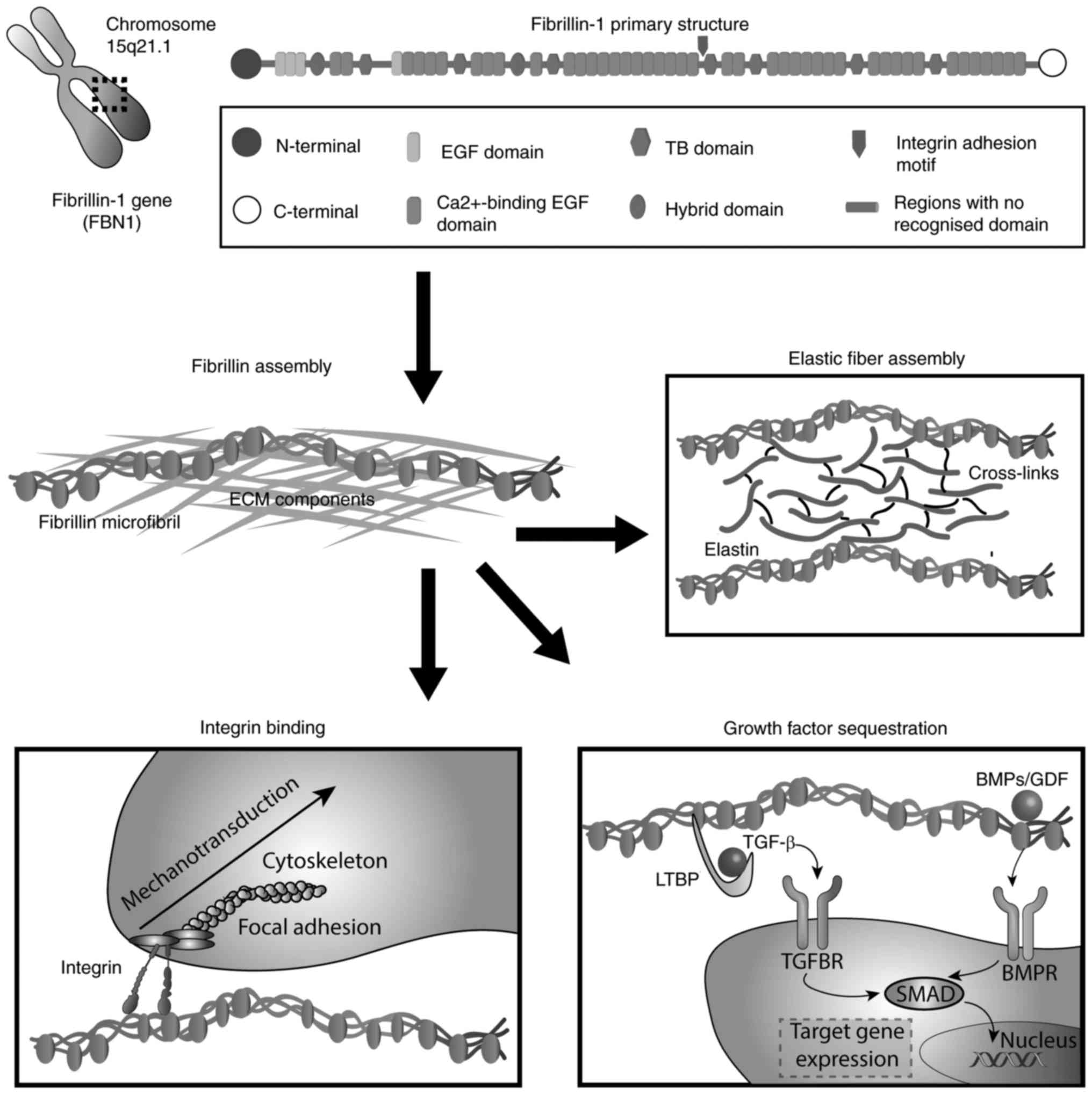
Fibrillin (FBN)-1 is a calcium-binding protein that
assembles to form 10–12 nm microfibrils in the extracellular matrix
(ECM) of elastic and non-elastic tissues. The human gene FBN-1
spans >230 kb (1) on
chromosome 15q15-21.1 (2) and is
highly fragmented into 65 exons. The primary protein structure
reveals multi-domains (3), which
primarily consist of epidermal growth factor (EGF)-like and certain
other modules (4). Out of a total
of 47 EGF domains (5), 43 modules
contain the calcium binding (cbEGF) consensus sequence
D/N-XD/N-E/Q-Xm-D/N-Xn-Y/F (6),
which provides structural stabilization (7), a characteristic rigid rod-like shape
(8–10) and protection against proteolysis
(11), and allows the control of
self- or FBN-2-interaction (12,13) and interactions with ECM
components, including fibulin-2, heparin/heparan sulphate and
microfibril-associated glycoprotein (MAGP)-1 (14–17). Disulphide bonds formed among the
six cysteine residues in EGF and cbEGF, in a C1–C3, C2–C4 and C5–C6
pattern (9), contribute to
further stabilize FBN-1. EGF-like domains are interspersed by seven
transforming growth factor (TGF)-β binding protein (TB)-like
modules and structurally related latent TGF-β-binding proteins
(LTBPs) (18). Characterized by
eight cysteine residues that form four disulphide bonds (C1–C3,
C2–C6, C4–C7 and C5–C8 arrangement), TB domains occur seven times
in FBN-1. Among them, the fourth TB module is of particular
interest due to the presence of the cell binding site RGD
(arginine-glycine-aspartic acid), which mediates interactions with
integrins (19). Additionally, as
with other FBNs, 'hybrid domains' are repeated twice in FBN-1 and
are stabilized by four intradomain disulphide bonds in a C1–C3,
C2–C5, C4–C6 and C7–C8 formation (20). The unique N- and C-terminal
domains of FBN-1 include four and two cysteine residues,
respectively, and contain the basic consensus sequence for
processing by furin-type enzymes (21–23). A distinguishing feature of FBN-1
is the presence of a proline-rich domain close to its N-terminus
(4,24). A summary of the chromosomal
location, domain organisation and primary functions of FBN-1 is
presented in Fig. 1.
FBN-1 is synthesized as an ~350 kDa precursor
molecule, profibrillin-1, which requires proteolytic processing by
furin proteases into its biologically active form (~320 kDa) prior
to incorporation into microfibrils (22,25). Accounting for all microfibril
structural features, FBN alignment models predict the initial
interactions between the N- and C-terminal sequences, which cause a
head-to-tail alignment and an approximate one-third stagger that is
stable as a 56 nm folded form (26–28). FBN bundles are stabilized by
transglutaminase-derived cross-links (29). Microfibril assembly has been
reported to be dependent and fine-tuned by a variety of
FBN-associated proteins. When visualized by rotary electron
microscopy (30), the extracted
microfibrils exhibit a beaded string morphology with dark areas,
which are termed 'bead' regions and appear in an average
periodicity of 56 nm (31).
Highlighting their important structural role, FBN microfibrils are
essential for the process of elastogenesis, acting as a scaffold
for the soluble precursor of elastin (tropoelastin) (32). Tropoelastin molecules are secreted
and deposited extracellularly onto a preformed, organized FBN
microfibril network, which gives rise to mature, elastic fibres
that are subsequently processed by the lysyl oxidase enzyme for the
formation of desmosine cross-links. The importance of FBN in the
formation of elastic fibres is highlighted by the inability of
FBN-1 knockout mice to form functioning elastic fibres, in addition
to a disorganization of elastic fibres (33) and a reduction of tissue
flexibility and extensibility, primarily in the arteries, lungs,
skin and other dynamic connective tissues (17). Unlike cbEGF-cbEGF, EGF1-EGF2 and
TB6-cbEGF32 are flexible domain interfaces (34,35).
FBN microfibrils interact with a large variety of
ligands. The binding with ECM components involves the C-terminal
regions of FBNs (36) and is
essential for regulating protein assembly and functionality.
Depending on the cell type, the FBN network (36–39) and MAGP (40–42) contribute to micro-fibril
biogenesis. Additionally, fibulin-2 appears to colocalise with
microfibrils in certain tissues at the interface between
microfibrils and elastin (14).
Fibulin-2 specifically binds to the N-terminal region of FBN-1,
while it also interacts with fibronectin and exhibits a connecting
role with other ECM molecules. As with fibulin-2, fibulin-1
localizes with elastin providing connective bridges to other ECM
components and to cells through laminin, fibronectin, nidogen or
fibrinogen (43). Contributing to
elastic fibre assembly, fibulin-5 interacts with FBN and
tropoelastin (44). According to
experimental data, fibulin-5 null mice exhibit structural
abnormalities due to disrupted elastogenesis (45,46). As they may be absent in tissues
exerting strong tensional forces, such as tendons, fibulins are
associated with elastic fibre assembly rather than the mechanical
properties of microfibrils. Furthermore, studies have demonstrated
that A disintegrin-like and metalloprotease (reprolysin-type) with
thrombospondin type-1 motif (ADAMTS) and ADAMTS-like (ADAMTSL)
proteins, including ADAMTSL4 (47), ADAMTSL6 (48) and ADAMTSL10 (49), bind to FBN and modulate
microfibril assembly (49,50).
If mutations occur in these genes, pathologies similar to
fibrillinopathies are observed. Direct interaction of FBN with
various proteoglycans are reported to be essential for network
assembly and the maintenance of basement membranes (51,52). The proteoglycans decorin and
biglycan are able to bind to tropoelastin, while only decorin
directly interacts with FBN-1 (41,53). However, biglycan forms a ternary
complex with tropoelastin and MAGP-1, indicating a potential role
during elastogenesis (53).
Notably, alterations in decorin expression have been observed in
neonatal Marfan syndrome, which is connective tissue disorder
(54,55). The heparan sulphate proteoglycan
(HSPG) perlecan, also termed HSPG-2, colocalises with FBN and
elastin (56), and binds to the
central region of FBN-1 (57).
