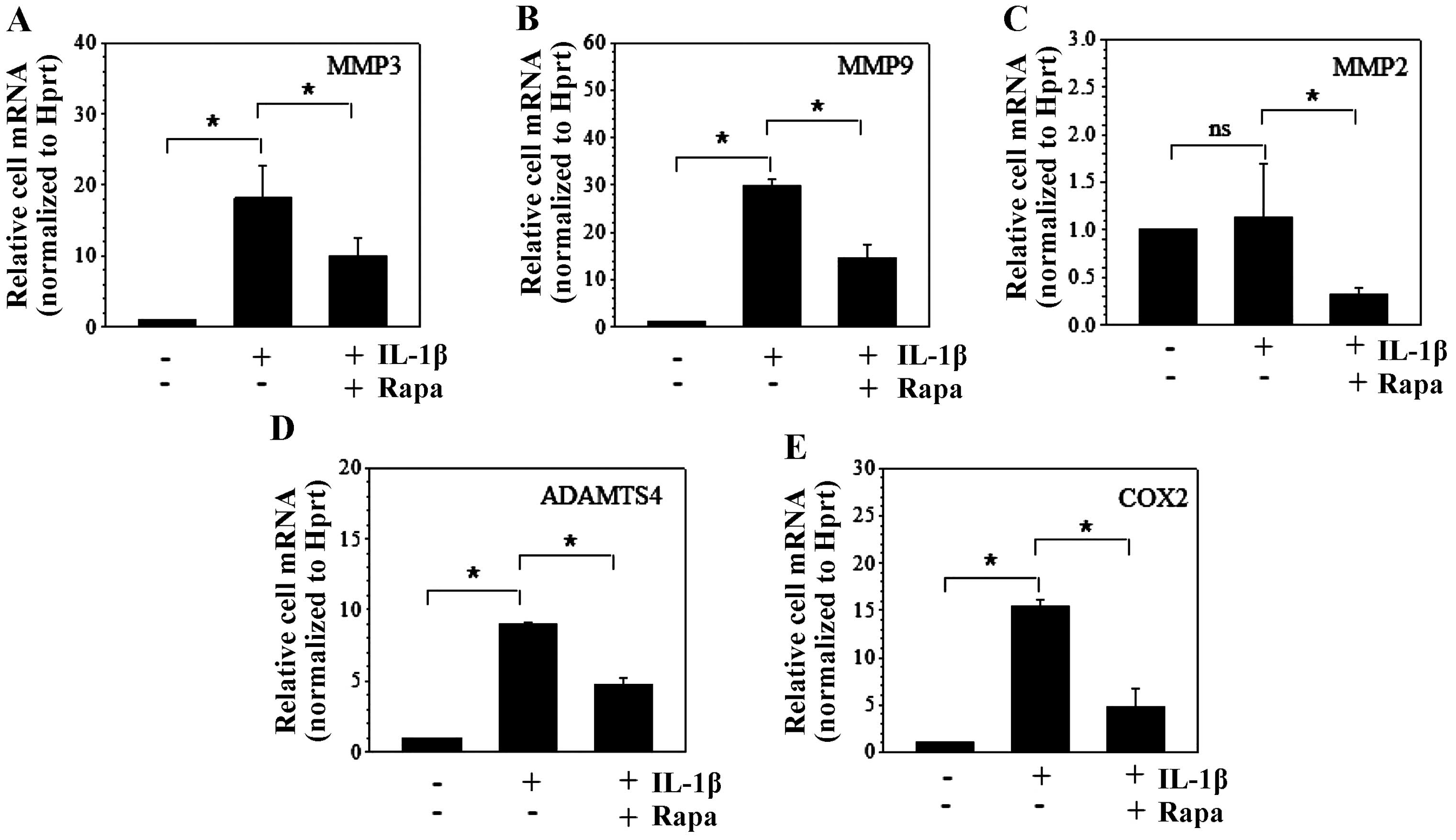
added 1 h before 10 ng/ml IL-1β. Compared to the control group,
IL-1β stimulation led to ~18- and 30-fold increases of MMP3
and MMP9 mRNA expression in NP cells, respectively, and
additional rapamycin treatment resulted in decreases of ~10- and
16-fold, respectively (Fig. 1A and
B). Although IL-1β had no effect on MMP2 mRNA
expression, rapamycin significantly suppressed MMP2 mRNA
expression when IL-1β was present (Fig. 1C). In addition, ADAMTS4 and
COX2 mRNA expression were induced to ~9- and 15-fold by
IL-1β treatment and reduced to ~4.5- and 5-fold following the
additional rapamycin stimulation, respectively (Fig. 1D and E). Since the baseline
expression of catabolic factors, such as MMP3 and ADAMTS5, was not
too high, we did not observe the effects of rapamycin alone.
