Introduction
Myocardial infarction (MI), also known as a 'heart
attack', results from a reduction in coronary blood flow which is
extensive enough to render the oxygen supply to myocardial tissues
insufficient (1,2). Ischemia and consequent oxygen
shortage, if left untreated for a long period of time, can cause
myocardial cell death and necrosis (3–5).
MI remains a leading cause of morbidity and mortality among all
cardiovascular diseases in both developed and developing countries
(6–8). Successful reperfusion therapy for MI
can prevent heart failure, leading to reduced mortality (9–11).
However, a side-effect of this therapy is myocardial
ischemia/reperfusion (I/R), which can lead to tissue damage and
pathological remodeling (9,10).
Furthermore, I/R injury is a critical factor in the pathogenesis of
tissue injury following MI, multiple organ failure and other acute
ischemic events (11). Therefore,
it is necessary to try and prevent myocardial I/R injury as an
adjunct therapy for the treatment of MI. As an important in
vitro model for myocardial I/R injury, hypoxia/reoxygenation
(H/R) is similar to myocar dial I/R (12), and is one of the cellular stresses
in pathological conditions, such as MI (13). Thus, it is imperative to explore
the molecular mechanisms responsible for H/R injury and to identify
possible treatment targets with which to prevent H/R injury in
cardiomyocytes.
High mobility group box 1 (HMGB1, also known as
amphoterin) is an abundantly occurring parental form of HMG
proteins and an exceptional member of the family of HMG-box
proteins (14,15). Dependent on the cell type and its
activation state, HMGB1 exhibits a non-nuclear localization and is
secreted from cells, in contrast to the majority of HMG-box
proteins that are strictly bound to the cell nuclei (16,17). HMGB1 can be passively released
from injured cells (15,16), and has been implicated in the
development of MI by a number of studies (18–21). Over the past few years, studies
have demonstrated that HMGB1 functions as an extracellular
signaling molecule (22) that can
promote autophagy in multiple biological processes, such as under
conditions of oxidative stress (23,24). It has been reported that autophagy
is enhanced during myocardial I/R injury (25). However, another study suggested
that although autophagy exerts protective effects during ischemia,
it plays a detrimental role during reperfusion (26). Thus, the ambiguous role of
autophagy in myocardial H/R injury remains to be elucidated, and
whether HMGB1-mediated autophagy is involved in the development of
myocardial H/R injury remains largely unclear. Therefore, in the
present study, we evaluated the effects of HMGB1-mediated autophagy
on the apoptosis of H9c2 cells and on epithelial-to-mesenchymal
transition (EMT). In addition, we investigated the molecular
mechanisms responsible for these effects during myocardial H/R
injury.
Materials and methods
Cell culture and treatment
The cardiomyocyte cell line, H9c2 cells was obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). The cells were cultured in DMEM medium containing 10% fetal
bovine serum (FBS) and 1% antibiotic-antimycotic solution (100 U/ml
penicillin and 100 μg/ml streptomycin). The cells were
maintained at 37°C in a humidified atmosphere containing 5%
CO2. Furthermore, the cells were treated with 3-MA
(Selleck Chemicals, TX, USA), an autophagy inhibitor, in order to
determine whether the autophagy process is involved in H/R injury.
In addition, as an inhibitor of mammalian target of rapamycin
(mTOR), OSI-027 (Selleck Chemicals, TX, USA) was added to the cells
to observe the function of mTOR signaling.
Construction of a cell model of H/R
injury using H9c2 cells
The exposure of the cells to H/R was performed as
previously described in the study by Cao et al (9). Hypoxia was achieved by placing the
H9c2 cells in a hypoxia chamber filled with 5% CO2 and
95% N2 at 37°C for 4 h. Following exposure to hypoxia, the cells
were reoxygenated with 5% CO2 and 95% air for 3 h in
DMEM with 10% FBS. Cells not subjected to H/R were used as the
negative controls.
Construction of rat animal model of H/R
injury
A total of 40 male Sprague-Dawley rats (weighing
320±20 g) were obtained from the Experimental Animal Center of the
Second Xiangya Hospital of Central South University, Changsha,
China. All animal experiments were performed in accordance with the
National Institutes of Health Guidelines on the Use of Laboratory
Animals and were approved by the Ethics Committee of the Second
Xiangya Hospital. The rats were kept in plastic cages and had
access to pelleted food and water ad libitum. They were kept
under standard laboratory conditions (12-h light/dark cycle,
controlled temperature of 20–22°C). The rats were anesthetized with
pentobarbital sodium (50 mg, intraperitoneal), and intu-bated and
ventilated with 100% oxygen. The chest was opened via a left
thoracotomy through the fourth or fifth intercostals space and the
hearts were exposed. An 8-0 silk ligature was placed under the left
coronary artery (LCA) and tied using a shoestring knot. MI was
confirmed by the presence of discoloration of the ischemic area,
left ventricular (Lv) dyskinesia and ST-segment elevation on an
electrocardiogram. Following occlusion for 30 min, reperfusion was
initiated by releasing the knot. Reperfusion was confirmed by the
return of color to the ischemic area. The loosened suture was left
in place and then retied for the purpose of evaluating the ischemic
area. The chest wall was closed, the animal extubated and body
temperature maintained by the use of a 37°C warm plate. In the
sham-operated animals, the same procedure was performed with the
exception of the coronary artery ligation. Following reperfusion,
the rats were sacrificed by femoral artery bloodletting following
anesthetization, and the rat hearts were harvested using surgical
scissors. The cardiac base and cardiac apex tissue was separated
for morphological analysis and molecular biological detection,
respectively at 0, 1 and 2 weeks post-MI.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the cells and heart
tissue using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and the
cDNA was reverse transcribed as the protocol for the reverse
transcription system (Fermentas, Burlington, ON, Canada). The
amplification primers were designed using Primer 5.0 with melting
temperatures at 58°C. The forward and reverse sequences of the
primers used are presented in Table
I. Quantitative PCR (qPCR) was performed using SYBR-Green qRCR
(Toyobo, Osaka, Japan) in order to determine the mRNA expression
levels of various genes [HMGB1, miR-210, cleaved caspase-3, Bcl-2
and discoidin domain receptor 1 (DDR1)] according to the
manufacturer's instructions. Three repetitions were performed for
qRT-PCR. The relative mRNA levels were normalized to those of
β-actin mRNA, and were evaluated using the 2−ΔΔCt
method.
 | Table IForward and reverse primer sequences
of targeted genes. |
Table I
Forward and reverse primer sequences
of targeted genes.
| Gene | Name | Sequence |
|---|
| HMGB1 | Sense |
GATGGGCAAAGGAGATCCTA |
| Antisense |
CTTGGTCTCCCCTTTGGGGG |
| DDR1 | Sense |
ATGGAGCAACCACAGCTTCTC |
| Antisense |
CTCAGCCGGTCAAACTCAAACT |
| Cleaved
caspase-3 | Sense |
GAGCTGCCTGTAACTTG |
| Antisense |
ACCTTTAGAACATTTCCACT |
| Bcl-2 | Sense |
TTGCCACGGTGGTGGAGGAAC |
| Antisense |
GACAGCCAGGAGAAATCAAACAGA |
| β-actin | Sense |
AGGGGCCGGACTCGTCATACT |
| Antisense |
GGCGGCACCACCATGTACCCT |
Western blot analysis
Protein extracts from the H9c2 cells were prepared
using RIPA Lysis buffer (Auragene Bioscience Co., Changsha, China)
following the manufacturer's instructions. The protein
concentration was determined according to the Bradford Protein
assay reagent (Beyotime Institute of Biotechnology, Shanghai,
China) using bovine serum albumin as a standard. Western blot
analysis was subsequently carried out in order to detect the
protein levels of HMGB1, cleaved caspase-3, Bcl-2, Beclin 1, light
chain 3 (LC3)-II/I and DDR1. Equal amounts of lysate were resolved
by sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto PVDF membranes (Millipore, Bedford,
MA, USA) using a semi-dry transfer method. The membranes were
blocked with 5% non-fat milk in TBST buffer at room temperature for
2 h and then incubated overnight with the following primary
antibodies: HMGB rabbit polyclonal antibody (1:1,000; sc-33199;
Santa Cruz Biotechnology, Inc. Dallas, TX, USA), cleaved caspase-3
rabbit polyclonal antibody (1:1,000; sc-22171-R, Santa Cruz
Biotechnology, Inc.), Bcl-2 rabbit polyclonal antibody (1:800;
sc-492; Santa Cruz Biotechnology, Inc.), E-cadherin mouse
monoclonal antibody (1:800; BM0339; Abzoom, Dallas, TX, USA),
vimentin mouse monoclonal antibody (1:1,000; sc-32322; Santa Cruz
Biotechnology, Inc.), fibroblast-specific protein (FSP) rabbit
polyclonal antibody (1:1,500; orb89918; Biocompare, South San
Francisco, CA, USA), Beclin rabbit monoclonal antibody (1:1,000;
2026-1; Epitomics, Burlingame, CA, USA), LC3B rabbit polyclonal
antibody (1:1,000; ab51520; Abcam, Hong Kong, China), p62 rabbit
polyclonal antibody (1:1,000; ab96134; Abcam) and DDR1 rabbit
polyclonal antibody (1:1,000; sc-532; Santa Cruz Biotechnology,
Inc.). The membranes were then incubated for 1 h with the
appropriate secondary antibodies (goat anti-rabbit antibody,
1:2,000; 111-035-003; goat anti-mouse antibody, 1:2,000;
111-035-008; Jackson Immunoresearch, Inc., West Grove, PA, USA; and
donkey anti-goat antibody, 1:2,000; CW0214; CWBio, Beijing, China).