Additionally, these HSPGs bind to cell surface molecules and growth
factors (58), such as basic
fibroblast growth factor, indicating an indirect involvement of FBN
in the regulation of cell functions and stem cell niches (59,60). The chondroitin sulphate
proteoglycan versican controls the genesis of elastic fibres
(61,62) and acts as a key factor in
inflammation by interacting with the adhesion molecules of
activated leukocytes, including L-selectin, CD44 and chemokines, to
recruit inflammatory cells (63,64). FBN-associated collagen with
interrupted triple helices type XVI is associated with microfibrils
in various tissues, including the upper papillary dermis (65) and dorsal root ganglia (66), indicating a potential association
between FBN assembly and neuronal regeneration. LTBPs interact with
FBN at the N-terminal region (16,67) while they are also anchored to
other ECM components, such as fibronectin (68–70). These interactions are important in
regulating the availability and the activation of TGF-β deposited
in the ECM. LTBP 1, 3 and 4 covalently bind to the small latent
TGF-β complex with their third TB domain and control the local
TGF-β bioavailability (71). In
addition to TGF-β via LTBPs, a number of bone morphogenetic
proteins (BMPs), and growth and differentiation factors, directly
bind to FBN at the N-terminal region (72–75). Furthermore, through the RGD
binding site in the TB4 domain, FBN-1 interacts with different
integrins that are responsible for cell-matrix communication.
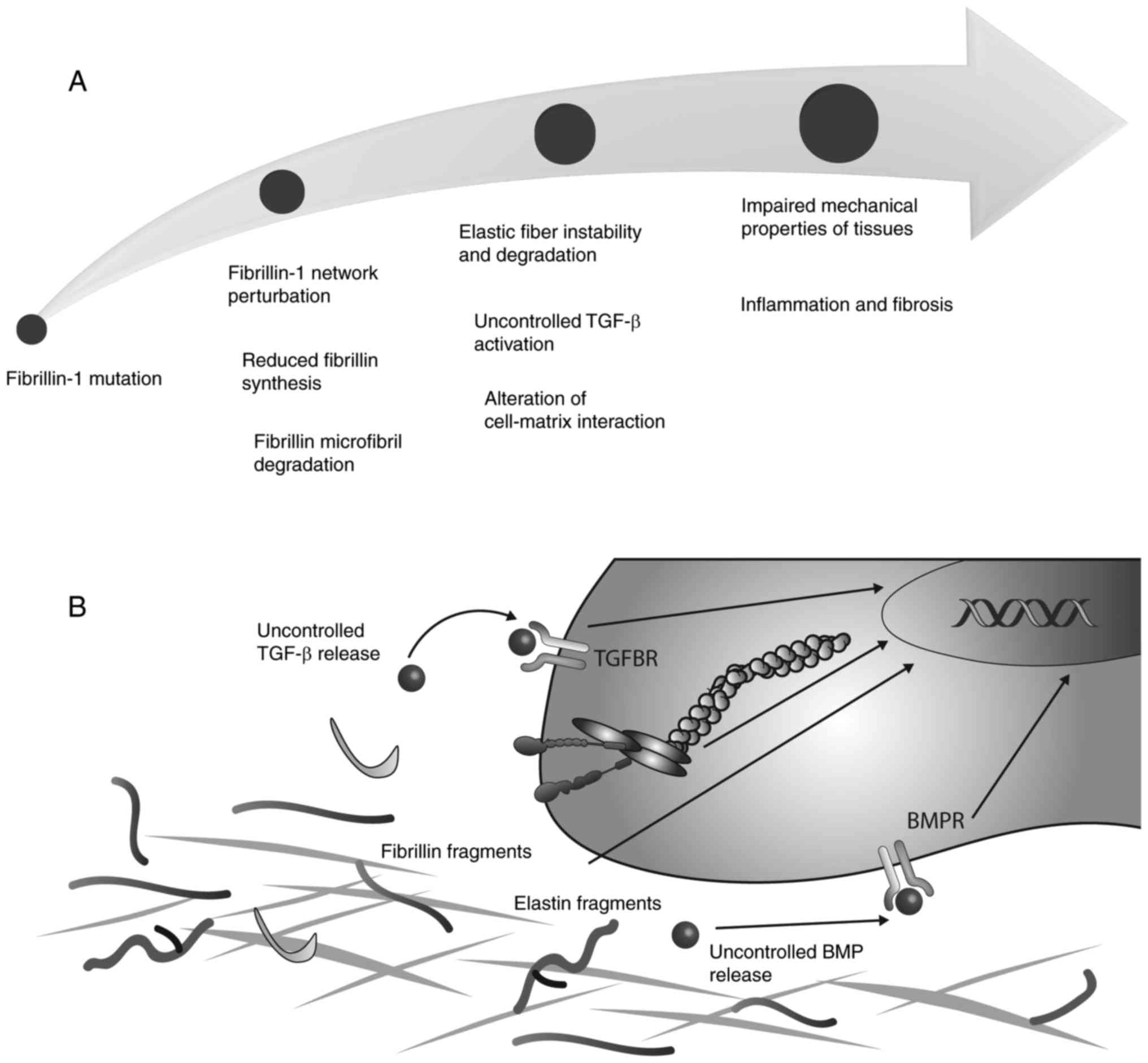
The FBN network is an important constituent of
connective tissues that interacts with the cellular compartment. It
controls the bioavailability and activity of the TGF-β superfamily,
which activates specific cellular signalling pathways for
preserving tissue homeostasis. The loss of cell matrix interactions
is a factor implicated in the pathological manifestations observed
in microfibrillinopathies (Fig.
2). By indirect interaction with FBN through LTBPs, as with
TGF-β, or direct interaction, for example BMPs (76), growth factors regulate the
cellular behaviour and control cell survival, differentiation and
response to injury (77). TGF-β
isoforms (TGF-β1, 2 and 3) are synthesized as precursor proteins
that comprise a growth factor domain at the C-terminal end and a
latency-associated peptide (LAP) at the N-terminus (78). Two precursor proteins homodimerize
and, following cleavage by furin-like endoproteases, form a complex
that is termed the small latent complex (SLC) (79), in which LAP is non-covalently
bound to the active TGF-β dimer. The SLC binds covalently to the
penultimate TB domain in LTBPs, which together form a complex
termed the large latent complex (LLC). The C-terminal region of
LTBP-1 and -4 exhibit non-covalent interactions with the N-terminus
of FBN-1 within the core of beaded microfibrils, while the
N-terminal regions bind to fibronectin. LTBP-3 localizes to
microfibrils using a different mechanism (80). The LLC is biologically inactive
and TGF-βs are accessible to its receptors following proteolytic
degradation or conformational changes (81,82) induced by integrin binding or
cell-mediated force transmission (79,83,84) The enzymatic activation followed by
TGF-β release is reported to be mediated by matrix metalloprotease
(MMP)-2 and -9 (85), the serine
protease plasmin (85–88), thrombospondin-1 (89) and reactive oxygen species
(90). Following cleavage and
activation, TGF-β binds to its serine and threonine kinase
receptors (TβRI and TβRII) on cell membranes, forming a receptor
heterocomplex (77,91) that, through Smad signalling
activation (92,93), promotes the expression of target
genes (94,95), including collagen type 1 α1 chain,
collagen type 3 α1 chain and TIMP metallopeptidase inhibitor 1, in
addition to another 60 ECM-associated genes (96). The direct binding of FBN-1 to
different BMPs, including BMP-2, -4, -5, -7 and -10, has been
previously reported (73,75,97). In addition, there is increasing
evidence that other growth factors are indirectly controlled
through targeting to other FBN binding partners within the ECM,
such as perlecan (57).
Due to the number of functions that are controlled
to a certain degree by FBN, it is clear that mutations in FBN genes
lead to a number of diseases that affect multiple organs, which are
collectively termed fibrillinopathies. Mutations in the FBN-1 gene
have been demonstrated to cause Marfan syndrome, an autosomal
dominant disorder of the connective tissue that is characterized by
pleiotropic manifestations in ocular, skeletal and cardiovascular
systems. Since the identification of the first mutation in 1991
(111), at present, >1,800
genetic abnormalities have been identified throughout the entire
length of FBN-1 (112).
Unfortunately, due to phenotypic variability and disease severity,
a phenotype-genotype correlation remains to be established
(113,114). Mutations in the central region
of the FBN-1 gene, comprising exons 24–32, are commonly associated
with severe myocardial dysfunctions, neonatal Marfan syndrome and
mortality within the first two years of postnatal life (115–117). It is reported that approximately
two-thirds of missense mutations involve cysteine residues and lead
to ocular complications, while premature terminations are
associated with severe skeletal and skin anomalies (115). A growing body of evidence
indicates that not all mutations in FBN-1 result in Marfan
syndrome; however, those that are not are associated with
Marfan-like disorders (118),
including MASS phenotype (119),
familial thoracic aortic aneurysm (120,121), Shprintzen-Goldberg syndrome
(122) and ectopia lentis
(123). It has also been
established that mutations in FBN-1 may lead to acromelic
dysplasias, such as Weill-Marchesani syndrome (WMS), geleophysic
dysplasia, acromicric dysplasia and Myhre syndrome (74,124,125). The patients affected by these
syndromes generally exhibit short statue, short hands and feet,
stiff joints and a hypermuscular build, which is unlike patients
with Marfan syndrome, who present with a tall stature,
arachnodactyly, hypermobile joints and a thin hypomuscular
structure. By contrast to Marfan syndrome, the mutations in FBN-1
that cause acromelic dysplasias, such as WMS, are located in a hot
spot within the FBN-1 gene (126) and are in-frame deletions of 24
nucleotides in exon 41 and 42, which encode the fifth TB (124,126,127). An in-frame deletion of exons
9–11, encoding the first TB domain, the proline rich region and the
fourth EGF-like domain, have been identified in WMS (74). Notably, while FBN-1 mutations
account for the dominant form of WMS, the recessive form is
reported to be caused by mutations in ADAMTS10 (128). According to experimental
evidence from mouse models expressing RGD sequence mutations and
the ability of integrin-modulating therapy to prevent fibrosis and
autoimmunity (129), the primary
cause of SSS may be the loss of integrin binding sites. A mutation
in the TB4 domain has also been reported in patients affected by
this syndrome (107). A summary
of the structural and signalling effects of mutations in FBN-1 is
presented in Fig. 2.