Autophagy decreases the catabolic effect
of TNF-α
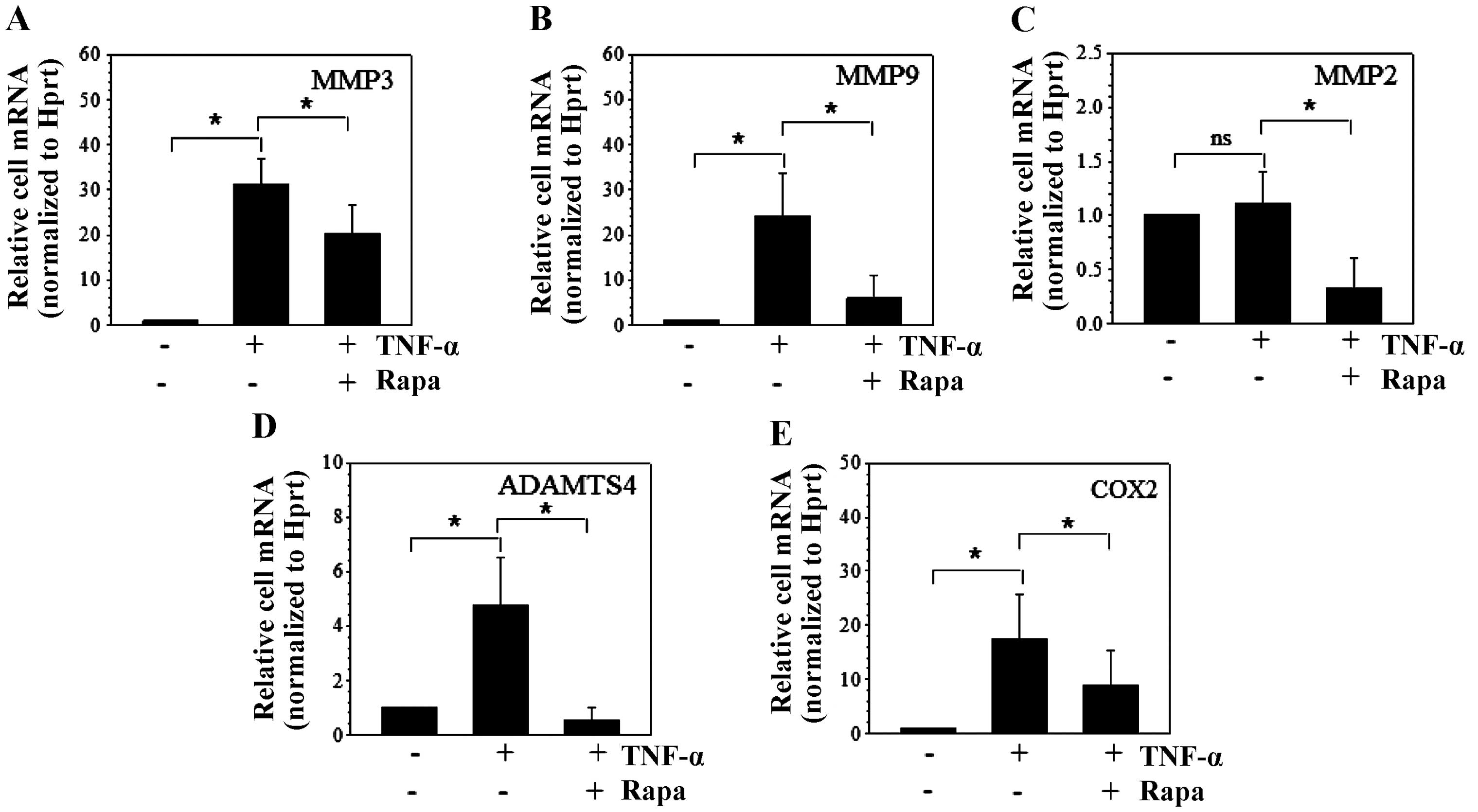
Similarly, rapamycin (1 µM) or 3-MA (5 mM)
was used to observe the effect of autophagy on the catabolic effect
induced by 50 ng/ml TNF-α. TNF-α stimulation led to the increase of
MMP3 and MMP9 mRNA expression in NP cells (Fig. 2A–C). However, additional rapamycin
treatment decreased MMP3 and MMP9 mRNA expression
(Fig. 2A and B). As with IL-1β
treatment, MMP2 mRNA expression was not regulated by TNF-α,
however, rapamycin reduced MMP2 mRNA expression when TNF-α
was present (Fig. 2C).
Additionally, ADAMTS4 and COX2 mRNA expression was
upregulated by TNF-α, and was markedly suppressed by additional
rapamycin treatment (Fig. 2D and
E).