Electrochemiluminescence was performed with an IPP6.0 system.
Immunocytochemistry (ICC)
The H9c2 cells were washed with phosphate-buffered
saline (PBS) 3 times and fixed for 30 min in 4% paraformaldehyde.
At that point, the cells were permeabilized for 15 min with PBS
containing 0.3% Triton X-100, and treated with 3%
H2O2 for 30 min followed by goat serum
(Auragene Bioscience Co.) for 10 min, and then incubated with
Beclin 1 monoclonal antibody (1:500; 2026-1; Epitomics) overnight
at 4°C. Following 3 rinses with PBST, the cells were incubated for
30 min with the secondary antibody (donkey anti-goat, 1:1,000;
CW0218, CWBio) and for a further 30 min with SABC complex (both
from Auragene Bioscience Co.) at room temperature. Following 3
additional PBST rinses, the immunoreactive cultured cells were
observed.
Construction of HMGB1 overexpression
lentiviral vector
Primers were designed using the HMGB1 sequence in
GenBank, which contained the HindIII and EcoRI
restriction enzymes sites. The forward primer was 5′-cccAAGCTTA
TGGGCAAAGGAGATCCTA-3′, and the reverse primer was
5′-ccgGAATTCTTATTCATCATCATCATCT-3′. Rat myocardium genomic DNA was
used as the template and RT-PCR was used to clone the HMGB1 gene.
The RT-PCR products were resolved by 1% agarose gel
electrophoresis, and then were purified, sequenced and cloned into
the lentiviral vector, pCD513B-1, to produce the HMGB1
overexpression lentiviral vector (Lv-HMGB1). An empty vector was
used as the negative control for HMGB1 (Lv-NC).
Construction of HMGB1 shRNA lentiviral
vector
The mRNA sequence of HMGB1 shRNA was designed using
online software (https://rnaidesigner.invitrogen.com/rnaiexpress/).
The RNAi candidate target sequence was CCGGCTGCTTAGTTTAGGGAA
CACTCGAGTGTTCCCTAAACTAAGCAGTTTTTTg, AATT
CAAAAAACTGCTTAGTTTAGGGAACACTCGAGTGTT CCCTAAACTAAGCAG. A negative
control shRNA containing a scrambled sequence with the same
nucleotide composition was also selected and termed Lv-shRNA-scr.
The shRNA-annealed oligonucleotides were ligated into the
lentiviral vector, pCD513B-1, to construct the HMGB1 shRNA
lentiviral vector (Lv-HMGB1-shRNA) by T4 DNA ligase (Takara,
Dalian, China). All constructs were verified by sequence
analysis.
Cell transfection
The cells were transfected with the lentiviral
vector carrying HMGB1 shRNA (Lv-HMGB1-shRNA) or with the HMGB1
overexpression lentiviral vector (Lv-HMGB1), or with the respective
negative control vectors [Lv-NC or the scramble shRNA vector
(Lv-shRNA-scr)]. The cells were transfected using Lipofectamine
2000 (Life Technologies/Thermo Fisher Scientific, Grand Island, NY,
USA) according to manufacturer's instructions. Untransfected cells
were used as the controls (Con).
Transcription activator-like effector
nuclease (TALEN)-mediated knockdown of the DDR1 gene in H9c2
cells
We also used TALEN technology to knockdown the DDR1
gene in the H9c2 cells. TALENs designed to target the DDR1 gene
were purchased from Sidansai Biotechnology Co., Ltd. (Shanghai,
China). The cells in 24-well plates were transfected with 400 ng
TALEN expression plasmids (DDR1-TALEN) using Lipofectamine 2000
(Invitrogen Life Technologies), according to the manufacturer's
instructions. Western blot analysis was performed to measure the
protein expression levels of DDR1 and to confirm the efficiency of
the TALEN-mediated knockdown.
Flow cytometry
The H9c2 cells were washed twice with cold PBS and