Pathophysiological mechanisms accounting for the
clinical manifestation of Marfan syndrome and similar disorders are
associated with an altered FBN network. Early immunofluo-rescent
studies using anti-FBN antibodies revealed qualitative and
quantitative abnormalities of the dermal microfibrils, with a
fragmented appearance in tissues extracted from patients with
Marfan syndrome. Isolated dermal fibroblasts exhibited a reduced
expression of FBN fibres and an abnormal morphology in
immunofluorescent analyses (130,131). Differences in microfibril
morphology have also been observed in neonatal Marfan syndrome
fibroblast cultures (132). In
contrast to the fragmented FBN networks observed in Marfan syndrome
(130,133), the FBN network in WMS is
abnormal for a different reason, as large FBN aggregate
accumulations (74) have been
reported in the skin of patients with SSS (107). Several in vitro and in
vivo studies of FBN-1 disorders have been performed in the last
two decades. The dominant negative model is supported by an in
vitro study in which the wild-type protein function is
disrupted by the mutant FBN, indicating that one FBN-1 mutant
allele is sufficient to diminish microfibril assembly (131). Furthermore, data from this model
are consistent with published data that reported that low levels of
mutant FBN-1 expression in patients with Marfan syndrome is
associated with a less severe phenotype (134). On the other hand,
haplosufficient models have demonstrated that selected mutations,
such as C1039G, lead to a disorganization of the microfibril
network, while the C1663R FBN-1 mutation participates in productive
microfibril assembly (135).
Based on this body of evidence, it is clear that FBN-1 disorders
are caused by mechanisms that are dependent on the position and
type of mutation. In vivo studies of mutant FBN have
indicated that abnormalities within the first hybrid domain do not
affect microfibril stability (133), while mutations in cbEGF-like
domains perturb microfibril assembly (136). Certain FBN-1 mutations also lead
to a gene product that, although it may be assembled into
microfibrils with a normal appearance, the mutation destabilizes
the structure of FBN-1 and renders it more susceptible to
proteolysis, leading to a gradual degradation (137,138). As reported by several studies,
the regulation of MMPs is implicated in the pathogenesis of Marfan
syndrome and other fibrillinopathies (139,140). In particular, MMP-1, -2, -3 and
-9 appear to exert a pivotal role in FBN fragmentation, as
demonstrated by the increased concentration of FBN fragments in the
aortic specimens of patients with Marfan syndrome (140–142). Studies concerning connective
tissue disorders caused by FBN-1 mutations have also revealed
alterations in the targeting and activation of growth factors. In
addition, an association between FBN-1 mutations and the altered
release of TGF-β has been associated with the development of
fibrillinopathies (143). In
support of this hypothesis, the administration of TGF-β antagonists
led to anti-apoptotic effects in the lungs of FBN-1-deficient mice
(144). Additionally,
neutralizing TGF-β antibodies successfully prevented the
development of aortic aneurysm by normalizing the levels of TGF-β
in Marfan syndrome mouse models (145). Furthermore, TGF-β antagonists
have been reported to reduce the levels of circulating TGF-β in
patients with Marfan syndrome (146). Notably, mutations in LTBPs or
TGF-β receptors, as observed in Loyes-Dietz syndrome, may lead to
the uncontrolled release of TGF-β. A perturbation of TGF-β
signalling is also observed in other fibrillinopathies, including
SSS (107) and acromicric or
geleophysic dysplasia (124).
Scleroderma is a heterogeneous connective tissue
disease that is characterized by excessive cutaneous and visceral
fibrosis, Raynaud's phenomenon, vascular lesions and
gastrointestinal manifestations (147). A widely used mouse model of
systemic sclerosis is the tight skin (Tsk) mouse, which exhibits an
in-frame tandem duplication of FBN-1 (148). While homozygotes suffer
embryonic lethality at day 7–8 of gestation, heterozygotes (Tsk/+)
have a normal life span but manifest myocardial, skeletal, and
pulmonary abnormalities. Furthermore, heterozygotes also present
with abnormal/altered fibrotic, inflammatory and autoimmune
function. Comparable levels of normal and mutant FBN-1 transcripts
in Tsk/+ tissues, and the presence of abundant tissue microfibrils,
indicates that the mutant FBN-1 is regularly synthesized and
assembled (148). Mutant FBN
copolymerizes with wild-type FBN-1, which leads to an unstable
structure (149) that is more
sensitive to proteolysis (150).
Briefly, Tsk/+ mice synthesize two types of microfibrils that
present with a normal morphology and a well-organized periodicity,
or diffuse interbeads, a longer periodicity and a tendency to
aggregate (151). The
instability of Tsk microfibrils leads to a disorganization and
fragmentation of elastic fibres, subsequently leading to reduced
ECM integrity (152,153) and increased cellular processing,
followed by an autoimmune response and the development of
autoantibodies (154). The
autoimmune phenotype, however, is not required for the development
of dermal thickening observed in Tsk/+ mice, and the Tsk phenotype
appears to be independent of the immune system, as this phenotype
has also been reported in mice lacking mature T and B cells
(155,156). A potential mechanism involved in
the promotion of the fibrotic phenotype may be driven by altered
TGF-β signalling (157).
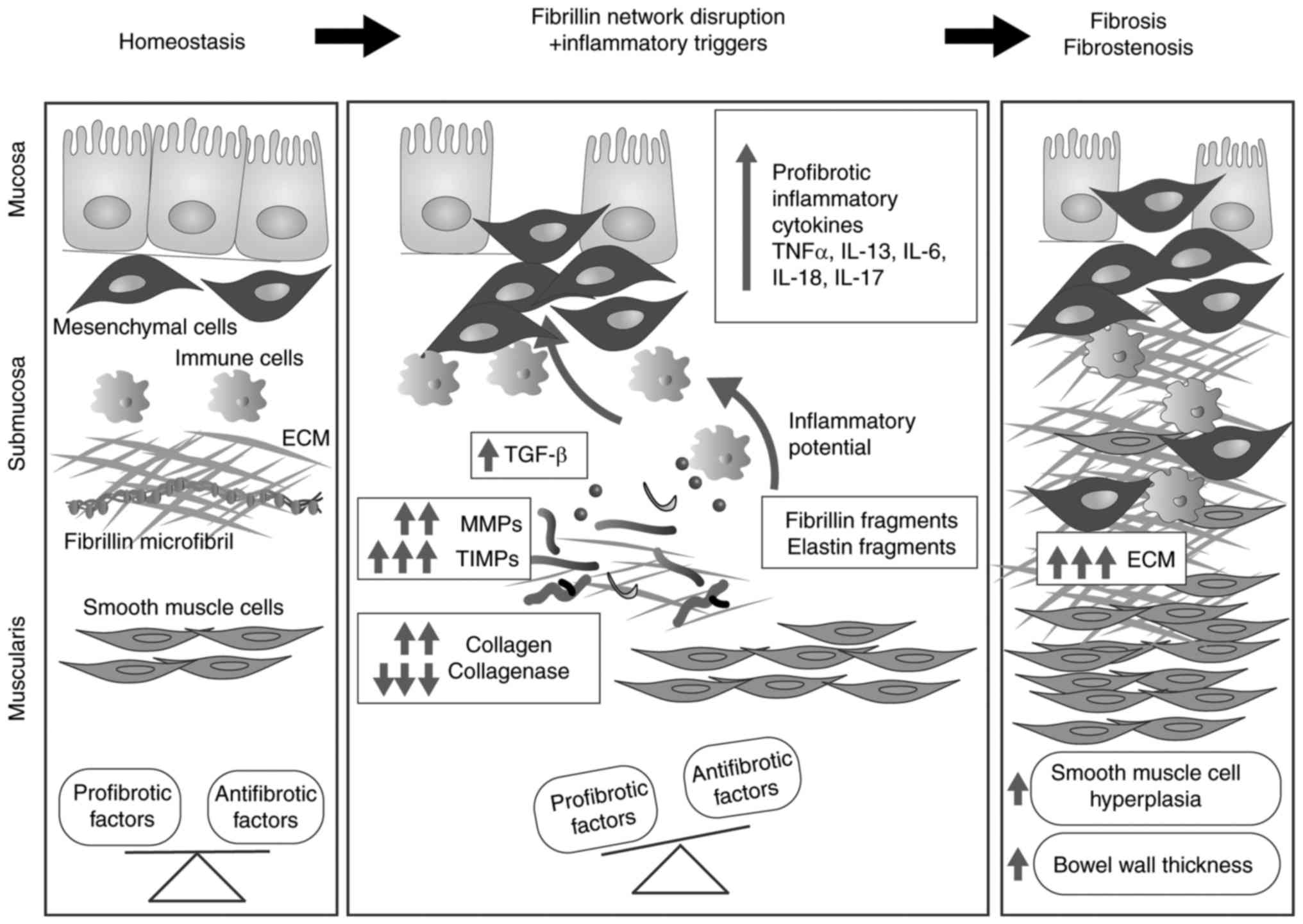
Inflammatory bowel disease (IBD) comprises a group
of gut immunopathological conditions that are a result of genetic,
environmental and cellular cues (158). ECM components have important
immunoregulatory roles, and the composition and ultrastructure of
the ECM are involved in intestinal immune responses, pathological
signalling, and chronic inflammation (159). Uncontrolled alterations in ECM
composition are reported in IBD and involve collagen I (160), collagen III (161,162), collagen V (163), collagen XVI (164), laminin (165,166), hyaluronan (167) and, recently, FBN-1 (164). FBN and elastic fibre networks
have important structural and biomechanical roles within the
intestinal tract as they are essential for the peristaltic movement
of the gastrointestinal tract. Notably, in up to 90% of patients
with SSS (168), FBN network
perturbations are reported to lead to excessive fibrosis,
inflammation and vascular dysfunction (169–175). Reinforcing the hypothesis that
the FBN network is involved in intestinal homeostasis, a previous
study reported the downregulation of FBN in the lamina propria of
patients with IBD compared with healthy donors (164). The development of gut fibrosis
(176) involves multiple cell
types and a large number of soluble factors (Fig. 3). Among soluble factors, TGF-β1,
which is generally considered to be the key mediator of fibrosis
(177), is overexpressed in IBD
(178), while under
physiological conditions TGF-β1 regulates the immune homeostasis by
preventing abnormal proinflammatory responses, as demonstrated by
the development of severe and lethal systematic inflammation in
TGF-β1 knockout mice (179) or
animals expressing T cells that do not respond to TGF-β1 (180). As observed in other organs, FBN
and elastin fragments deriving from unstable networks lead to the
upregulated expression of MMPs, including MMP-1, -2, -3, -7, -9,
-10, -12 and -13 (181–183), which results in disturbed ECM
turnover and subsequent fibrosis (184,185).
