Introduction
Soft tissue sarcomas (STS) are a heterogeneous group
of malignant neoplasms. They represent 15% of all malignancies in
children and 1% in adults (1). The
therapy of choice involves surgical resection with a wide margin of
healthy tissue, usually followed by radiation treatment in order to
decrease local recurrence (2,3).
Unfortunately, about 50% of all patients develop distant metastases
and are ineligible for surgical treatment (4,5). In
cases of advanced metastatic disease the median survival time from
the time of diagnosis with and without chemotherapy treatment is
less than 12 months (6,7). Few agents such as doxorubicin,
ifosfamide and dacarbazine have proven to be effective in the
therapy of soft tissue sarcomas (2). However, the results of these
treatments are poor and often exhibit no significant improvements
in overall survival (8).
Doxorubicin, which has been the most frequently used
chemotherapeutic agent in the treatment of soft tissue sarcomas,
demonstrates response rates of 20 to 30% in disseminated disease
(9,10). The combination of doxorubicin with
ifosfamide is more effective, exhibiting slightly higher response
rates than doxorubicin alone, but is associated with severe short-
and long-term toxicities, including bone marrow suppression
(11,12). To date, most large trials have not
distinguished between histological subtypes of soft tissue
sarcomas. One example of such an early trial is the EORTC 62771
trial which was conducted in 1994 and involved 317 patients with
several histological types of sarcomas (malignant fibrous
histiocytoma, synovial sarcoma, liposarcoma and leiomyosarcoma)
(13). Local recurrence was
reduced in the chemotherapy arm, but there was no significant
benefit in overall survival. The Sarcoma Meta-Analysis
Collaboration in 1997 published a quantitative meta-analysis of
1,568 patients with localized resectable soft tissue sarcomas and
reported that doxorubicin-based adjuvant chemotherapy significantly
improved the time to local and distant recurrence and overall
recurrence-free survival. However, there was no significant overall
survival benefit at 10 years (14). Unfortunately, most of the large
meta-analyses did not differentiate between histological subtypes
because of the overall rarity of soft tissue sarcomas. However,
differentiation is an important prognostic factor because the soft
tissue sarcoma subtypes are differentially sensitive to several
agents (2). In recent phase II
trials, paclitaxel has proven to be effective in the treatment of
angiosarcomas, whereas trabectedin has demonstrated promising
activity in leiomyosarcomas and liposarcomas (2,15,16).
Within the scope of this trial, we investigated the
effects of TRAIL and taurolidine on three different STS cell lines.
Two common subtypes of soft tissue sarcomas, rhabdomyosarcoma
(A-204) in children and leiomyosarcoma (SK-LMS-1) in adults
(1), and epithelioid cell sarcoma
(VA-ES-BJ), which is a rare subtype affecting mostly young patients
and has a poor long-term prognosis (17), were examined.
TRAIL and taurolidine are promising combination
partners that exhibit synergistic apoptotic effects on a wide range
of malignant cells in vitro, including carcinoma cells of
the oesophagus, pancreas, colon and liver (18–20)
as well as fibrosarcoma (21).
Since the discovery of TRAIL, a member of the
TNF-superfamily, its apoptosis-inducing effects were documented in
several types of malignant cells (22–25).
TRAIL binds to its receptors DR4 and DR5 (death receptor 4 and 5)
resulting in receptor oligomerization and recruitment of
FAS-associated protein with death domain (FADD) and caspase 8,
forming a functional death-inducing signalling complex (DISC).
Subsequently, DISC leads to the activation of the extrinsic pathway
of apoptosis via caspase 8 (26–28).
However, associations between TRAIL and the intrinsic mitochondrial
pathway have been also described (20). In this pathway, formation of the
apoptosome is a key regulatory point following the release of
mitochondrial cytochrome c and thus leading to apoptosis
(29).
Taurolidine (TRD) is an antiseptic agent derived
from the amino acid taurine. It has been used to treat peritonitis
and catheter-related infections (18). Recently, TRD was used effectively
to treat malignant diseases (30–33).
In a variety of malignant cell lines including carcinomas of the
gastrointestinal tract as well as glioblastoma, fibrosarcoma,
prostate and melanoma cancer cell lines, TRD caused the inhibition
of proliferation (31,34–36),
the inhibition of angiogenesis (30) and the induction of cell death
(20,30–32,37–39).
The precise mechanism of action is still not clear, but
translational inhibition (35) and
several pathways of programmed cell death (38) have been implicated. Some groups
suggest that the extrinsic pathway (19,20,31,39,40)
is involved, whereas others report involvement of the intrinsic
pathway (37,41). Furthermore, first clinical use of
TRD was associated with remarkable low toxicity which could be a
decided advantage over established chemotherapeutic agents
(30,42). TRD was well tolerated after
intravenous application in patients with advanced melanoma or
glioblastoma (43,44).
Recent in vitro studies have revealed that
the combination of TRAIL and TRD resulted in sustained cell death,
which was superior to single application of TRAIL or TRD. This is
despite the use of lower concentrations of both substances in the
combination trials (19,20,40).
Experimental findings have demonstrated that combined treatment
with taurolidine reduces the potential toxic side-effects of TRAIL,
not only by reducing the required optimal dose of TRAIL but also by
modulating TRAIL’s effector pathways without affecting its
antitumour efficacy (20).
Inspired by these results, we examined the effects of TRAIL and TRD
on rhabdomyosarcoma (A-204), leiomyosarcoma (SK-LMS-1) and
epithelioid cell sarcoma (VA-ES-BJ) cells.
Materials and methods
Cell lines and cell culture
Three different cell lines were used for this study.
The human rhabdomyosarcoma cell line A-204 was purchased from DSMZ
(Braunschweig, Germany, DSMZ no. ACC 250) and cultivated in McCoy’s
5A with glutamine and 10% fetal bovine serum (FBS). Human
leiomyosarcoma cells, SK-LMS-1, were purchased from ATCC (Manassas,
USA) and maintained in MEM with Earle’s Salts supplemented with 10%
FBS, 1% non-essential amino acids, 1 mM sodium pyruvate and 2 mM
L-glutamine. The human epithelioid sarcoma cell line VA-ES-BJ also
was purchased from DSMZ (DSMZ no. ACC 328) and was cultured in
Dulbecco’s MEM with sodium pyruvate, supplemented with 20% FBS, 1%
non-essential amino acids and 2 mM L-glutamine. All culture media
were supplemented with 100 U/ml penicillin and 100 μg/ml
streptomycin. The cells were grown to a sub-confluent monolayer and
maintained at 37°C and 5% CO2 in a humidified
atmosphere.