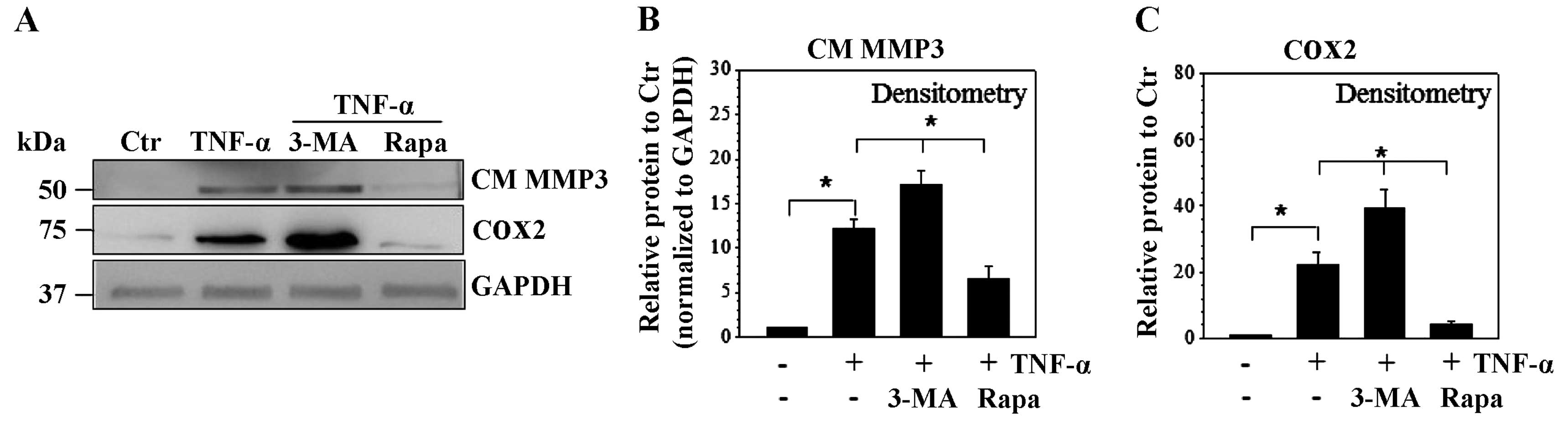
To confirm the effect of autophagy on the catabolic
effect induced by cytokines, western blot analysis was used to
demonstrate the protein expression of the catabolic factors. The
results showed that following TNF-α treatment the expression of
medium protein, MMP3, and cell protein, COX2, increased to ~12- and
22-fold, respectively. Similar to the mRNA results, additional
rapamycin repressed the expression to ~6- and 4-fold, respectively.
However, the autophagy inhibitor, 3-MA, significantly upregulated
the protein expression to ~18- and 39-fold, respectively (Fig. 3A–C).
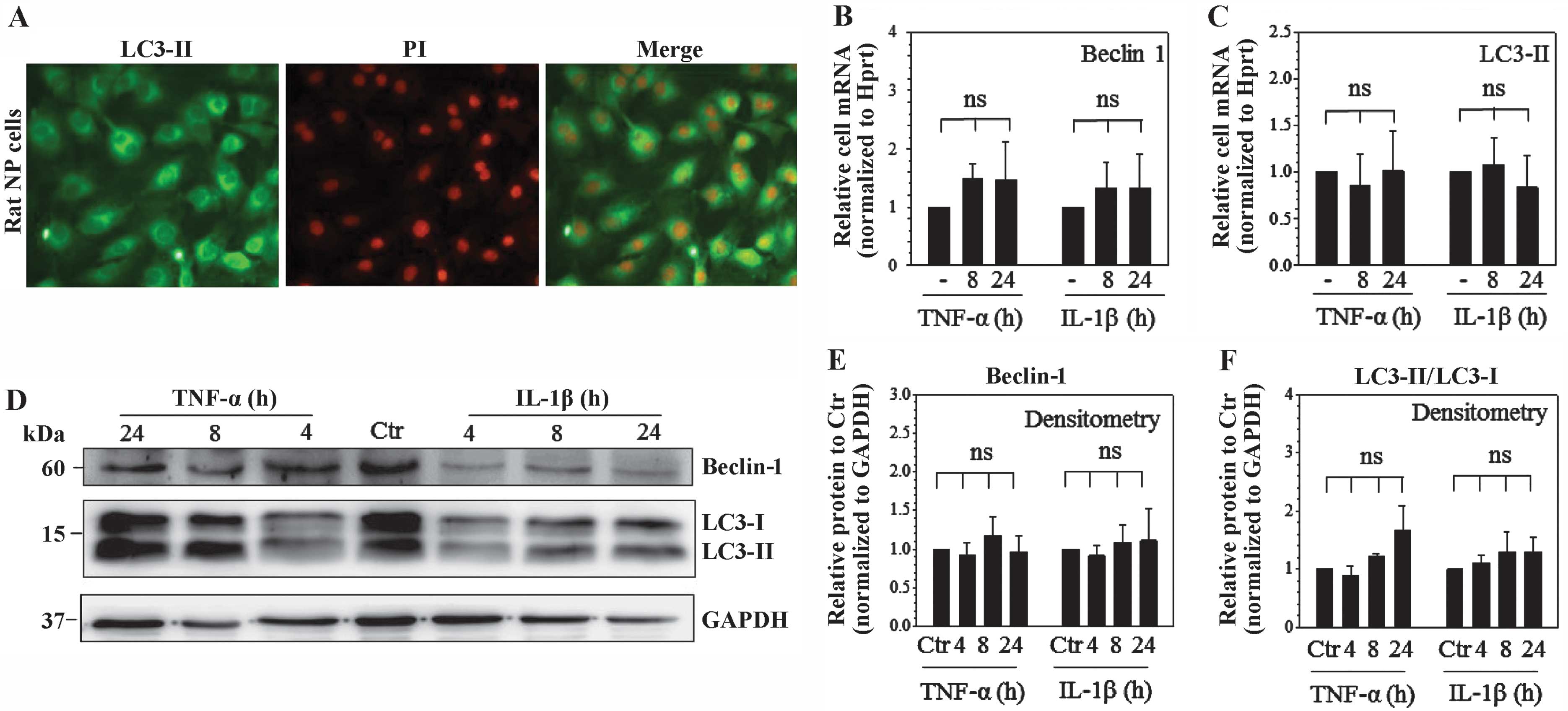
Autophagy is not regulated by TNF-α and
IL-1β in rat NP cells
There are numerous methods to identify the presence
of autophagy in the cultured experimental cells. Among them,
electron microscopy, LC3 turnover (LC3-I to LC3-II) and LC3-II
expression are frequently used. LC3B immunofluorescence, acridine
orange, monodansylcadav-erine staining and other methods are
auxiliary methods to identify the presence of autophagy (29–35). To determine the effect of TNF-α
and IL-1β on the autophagy of NP cells, immunofluorescence, RT-qPCR
and western blot analyses were performed. Immunofluorescence