FBN-1 is an important ECM component that integrates
the biological network of structural and instructive information
for the modulation of cell-cell and cell-matrix interactions.
Acting as a key relay molecule for the transmission of
extracellular information into cellular signalling and function,
FBN-1 contributes to the accumulation of latent forms of growth
factors, such as TGF-β and BMPs, and regulates their
bioavailability and activity. Regulating the expression of MMPs,
fragmented microfibrils are associated with the development of
multiorgan inflammation and fibrosis. At present, the
characterization of FBN-1 dysfunction has improved the
characterization of the pathological pattern of connective tissue
diseases and the identification of novel therapeutic biological
approaches for the treatment of inflammation-associated states.
The present study was financially supported by a
grant (PRID-2016, to Professor Rosa Di Liddo) from the University
of Padova (Padova, Italy).
|
1
|
Biery NJ, Eldadah ZA, Moore CS, Stetten G,
Spencer F and Dietz HC: Revised genomic organization of FBN1 and
significance for regulated gene expression. Genomics. 56:70–77.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kainulainen K, Pulkkinen L, Savolainen A,
Kaitila I and Peltonen L: Location on chromosome 15 of the gene
defect causing Marfan syndrome. N Engl J Med. 323:935–939. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robertson I, Jensen S and Handford P: TB
domain proteins: Evolutionary insights into the multifaceted roles
of fibrillins and LTBPs. Biochem J. 433:263–276. 2011. View Article : Google Scholar
|
|
4
|
Corson GM, Chalberg SC, Dietz HC,
Charbonneau NL and Sakai LY: Fibrillin binds calcium and is coded
by cDNAs that reveal a multidomain structure and alternatively
spliced exons at the 5′end. Genomics. 17:476–484. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maslen CL, Corson GM, Maddox BK, Glanville
RW and Sakai LY: Partial sequence of a candidate gene for the
Marfan syndrome. Nature. 352:334–337. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Handford PA, Mayhew M and Brownlee GG:
Calcium binding to fibrillin? Nature. 353:3951991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Werner JM, Knott V, Handford PA, Campbell
ID and Downing AK: Backbone dynamics of a cbEGF domain pair in the
presence of calcium. J Mol Biol. 296:1065–1078. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Downing AK, Knott V, Werner JM, Cardy CM,
Campbell ID and Handford PA: Solution structure of a pair of
calcium-binding epidermal growth factor-like domains: Implications
for the Marfan syndrome and other genetic disorders. Cell.
85:597–605. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smallridge RS, Whiteman P, Werner JM,
Campbell ID, Handford PA and Downing AK: Solution structure and
dynamics of a calcium binding epidermal growth factor-like domain
pair from the neonatal region of human fibrillin-1. J Biol Chem.
278:12199–12206. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reinhardt DP, Mechling DE, Boswell BA,
Keene DR, Sakai LY and Bächinger HP: Calcium determines the shape
of fibrillin. J Biol Chem. 272:7368–7373. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reinhardt DP, Ono RN and Sakai LY: Calcium
stabilizes fibrillin-1 against proteolytic degradation. J Biol
Chem. 272:1231–1236. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin G, Tiedemann K, Vollbrandt T, Peters
H, Batge B, Brinckmann J and Reinhardt DP: Homo- and heterotypic
fibrillin-1 and -2 interactions constitute the basis for the
assembly of microfibrils. J Biol Chem. 277:50795–50804. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marson A, Rock MJ, Cain SA, Freeman LJ,
Morgan A, Mellody K, Shuttleworth CA, Baldock C and Kielty CM:
Homotypic fibrillin-1 interactions in microfibril assembly. J Biol
Chem. 280:5013–5021. 2005. View Article : Google Scholar
|
|
14
|
Reinhardt DP, Sasaki T, Dzamba BJ, Keene
DR, Chu ML, Göhring W, Timpl R and Sakai LY: Fibrillin-1 and
fibulin-2 interact and are colocalized in some tissues. J Biol
Chem. 271:19489–19496. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jensen SA, Reinhardt DP, Gibson MA and
Weiss AS: Protein interaction studies of MAGP-1 with tropoelastin
and fibrillin-1. J Biol Chem. 276:39661–39666. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Isogai Z, Ono RN, Ushiro S, Keene DR, Chen
Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB and Sakai
LY: Latent transforming growth factor beta-binding protein 1
interacts with fibrillin and is a microfibril-associated protein. J
Biol Chem. 278:2750–2757. 2003. View Article : Google Scholar
|
|
17
|
Rock MJ, Cain SA, Freeman LJ, Morgan A,
Mellody K, Marson A, Shuttleworth CA, Weiss AS and Kielty CM:
Molecular basis of elastic fiber formation. Critical interactions
and a tropoelastin-fibrillin-1 cross-link. J Biol Chem.
279:23748–23758. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Robertson IB, Horiguchi M, Zilberberg L,
Dabovic B, Hadjiolova K and Rifkin DB: Latent TGF-β-binding
proteins. Matrix Biol. 47:44–53. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jovanovic J, Takagi J, Choulier L,
Abrescia NG, Stuart DI, van der Merwe PA, Mardon HJ and Handford
PA: alphaVbeta6 is a novel receptor for human fibrillin-1.
Comparative studies of molecular determinants underlying
integrin-rgd affinity and specificity. J Biol Chem. 282:6743–6751.
2007. View Article : Google Scholar
|
|
20
|
Jensen SA, Iqbal S, Lowe ED, Redfield C
and Handford PA: Structure and interdomain interactions of a hybrid
domain: A disulphide-rich module of the fibrillin/LTBP superfamily
of matrix proteins. Structure. 17:759–768. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lönnqvist L, Reinhardt D, Sakai L and
Peltonen L: Evidence for furin-type activity-mediated C-terminal
processing of profibrillin-1 and interference in the processing by
certain mutations. Hum Mol Genet. 7:2039–2044. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Raghunath M, Putnam EA, Ritty T, Hamstra
D, Park ES, Tschödrich-Rotter M, Peters R, Rehemtulla A and
Milewicz DM: Carboxy-terminal conversion of profibrillin to
fibrillin at a basic site by PACE/furin-like activity required for
incorporation in the matrix. J Cell Sci. 112:1093–1100.
1999.PubMed/NCBI
|
|
23
|
Trask TM, Ritty TM, Broekelmann T, Tisdale
C and Mecham RP: N-terminal domains of fibrillin 1 and fibrillin 2
direct the formation of homodimers: A possible first step in
microfibril assembly. Biochem J. 340:693–701. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang H, Apfelroth SD, Hu W, Davis EC,
Sanguineti C, Bonadio J, Mecham RP and Ramirez F: Structure and
expression of fibrillin-2, a novel microfibrillar component
preferentially located in elastic matrices. J Cell Biol.
124:855–863. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wallis DD, Putnam EA, Cretoiu JS, Carmical
SG, Cao SN, Thomas G and Milewicz DM: Profibrillin-1 maturation by
human dermal fibroblasts: Proteolytic processing and molecular
chaperones. J Cell Biochem. 90:641–652. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reinhardt DP, Keene DR, Corson GM, Pöschl
E, Bächinger HP, Gambee JE and Sakai LY: Fibrillin-1: Organization
in microfibrils and structural properties. J Mol Biol. 258:104–116.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baldock C, Siegler V, Bax DV, Cain SA,
Mellody KT, Marson A, Haston JL, Berry R, Wang MC, Grossmann JG, et
al: Nanostructure of fibrillin-1 reveals compact conformation of
EGF arrays and mechanism for extensibility. Proc Natl Acad Sci USA.
103:11922–11927. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuo CL, Isogai Z, Keene DR, Hazeki N, Ono
RN, Sengle G, Bächinger HP and Sakai LY: Effects of fibrillin-1
degradation on microfibril ultrastructure. J Biol Chem.
282:4007–4020. 2007. View Article : Google Scholar
|
|
29
|
Qian RQ and Glanville RW: Alignment of
fibrillin molecules in elastic microfibrils is defined by
transglutaminase-derived cross-links. Biochemistry. 36:15841–15847.