Reagents
TRD (Taurolin® 2%, Boehringer Ingelheim,
Ingelheim, Germany) containing 5% Povidon was used as supplied by
the manufacturer. A 5% Povidon solution (K16 Povidon, generously
provided by Geistlich Pharma AG, Wolhusen, Switzerland) was applied
in equal volume and served as a control for the TRD group.
Recombinant human TRAIL/Apo2L (Bender MedSystems GmbH, Vienna,
Austria) was dissolved in distilled water according to the
manufacturer’s instructions. Distilled water served as a control
for TRAIL and was applied in equal volume.
Dose-finding study and application of
reagents
To determine the most effective single
concentrations and the time dependency of the effects, cells were
incubated with various concentrations of TRAIL (50, 100, 250 and
500 ng/ml), TRD (50, 100, 250 and 500 μmol/l) and the
respective controls (distilled water or Povidon) for 2, 6, 12, 24
and 48 h. All experiments were repeated with three consecutive cell
passages. All cell lines showed highest apoptotic response to 250
ng/ml TRAIL and 250 μmol/l TRD as single substances. These
most effective single concentrations of TRD and TRAIL were then
used as single substances and in combination to identify a possibly
synergistic effect. We chose 2, 6, 12, 24 and 48-h time points.
Flow cytometry analysis
After the defined incubation time, the supernatant
with floating cells was collected and the adherent cells were
harvested by trypsinisation. These cells were centrifuged and
subsequently resolved with Binding Buffer (Bender MedSystems GmbH)
to an absolute cell count of 1×105. Next, the cells were
incubated with Annexin V-FITC (BD Biosciences, Heidelberg, Germany)
and propidium iodide (PI, Bender MedSystems GmbH) following the
manufacturer’s instructions. Cells were analysed using a FACS flow
cytometer (BD FACSCalibur, BD Biosciences). Cells (20,000) were
counted for each measurement. Dot plots and histograms were
analysed by CellQuest Pro Software (BD Biosciences). Annexin V
binds phosphatidylserine on the outer membranes of cells, and
phosphatidylserine becomes exposed on the surfaces of apoptotic
cells. Thus, the Annexin V-positive cells were considered
apoptotic. PI is an intercalating agent that cannot permeate
through the cell membranes of viable or early apoptotic cells.
Therefore, PI stains only the DNA of necrotic or very late
apoptotic cells. In this study, Annexin V- and PI-positive cells
were termed necrotic. Annexin V- and PI-negative cells were counted
as viable.
Cell morphology
Morphology of cultured cells was observed and
documented using a phase contrast microscope (Zeiss Axiovert 25,
Carl Zeiss, Göttingen, Germany).
TUNEL assay
To stain apoptotic cells, we used terminal
deoxynucleotidyl transferase-mediated dUTP nick end-labelling
consistent with the manufacturer’s protocol (In Situ Cell Death
Detection Kit, Fluorescein, Roche Applied Science, Mannheim,
Germany). Cells were incubated with the appropriate reagents
(TRD/TRAIL) for 12 h.
Proliferation assay
To evaluate the proliferation of the cells, we used
a colorimetric cell proliferation BrdU-ELISA (Roche Applied
Science) according to the manufacturer’s instructions. An
ELISA-Reader (Sunrise™, Tecan Trading AG, Männedorf, Switzerland)
was used to detect the amount of newly synthesised DNA. We plated
10,000 cells per well in a 96-well plate. The incubation time was 6
h.
Statistical analysis
SPSS Version 17.0 for Windows was used for
statistical analysis. The results of FACS analysis for percentages
of viable, apoptotic and necrotic cells are given as the means ±
SEM of three independent experiments with consecutive passages. In
this study, comparisons between experimental groups were performed
using one-way analysis of variance (one-way ANOVA) and a post hoc
test (Tukey’s) over all time points and at singular time points.
P-values ≤0.05 were considered statistically significant and
indicated in the figures as follows: ***p≤0.001,
**p≤0.005 and *p≤0.05.
Oligonucleotide microarray analysis
To identify the changes in gene expression levels
caused by the treatment with TRAIL and TRD, total-RNA was purified
from the cells after incubation with the appropriate agent for 6 h
using a RNeasy kit from Qiagen (Hilden, Germany) as specified by
the manufacturer. RNA integrity was assessed using an Agilent 2100
Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). For
microarray analyses, we applied the methods previously described by
Daigeler et al(40). We
used the Affymetrix GeneChip platform, employing a standard
protocol for sample preparation and microarray hybridisation. A
one-way ANOVA model followed by Tukey’s HSD (Honestly Significant
Difference)-test was used to verify the hypothesis that there were
no differences in expression between the drug-treated group and the
control group. The multiplicity correction was performed using
Benjamini and Hochberg procedure to control the false discovery
rate (FDR) at 0.05%. In a pair-wise comparison of the
differentially expressed genes between the control and the
drug-treated cells identified by the ANOVA analyses, a subset of
genes was identified that displayed a conjoint regulation in the
treated cells. Genes were placed in this latter group if they
exhibited a mean ≥2-fold increase or decrease compared to the
control cells. This subset of genes was subjected to the GeneTrail
(45) software to identify any
over-representation of genes associated with the regulatory
pathways that are represented in the Kyoto Encyclopaedia of Genes
and Genomes (KEGG) and TRANSPATH databases. Microarray data are
deposited in the GEO public database (accession no. GSE36572).
These methods fulfilled the MIAME criteria (http://www.mged.org/miame).
Real-time PCR for microarray data
validation
Microarray data validation was performed for
selected candidate genes (BAG5, EGFR, FADD, FYN, GADD45A,
HSPA1B/HSP70, NEU1, PPM1D, PPP1R15A/GADD34, PPP1R3D, SIAH1, WEE1).
RNA isolation was performed from cells harvested after 6 h of
treatment. Total-RNA (2 μg) was reverse transcribed using
the High Capacity cDNA Archive kit (Applied Biosystems, Carslbad,
CA, USA). Real-time PCR was performed with a 7900 HT SDS system
(Applied Biosystems) in a 20 μl reaction volume containing
1X Master Mix, 1 μl assay and cDNA equivalent to 2 ng
total-RNA. All reagents and real-time PCR assays (BAG5
Hs00191644_m1, EGFR Hs01076092_m1, FADD Hs00538709_m1, FYN
Hs00941604_ m1, GADD45A Hs00169255_m1, HSPA1B Hs01040501_sH, NEU1
Hs00166421_m1, PPM1D Hs01013293_m1, PPP1R15A Hs00169585_m1, PPP1R3D
Hs00901540_s1, SIAH1 Hs00361785_m1, WEE1 Hs01119388_m1) were
purchased from Applied Biosystems. Reactions were performed in
duplicate and analysed by the ΔΔCt method. Human GAPD was used for
normalisation.