microscopy showed the LC3-II expressed in rat NP cells (Fig. 4A) and no difference was observed
following treatment with TNF-α and IL-1β. RT-qPCR and western blot
analysis showed 8–24 h TNF-α and IL-1β treatment had no significant
effect on the mRNA expression of Beclin-1, a marker of early
phagophore formation or autophagy initiation, and LC3-II, the
categorical autophagy-specific marker and indicator of
autophagosome maturation (Fig. 4B and
C). In addition, the Beclin-1 and LC3-II protein turnover
showed no significant change after 4–24 h of TNF-α and IL-1β
stimulation (Fig. 4D–F).
Autophagy of rat NP cells is induced by
NF-κB and JNK inhibition in inflammatory conditions
To investigate the molecular mechanism of NP cell
autophagy in inflammatory conditions, TNF-α was used to mimic the
inflammatory condition and the NP cells were treated with NF-κB or
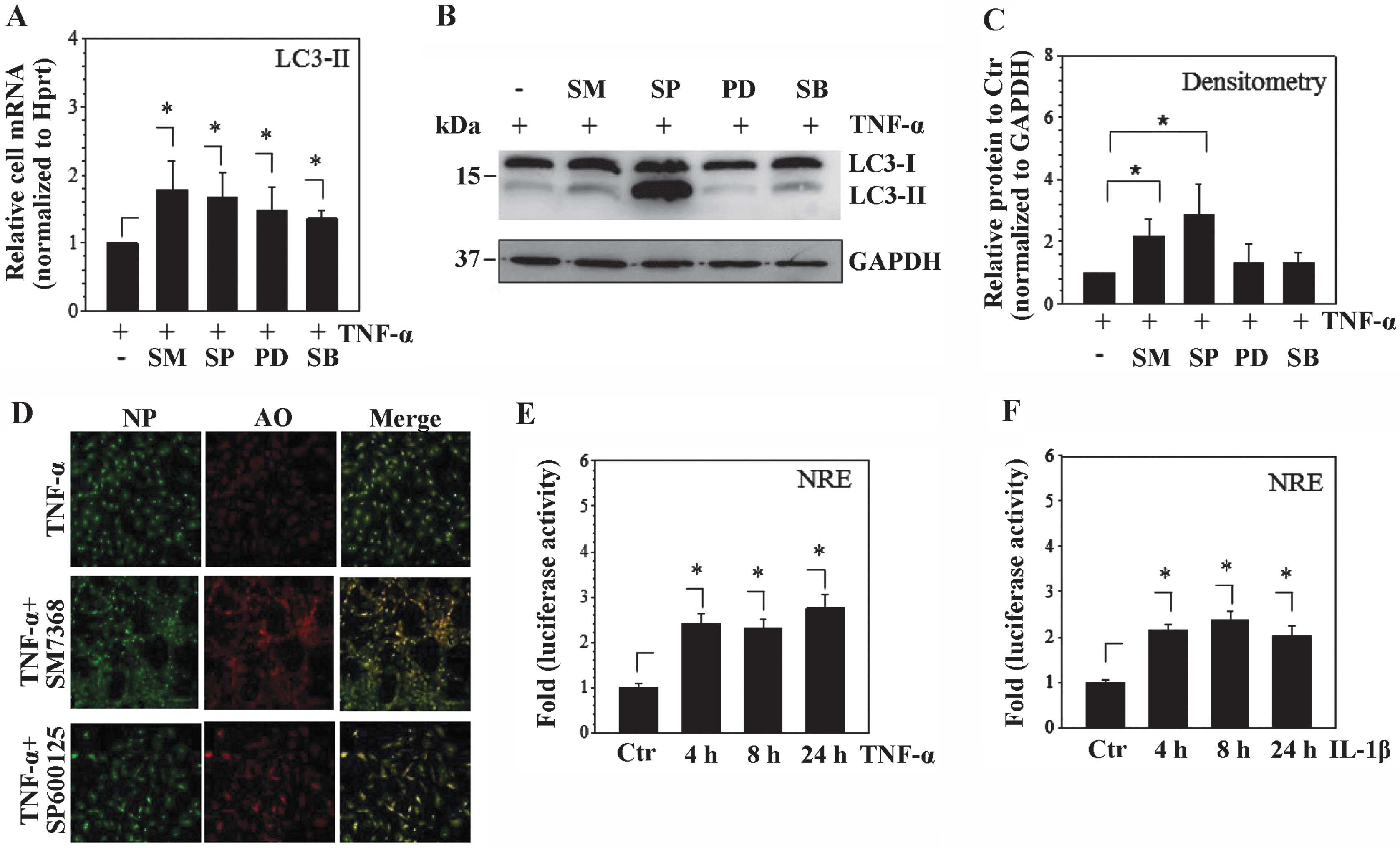
MAPK inhibitors prior to TNF-α. SM7368, PD98059, SP600125 and
SB203580 significantly increased the Beclin-1 and LC3-II mRNA
expression in inflammatory conditions (Fig. 5A). However, only SM7368 and
SP600125 treatment increased the LC3 protein turnover
(LC3-II/LC3-I) (Fig. 5B and C).