then resuspended in 500 μl pre-cooled binding buffer. The
cells were subsequently stained with Annexin V-FITC and propidium
iodide (PI) according to the protocols provided with the kit and
were then analyzed using a flow cytometer (MoFlo™ XDP; Beckman
Coulter, Brea, CA, USA).
Statistical analysis
All statistical analyses were performed using SPSS
17.0 software. The differences between 2 groups were compared using
the Student's t-test. Differences between nultiple groups were
compared using one-way analysis of variance (ANOVA). Data are
expressed as the means ± standard deviation (SD). A p-value
<0.05 was considered to indicate a statistically significant
difference.
Results
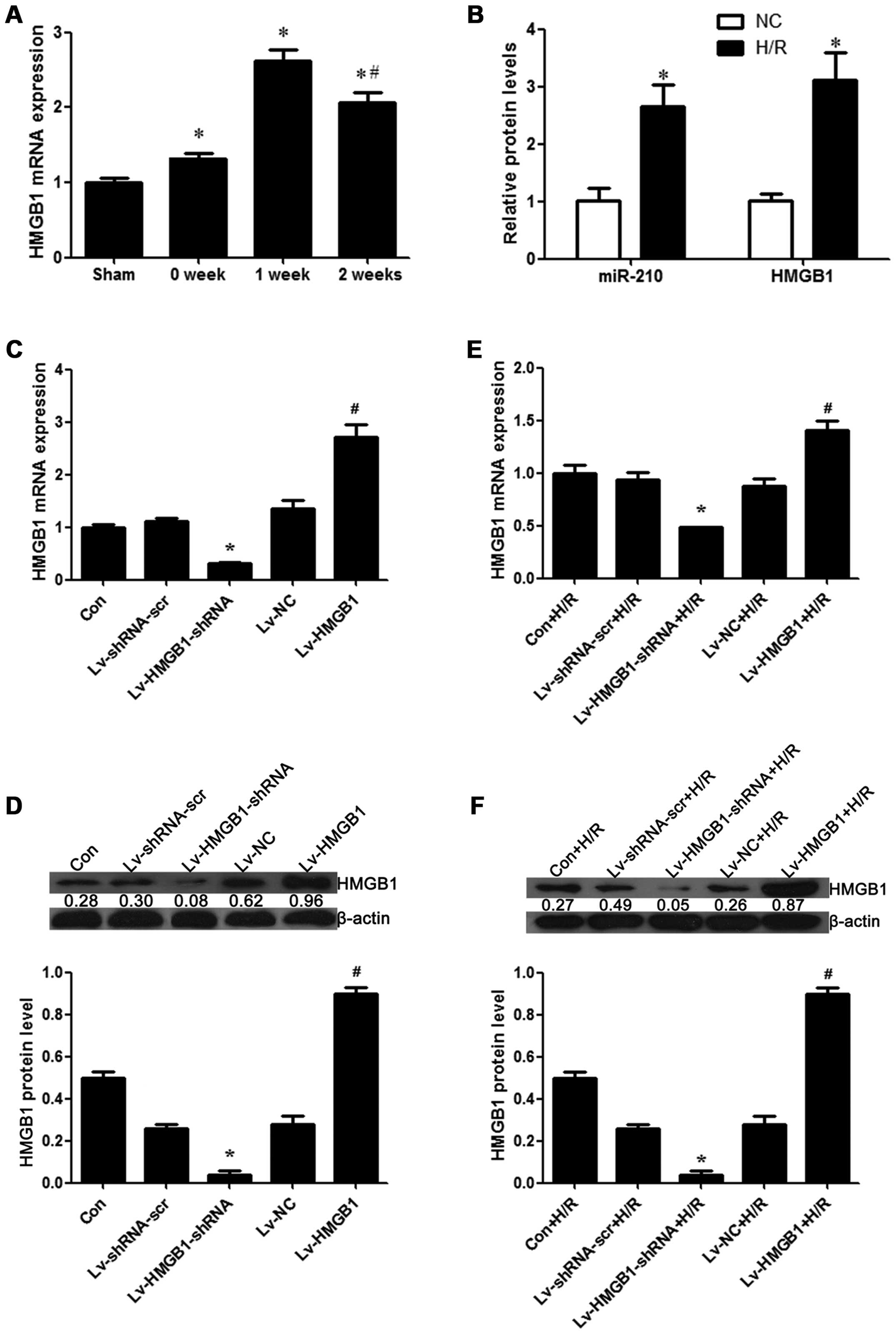
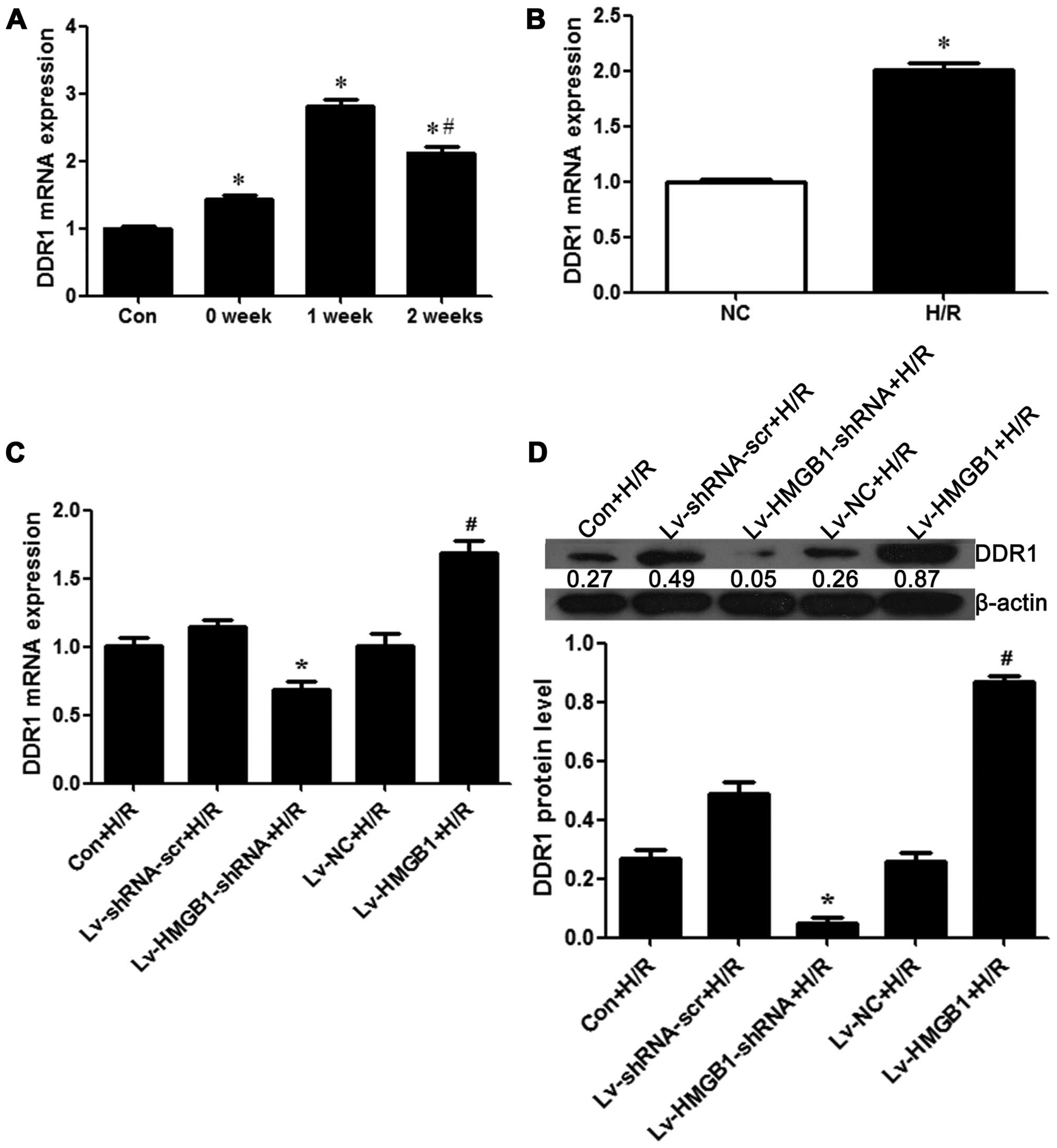
Expression of HMGB1 in myocardial tissue
and cardiomyocytes
In order to determine the role of the expression of
HMGB1 in myocardial necrosis, the differential expression of HMGB1
in normal and myocardial tissue was detected. We found that the
mRNA expression of HMGB1 was significantly increased at weeks 1 and
2 post-MI in the tissue with myocardial necrosis from the rats with
I/R injury when compared with the control heart tissue from the
sham-operated rats (p<0.05; Fig.
1A). In addition, a cell model of H/R injury using H9c2
cells was established, and we detected the expression of
miR-210, which is a hypoxia-inducible factor and its overexpression
is regarded as a biomarker for the successful induction of hypoxia
(27–29). Our result revealed that the
expression of miR-210 was significantly increased in the cells
exposed to H/R (p<0.05; Fig.
1B), which indicated that the model of hypoxia was successfully
established. In comparison to the control group, the mRNA
expression of HMGB1 was increased during H/R in the H9c2 cells
(p<0.05; Fig. 1B).
Furthermore, we constructed a series of lentiviral vectors
containing Lv-HMGB1-shRNA and Lv-HMGB1, and examined the efficiency
of the silencing or the overexpression of HMGB1. We found that the
mRNA and protein expression level of HMGB1 was decreased when the
H9c2 cells were transfected with Lv-HMGB1-shRNA, while it was
increased when the cells were transfected with Lv-HMGB1 (p<0.05;
Fig. 1C and D), suggesting that
the lentiviral vectors containing Lv-HMGB1-shRNA and Lv-HMGB1 were
effectively transfected into the cells. In order to further examine
the effects of H/R on the expression of HMGB1, the mRNA and protein
expression levels of HMGB1 were measured. The results revealed that
the levels were decreased when the H9c2 cells were transfected with
Lv-HMGB1-shRNA during H/R, while they were increased when the cells
were transfected with Lv-HMGB1 during H/R (p<0.05; Fig. 1E and F).
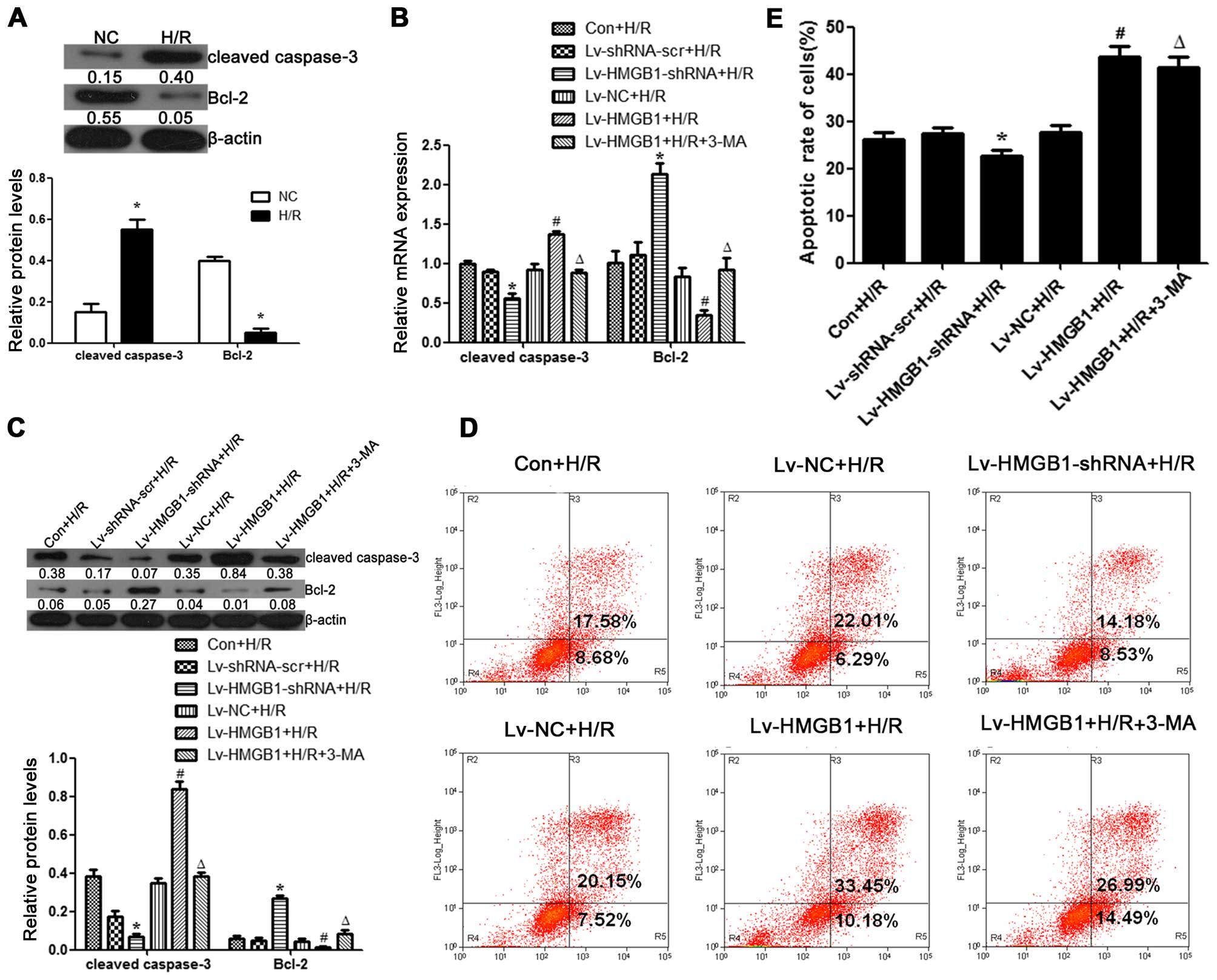
HMGB1 induces apoptosis during H/R in
cardiomyocytes
To determine the effects of HMGB1 on apoptosis
during H/R in cardiomyocytes, we measured the expression levels of
caspase-3 and Bcl-2 (apoptosis-related proteins) in the cells. The
results revealed that the protein expression level of cleaved
caspase-3 was increased following H/R in the H9c2 cells, whereas
the protein level of Bcl-2 was decreased (p<0.05; Fig. 2A). The mRNA and protein level of
cleaved caspase-3 was downregulated when the H9c2 cells were
transfected with Lv-HMGB1-shRNA during H/R, while it was
upregulated when the cells were transfected with Lv-HMGB1 during
H/R (p<0.05; Fig. 2B and C).