1997. View Article : Google Scholar
|
|
30
|
Keene DR, Maddox BK, Kuo HJ, Sakai LY and
Glanville RW: Extraction of extendable beaded structures and their
identification as fibrillin-containing extracellular matrix
microfibrils. J Histochem Cytochem. 39:441–449. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kielty CM and Shuttleworth CA:
Fibrillin-containing microfibrils: Structure and function in health
and disease. Int J Biochem Cell Biol. 27:747–760. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kewley MA, Williams G and Steven FS:
Studies of elastic tissue formation in the developing bovine
ligamentum nuchae. J Pathol. 124:95–101. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carta L, Pereira L, Arteaga-Solis E,
Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, Sukoyan M, Kerkis
A, Hazeki N, et al: Fibrillins 1 and 2 perform partially
overlapping functions during aortic development. J Biol Chem.
281:8016–8023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan X, Werner JM, Lack J, Knott V,
Handford PA, Campbell ID and Downing AK: Effects of the N2144S
mutation on backbone dynamics of a TB-cbEGF domain pair from human
fibrillin-1. J Mol Biol. 316:113–125. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yadin DA, Robertson IB, McNaught-Davis J,
Evans P, Stoddart D, Handford PA, Jensen SA and Redfield C:
Structure of the fibrillin-1 N-terminal domains suggests that
heparan sulfate regulates the early stages of microfibril assembly.
Structure. 21:1743–1756. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sabatier L, Chen D, Fagotto-Kaufmann C,
Hubmacher D, McKee MD, Annis DS, Mosher DF and Reinhardt DP:
Fibrillin assembly requires fibronectin. Mol Biol Cell. 20:846–858.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kinsey R, Williamson MR, Chaudhry S,
Mellody KT, McGovern A, Takahashi S, Shuttleworth CA and Kielty CM:
Fibrillin-1 microfibril deposition is dependent on fibronectin
assembly. J Cell Sci. 121:2696–2704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sabatier L, Djokic J, Fagotto-Kaufmann C,
Chen M, Annis DS, Mosher DF and Reinhardt DP: Complex contributions
of fibronectin to initiation and maturation of microfibrils.
Biochem J. 456:283–295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baldwin AK, Cain SA, Lennon R, Godwin A,
Merry CL and Kielty CM: Epithelial-mesenchymal status influences
how cells deposit fibrillin microfibrils. J Cell Sci. 127:158–171.
2014. View Article : Google Scholar :
|
|
40
|
Gibson MA, Kumaratilake JS and Cleary EG:
The protein components of the 12-nanometer microfibrils of elastic
and nonelastic tissues. J Biol Chem. 264:4590–4598. 1989.PubMed/NCBI
|
|
41
|
Trask BC, Trask TM, Broekelmann T and
Mecham RP: The microfibrillar proteins MAGP-1 and fibrillin-1 form
a ternary complex with the chondroitin sulfate proteoglycan
decorin. Mol Biol Cell. 11:1499–1507. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mecham RP and Gibson MA: The
microfibril-associated glycoproteins (MAGPs) and the microfibrillar
niche. Matrix Biol. 47:13–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kostka G, Giltay R, Bloch W, Addicks K,
Timpl R, Fässler R and Chu ML: Perinatal lethality and endothelial
cell abnormalities in several vessel compartments of
fibulin-1-deficient mice. Mol Cell Biol. 21:7025–7034. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Freeman LJ, Lomas A, Hodson N, Sherratt
MJ, Mellody KT, Weiss AS, Shuttleworth A and Kielty CM: Fibulin-5
interacts with fibrillin-1 molecules and microfibrils. Biochem J.
388:1–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yanagisawa H, Davis EC, Starcher BC, Ouchi
T, Yanagisawa M, Richardson JA and Olson EN: Fibulin-5 is an
elastin-binding protein essential for elastic fibre development in
vivo. Nature. 415:168–171. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hirai M, Ohbayashi T, Horiguchi M, Okawa
K, Hagiwara A, Chien KR, Kita T and Nakamura T: Fibulin-5/DANCE has
an elastogenic organizer activity that is abrogated by proteolytic
cleavage in vivo. J Cell Biol. 176:1061–1071. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gabriel LA, Wang LW, Bader H, Ho JC,
Majors AK, Hollyfield JG, Traboulsi EI and Apte SS: ADAMTSL4, a
secreted glycoprotein widely distributed in the eye, binds
fibrillin-1 microfibrils and accelerates microfibril biogenesis.
Invest Ophthalmol Vis Sci. 53:461–469. 2012. View Article : Google Scholar :
|
|
48
|
Tsutsui K, Manabe R, Yamada T, Nakano I,
Oguri Y, Keene DR, Sengle G, Sakai LY and Sekiguchi K: ADAMTSL-6 is
a novel extracellular matrix protein that binds to fibrillin-1 and
promotes fibrillin-1 fibril formation. J Biol Chem. 285:4870–4882.
2010. View Article : Google Scholar :
|
|
49
|
Kutz WE, Wang LW, Bader HL, Majors AK,
Iwata K, Traboulsi EI, Sakai LY, Keene DR and Apte SS: ADAMTS10
protein interacts with fibrillin-1 and promotes its deposition in
extracellular matrix of cultured fibroblasts. J Biol Chem.
286:17156–17167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hubmacher D and Apte SS: ADAMTS proteins
as modulators of microfibril formation and function. Matrix Biol.
47:34–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Iozzo RV: Basement membrane proteoglycans:
From cellar to ceiling. Nat Rev Mol Cell Biol. 6:646–656. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Murdoch AD, Liu B, Schwarting R, Tuan RS
and Iozzo RV: Widespread expression of perlecan proteoglycan in
basement membranes and extracellular matrices of human tissues as
detected by a novel monoclonal antibody against domain III and by
in situ hybridization. J Histochem Cytochem. 42:239–249. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Reinboth B, Hanssen E, Cleary EG and
Gibson MA: Molecular interactions of biglycan and decorin with
elastic fiber components: Biglycan forms a ternary complex with
tropoelastin and microfibril-associated glycoprotein 1. J Biol
Chem. 277:3950–3957. 2002. View Article : Google Scholar
|
|
54
|
Raghunath M, Superti-Furga A, Godfrey M
and Steinmann B: Decreased extracellular deposition of fibrillin
and decorin in neonatal Marfan syndrome fibroblasts. Hum Genet.
90:511–515. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Superti-Furga A, Raghunath M and Willems
PJ: Deficiencies of fibrillin and decorin in fibroblast cultures of
a patient with neonatal Marfan syndrome. J Med Genet. 29:875–878.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hayes AJ, Lord MS, Smith SM, Smith MM,
Whitelock JM, Weiss AS and Melrose J: Colocalization in vivo and
association in vitro of perlecan and elastin. Histochem Cell Biol.
136:437–454. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tiedemann K, Sasaki T, Gustafsson E,
Göhring W, Bätge B, Notbohm H, Timpl R, Wedel T,
Schlötzer-Schrehardt U and Reinhardt DP: Microfibrils at basement
membrane zones interact with perlecan via fibrillin-1. J Biol Chem.
280:11404–11412. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Whitelock JM, Melrose J and Iozzo RV:
Diverse cell signaling events modulated by perlecan. Biochemistry.
47:11174–11183. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kerever A, Mercier F, Nonaka R, de Vega S,
Oda Y, Zalc B, Okada Y, Hattori N, Yamada Y and Arikawa-Hirasawa E:
Perlecan is required for FGF-2 signaling in the neural stem cell
niche. Stem Cell Res. 12:492–505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Thisse B and Thisse C: Functions and
regulations of fibroblast growth factor signaling during embryonic
development. Dev Biol. 287:390–402. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Murasawa Y, Watanabe K, Yoneda M, Zako M,
Kimata K, Sakai LY and Isogai Z: Homotypic versican G1 domain
interactions enhance hyaluronan incorporation into fibrillin
microfibrils. J Biol Chem. 288:29170–29181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wight TN and Merrilees MJ: Proteoglycans
in atherosclerosis and restenosis: Key roles for versican. Circ
Res. 94:1158–1167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wu YJ, La Pierre DP, Wu J, Yee AJ and Yang
BB: The interaction of versican with its binding partners. Cell
Res. 15:483–494. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zheng PS, Vais D, Lapierre D, Liang YY,
Lee V, Yang BL and Yang BB: PG-M/versican binds to P-selectin
glycoprotein ligand-1 and mediates leukocyte aggregation. J Cell
Sci. 117:5887–5895. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Grässel S, Unsöld C, Schäcke H,
Bruckner-Tuderman L and Bruckner P: Collagen XVI is expressed by
human dermal fibroblasts and keratinocytes and is associated with
the microfibrillar apparatus in the upper papillary dermis. Matrix
Biol. 18:309–317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hubert T, Grimal S, Ratzinger S, Mechaly
I, Grassel S and Fichard-Carroll A: Collagen XVI is a neural
component of the developing and regenerating dorsal root ganglia
extracellular matrix. Matrix Biol. 26:206–210. 2007. View Article : Google Scholar
|
|
67
|
Ono RN, Sengle G, Charbonneau NL, Carlberg
V, Bächinger HP, Sasaki T, Lee-Arteaga S, Zilberberg L, Rifkin DB,
Ramirez F, et al: Latent transforming growth factor beta-binding
proteins and fibulins compete for fibrillin-1 and exhibit exquisite
specificities in binding sites. J Biol Chem. 284:16872–16881. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Dallas SL, Sivakumar P, Jones CJ, Chen Q,
Peters DM, Mosher DF, Humphries MJ and Kielty CM: Fibronectin
regulates latent transforming growth factor-beta (TGF beta) by
controlling matrix assembly of latent TGF-beta binding protein-1. J
Biol Chem. 280:18871–18880. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Fontana L, Chen Y, Prijatelj P, Sakai T,
Fässler R, Sakai LY and Rifkin DB: Fibronectin is required for
integrin alphavbeta6-mediated activation of latent TGF-beta
complexes containing LTBP-1. FASEB J. 19:1798–1808. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kantola AK, Keski-Oja J and Koli K:
Fibronectin and heparin binding domains of latent TGF-beta binding
protein (LTBP)-4 mediate matrix targeting and cell adhesion. Exp
Cell Res. 314:2488–2500. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Saharinen J, Hyytiäinen M, Taipale J and
Keski-Oja J: Latent transforming growth factor-beta binding
proteins (LTBPs)-structural extracellular matrix proteins for
targeting TGF-beta action. Cytokine Growth Factor Rev. 10:99–117.