To further verify the effect of the NF-κB and JNK inhibitor,
acridine orange staining was used to detect the autophagosome
formation following SM7368 or SP600125 stimulation. The data showed
that SM7368 or SP600125 treatment increased the autophagosome
formation (Fig. 5D). To verify
the efficacy of cytokines TNF-α and IL-1β on rat NP cells, the
activity of the NF-κB promoter construct was measured following
cytokine stimulation. As expected, the results showed that TNF-α or
IL-1β significantly increased the activity of the NF-κB promoter
construct (Fig. 5E and F).
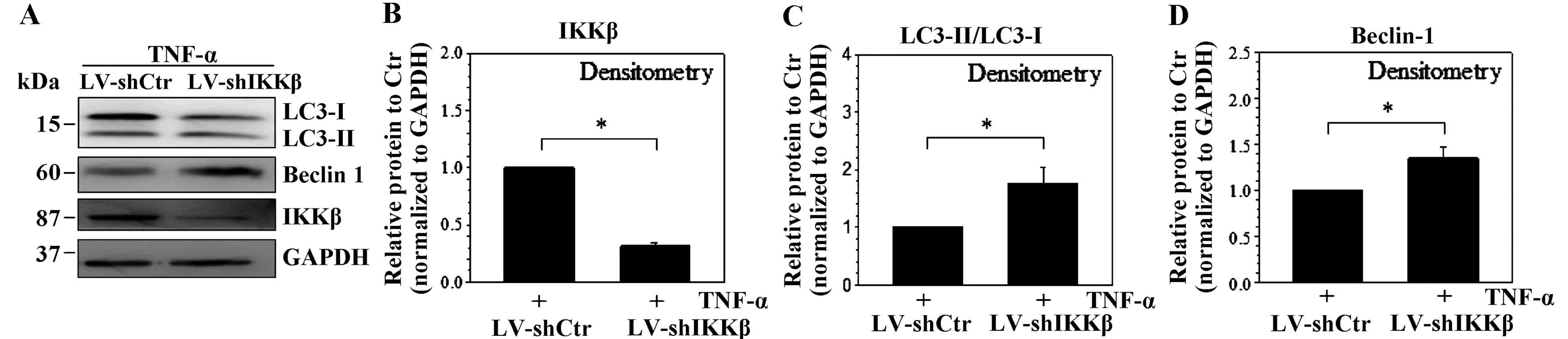
Autophagy of NP cells is induced by IKKβ
knockdown
To further confirm the involvement of the NF-κB
signaling pathway in controlling NP cells autophagy, an IKKβ
loss-of-function study in TNF-α conditions using shRNA transduction
was performed. As expected, compared to the transduction with
control shRNA, densitometric analysis showed transduction with
shIKKβ led to ~80% decrease of IKKβ protein in TNF-α conditions
(Fig. 6A and B). Accordingly,
transduction with shIKKβ in human NP cells resulted in a
significant increase of the LC3 protein turnover and Beclin-1
protein in the inflammatory conditions (Fig. 6C and D).