Western blot analysis
Western blot analyses were performed to validate the
effects of alterations in gene expression on protein levels using
an SDS-PAGE gel and the following antibodies (except BAG5, which
was purchased from Abcam PLC, Cambridge, UK); all other antibodies
were purchased from Santa Cruz Biotechnology Inc. (Heidelberg,
Germany): BAG5 (mouse, ab56738), EGFR (rabbit, 1005), FADD (rabbit,
H-181), GADD34/PPP1R15A (rabbit, S-20), GADD45α (rabbit, H-165),
HSP70 (mouse, C92F3A-5) and Wee1 (rabbit, C-20). Western blot
analyses were not performed for PPPM1D, FYN and PPP1R3D because
functional antibodies were not available. Total protein was
purified from the cells after incubation with the appropriate
substances for 8 h. Floating cells were collected together with the
supernatant; adherent cells were harvested using a cell scraper and
added to the solution. Cells were gathered by centrifugation. After
removal of the supernatant, pellets were incubated with 100
μl Cell Culture Lysis Reagent (Promega Corporation,
Mannheim, Germany) each for 1 h on ice. The cell remnants were then
separated by centrifugation, and the supernatant containing the
purified protein was frozen at −80°C until further use.
Results
The combination of TRAIL and TRD
amplifies apoptotic effects in A-204 human rhabdomyosarcoma and
VA-ES-BJ human epithelioid sarcoma cells
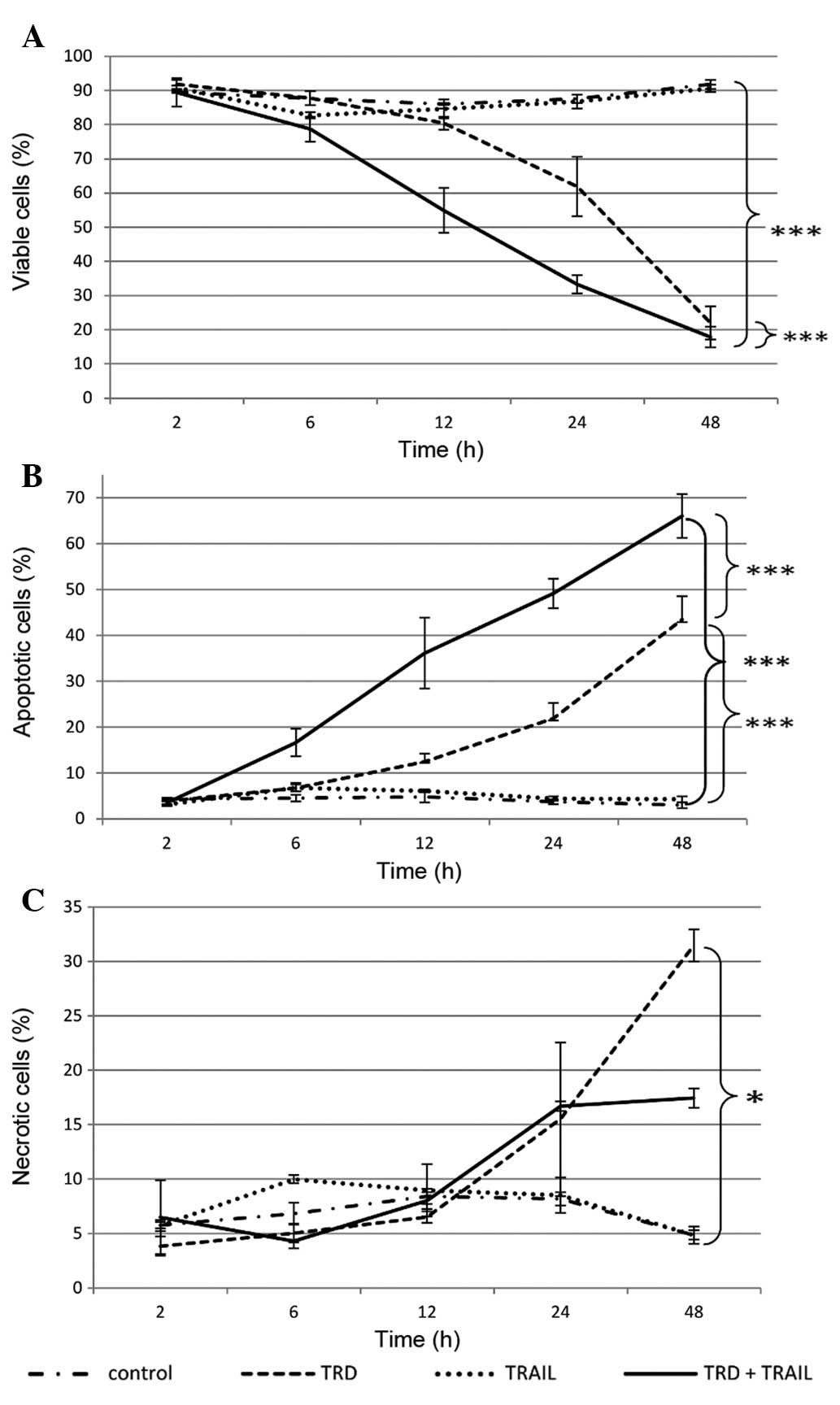
Single application of TRD induced significantly
apoptotic and necrotic cell death in A-204 human rhabdomyosarcoma
cells. Viable cells were decreased to 22.0% after 48 h of
incubation compared to 91.9% in the control and 90.6% in the TRAIL
group (p<0.001) (Fig. 1).
Addition of TRAIL promoted the apoptotic influence of TRD in
rhabdomyosarcoma cells. The combined treatment of TRD and TRAIL
resulted in a marked increase in cells undergoing apoptosis over
all time points (p≤0.001). After 6 h of incubation first apoptotic
effects were seen. The combination with TRAIL and TRD exhibited
highest apoptosis rates after 48 h with 66.1% apoptotic cells (3.0%
in control group, p≤0.001). Thus, combination treatment was most
effective in reducing cell viability with 17.9% remaining viable
cells after 48 h (vs. 91.9% in the control group, p<0.001)
(Fig. 1A).