1999. View Article : Google Scholar
|
|
72
|
Gregory KE, Ono RN, Charbonneau NL, Kuo
CL, Keene DR, Bachinger HP and Sakai LY: The prodomain of BMP-7
targets the BMP-7 complex to the extracellular matrix. J Biol Chem.
280:27970–27980. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sengle G, Charbonneau NL, Ono RN, Sasaki
T, Alvarez J, Keene DR, Bächinger HP and Sakai LY: Targeting of
bone morphogenetic protein growth factor complexes to fibrillin. J
Biol Chem. 283:13874–13888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sengle G, Tsutsui K, Keene DR, Tufa SF,
Carlson EJ, Charbonneau NL, Ono RN, Sasaki T, Wirtz MK, Samples JR,
et al: Microenvironmental regulation by fibrillin-1. PLoS Genet.
8:e10024252012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wohl AP, Troilo H, Collins RF, Baldock C
and Sengle G: Extracellular regulation of bone morphogenetic
protein activity by the microfibril component fibrillin-1. J Biol
Chem. 291:12732–12746. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Charbonneau NL, Ono RN, Corson GM, Keene
DR and Sakai LY: Fine tuning of growth factor signals depends on
fibrillin microfibril networks. Birth Defects Res Part C Embryo
Today. 72:37–50. 2004. View Article : Google Scholar
|
|
77
|
Massagué J and Chen YG: Controlling
TGF-beta signaling. Genes Dev. 14:627–644. 2000.PubMed/NCBI
|
|
78
|
Lawrence DA, Pircher R, Krycève-Martinerie
C and Jullien P: Normal embryo fibroblasts release transforming
growth factors in a latent form. J Cell Physiol. 121:184–188. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T
and Springer TA: Latent TGF-β structure and activation. Nature.
474:343–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zeyer KA and Reinhardt DP:
Fibrillin-containing microfibrils are key signal relay stations for
cell function. J Cell Commun Signal. 9:309–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Dubois CM, Laprise MH, Blanchette F,
Gentry LE and Leduc R: Processing of transforming growth factor
beta 1 precursor by human furin convertase. J Biol Chem.
270:10618–10624. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Nunes I, Munger J, Harpel JG, Nagano Y,
Shapiro R, Gleizes PE and Rifkin DB: Structure and activation of
the large latent transforming growth factor-Beta complex. J Am
Optom Assoc. 69:643–648. 1998.PubMed/NCBI
|
|
83
|
Annes JP, Munger JS and Rifkin DB: Making
sense of latent TGFbeta activation. J Cell Sci. 116:217–224. 2003.
View Article : Google Scholar
|
|
84
|
Hinz B: It has to be the αv: Myofibroblast
integrins activate latent TGF-β1. Nat Med. 19:1567–1568. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sato Y and Rifkin DB: Inhibition of
endothelial cell movement by pericytes and smooth muscle cells:
Activation of a latent transforming growth factor-beta 1-like
molecule by plasmin during co-culture. J Cell Biol. 109:309–315.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yu Q and Stamenkovic I: Cell
surface-localized matrix metalloproteinase-9 proteolytically
activates TGF-beta and promotes tumor invasion and angiogenesis.
Genes Dev. 14:163–176. 2000.PubMed/NCBI
|
|
87
|
Jenkins G: The role of proteases in
transforming growth factor-beta activation. Int J Biochem Cell
Biol. 40:1068–1078. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lyons RM, Gentry LE, Purchio AF and Moses
HL: Mechanism of activation of latent recombinant transforming
growth factor beta 1 by plasmin. J Cell Biol. 110:1361–1367. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Schultz-Cherry S and Murphy-Ullrich JE:
Thrombospondin causes activation of latent transforming growth
factor-beta secreted by endothelial cells by a novel mechanism. J
Cell Biol. 122:923–932. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Barcellos-Hoff MH, Derynck R, Tsang ML and
Weatherbee JA: Transforming growth factor-beta activation in
irradiated murine mammary gland. J Clin Invest. 93:892–899. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Schmierer B and Hill CS: TGFbeta-SMAD
signal transduction: Molecular specificity and functional
flexibility. Nat Rev Mol Cell Biol. 8:970–982. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chen X and Xu L: Mechanism and regulation
of nucleocytoplasmic trafficking of smad. Cell Biosci. 1:402011.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Tang LY and Zhang YE: Non-degradative
ubiquitination in Smad-dependent TGF-β signaling. Cell Biosci.
1:432011. View Article : Google Scholar
|
|
94
|
Feng XH and Derynck R: Specificity and
versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev
Biol. 21:659–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Massagué J, Seoane J and Wotton D: Smad
transcription factors. Genes Dev. 19:2783–2810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Verrecchia F, Chu ML and Mauviel A:
Identification of novel TGF-beta/Smad gene targets in dermal
fibroblasts using a combined cDNA microarray/promoter
transactivation approach. J Biol Chem. 276:17058–17062. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sengle G, Ono RN, Sasaki T and Sakai LY:
Prodomains of transforming growth factor beta (TGFbeta) superfamily
members specify different functions: Biglycan forms a ternary
complex with tropoelastin and microfibril-associated glycoprotein
1. J Biol Chem. 286:5087–5099. 2011. View Article : Google Scholar
|
|
98
|
Pereira L, D'Alessio M, Ramirez F, Lynch
JR, Sykes B, Pangilinan T and Bonadio J: Genomic organization of
the sequence coding for fibrillin, the defective gene product in
Marfan syndrome. Hum Mol Genet. 2:17621993. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Bax DV, Bernard SE, Lomas A, Morgan A,
Humphries J, Shuttleworth CA, Humphries MJ and Kielty CM: Cell
adhesion to fibrillin-1 molecules and microfibrils is mediated by
alpha 5 beta 1 and alpha v beta 3 integrins. J Biol Chem.
278:34605–34616. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Marek I, Volkert G, Hilgers KF, Bieritz B,
Rascher W, Reinhardt DP and Hartner A: Fibrillin-1 and alpha8
integrin are co-expressed in the glomerulus and interact to convey
adhesion of mesangial cells. Cell Adh Migr. 8:389–395. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lee SS, Knott V, Jovanović J, Harlos K,
Grimes JM, Choulier L, Mardon HJ, Stuart DI and Handford PA:
Structure of the integrin binding fragment from fibrillin-1 gives
new insights into microfibril organization. Structure. 12:717–729.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Bouzeghrane F, Reinhardt DP, Reudelhuber
TL and Thibault G: Enhanced expression of fibrillin-1, a
constituent of the myocardial extracellular matrix in fibrosis. Am
J Physiol Heart Circ Physiol. 289:H982–H991. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Bax DV, Mahalingam Y, Cain S, Mellody K,
Freeman L, Younger K, Shuttleworth CA, Humphries MJ, Couchman JR
and Kielty CM: Cell adhesion to fibrillin-1: Identification of an
Arg-Gly-Asp-dependent synergy region and a heparin-binding site
that regulates focal adhesion formation. J Cell Sci. 120:1383–1392.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Tiedemann K, Bätge B, Müller PK and
Reinhardt DP: Interactions of fibrillin-1 with heparin/heparan
sulfate, implications for microfibrillar assembly. J Biol Chem.
276:36035–36042. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Cain SA, Baldwin AK, Mahalingam Y, Raynal
B, Jowitt TA, Shuttleworth CA, Couchman JR and Kielty CM: Heparan
sulfate regulates fibrillin-1 N- and C-terminal interactions. J
Biol Chem. 283:27017–27027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Alexopoulou AN, Multhaupt HA and Couchman
JR: Syndecans in wound healing, inflammation and vascular biology.
Int J Biochem Cell Biol. 39:505–528. 2007. View Article : Google Scholar
|
|
107
|
Loeys BL, Gerber EE, Riegert-Johnson D,
Iqbal S, Whiteman P, McConnell V, Chillakuri CR, Macaya D, Coucke
PJ, De Paepe A, et al: Mutations in fibrillin-1 cause congenital
scleroderma: Stiff skin syndrome. Sci Transl Med. 2:23ra202010.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zou Y, Akazawa H, Qin Y, Sano M, Takano H,
Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, et al: Mechanical
stress activates angiotensin II type 1 receptor without the
involvement of angiotensin II. Nat Cell Biol. 6:499–506. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cook JR, Carta L, Bénard L, Chemaly ER,
Chiu E, Rao SK, Hampton TG, Yurchenco P; GenTAC Registry
Consortium; Costa KD, et al: Abnormal muscle mechanosignaling
triggers cardiomyopathy in mice with Marfan syndrome. J Clin
Invest. 124:1329–1339. 2014.PubMed/NCBI
|
|
110
|
Weber E, Rossi A, Solito R, Sacchi G,
Agliano' M and Gerli R: Focal adhesion molecules expression and
fibrillin deposition by lymphatic and blood vessel endothelial
cells in culture. Microvasc Res. 64:47–55. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Dietz HC, Cutting CR, Pyeritz RE, Maslen
CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ,
Curristin SM, et al: Marfan syndrome caused by a recurrent de novo
missense mutation in the fibrillin gene. Nature. 352:337–339. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Collod-Béroud G, Le Bourdelles S, Ades L,
Ala-Kokko L, Booms P, Boxer M, Child A, Comeglio P, De Paepe A,
Hyland JC, et al: Update of the UMD-FBN1 mutation database and
creation of an FBN1 polymorphism database. Hum Mutat. 22:199–208.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ramirez F and Dietz HC: Marfan syndrome:
From molecular pathogenesis to clinical treatment. Curr Opin Genet
Dev. 17:252–258. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sakai LY, Keene DR, Renard M and De Backer
J: FBN1: The disease-causing gene for Marfan syndrome and other
genetic disorders. Gene. 591:279–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Faivre L, Collod-Beroud G, Loeys BL, Child
A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K,
Arslan-Kirchner M, et al: Effect of mutation type and location on
clinical outcome in 1,013 probands with marfan syndrome or related
phenotypes and fbn1 mutations: An international study. Am J Hum
Genet. 81:454–466. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
116
|
Booms P, Cisler J, Mathews KR, Godfrey M,
Tiecke F, Kaufmann UC, Vetter U, Hagemeier C and Robinson PN: Novel
exon skipping mutation in the fibrillin-1 gene: Two 'hot spots' for
the neonatal Marfan syndrome. Clin Genet. 55:110–117. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Morse RP, Rockenmacher S, Pyeritz RE,
Sanders SP, Bieber FR, Lin A, MacLeod P, Hall B and Graham JM Jr:
Diagnosis and management of infantile marfan syndrome. Pediatrics.