Discussion
The experiments described in the present
investigation demonstrated that autophagy activation suppressed,
while autophagy inhibition promoted, the catabolic effects induced
by TNF-α and IL-1β. In addition, although autophagy-related mRNA
and protein expression in NP cells was refractory to TNF-α and
IL-1β, NF-κB and JNK inhibition increased the autophagy expression
in inflammatory conditions.
Proteoglycan degradation contributes to the
pathogenesis of IVD degeneration. TNF-α and IL-1β, significant
inflammatory cytokines in IVDs, increased the proteoglycan
degradation through the regulation of catabolic factors, such as
MMP3, MMP9, ADAMTS4, ADAMTS5 and COX2 (7,36).
In chondrocyte cells, autophagy suppressed the expression of
catabolic factors induced by TNF-α and IL-1β (16,17). The present data also showed that
TNF-α and IL-1β increased the mRNA expression of catabolic factors,
MMP3, MMP9, ADAMTS4 and COX2.
Additionally, autophagy repressed the effect of TNF-α and IL-1β on
MMP3, MMP9, ADAMTS4 and COX2 mRNA
expression. Autophagy also suppressed MMP2 mRNA expression
in inflammatory conditions. Furthermore, the present results
demonstrated autophagy inhibition could promote, while autophagy
activation could repress, the catabolic effect of TNF-α on MMP3 and
COX2 protein expression. Therefore, autophagy decreased the effect
of cytokines, TNF-α and IL-1β, and thus, autophagy should be a
protective factor in the process of IVD degeneration.
Of note, although TNF-α and IL-1β increased the
proteoglycan degradation (7,36),
autophagy was induced by TNF-α and IL-1β in chondrocytes, AF cells,
fibrosarcoma L929 cells and breast cancer cells (14,16,18,30). The present data showed that, as
opposed to in chondrocyte or cancer cells, the mRNA and protein
expression of autophagy-related genes, LC3 and Beclin-1, showed no
change in response to TNF-α and IL-1β. Therefore, cytokines
appeared to be less critical for autophagy in NP cells compared
with the other types of cells.
Inflammatory conditions were present in degenerative
discs (37). Ye et al
(18) showed that NF-κB and p38
inhibitor enhanced the TNF-α-induced autophagy in inflammatory
conditions. Djavaheri-Mergny et al (38) also demonstrated that NF-κB
activation represses TNF-α-induced autophagy. However, Copetti
et al (39) showed that
NF-κB can induce autophagy by transactivating Beclin-1. The
autophagy regulation by MAPK is similar to NF-κB (19,40,41). The present data showed that in the
inflammatory conditions, NF-κB, JNK, ERK and P38 MAPK inhibitor
upregulated LC3-II mRNA expression; however, only the NF-κB and JNK
inhibitor promoted the LC3 protein turnover. Furthermore, IKKβ loss
of function experiments showed knockdown of IKKβ led to the
increase of LC3 protein turnover even though TNF-α was present.
Taken together, the NF-κB and JNK signaling pathway inhibition
sustained the autophagy of NP cells in the presence of TNF-α.
In conclusion, inhibition of autophagy could
diminish, and activation of autophagy could enhance, the catabolic
effect of cytokines. Although autophagy is refractory to cytokines
in NP cells, the NF-κB and JNK signaling pathway inhibition
sustained the autophagy of NP cells in inflammatory conditions.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81101385), the
Natural Science Foundation of Guangdong Province, China (no.
10151008901000084) and the Science and Technology Planning Project
of Guangdong Province, China (nos. 2009B060700097, 2012B031800359,
2012B031800360 and 2014A020212058).
References
|
1
|
Luoma K, Riihimäki H, Luukkonen R,
Raininko R, Viikari-Juntura E and Lamminen A: Low back pain in
relation to lumbar disc degeneration. Spine. 25:487–492. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pockert AJ, Richardson SM, Le Maitre CL,
Lyon M, Deakin JA, Buttle DJ, Freemont AJ and Hoyland JA: Modified
expression of the ADAMTS enzymes and tissue inhibitor of
metalloproteinases 3 during human intervertebral disc degeneration.