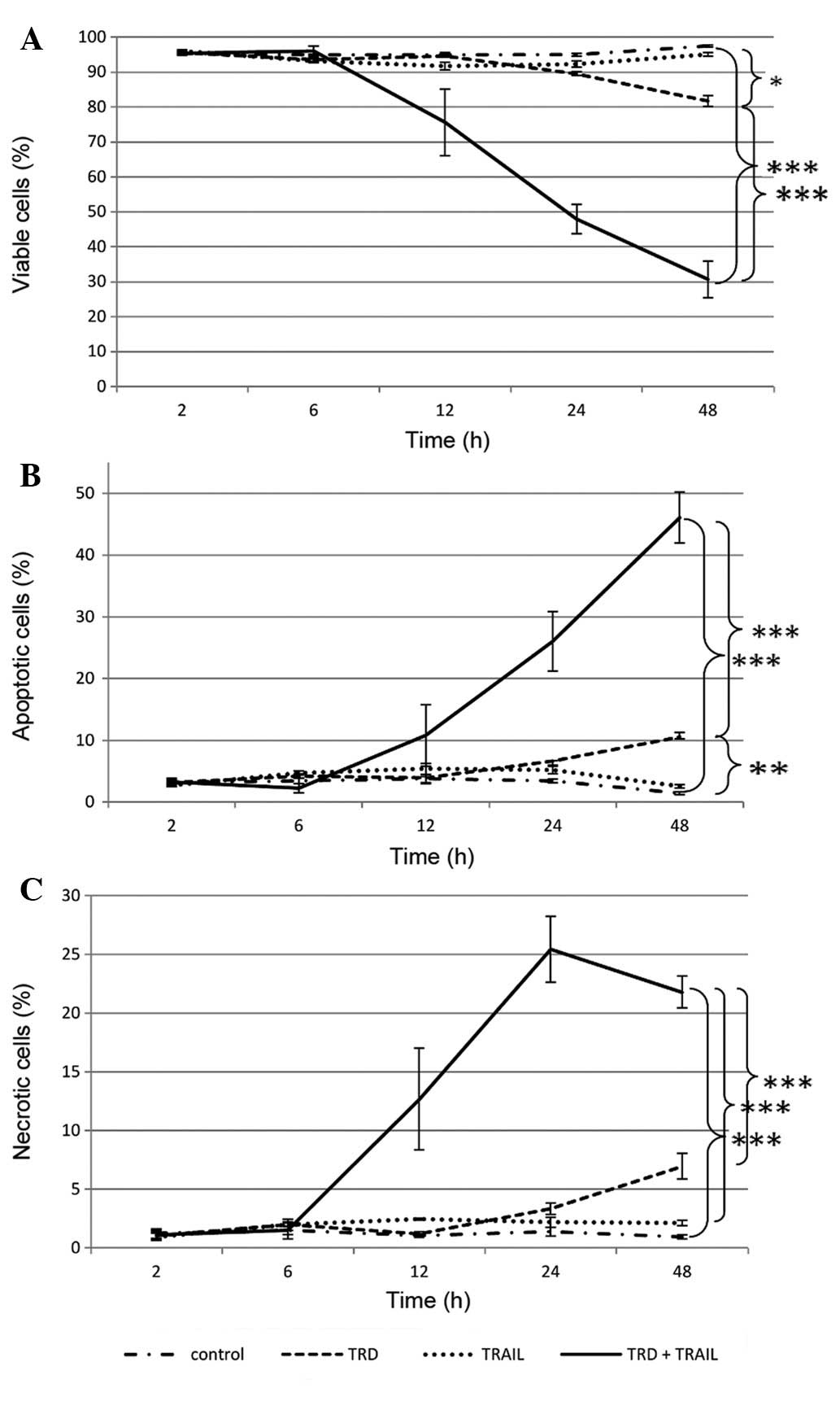
The viability of the VA-ES-BJ human epithelioid
sarcoma cells was moderately but significantly reduced by single
treatment with TRD (Fig. 2). A
total of 81.8% of the cells was detected as viable after 48 h
treatment with TRD (vs. 97.5% in the control group, p=0.015).
Single application of TRAIL had no significant effect on cell
viability. The combined treatment with TRAIL and TRD led to a
significant increased apoptotic cell death after 12 h. The
population of viable cells was reduced to 30.7% after 48 h of
incubation whereas 97.5% were left viable in the control group
(p<0.001). Apoptosis also peaked at this time point, reaching a
maximum of 46.1% in the combination group compared to only 1.5% in
the control group (p<0.001) (Fig.
2A).
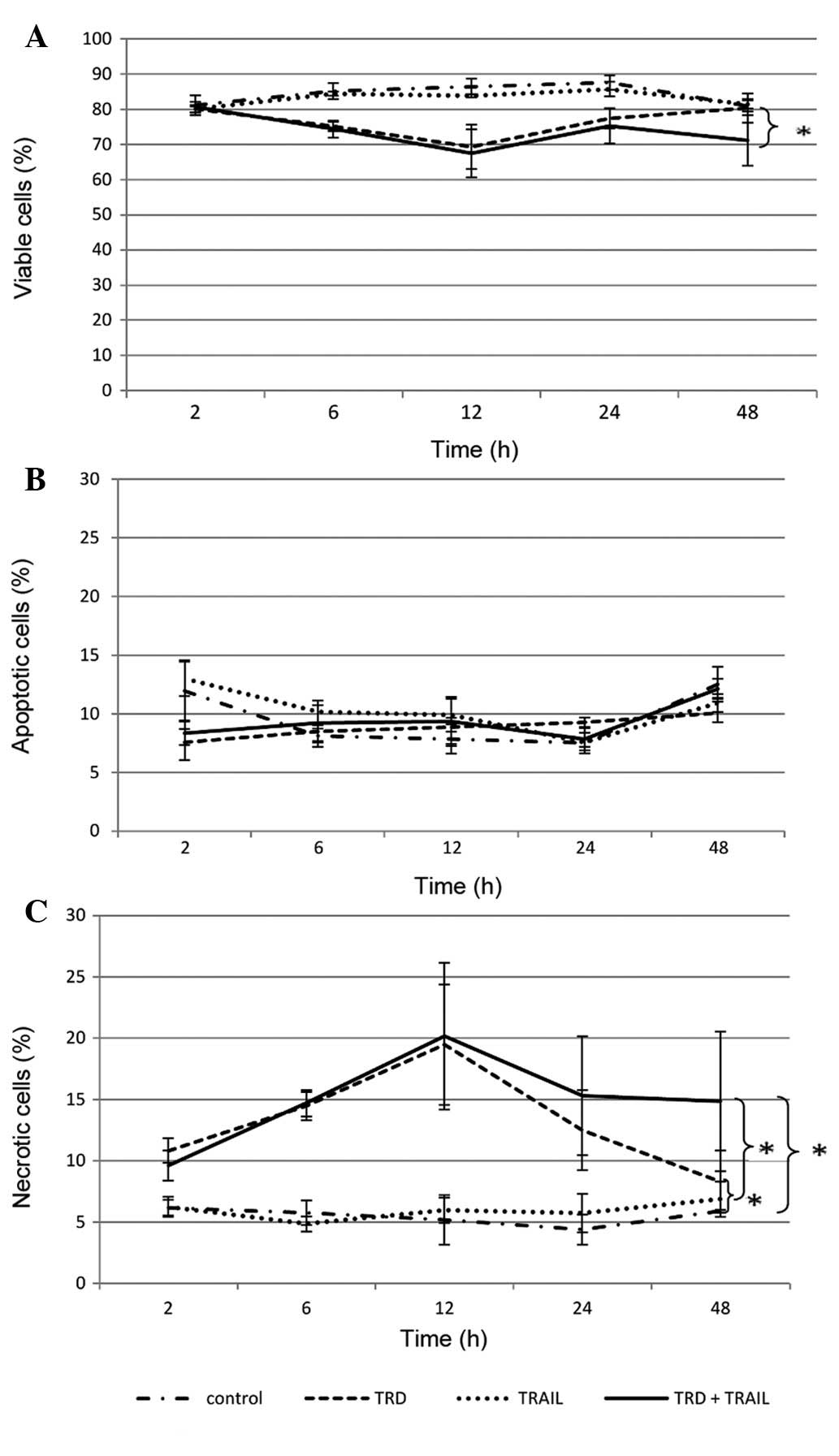
Neither single application nor
combination treatment with TRAIL and TRD affected viability of
SK-LMS-1 leiomyosarcoma cells significantly
The effects when treating SK-LMS-1 leiomyosarcoma
cells with TRD and TRAIL were only moderate (Fig. 3). The combined application of TRAIL
and TRD led to a slight decrease of viable cells to 67,5% after 12
h (vs. 86.5% in the control group, p=0.088). Single application of
TRD had a similar effect on SK-LMS-1 cells (69.4% viable cells
after 12 h). Neither single application nor combination treatment
with TRAIL and TRD had a significant influence on apoptosis.