86:888–895. 1990.PubMed/NCBI
|
|
118
|
Loeys BL, Dietz HC, Braverman AC,
Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y,
Jondeau G, Faivre L, Milewicz DM, et al: The revised Ghent nosology
for the Marfan syndrome. J Med Genet. 47:476–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Dietz HC and Pyeritz RE: Mutations in the
human gene for fibrillin-1 (FBN1) in the Marfan syndrome and
related disorders. Hum Mol Genet. 4(Spec No): 1799–1809. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Francke U, Berg MA, Tynan K, Brenn T, Liu
W, Aoyama T, Gasner C, Miller DC and Furthmayr H: A Gly1127Ser
mutation in an EGF-like domain of the fibrillin-1 gene is a risk
factor for ascending aortic aneurysm and dissection. Am J Hum
Genet. 56:1287–1296. 1995.PubMed/NCBI
|
|
121
|
Yamawaki T, Nagaoka K, Morishige K,
Sadamatsu K, Tashiro H, Yasunaga H, Morisaki H and Morisaki T:
Familial thoracic aortic aneurysm and dissection associated with
Marfan-related gene mutations: Case report of a family with two
gene mutations. Intern Med. 48:555–558. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Sood S, Eldadah ZA, Krause WL, McIntosh I
and Dietz HC: Mutation in fibrillin-1 and the
Marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nat
Genet. 12:209–211. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kainulainen K, Karttunen L, Puhakka L,
Sakai L and Peltonen L: Mutations in the fibrillin gene responsible
for dominant ectopia lentis and neonatal Marfan syndrome. Nat
Genet. 6:64–69. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Le Goff C, Mahaut C, Wang LW, Allali S,
Abhyankar A, Jensen S, Zylberberg L, Collod-Beroud G, Bonnet D,
Alanay Y, et al: Mutations in the TGFβ Binding-protein-like domain
5 of FBN1 are responsible for acromicric and geleophysic
dysplasias. Am J Hum Genet. 89:7–14. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Faivre L, Dollfus H, Lyonnet S, Alembik Y,
Mégarbané A, Samples J, Gorlin RJ, Alswaid A, Feingold J, Le Merrer
M, et al: Clinical homogeneity and genetic heterogeneity in
Weill-Marchesani syndrome. Am J Med Genet A. 123A:204–207. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Cecchi A, Ogawa N, Martinez HR, Carlson A,
Fan Y, Penny DJ, Guo DC, Eisenberg S, Safi H, Estrera A, et al:
Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani
syndrome with thoracic aortic disease and Marfan syndrome. Am J Med
Genet Part A. 161A:2305–2310. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Faivre L, Gorlin RJ, Wirtz MK, Godfrey M,
Dagoneau N, Samples JR, Le Merrer M, Collod-Beroud G, Boileau C,
Munnich A and Cormier-Daire V: In frame fibrillin-1 gene deletion
in autosomal dominant Weill-Marchesani syndrome. J Med Genet.
40:34–36. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Dagoneau N, Benoist-Lasselin C, Huber C,
Faivre L, Mégarbané A, Alswaid A, Dollfus H, Alembik Y, Munnich A,
Legeai-Mallet L and Cormier-Daire V: ADAMTS10 mutations in
autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet.
75:801–806. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
129
|
Gerber EE, Gallo EM, Fontana SC, Davis EC,
Wigley FM, Huso DL and Dietz HC: Integrin-modulating therapy
prevents fibrosis and autoimmunity in mouse models of scleroderma.
Nature. 503:126–130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Hollister DW, Godfrey M, Sakai LY and
Pyeritz RE: Immunohistologic abnormalities of the
Microfibrillar-fiber system in the marfan syndrome. N Engl J Med.
323:152–159. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Eldadah ZA, Brenn T, Furthmayr H and Dietz
HC: Expression of a mutant human fibrillin allele upon a normal
human or murine genetic background recapitulates a Marfan cellular
phenotype. J Clin Invest. 95:874–880. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Godfrey M, Raghunath M, Cisler J, Bevins
CL, DePaepe A, Di Rocco M, Gregoritch J, Imaizumi K, Kaplan P,
Kuroki Y, et al: Abnormal morphology of fibrillin microfibrils in
fibroblast cultures from patients with neonatal Marfan syndrome. Am
J Pathol. 146:1414–1421. 1995.PubMed/NCBI
|
|
133
|
Charbonneau NL, Carlson EJ, Tufa S, Sengle
G, Manalo EC, Carlberg VM, Ramirez F, Keene DR and Sakai LY: In
vivo studies of mutant Fibrillin-1 microfibrils. J Biol Chem.
285:24943–24955. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Aoyama T, Tynan K, Dietz HC, Francke U and
Furthmayr H: Missense mutations impair intracellular processing of
fibrillin and microfibril assembly in Marfan syndrome. Hum Mol
Genet. 2:2135–2140. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Judge DP, Biery NJ, Keene DR, Geubtner J,
Myers L, Huso DL, Sakai LY and Dietz HC: Evidence for a critical
contribution of haploinsufficiency in the complex pathogenesis of
Marfan syndrome. J Clin Invest. 114:172–181. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Arbustini E, Grasso M, Ansaldi S, Malattia
C, Pilotto A, Porcu E, Disabella E, Marziliano N, Pisani A,
Lanzarini L, et al: Identification of sixty-two novel and twelve
known FBN1 mutations in eighty-one unrelated probands with Marfan
syndrome and other fibrillinopathies. Hum Mutat. 26:4942005.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Reinhardt DP, Ono RN, Notbohm H, Müller
PK, Bächinger HP and Sakai LY: Mutations in calcium-binding
epidermal growth factor modules render fibrillin-1 susceptible to
proteolysis. A potential disease-causing mechanism in Marfan
syndrome. J Biol Chem. 275:12339–12345. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Booms P, Tiecke F, Rosenberg T, Hagemeier
C and Robinson PN: Differential effect of FBN1 mutations on in
vitro proteolysis of recombinant fibrillin-1 fragments. Hum Genet.
107:216–224. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Hindson VJ, Ashworth JL, Rock MJ, Cunliffe
S, Shuttleworth CA and Kielty CM: Fibrillin degradation by matrix
metalloproteinases: Identification of amino- and carboxy-terminal
cleavage sites. FEBS Lett. 452:195–198. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ikonomidis JS, Jones JA, Barbour JR,
Stroud RE, Clark LL, Kaplan BS, Zeeshan A, Bavaria JE, Gorman JH
III, Spinale FG and Gorman RC: Expression of matrix
metalloproteinases and endogenous inhibitors within ascending
aortic aneurysms of patients with Marfan syndrome. Circulation.
114(Suppl 1): I365–I370. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Segura AM, Luna RE, Horiba K,
Stetler-Stevenson WG, McAllister HA Jr, Willerson JT and Ferrans
VJ: Immunohistochemistry of matrix metalloproteinases and their
inhibitors in thoracic aortic aneurysms and aortic valves of
patients with Marfan's syndrome. Circulation. 98(Suppl 19):
II331–II338. 1998.PubMed/NCBI
|
|
142
|
Fleischer KJ, Nousari HC, Anhalt GJ, Stone
CD and Laschinger JC: Immunohistochemical abnormalities of
fibrillin in cardiovascular tissues in Marfan's syndrome. Ann
Thorac Surg. 63:1012–1017. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Granata A, Serrano F, Bernard WG, McNamara
M, Low L, Sastry P and Sinha S: An iPSC-derived vascular model of
Marfan syndrome identifies key mediators of smooth muscle cell
death. Nat Genet. 49:97–109. 2017. View Article : Google Scholar
|
|
144
|
Neptune ER, Frischmeyer PA, Arking DE,
Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY and Dietz HC:
Dysregulation of TGF-beta activation contributes to pathogenesis in
Marfan syndrome. Nat Genet. 33:407–411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ng CM, Cheng A, Myers LA, Martinez-Murillo
F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP
and Dietz HC: TGF-beta-dependent pathogenesis of mitral valve
prolapse in a mouse model of Marfan syndrome. J Clin Invest.