The mRNA and protein level of Bcl-2 was upregulated when the H9c2
cells were transfected with Lv-HMGB1-shRNA during H/R, while it
was downregulated when the cells were transfected with Lv-HMGB1
during H/R (p<0.05; (Fig. 2B and
C). In addition, we examined whether HMGB1 induced apoptosis by
flow cytometry. A decrease in the percentage of cells in early
apoptosis (quadrant 2) plus late apoptosis (quadrant 3) was
observed following transfection of the H9c2 cells with
Lv-HMGB1-shRNA during H/R (p<0.05; Fig. 2D and E). However, an increase in
the percentage of cells in early apoptosis (quadrant 2) plus late
apoptosis (quadrant 3) was observed following transfection of the
H9c2 cells with Lv-HMGB1 during H/R (p<0.05; Fig. 2D and E). Furthermore, 3-MA, an
autophagy inhibitor, markedly decreased the mRNA and protein
expression levels of cleaved caspase-3, while it increased those of
Bcl-2 during H/R in the H9c2 cells transfected with Lv-HMGB1
(p<0.05; Fig. 2B and C). In
addition, treatment with 3-MA decreased the percentage of apoptotic
cells following transfection of the H9c2 cells with Lv-HMGB1 during
H/R (p<0.05; Fig. 2D and E).
These results suggest that the induction of apoptosis by HMGB1 may
be associated with autophagy during H/R in cardiomyocytes.
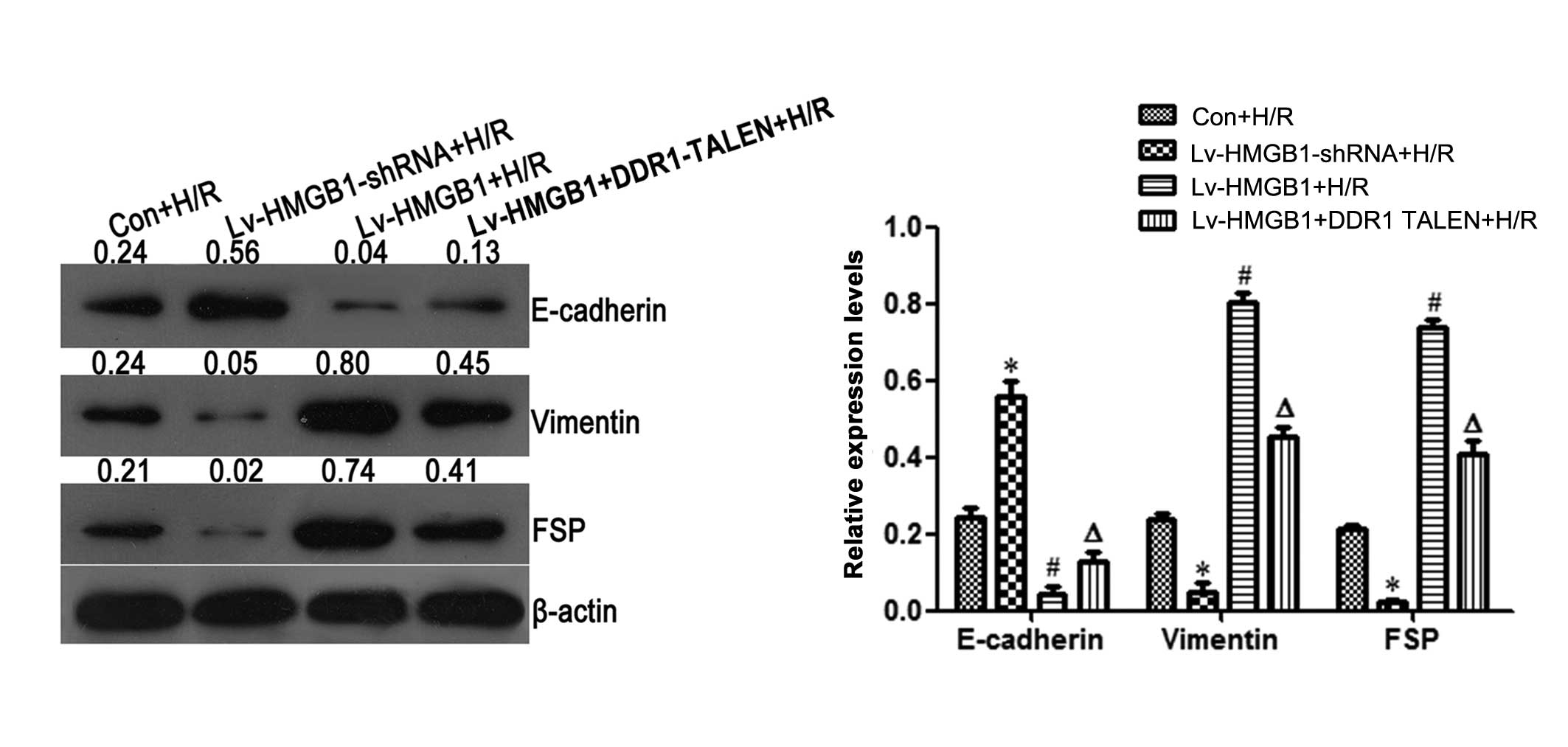
HMGB1 promotes EMT in coordination with
DDR1 during H/R in cardiomyocytes
To determine the effects of HMGB1 on EMT during H/R
in cardiomyocytes, the protein levels of the EMT biomakers,
E-cadherin, vimentin and FSP, were detected by western blot
analysis. In comparison to the Con + H/R group, the protein level
of the epithelial marker, E-cadherin, was upregulated when the H9c2
cells were transfected with Lv-HMGB1-shRNA during H/R, while it was
downregulated when the cells were transfected with Lv-HMGB1 during
H/R (p<0.05; Fig. 3). Compared
to the Con + H/R group, the protein levels of the mesenchymal
markers, vimentin and FSP, were downregulated when the H9c2 cells
were transfected with Lv-HMGB1-shRNA during H/R. However, the
levels of these markers were upregulated when the cells were
transfected with Lv-HMGB1 during H/R (p<0.05; Fig. 3).
Furthermore, in comparison to the Lv-HMGB1-shRNA
group, the protein level of the epithelial marker, E-cadherin, was
decreased when the H9c2 cells were transfected with Lv-HMGB1 during
H/R. However, compared to the Lv-HMGB1 +H/R group, the level was
increased when the cells were transfected with both Lv-HMGB1-shRNA
and DDR1 TALEN during H/R (p<0.05; Fig. 3). Compared to the Lv-HMGB1-shRNA
group, the protein levels of the mesenchymal markers, vimentin and
FSP, were increased when the cells were transfected with Lv-HMGB1
during H/R. However, compared to the Lv-HMGB1 +H/R group, the
levels were decreased when the H9c2 cells were transfected with
Lv-HMGB1 and DDR1 TALEN during H/R (p<0.05; Fig. 3).