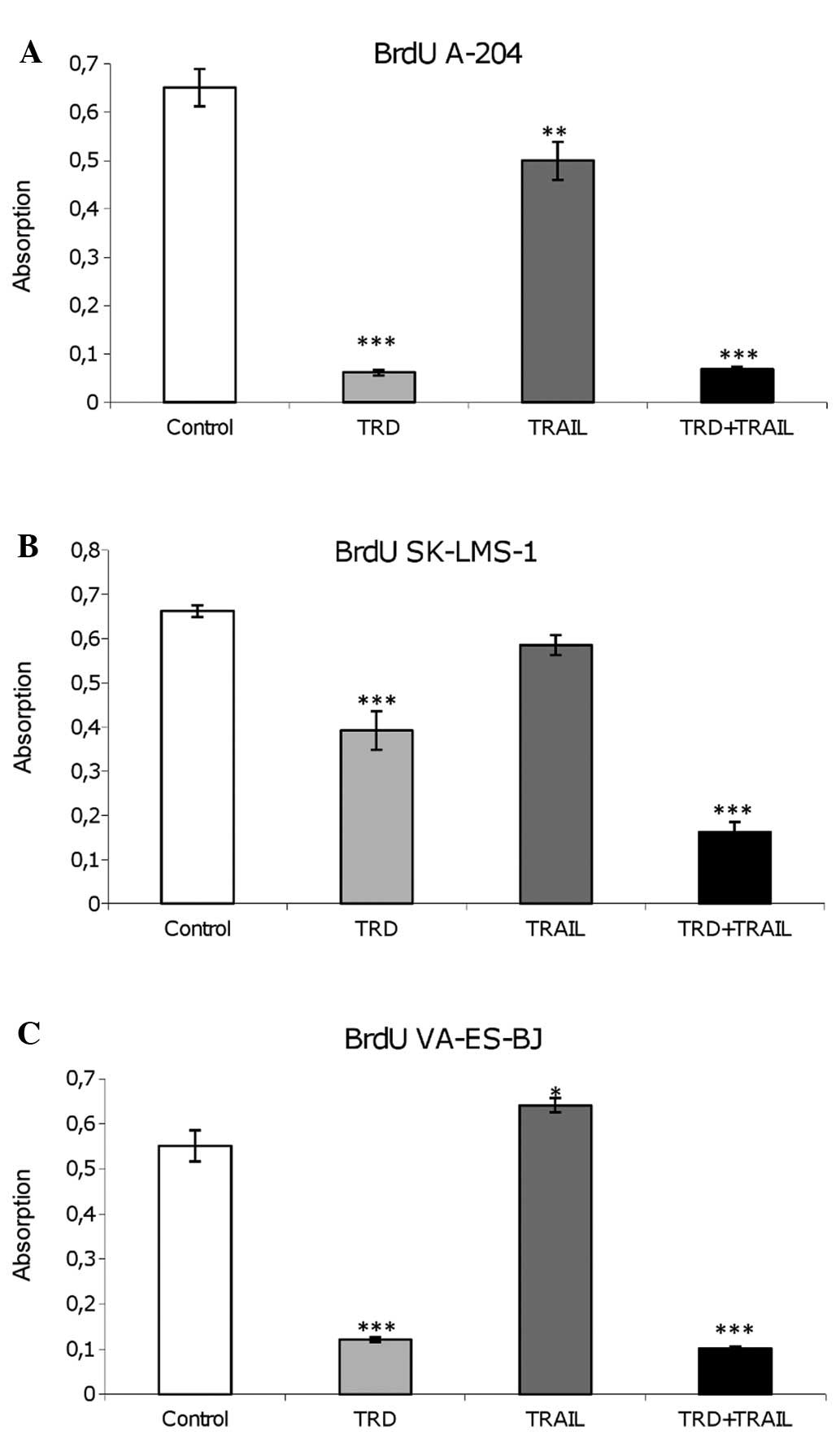
TRD significantly inhibited proliferation
of A-204 human rhabdomyosarcoma, VA-ES-BJ human epithelioid sarcoma
and SK-LMS-1 leiomyosarcoma cells
TRD was able to inhibit cell proliferation in all
examined cell lines (p<0.001) as indicated by the BrdU-Assay
(Fig. 4). In SK-LMS-1 cells,
combination with TRAIL resulted in a stronger effect compared to
incubation with TRD alone (p<0.001). For the other cell lines,
combination therapy did not increase the inhibition of
proliferation. Administration of TRAIL as a single agent reduced
proliferation significantly only in A-204 cells. Strikingly,
proliferation was increased in VA-ES-BJ cells after single
application of TRAIL.