Arthritis Rheum. 60:482–491. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Séguin CA, Pilliar RM, Roughley PJ and
Kandel RA: Tumor necrosis factor-alpha modulates matrix production
and catabolism in nucleus pulposus tissue. Spine. 30:1940–1948.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Studer RK, Gilbertson LG, Georgescu H,
Sowa G, Vo N and Kang JD: p38 MAPK inhibition modulates rabbit
nucleus pulposus cell response to IL-1. J Orthop Res. 26:991–998.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cui LY, Liu SL, Ding Y, Huang DS, Ma RF,
Huang WG, Hu BS and Pan QH: IL-1beta sensitizes rat intervertebral
disc cells to Fas ligand mediated apoptosis in vitro. Acta
Pharmacol Sin. 28:1671–1676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Genevay S, Finckh A, Mezin F, Tessitore E
and Guerne PA: Influence of cytokine inhibitors on concentration
and activity of MMP-1 and MMP-3 in disc herniation. Arthritis Res
Ther. 11:R1692009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tian Y, Yuan W, Fujita N, Wang J, Wang H,
Shapiro IM and Risbud MV: Inflammatory cytokines associated with
degenerative disc disease control aggrecanase-1 (ADAMTS-4)
expression in nucleus pulposus cells through MAPK and NF-κB. Am J
Pathol. 182:2310–2321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ogier-Denis E and Codogno P: Autophagy: A
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
9
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scheper W, Nijholt DA and Hoozemans JJ:
The unfolded protein response and proteostasis in Alzheimer
disease: Preferential activation of autophagy by endoplasmic
reticulum stress. Autophagy. 7:910–911. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caramés B, Hasegawa A, Taniguchi N, Miyaki
S, Blanco FJ and Lotz M: Autophagy activation by rapamycin reduces
severity of experimental osteoarthritis. Ann Rheum Dis. 71:575–581.
2012. View Article : Google Scholar :
|
|
12
|
Ye W, Xu K, Huang D, Liang A, Peng Y, Zhu
W and Li C: Age-related increases of macroautophagy and
chaperone-mediated autophagy in rat nucleus pulposus. Connect
Tissue Res. 52:472–478. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye W, Zhu W, Xu K, Liang A, Peng Y, Huang
D and Li C: Increased macroautophagy in the pathological process of
inter-vertebral disc degeneration in rats. Connect Tissue Res.
54:22–28. 2013. View Article : Google Scholar
|
|
14
|
Shen C, Yan J, Jiang LS and Dai LY:
Autophagy in rat annulus fibrosus cells: Evidence and possible
implications. Arthritis Res Ther. 13:R1322011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma KG, Shao ZW, Yang SH, Wang J, Wang BC,
Xiong LM, Wu Q and Chen SF: Autophagy is activated in
compression-induced cell degeneration and is mediated by reactive
oxygen species in nucleus pulposus cells exposed to compression.
Osteoarthritis Cartilage. 21:2030–2038. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin NY, Beyer C, Giessl A, Kireva T,
Scholtysek C, Uderhardt S, Munoz LE, Dees C, Distler A, Wirtz S, et
al: Autophagy regulates TNFα-mediated joint destruction in
experimental arthritis. Ann Rheum Dis. 72:761–768. 2013. View Article : Google Scholar
|
|
17
|
Sasaki H, Takayama K, Matsushita T, Ishida
K, Kubo S, Matsumoto T, Fujita N, Oka S, Kurosaka M and Kuroda R:
Autophagy modulates osteoarthritis-related gene expression in human
chondrocytes. Arthritis Rheum. 64:1920–1928. 2012. View Article : Google Scholar
|
|
18
|
Ye YC, Yu L, Wang HJ, Tashiro S, Onodera S
and Ikejima T: TNFα-induced necroptosis and autophagy via
supression of the p38-NF-κB survival pathway in L929 cells. J
Pharmacol Sci. 117:160–169. 2011. View Article : Google Scholar
|
|
19
|
Xu P, Das M, Reilly J and Davis RJ: JNK
regulates FoxO-dependent autophagy in neurons. Genes Dev.