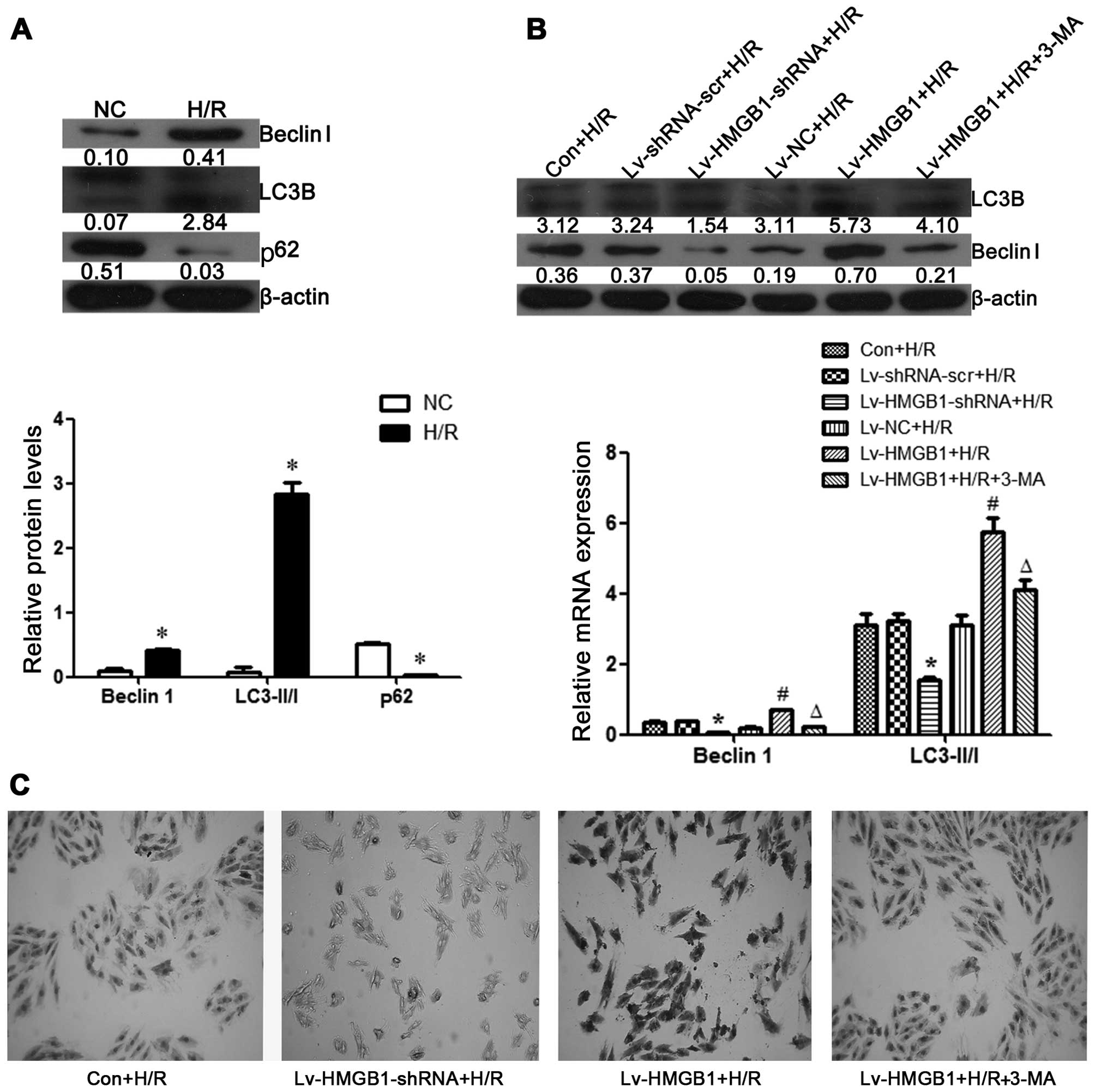
H/R induces autophagy in
cardiomyocytes
The H9c2 cells were subjected to H/R, and the
protein expression levels of Beclin 1, LC3-II/LC3-I and p62,
markers of autophagy, were then examined by western blot analysis.
The results revealed that the levels of Beclin 1 and the ratio of
LC3-II/LC3-I were increased, whereas the expression of p62 was
decreased in the H9c2 cells following H/R, suggesting that H/R
induces autophagy in H9c2 cells (p<0.05; Fig. 4A).
HMGB1 mediates autophagy following H/R
injury in cardiomyocytes
In this study, we also examined the effects of HMGB1
on H9c2 cell autophagy following H/R. We found that the protein
levels of Beclin 1 and LC3-II/I were decreased when the cells were
transfected with Lv-HMGB1-shRNA, whereas these levels were
increased when the H9c2 cells were transfected with Lv-HMGB1 during
H/R (p<0.05; Fig. 4B).
Moreover, the content of Beclin 1 during H/R in the H9c2 cells was
also determined by performing ICC. The results revealed that the
content of Beclin 1 was decreased when the cells were transfected
with Lv-HMGB1-shRNA, whereas it was increased when the H9c2 cells
were transfected with Lv-HMGB1 during H/R. The Beclin 1 content was
also decreased following treatment of the cells with 3-MA (an
autophagy inhibitor) (p<0.05; Fig.
4C).
HMGB1 induces autophagy through the
upregulation of the expression of DDR1 and the downregulation of
the phosphorylation of mTOR
In order to further explore the mechanisms of action
of HMGB1 and its effects on autophagy during H/R in cardiomyocytes,
the expression of DDR1 in myocardial tissue from rats with H/R
injury and in H9c2 cells subjected to H/R injury was determined by
RT-qPCR and western blot analysis. The results revealed that the
mRNA expression of DDR1 was significantly increased at weeks 1 and
2 post-MI in the tissue with myocardial necrosis from the rats with
I/R injury when compared with the control heart tissue from the
sham-operated rats (p<0.05; Fig.
5A). Compared to the control group, the mRNA expression of DDR1
was increased during H//R in the H9c2 cells (p<0.05; Fig. 5B). For further investigations on
the regulatory effects of HMGB1 on the expression of DDR1 during
H/R in cardiomyocytes, the mRNA and protein levels of DDR1 were
measured following transfection of the H9c2 cells with either
Lv-HMGB1-shRNA or Lv-HMGB1. The results revealed that DDR1
expression was decreased when the H9c2 cells were transfected with
Lv-HMGB1-shRNA during H/R, whereas it was increased when the cells
were transfected with Lv-HMGB1 during H/R (p<0.05; Fig. 5C and D).
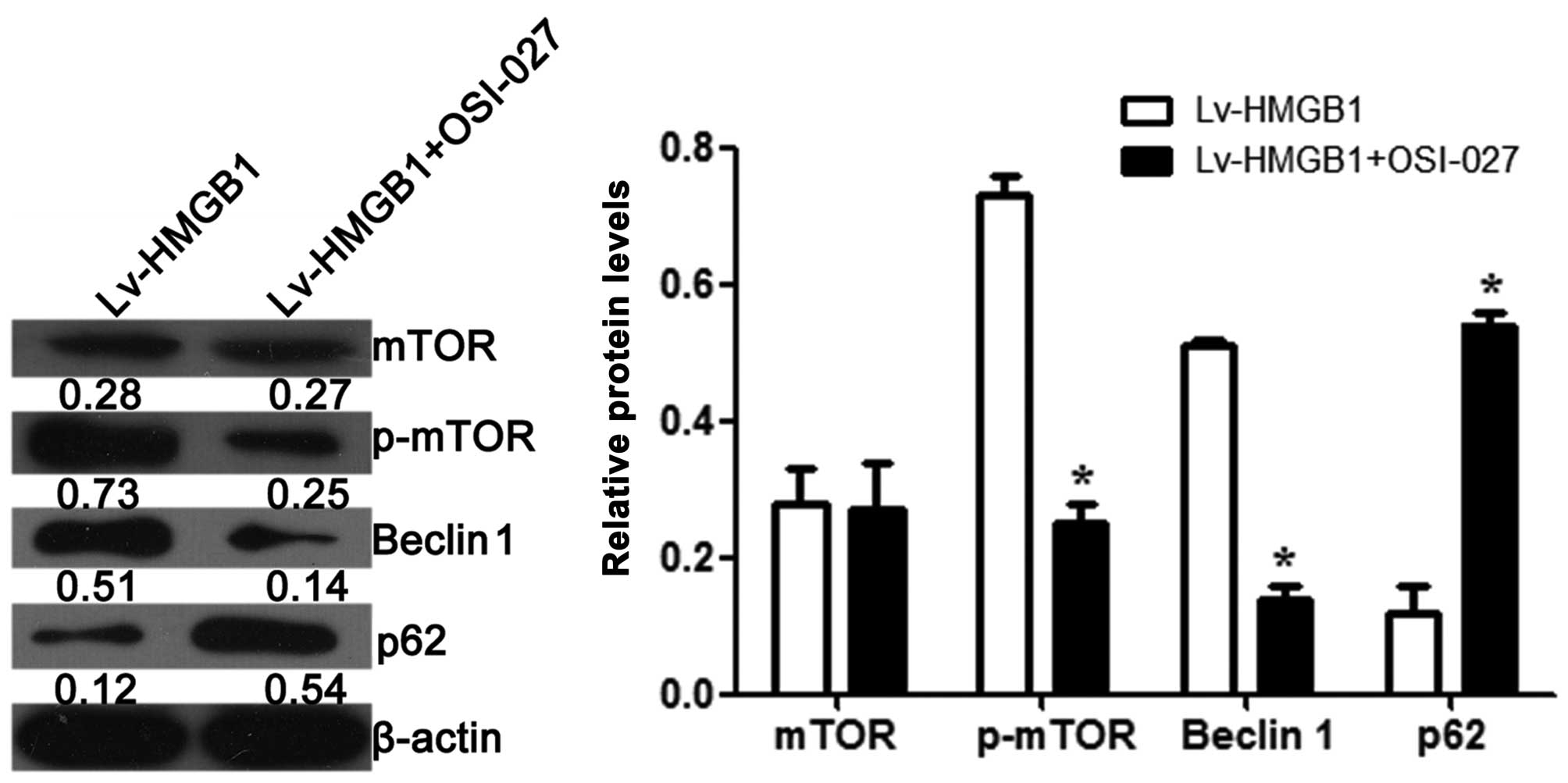
We also examined the effects of HMGB1 on the
expression and phosphorylation of mTOR. The results revealed that
OSI-027, an inhibitor of mTOR, decreased the phosphorylation level
of mTOR (p<0.05), but had no effect on the protein level of mTOR
when the cells were transfected with Lv-HMGB1 (p>0.05; Fig. 6). The protein level of Beclin 1
was also decreased, while the expression level of p62 was increased
when the H9c2 cells were treated with OSI-027 and transfected with
Lv-HMGB1.