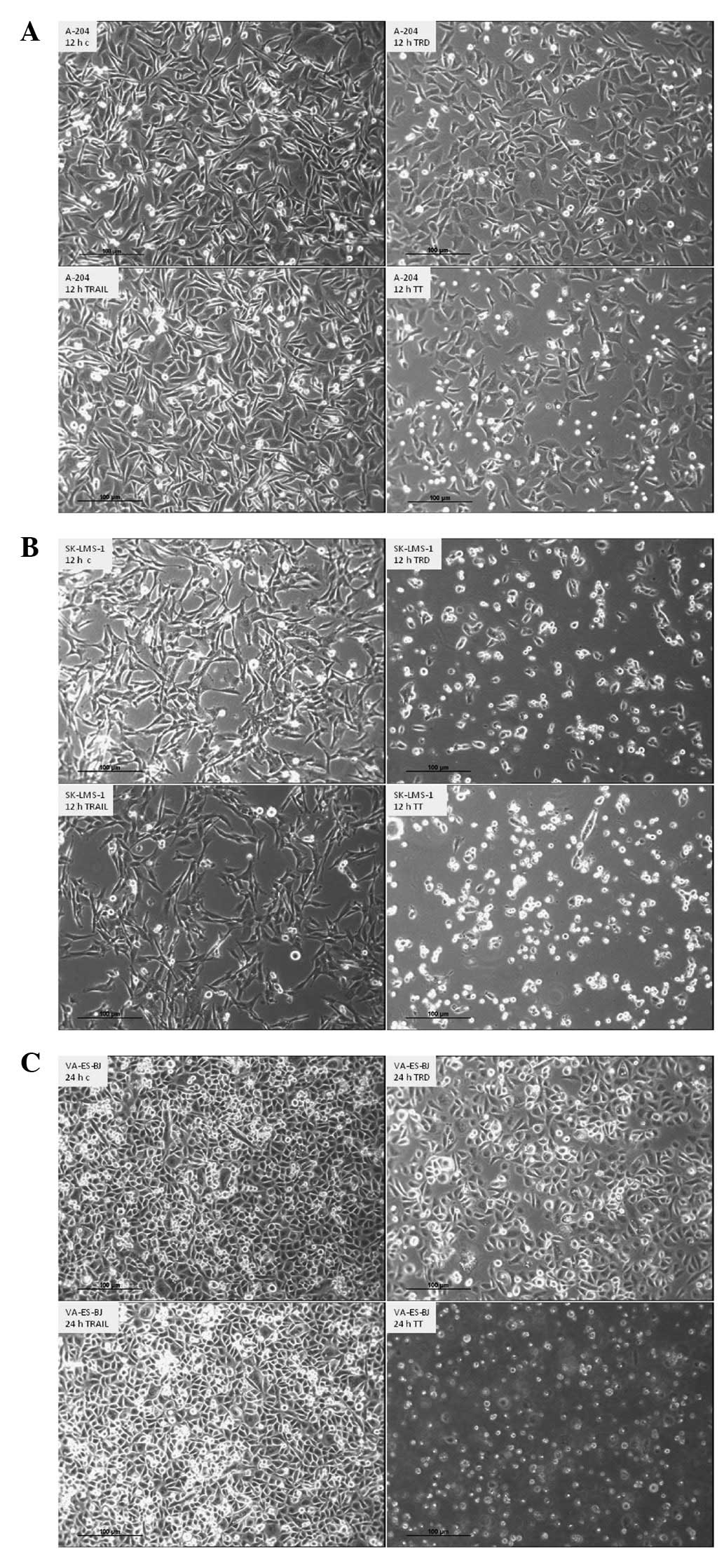
The addition of TRD induced morphological
changes and cell detachment
As demonstrated in Fig.
5, addition of TRD resulted in morphological changes in all
cell lines. TRD led to shrinkage of cells and dissolution of
confluent cells groups. Longer incubation with TRD resulted in
marked cell detachment.
Microarray analysis revealed differential
gene expression patterns in all examined cell lines after the
treatment with TRAIL and TRD
With respect to the overall gene expression
patterns, there were differences between the individual cell lines
(Fig. 6). In our gene expression
study, we focused on apoptosis-related genes. For all three cell
lines, we tested 621 probe sets as previously described by Daigeler
et al(20,40), which corresponded to 349 genes.
TRAIL caused only a few differences in the expression of the
analysed genes compared to the control, whereas TRD alone and in
combination with TRAIL led to expression changes in a wide range of
apoptosis-related genes. The number of altered genes with at least
a ≥2-fold change is shown in Table
I.
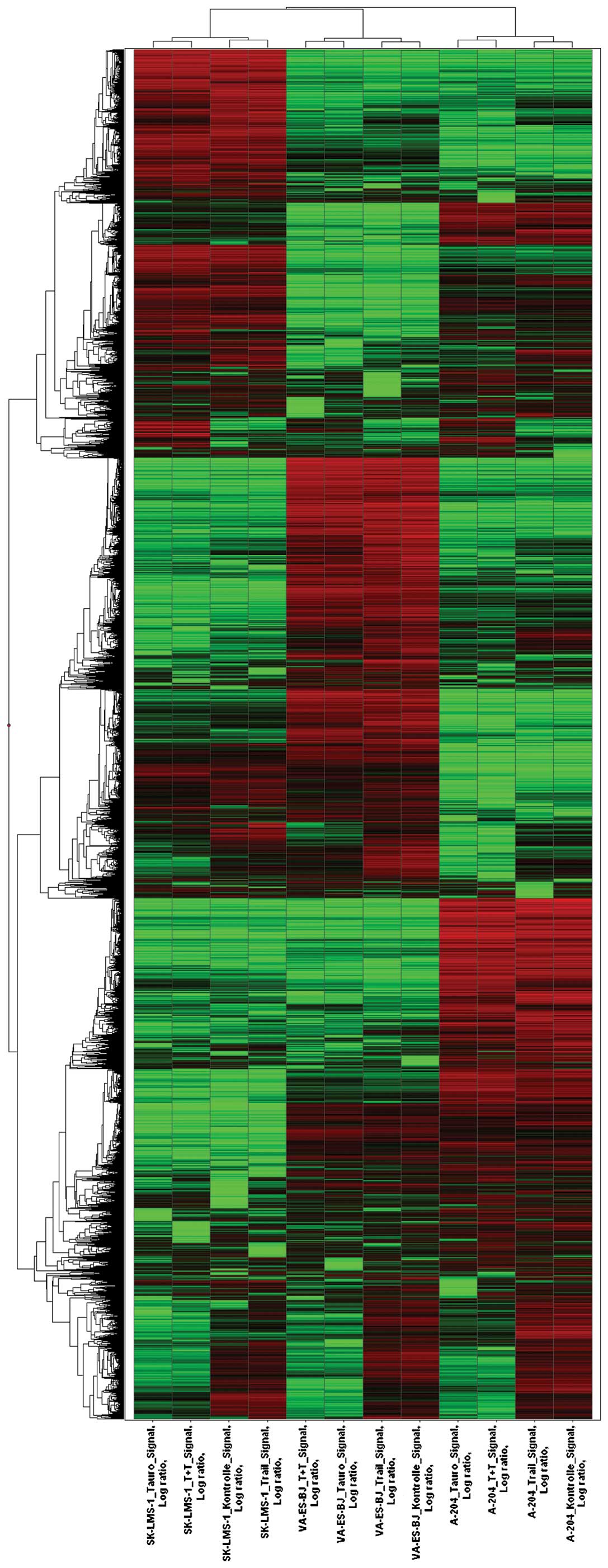 | Figure 6Hierarchical clustering: overall gene
expression pattern of reliably measured probesets (24797).