114:1586–1592. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Franken R, den Hartog AW, de Waard V,
Engele L, Radonic T, Lutter R, Timmermans J, Scholte AJ, van den
Berg MP, Zwinderman AH, et al: Circulating transforming growth
factor-β as a prognostic biomarker in Marfan syndrome. Int J
Cardiol. 168:2441–2446. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Pattanaik D, Brown M and Postlethwaite AE:
Vascular involvement in systemic sclerosis (scleroderma). J Inflamm
Res. 4:105–125. 2011.PubMed/NCBI
|
|
148
|
Siracusa LD, McGrath R, Ma Q, Moskow JJ,
Manne J, Christner PJ, Buchberg AM and Jimenez SA: A tandem
duplication within the fibrillin 1 gene is associated with the
mouse tight skin mutation. Genome Res. 6:300–313. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Lemaire R, Bayle J and Lafyatis R:
Fibrillin in Marfan syndrome and tight skin mice provides new
insights into transforming growth factor-beta regulation and
systemic sclerosis. Curr Opin Rheumatol. 18:582–587. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Gayraud B, Keene DR, Sakai LY and Ramirez
F: New insights into the assembly of extracellular microfibrils
from the analysis of the fibrillin 1 mutation in the tight skin
mouse. J Cell Biol. 150:667–680. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kielty CM, Raghunath M, Siracusa LD,
Sherratt MJ, Peters R, Shuttleworth CA and Jimenez SA: The tight
skin mouse: Demonstration of mutant fibrillin-1 production and
assembly into abnormal microfibrils. J Cell Biol. 140:1159–1166.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Saito S, Nishimura H, Brumeanu TD, Casares
S, Stan AC, Honjo T and Bona CA: Characterization of mutated
protein encoded by partially duplicated fibrillin-1 gene in tight
skin (TSK) mice. Mol Immunol. 36:169–176. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Gardi C, Martorana PA, de Santi MM, van
Even P and Lungarella G: A biochemical and morphological
investigation of the early development of genetic emphysema in
tight-skin mice. Exp Mol Pathol. 50:398–410. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Tan FK, Arnett FC, Antohi S, Saito S,
Mirarchi A, Spiera H, Sasaki T, Shoichi O, Takeuchi K, Pandey JP,
et al: Autoantibodies to the extracellular matrix microfibrillar
protein, fibrillin-1, in patients with scleroderma and other
connective tissue diseases. J Immunol. 163:1066–1072.
1999.PubMed/NCBI
|
|
155
|
Siracusa LD, McGrath R, Fisher JK and
Jimenez SA: The mouse tight skin (Tsk) phenotype is not dependent
on the presence of mature T and B lymphocytes. Mamm Genome.
9:907–909. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Dodig TD, Mack KT, Cassarino DF and Clark
SH: Development of the tight-skin phenotype in immune-deficient
mice. Arthritis Rheum. 44:723–727. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Kissin EY, Lemaire R, Korn JH and Lafyatis
R: Transforming growth factor beta induces fibroblast fibrillin-1
matrix formation. Arthritis Rheum. 46:3000–3009. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Podolsky DK: Inflammatory bowel disease. N
Engl J Med. 347:417–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Shimshoni E, Yablecovitch D, Baram L,
Dotan I and Sagi I: ECM remodelling in IBD: Innocent bystander or
partner in crime? The emerging role of extracellular molecular
events in sustaining intestinal inflammation. Gut. 64:367–372.
2015. View Article : Google Scholar :
|
|
160
|
Stumpf M, Cao W, Klinge U, Klosterhalfen
B, Junge K, Krones CJ and Schumpelick V: Reduced expression of
collagen type I and increased expression of matrix
metalloproteinases 1 in patients with Crohn's disease. J Invest
Surg. 18:33–38. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Stumpf M, Cao W, Klinge U, Klosterhalfen
B, Kasperk R and Schumpelick V: Increased distribution of collagen
type III and reduced expression of matrix metalloproteinase 1 in
patients with diverticular disease. Int J Colorectal Dis.
16:271–275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Stallmach A, Schuppan D, Riese HH, Matthes
H and Riecken EO: Increased collagen type III synthesis by
fibroblasts isolated from strictures of patients with Crohn's
disease. Gastroenterology. 102:1920–1929. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Graham MF, Diegelmann RF, Elson CO,
Lindblad WJ, Gotschalk N, Gay S and Gay R: Collagen content and
types in the intestinal strictures of Crohn's disease.
Gastroenterology. 94:257–265. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Ratzinger S, Eble JA, Pasoldt A, Opolka A,
Rogler G, Grifka J and Grässel S: Collagen XVI induces formation of
focal contacts on intestinal myofibroblasts isolated from the
normal and inflamed intestinal tract. Matrix Biol. 29:177–193.
2010. View Article : Google Scholar
|
|
165
|
Koutroubakis IE, Petinaki E, Dimoulios P,
Vardas E, Roussomoustakaki M, Maniatis AN and Kouroumalis EA: Serum
laminin and collagen IV in inflammatory bowel disease. J Clin
Pathol. 56:817–820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Spenlé C, Lefebvre O, Lacroute J,
Méchine-Neuville A, Barreau F, Blottière HM, Duclos B, Arnold C,
Hussenet T, Hemmerlé J, et al: The laminin response in inflammatory
bowel disease: Protection or malignancy? PLoS One. 9:e1113362014.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
de la Motte CA: Hyaluronan in intestinal
homeostasis and inflammation: Implications for fibrosis. Am J
Physiol Gastrointest Liver Physiol. 301:G945–G949. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Sallam H, McNearney TA and Chen JD:
Systematic review: Pathophysiology and management of
gastrointestinal dysmotility in systemic sclerosis (scleroderma).
Aliment Pharmacol Ther. 23:691–712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Sjogren RW: Gastrointestinal motility
disorders in scleroderma. Arthritis Rheum. 37:1265–1282. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Marie I, Ducrotté P, Denis P, Hellot MF
and Levesque H: Outcome of small-bowel motor impairment in systemic
sclerosis-a prospective manometric 5-yr follow-up. Rheumatology
(Oxford). 46:150–153. 2007. View Article : Google Scholar
|
|
171
|
Greydanus MP and Camilleri M: Abnormal
postcibal antral and small bowel motility due to neuropathy or
myopathy in systemic sclerosis. Gastroenterology. 96:110–115. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Iovino P, Valentini G, Ciacci C, De Luca
A, Tremolaterra F, Sabbatini F, Tirri E and Mazzacca G: Proximal
stomach function in systemic sclerosis: Relationship with autonomic
nerve function. Dig Dis Sci. 46:723–730. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Ibba-Manneschi L, Del Rosso A, Pacini S,
Tani A, Bechi P and Matucci Cerinic M: Ultrastructural study of the
muscle coat of the gastric wall in a case of systemic sclerosis.
Ann Rheum Dis. 61:754–756. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Manetti M, Neumann E, Milia AF, Tarner IH,
Bechi P, Matucci-Cerinic M, Ibba-Manneschi L and Müller-Ladner U:
Severe fibrosis and increased expression of fibrogenic cytokines in
the gastric wall of systemic sclerosis patients. Arthritis Rheum.
56:3442–3447. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Pedersen J, Gao C, Egekvist H, Bjerring P,
Arendt-Nielsen L, Gregersen H and Drewes AM: Pain and biomechanical
responses to distention of the duodenum in patients with systemic
sclerosis. Gastroenterology. 124:1230–1239. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Latella G, Di Gregorio J, Flati V, Rieder
F and Lawrance IC: Mechanisms of initiation and progression of
intestinal fibrosis in IBD. Scand J Gastroenterol. 50:53–65. 2015.
View Article : Google Scholar
|
|
177
|
LeRoy EC, Trojanowska MI and Smith EA:
Cytokines and human fibrosis. Eur Cytokine Netw. 1:215–219.
1990.PubMed/NCBI
|
|
178
|
Babyatsky MW, Rossiter G and Podolsky DK:
Expression of transforming growth factors alpha and beta in colonic
mucosa in inflammatory bowel disease. Gastroenterology.
110:975–984. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Kulkarni AB and Karlsson S: Transforming
growth factor-beta 1 knockout mice. A mutation in one cytokine gene
causes a dramatic inflammatory disease. Am J Pathol. 143:3–9.
1993.PubMed/NCBI
|
|
180
|
Gorelik L and Flavell RA: Transforming
growth factor-beta in T-cell biology. Nat Rev Immunol. 2:46–53.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
181
|
Meijer MJ, Mieremet-Ooms MA, van der Zon
AM, van Duijn W, van Hogezand RA, Sier CF, Hommes DW, Lamers CB and
Verspaget HW: Increased mucosal matrix metalloproteinase-1, -2, -3
and -9 activity in patients with inflammatory bowel disease and the
relation with Crohn's disease phenotype. Dig Liver Dis. 39:733–739.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Lakatos G, Hritz I, Varga MZ, Juhász M,
Miheller P, Cierny G, Tulassay Z and Herszényi L: The impact of
matrix metalloproteinases and their tissue inhibitors in
inflammatory bowel diseases. Dig Dis. 30:289–295. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Rath T, Roderfeld M, Graf J, Wagner S,
Vehr AK, Dietrich C, Geier A and Roeb E: Enhanced expression of
MMP-7 and MMP-13 in inflammatory bowel disease: A precancerous
potential? Inflamm Bowel Dis. 12:1025–1035. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Booms P, Pregla R, Ney A, Barthel F,
Reinhardt DP, Pletschacher A, Mundlos S and Robinson PN:
RGD-containing fibrillin-1 fragments upregulate matrix
metallopro-teinase expression in cell culture: A potential factor
in the pathogenesis of the Marfan syndrome. Hum Genet. 116:51–61.
2005. View Article : Google Scholar
|
|
185
|
Booms P, Ney A, Barthel F, Moroy G,
Counsell D, Gille C, Guo G, Pregla R, Mundlos S, Alix AJ and
Robinson PN: A fibrillin-1-fragment containing the
elastin-binding-protein GxxPG consensus sequence upregulates matrix
metallopro-teinase-1: Biochemical and computational analysis. J Mol
Cell Cardiol. 40:234–246. 2006. View Article : Google Scholar : PubMed/NCBI
|