Discussion
H//R injury occurs in a number of of important
clinical conditions, such as MI (30). Severe hypoxia threatens the
viability of the myocardium and, ultimately, cardiac function.
Subsequent reoxygenation may subject cells to further damage
(31). H/R injury is a complex,
multifactorial pathophysiological process, which mainly involves
the actions of nitric oxide (NO) (32–34), reactive oxygen species (ROS)
production (35,36), and other molecules, such as HMGB1
(22,37). In this study, we found that HMGB1
was upregulated in the tissues of rats with MI and in H9c2 cells
following H/R injury.
As a protein considered to be representative of
damage-associated molecular patterns (38), HMGB1 is actively secreted by
immune cells and some non-immune cells or is passively released by
necrotic cells (39). HMGB1 is a
multifunctional, ubiquitous protein located inside and outside
cells that plays a critical role in various physiological and
pathological processes, including cell development,
differentiation, inflammation, immunity, metastasis, metabolism and
death (40). However, to the best
of our knowledge, there are no studies available to date on the
effects of HMGB1 on EMT in cardiomyocytes. EMT is a biological
process that is involved in tissue fibrosis and cancer metastasis.
Recent research has indicated that EMT is re-activated in the heart
following ischemic injury (41).
E-cadherin commonly serves as an epithelial marker, while vimentin
and FSP as mesenchymal markers (42–44). Thus, in this study, we observed
the biological roles of HMGB1 in apoptosis and those of EMT markers
in H/R injury to H9c2 cells, and found that HMGB1 induced apoptosis
by increasing the mRNA and protein expression of cleaved caspase-3
and the apoptotic rate of the cells, while decreasing the
expression of Bcl-2, and promoting EMT. It also decreased the
protein level of the epithelial marker, E-cadherin, and increased
the protein expression of the mesenchymal markers, vimentin and
FSP, during H/R in the H9c2 cells.
Autophagy is a highly evolutionarily conserved
cellular process through which long-lived proteins and damaged
organelles are recycled to maintain cellular homeostasis (45). These proteins and organelles are
sequestered into autophagosomes and are then delivered to lysosomes
for degradation (46). As a major
intracellular degradation and recycling pathway, autophagy is
crucial for maintaining homeostasis, as well as remodeling during
normal development, and dysfunctions in autophagy have been shown
to be associated with a variety of pathologies, including cancers,
inflammatory diseases and MI (47). Beclin 1, LC3-II/I and p62 are
usually considered as markers of autophagy. The results of our
present study demonstrated that the expression of Beclin 1 and
LC3-II/I was markedly increased, while that of p62 was decreased
during H/R injury, thus indicating that H/R injury induces
autophagy in cardiomyocytes. As an important mediator of systemic
autophagic syndrome, HMGB1 participates in the autophagy process at
several levels, including at the nuclear, cytosolic and
extracellular level (40).
HMGB1-induced autophagy plays a vital role in various diseases,
such as cancer (48–51), endotoxemia/bacterial infection
(52,53), diabetes mellitus (54); however, research on the role of
HMGB1-induced autophagy in H/R injury or MI is limited (22). Thus, we found that through
autophagy that HMGB1 promoted apoptosis and EMT during H/R injury
in cardiomyocytes.
It has been reported that the expression of DDR1 is
increased in the MI-affected area of rats with congestive heart
failure (55). It is thus worth
researching whether DDR1 is involved in H/R injury in MI. DDR-1 is
a receptor tyrosine kinase (RTK). In humans, the DDR-1 gene is
localized on chromosome 6, in the region 6p21.3 (56,57). DDR1 is uniquely positioned to
function as a sensor for the extracellular matrix and to regulate a
wide range of cellular functions from migration and proliferation
to cytokine secretion and extracellular matrix
homeostasis/remodeling (58).
While the activation of DDR1 by extracellular matrix collagens is
required for normal development and tissue homeostasis, the
aberrant activation of these receptors following injury or in
disease is detrimental, and has been implicated in several
diseases, such as fibrosis, atherosclerosis and cancer (59). However, to the best of our
knowledge, there are no available studies to date on the
association between DDR1 and H/R injury in MI. Thus, in the present
study, we examined whether DDR1 is involved in H/R injury, and
found that the expression of DDR1 was significantly elevated in
tissues of rats with MI and in cardio-myocytes following H/R
injury. We then examined the role of DDR1 in HMGB1-induced
autophagy following H/R injury in cardiomyocytes, and found that
HMGB1 induced autophagy by upregulating the expression of DDR1.
In order to further explore the mechanisms of action
of HMGB1 in apoptosis and EMT in H/R injury, we examined the
alteration of autopagy-related signaling, mTOR following treatment
with its inhibitor, OSI-027. The results revealed that OSI-027
decreased the protein level of Beclin 1, and increased the level of
p62. It did not alter the protein level of mTOR, but altered its
phosphorylation level in H9c2 cells following transfection with an
HMGB1 overexpression vector (Lv-HMGB1). This suggests that HMGB1
induces autophagy by downregulating the phosphorylation of
mTOR.