25:310–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sommermann TG, Mack HI and Cahir-McFarland
E: Autophagy prolongs survival after NFκB inhibition in B-cell
lymphomas. Autophagy. 8:265–267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JE, You DJ, Lee C, Ahn C, Seong JY and
Hwang JI: Suppression of NF-kappaB signaling by KEAP1 regulation of
IKKbeta activity through autophagic degradation and inhibition of
phosphorylation. Cell Signal. 22:1645–1654. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Colleran A, Ryan A, O'Gorman A, Mureau C,
Liptrot C, Dockery P, Fearnhead H and Egan LJ: Autophagosomal
IkappaB alpha degradation plays a role in the long term control of
tumor necrosis factor-alpha-induced nuclear factor-kappaB
(NF-kappaB) activity. J Biol Chem. 286:22886–22893. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li DD, Wang LL, Deng R, Tang J, Shen Y,
Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al: The pivotal role of
c-Jun NH2-terminal kinase-mediated Beclin 1 expression during
anticancer agents-induced autophagy in cancer cells. Oncogene.
28:886–898. 2009. View Article : Google Scholar
|
|
24
|
Comes F, Matrone A, Lastella P, Nico B,
Susca FC, Bagnulo R, Ingravallo G, Modica S, Lo Sasso G, Moschetta
A, et al: A novel cell type-specific role of p38alpha in the
control of autophagy and cell death in colorectal cancer cells.
Cell Death Differ. 14:693–702. 2007. View Article : Google Scholar
|
|
25
|
Tang G, Yue Z, Talloczy Z, Hagemann T, Cho
W, Messing A, Sulzer DL and Goldman JE: Autophagy induced by
Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK
and mTOR signaling pathways. Hum Mol Genet. 17:1540–1555. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang SC, Lin XL, Li J, Zhang TT, Wang HY,
Shi JW, Yang S, Zhao WT, Xie RY, Wei F, et al: MicroRNA-122
triggers mesenchymal-epithelial transition and suppresses
hepatocellular carcinoma cell motility and invasion by targeting
RhoA. PLoS One. 9:e1013302014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pfirrmann CW, Metzdorf A, Zanetti M,
Hodler J and Boos N: Magnetic resonance classification of lumbar
intervertebral disc degeneration. Spine. 26:1873–1878. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye W, Zhou J, Markova DZ, Tian Y, Li J,
Anderson DG and Shapiro IM: novel regulation by AP-1, Sp1, and Sp3.
Am J Pathol. 185:485–495. 2015. View Article : Google Scholar
|
|
29
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
30
|
Wang J, Kim TH, Ahn MY, Lee J, Jung JH,
Choi WS, Lee BM, Yoon KS, Yoon S and Kim HS: Sirtinol, a class III
HDAC inhibitor, induces apoptotic and autophagic cell death in
MCF-7 human breast cancer cells. Int J Oncol. 41:1101–1109.
2012.PubMed/NCBI
|
|
31
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Caramés B, Kiosses WB, Akasaki Y, Brinson
DC, Eap W, Koziol J and Lotz MK: Glucosamine activates autophagy in
vitro and in vivo. Arthritis Rheum. 65:1843–1852. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen JW, Ni BB, Li B, Yang YH, Jiang SD
and Jiang LS: The responses of autophagy and apoptosis to oxidative
stress in nucleus pulposus cells: Implications for disc
degeneration. Cell Physiol Biochem. 34:1175–1189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cao Y, Yang W, Tyler MA, Gao X, Duan C,
Kim SO, Aronson JF, Popov V, Takahashi H, Saito H, et al: Noggin
attenuates cerulein-induced acute pancreatitis and impaired
autophagy. Pancreas. 42:301–307. 2013. View Article : Google Scholar
|
|
35
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Millward-Sadler SJ, Costello PW, Freemont
AJ and Hoyland JA: Regulation of catabolic gene expression in
normal and degenerate human intervertebral disc cells: Implications
for the pathogenesis of intervertebral disc degeneration. Arthritis
Res Ther. 11:R652009. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vo NV, Hartman RA, Yurube T, Jacobs LJ,
Sowa GA and Kang JD: Expression and regulation of
metalloproteinases and their inhibitors in intervertebral disc
aging and degeneration. Spine J. 13:331–341. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Djavaheri-Mergny M, Amelotti M, Mathieu J,
Besançon F, Bauvy C, Souquère S, Pierron G and Codogno P: NF-kappaB
activation represses tumor necrosis factor-alpha-induced autophagy.
J Biol Chem. 281:30373–30382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Copetti T, Bertoli C, Dalla E, Demarchi F
and Schneider C: p65/RelA modulates BECN1 transcription and
autophagy. Mol Cell Biol. 29:2594–2608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sivaprasad U and Basu A: Inhibition of ERK
attenuates autophagy and potentiates tumour necrosis
factor-alpha-induced cell death in MCF-7 cells. J Cell Mol Med.
12:1265–1271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Insulin-like growth factor-1 and TNF-alpha regulate autophagy
through c-jun N-terminal kinase and Akt pathways in human
atherosclerotic vascular smooth cells. Immunol Cell Biol.
84:448–454. 2006. View Article : Google Scholar : PubMed/NCBI
|