Horizontal rows represent individual probe sets/genes; vertical
columns represent individual samples. Kontrolle, control; Tauro,
TRD; Trail, TRAIL; T+T, combination treatment. Color scale: black,
mean (indicates unchanged expression), brightest green, 0,25 x mean
(indicates expression level below mean), brightest red, 4 x mean
(indicates higher expression level than mean). The dendogram at the
top of the matrix indicates the degree of similarity between
examined groups; the dendogram at the left side indicates the
degree of similarity among the selected genes according to their
expression patterns. |
 | Table INumber of altered genes with at least
a ≥2-fold change in gene expression. |
Table I
Number of altered genes with at least
a ≥2-fold change in gene expression.
| TRD vs.
control | TRAIL vs.
control | TT vs. control | TT vs. TRD | TT vs. TRAIL |
|---|
| A-204 | 128 | 10 | 113 | 19 | 125 |
| SK-LMS-1 | 116 | 22 | 122 | 2 | 118 |
| VA-ES-BJ | 102 | 35 | 117 | 7 | 97 |
| All three cell
lines | 56 | 3 | 53 | 1 | 49 |
Additional evaluations with real-time PCR for
selected candidate genes yielded consistent results regarding
changes in expression of several genes. A consistent increase of
expression in all three cell lines was observed after TRAIL and TRD
treatment for GADD45A (growth arrest and DNA damage A), PPP1R15A
(protein phosphatase 1, regulatory subunit 15A) and HSPA1B (heat
shock 70 kDa protein 1B) (Table
II). Further, microarray analysis revealed a downregulation of
Wee1 (protein kinase wee1), FADD (Fas-associated protein with death
domain), Fyn (proto-oncogene tyrosine-protein kinase Fyn) and PPM1D
(protein phosphatase 1D) in several treatment groups.
| Table IISummary of the microarray and rtPCR
data of the selected candidate genes. |
Table II
Summary of the microarray and rtPCR
data of the selected candidate genes.
| Signal log ratio
control vs. TT |
|---|
| Gene symbol/cell
line | GADD45A | PPP1R15A | HSPA1B |
|---|
| Microarray
data | | | |
| A-204 | 1.77 | 2.03 | 0.68 |
| SK-LMS-1 | 1.77 | 1.96 | 2.13 |
| VA-ES-BJ | 1.19 | 2.17 | 1.04 |
| rtPCR data | | | |
| A-204 | 2.05 | 1.81 | 0.82 |
| SK-LMS-1 | 1.74 | 1.36 | 1.69 |
| VA-ES-BJ | 1.89 | 2.24 | 1.57 |
Western blot analyses demonstrated
consistent results for gene expression and protein levels for
GADD45A, HSPA1B, PPP1R15A and WEE1
For GADD45A, the results of the western blot
analyses corresponded to the gene expression changes in A-204 and
VA-ES-BJ cells. Specifically, combination treatment with TRD and
TRAIL caused increased protein expression compared to control and
treatment with TRAIL (Fig. 7A).
HSPA1B was upregulated in TRD-treated SK-LMS-1 and VA-ES-BJ cells
corresponding to the results of the microarray and PCR analyses
(Fig. 7B). Analogous to the gene
expression levels, protein expression of PPP1R15A was enhanced by
treatment with the different agents in all cell lines, in which
combination treatment led to the highest protein levels (Fig. 7C). In all treated cell lines, Wee1
was downregulated in the microarray analyses and in the protein
analyses after treatment with TRD (Fig. 7D).
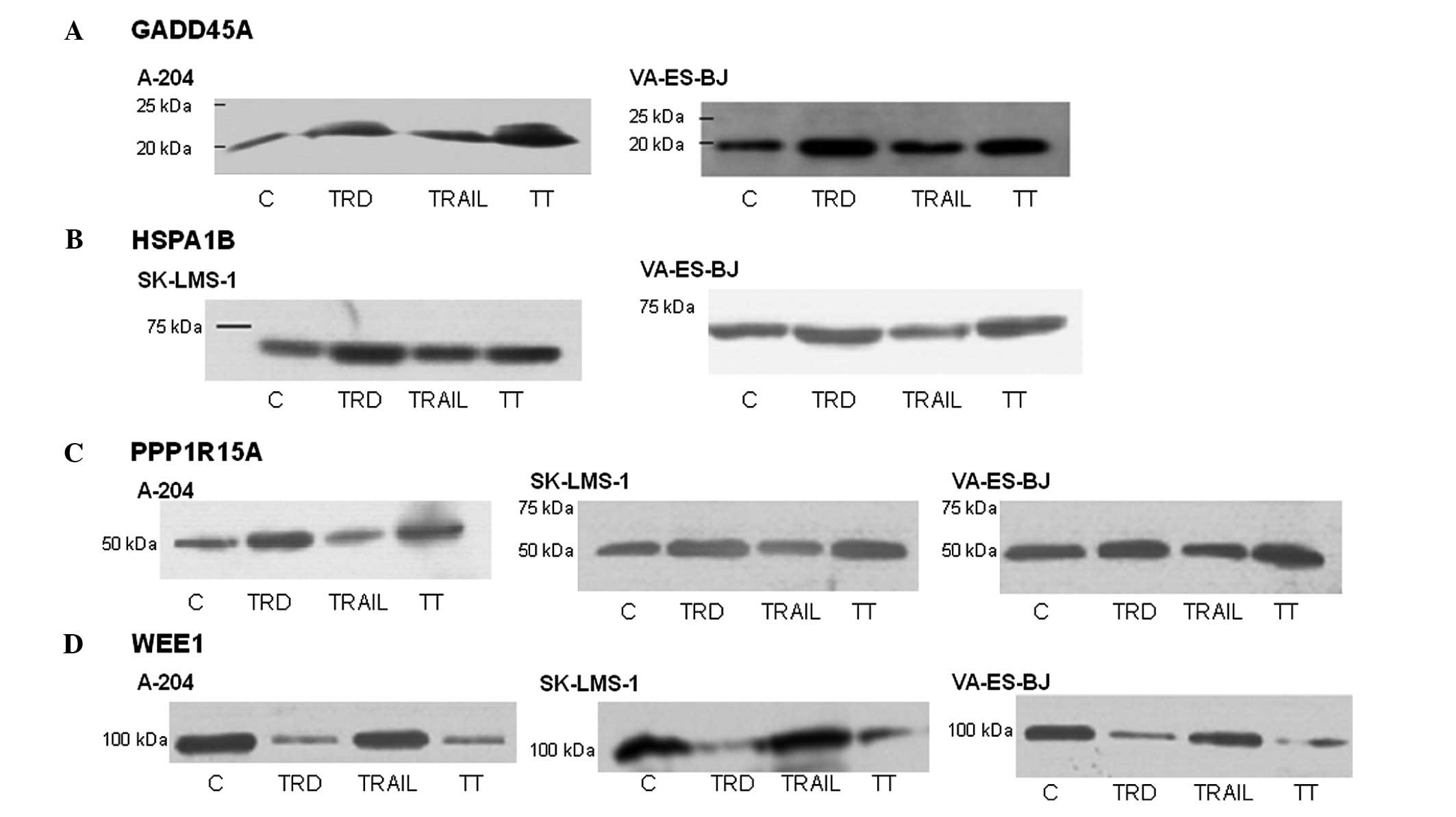 | Figure 7Selected western blot analysis
results for candidate proteins. Protein isolation was performed
after an incubation time of 8 h with the respective substances.