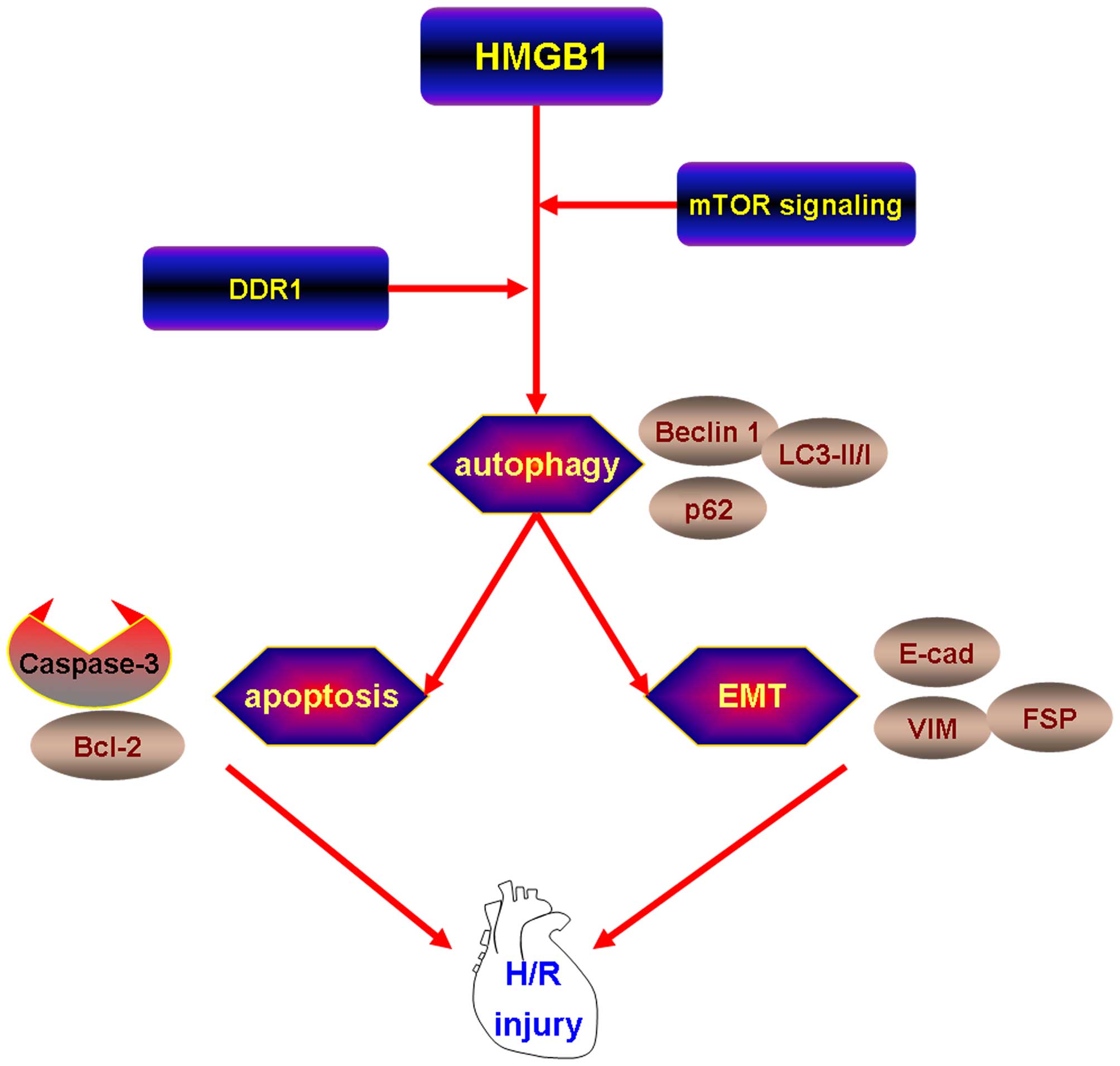
In conclusion, the findings of our study
demonstrated the following: i) the expression of HMGB1 was
upregulated in the myocardial tissue of rats with MI and following
H/R injury to H9c2 cells; ii) HMGB1 promoted apoptosis and EMT
during H/R in H9c2 cells; iii) in association with the induction of
autophagy, HMGB1 induces apoptosis and EMT following H/R in H9c2
cells; iv) HMGB1 induced autophagy by upregulating the expression
of DDR1 and downregulating the phosphorylation of mTOR (Fig. 7). Thus, it can be concluded that
HMGB1 promotes apoptosis and EMT in association with the induction
of autophagy through the upregulation of the expression of DDR1 and
the downregulation of the phosphorylation of mTOR following H/R
injury in cardiomyocytes.
References
|
1
|
Aydin S, Kuloglu T, Aydin S, Kalayci M,
Yilmaz M, Çakmak T and Eren MN: Elevated adropin: A candidate
diagnostic marker for myocardial infarction in conjunction with
troponin-I. Peptides. 58:91–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cabello JB, Burls A, Emparanza JI, Bayliss
S and Quinn T: Oxygen therapy for acute myocardial infarction.
Cochrane Database Syst Rev. 8:CD0071602013.PubMed/NCBI
|
|
3
|
Anderson JL, Adams CD, Antman EM, Bridges
CR, Califf RM, Casey DE Jr, Chavey WE II, Fesmire FM, Hochman JS,
Levin TN, et al American College of Cardiology Foundation/American
Heart Association Task Force on Practice Guidelines: 2012 ACCF/AHA
focused update incorporated into the ACCF/AHA 2007 guidelines for
the management of patients with unstable angina/non-ST-elevation
myocardial infarction: A report of the American College of
Cardiology Foundation/American Heart Association Task Force on
Practice Guidelines. Circulation. 127:e663–e828. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brown HF: Synopsis and Review of the
American College of Cardiology Foundation/American Heart
Association 2013 ST-Elevation Myocardial Infarction Guideline. AACN
Adv Crit Care. 25:142–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mitra A, Ray A, Datta R, Sengupta S and
Sarkar S: Cardioprotective role of p38 MAPK during myocardial
infarction via parallel activation of α-crystallin B and Nrf2. J
Cell Physiol. 229:1272–1282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sahoo S and Losordo DW: Exosomes and
cardiac repair after myocardial infarction. Circ Res. 114:333–344.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gai Y, Ma Z, Yu X, Qu S and Sui D: Effect
of ginsenoside Rh1 on myocardial injury and heart function in
isoproterenol-induced cardiotoxicity in rats. Toxicol Mech Methods.
22:584–591. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee: Heart disease and stroke statistics - 2013
update: A report from the American Heart Association. Circulation.
127:e6–e245. 2013. View Article : Google Scholar
|
|
9
|
Cao X, Chen A, Yang P, Song X, Liu Y, Li
Z, Wang X, Wang L and Li Y: Alpha-lipoic acid protects
cardiomyocytes against hypoxia/reoxygenation injury by inhibiting
autophagy. Biochem Biophys Res Commun. 441:935–940. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Verma S, Fedak PW, Weisel RD, Butany J,
Rao V, Maitland A, Li RK, Dhillon B and Yau TM: Fundamentals of
reperfusion injury for the clinical cardiologist. Circulation.
105:2332–2336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Feng Y, Hu L, Xu Q, Yuan H, Ba L, He Y and
Che H: Cytoprotective role of alpha1-antitrypsin in vascular
endothelial cell under hypoxia/reoxygenation condition. J
Cardiovasc Pharmacol. 66:96–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao X, Wang X, Ling Y, Song X, Yang P, Liu
Y, Liu L, Wang L, Guo J and Chen A: Comparison of the degree of
autophagy in neonatal rat cardiomyocytes and H9c2 cells exposed to
hypoxia/reoxygenation. Clin Lab. 60:809–814. 2014.PubMed/NCBI
|
|
13
|
Zhang Y, Hu S and Chen Y: Hepatocyte
growth factor suppresses hypoxia/reoxygenation-induced XO
activation in cardiac microvascular endothelial cells. Heart
Vessels. 2014.
|
|
14
|
Hock R, Furusawa T, Ueda T and Bustin M:
HMG chromosomal proteins in development and disease. Trends Cell
Biol. 17:72–79. 2007. View Article : Google Scholar
|
|
15
|
Zhao X, Kuja-Panula J, Rouhiainen A, Chen
YC, Panula P and Rauvala H: High mobility group box-1 (HMGB1;
amphoterin) is required for zebrafish brain development. J Biol
Chem. 286:23200–23213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rauvala H and Rouhiainen A: Physiological
and pathophysiological outcomes of the interactions of HMGB1 with
cell surface receptors. Biochim Biophys Acta. 1799:164–170. 2010.
View Article : Google Scholar
|
|
17
|
Stros M: HMGB proteins: Interactions with
DNA and chromatin. Biochim Biophys Acta. 1799:101–113. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou X, Hu X, Xie J, Xu C, Xu W and Jiang
H: Exogenous high-mobility group box 1 protein injection improves
cardiac function after myocardial infarction: Involvement of Wnt
signaling activation. J Biomed Biotechnol. 2012:7438792012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakamura Y, Suzuki S, Shimizu T, Miyata M,
Shishido T, Ikeda K, Saitoh S, Kubota I and Takeishi Y: High
mobility group box 1 promotes angiogenesis from bone marrow-derived
endothelial progenitor cells after myocardial infarction. J
Atheroscler Thromb. 22:570–581. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He YY, Wen Y, Zheng XX and Jiang XJ:
Intramyocardial delivery of HMGB1 by a novel thermosensitive
hydrogel attenuates cardiac remodeling and improves cardiac
function after myocardial infarction. J Cardiovasc Pharmacol.
61:283–290. 2013. View Article : Google Scholar
|
|
21
|
Andrassy M, Volz HC, Riedle N, Gitsioudis
G, Seidel C, Laohachewin D, Zankl AR, Kaya Z, Bierhaus A,
Giannitsis E, et al: HMGB1 as a predictor of infarct transmurality
and functional recovery in patients with myocardial infarction. J
Intern Med. 270:245–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu W, Jiang H, Hu X and Fu W: Effects of
high-mobility group box 1 on the expression of Beclin-1 and LC3
proteins following hypoxia and reoxygenation injury in rat
cardiomyocytes. Int J Clin Exp Med. 7:5353–5357. 2014.
|
|
23
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III,
et al: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang R, Livesey KM, Zeh HJ III, Lotze MT
and Tang D: HMGB1 as an autophagy sensor in oxidative stress.
Autophagy. 7:904–906. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma S, Wang Y, Chen Y and Cao F: The role
of the autophagy in myocardial ischemia/reperfusion injury. Biochim
Biophys Acta. 1852:271–276. 2015. View Article : Google Scholar
|
|
26
|
Sciarretta S, Hariharan N, Monden Y,
Zablocki D and Sadoshima J: Is autophagy in response to ischemia
and reperfusion protective or detrimental for the heart? Pediatr
Cardiol. 32:275–281. 2011. View Article : Google Scholar
|
|
27
|
Biswas S, Roy S, Banerjee J, Hussain SR,
Khanna S, Meenakshisundaram G, Kuppusamy P, Friedman A and Sen CK:
Hypoxia inducible microRNA 210 attenuates keratinocyte
proliferation and impairs closure in a murine model of ischemic
wounds. Proc Natl Acad Sci USA. 107:6976–6981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Devlin C, Greco S, Martelli F and Ivan M:
miR-210: More than a silent player in hypoxia. IUBMB Life.
63:94–100. 2011.PubMed/NCBI
|
|
29
|
Gee HE, Camps C, Buffa FM, Patiar S,
Winter SC, Betts G, Homer J, Corbridge R, Cox G, West CM, et al:
hsa-mir-210 is a marker of tumor hypoxia and a prognostic factor in
head and neck cancer. Cancer. 116:2148–2158. 2010.PubMed/NCBI
|
|
30
|
Feng GM, Chen JH, Lin CI and Yang JM:
Effect of docosa-hexaenoic acid on hypoxia/reoxygenation injury in
human coronary arterial smooth muscle cells. Eur J Nutr.
51:987–995. 2012. View Article : Google Scholar
|
|
31
|
McCord JM: Oxygen-derived free radicals in
postischemic tissue injury. N Engl J Med. 312:159–163. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jensen FB, Hansen MN, Montesanti G and
Wang T: Nitric oxide metabolites during anoxia and reoxygenation in
the anoxia-tolerant vertebrate Trachemys scripta. J Exp Biol.
217:423–431. 2014. View Article : Google Scholar
|
|
33
|
Robertson SJ, Mokgokong R, Kania KD, Guedj
AS, Hladky SB and Barrand MA: Nitric oxide contributes to
hypoxia-reoxygenation-induced P-glycoprotein expression in rat
brain endothelial cells. Cell Mol Neurobiol. 31:1103–1111. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rus A, Molina F, Peinado MA and Del Moral
ML: Nitric oxide averts hypoxia-induced damage during reoxygenation
in rat heart. Microsc Res Tech. 74:1093–1103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim JS, Wang JH and Lemasters JJ:
Mitochondrial permeability transition in rat hepatocytes after
anoxia/reoxygenation: Role of Ca2+-dependent
mitochondrial formation of reactive oxygen species. Am J Physiol
Gastrointest Liver Physiol. 302:G723–G731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kondoh M, Ohga N, Akiyama K, Hida Y,
Maishi N, Towfik AM, Inoue N, Shindoh M and Hida K: Hypoxia-induced
reactive oxygen species cause chromosomal abnormalities in
endothelial cells in the tumor microenvironment. PLoS One.
8:e803492013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu H, Yao Y, Su Z, Yang Y, Kao R, Martin
CM and Rui T: Endogenous HMGB1 contributes to
ischemia-reperfusion-induced myocardial apoptosis by potentiating
the effect of TNF-α/JNK. Am J Physiol Heart Circ Physiol.
300:H913–H921. 2011. View Article : Google Scholar
|
|
38
|
Okuma Y, Liu K, Wake H, Liu R, Nishimura
Y, Hui Z, Teshigawara K, Haruma J, Yamamoto Y, Yamamoto H, et al:
Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE
interaction. Neuropharmacology. 85:18–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Su Z, Yin J, Wang T, Sun Y, Ni P, Ma R,
Zhu H, Zheng D, Shen H, Xu W, et al: Up-regulated HMGB1 in EAM
directly led to collagen deposition by a PKCβ/Erk1/2-dependent
pathway: Cardiac fibroblast/myofibroblast might be another source
of HMGB1. J Cell Mol Med. 18:1740–1751. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun X and Tang D: HMGB1-dependent and
-independent autophagy. Autophagy. 10:1873–1876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Germani A, Foglio E, Capogrossi MC, Russo
MA and Limana F: Generation of cardiac progenitor cells through
epicardial to mesenchymal transition. J Mol Med Berl. 93:735–748.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bi WR, Xu GT, Lv LX and Yang CQ: The ratio
of transforming growth factor-β1/bone morphogenetic protein-7 in
the progression of the epithelial-mesenchymal transition
contributes to rat liver fibrosis. Genet Mol Res. 13:1005–1014.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wen SL, Gao JH, Yang WJ, Lu YY, Tong H,
Huang ZY, Liu ZX and Tang CW: Celecoxib attenuates hepatic
cirrhosis through inhibition of epithelial-to-mesenchymal
transition of hepatocytes. J Gastroenterol Hepatol. 29:1932–1942.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun S, Ning X, Zhai Y, Du R, Lu Y, He L,
Li R, Wu W, Sun W and Wang H: Egr-1 mediates chronic
hypoxia-induced renal interstitial fibrosis via the PKC/ERK
pathway. Am J Nephrol. 39:436–448. 2014.PubMed/NCBI
|
|
45
|
Hale AN, Ledbetter DJ, Gawriluk TR and
Rucker EB III: Autophagy: Regulation and role in development.
Autophagy. 9:951–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bánréti A, Sass M and Graba Y: The
emerging role of acetylation in the regulation of autophagy.
Autophagy. 9:819–829. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Guan JL, Simon AK, Prescott M, Menendez
JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A and Zhang
J: Autophagy in stem cells. Autophagy. 9:830–849. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang QY, Wu LQ, Zhang T, Han YF and Lin
X: Autophagy-mediated HMGB1 release promotes gastric cancer cell
survival via RAGE activation of extracellular signal-regulated
kinases 1/2. Oncol Rep. 33:1630–1638. 2015.PubMed/NCBI
|
|
49
|
Liu Y and Song L: HMGB1-induced autophagy
in Schwann cells promotes neuroblastoma proliferation. Int J Clin
Exp Pathol. 8:504–510. 2015.PubMed/NCBI
|
|
50
|
Liu W, Zhang Z, Zhang Y, Chen X, Guo S,
Lei Y, Xu Y, Ji C, Bi Z and Wang K: HMGB1-mediated autophagy
modulates sensitivity of colorectal cancer cells to oxaliplatin via
MEK/ERK signaling pathway. Cancer Biol Ther. 16:511–517. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li X, Wang S, Chen Y, Liu G and Yang X:
miR-22 targets the 3′UTR of HMGB1 and inhibits the HMGB1-associated
autophagy in osteosarcoma cells during chemotherapy. Tumour Biol.
35:6021–6028. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yanai H, Matsuda A, An J, Koshiba R,
Nishio J, Negishi H, Ikushima H, Onoe T, Ohdan H, Yoshida N, et al:
Conditional ablation of HMGB1 in mice reveals its protective
function against endotoxemia and bacterial infection. Proc Natl
Acad Sci USA. 110:20699–20704. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yang M, Cao L, Xie M, Yu Y, Kang R, Yang
L, Zhao M and Tang D: Chloroquine inhibits HMGB1 inflammatory
signaling and protects mice from lethal sepsis. Biochem Pharmacol.
86:410–418. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hagiwara S, Iwasaka H, Koga H, Hasegawa A,
Kudo K, Kusaka J, Oyama Y and Noguchi T: Stimulation of autophagy
in the liver by lipopolysaccharide-induced systemic inflammation in
a rat model of diabetes mellitus. Biomed Res. 31:263–271. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Andersson KB, Florholmen G, Winer LH,
Tonnessen T and Christensen G: Regulation of neuronal type genes in
congestive heart failure rats. Acta Physiol (Oxf). 186:17–27. 2006.
View Article : Google Scholar
|
|
56
|
Agarwal G, Mihai C and Iscru DF:
Interaction of discoidin domain receptor 1 with collagen type 1. J
Mol Biol. 367:443–455. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kothiwale S, Borza CM, Lowe EW Jr, Pozzi A
and Meiler J: Discoidin domain receptor 1 (DDR1) kinase as target
for structure-based drug discovery. Drug Discov Today. 20:255–261.
2015. View Article : Google Scholar :
|
|
58
|
Iwai LK, Luczynski MT and Huang PH:
Discoidin domain receptors: A proteomic portrait. Cell Mol Life
Sci. 71:3269–3279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vogel WF, Abdulhussein R and Ford CE:
Sensing extracellular matrix: An update on discoidin domain
receptor function. Cell Signal. 18:1108–1116. 2006. View Article : Google Scholar : PubMed/NCBI
|