Antibodies used: GADD34/PPP1R15A (rabbit, S-20), GADD45α (rabbit,
H-165), HSP70 (mouse, C92F3A-5) and Wee1 (rabbit, C-20). C,
control; TRD, 250 μmol/l taurolidine; TRAIL, 250 ng/ml
TRAIL; TT, 250 μmol/l TRD and 250 ng/ml TRAIL. |
Discussion
Since the discovery of TRAIL as a cell
death-inducing member of the TNF-superfamily, its effects on
apoptosis have been demonstrated for several malignancies,
including soft tissue sarcomas (46,47).
In order to overcome TRAIL-resistance or enhance its apoptotic
activity there is an increasing interest to find suitable
combination partners for TRAIL (48–50).
Recent trails unveiled the anti-neoplastic qualities of TRD
(31,36,38,51).
In this study, we chose to combine TRAIL with TRD to treat three
different soft tissue sarcoma cell lines. In our study the
combination of TRAIL and TRD induced apoptotic cell death in A-204
rhabdomyosarcoma and VA-ES-BJ epithelioid sarcoma cells but not in
SK-LMS-1 leiomyosarcoma cells. For both treated cell lines, the
apoptotic index could be increased significantly by addition of
TRAIL compared to treatment with TRD alone, suggesting that TRD and
TRAIL work synergistically in rhabdomyosarcoma and epithelioid
sarcoma cell lines beyond the mere additive effect. Earlier trials
also revealed synergistic effects of TRAIL and the chemotherapeutic
agent melphalan in the rhabdomyosarcoma cell line Te-671 (52). Likewise, the combined application
of TRAIL and doxorubicin was shown to be more effective than single
treatment of TRAIL in several rhabdomyosarcoma cell lines (53). To our knowledge, there are no
trials evaluating TRAIL in epithelioid sarcomas. Merely paclitaxel
and 5-FU are known to induce apoptosis in different epithelioid
sarcoma cell lines (54).
TRD inhibited proliferation in all tested sarcoma
cell lines. Strikingly, the anti-proliferative effect of TRD was
enhanced significantly in SK-LMS-1 leiomyosarcoma cells by addition
of TRAIL. Meanwhile, recent studies revealed TRD-induced
proliferation inhibition in a wide range of malignant cell lines,
including HT1080 human fibrosarcoma cells (30,34,35,40).
Remarkably, proliferation inhibition was accompanied by a
disruption of cell adherence and cytoskeleton which play a crucial
role in tumour growth, metastasis and development. However, the
exact mechanism of TRD-induced proliferation inhibition is still
unknown and the appealing hypothesis that TRD inhibits cell
proliferation by disruption of cell adhesion and cytoskeleton
requires further experimental support.
RNA-microarray technology showed high correlations
between apoptotic efficacy and upregulation of GADD45A (growth
arrest and DNA damage-inducible protein 45), PPP1R15A (protein
phosphatase 1 regulatory subunit 15A, synonym: GADD34) and HSPA1B
(heat shock 70 kDa protein 1B).
GADD45A and PPP1R15A were upregulated with at least
a ≥2-fold change in all three cell lines after single treatment
with TRD and combined treatment with TRD and TRAIL compared to
untreated control. Gene alteration could be confirmed by
quantitative real-time PCR and western blot analysis.
GADD45 proteins co-operate in the activation of S
and G2-M checkpoints following the exposure of cells to UV
irradiation and other genotoxic stresses, thereby inducing growth
arrest and apoptosis (55). The
mechanisms by which GADD45 proteins function in negative growth
control is not fully understood, although upregulation of these
proteins was reported to be associated with increased apoptosis and
p53-independent cell cycle arrest in a variety of soft tissue
sarcomas (56). In a recent
immunohistochemical study, high expression of GADD45 was associated
with reduced invasiveness of chondrosarcomas, suggesting its
potential diagnostic value in the histological grading of malignant
chondrogenic tumours (57).
However, the increased expression of GADD45B in our experiments
suggests a potential involvement of GADD45B in TRD-mediated cell
death in soft tissue sarcoma cells and has to be addressed in
further studies.
Stressful growth conditions and exposure to DNA
damaging agents lead to an upregulation of PPP1R15A in a wide range
of human cell lines (58,59). High expression of PPP1R15A is known
to promote apoptotic cell death in a p53-independent manner
(59–61). A recent in vitro study
detected enhanced gene expression of PPP1R15A during TRD-induced
cell death in different malignant cell lines including human
fibrosarcoma cells (62). Thus,
PPP1R15A should be considered as a potential target of TRD in
cancer cells.
Combination treatment with TRAIL and TRD enhanced
HSPA1B protein level in SK-LMS-1 leiomyosarcoma and VA-ES-BJ
epithelioid cell sarcoma cells. HSPA1B protein belongs to the heat
shock protein family with a molecular weight of 70 kDa which acts
as an important regulator of cell growth and survival in a wide
range of cancer cells (63–65).
These proteins are known to affect the intrinsic pathway by the
inhibition of mitochondrial cytochrome c release and also by
preventing apoptosome assembly (29). Upregulation of heat-shock proteins
in different malignant cell lines was associated with an enhanced
resistance towards hypoxia-induced apoptosis (66). More particularly, heat-shock 70 kDa
proteins protected nucleus integrity in non-small cell lung
carcinoma cells which were subjected to heat shock (67). In patients with urothelial
carcinoma high levels of HSPA1B protein were associated with early
tumour progression and invasion (68). Furthermore, recent in vitro
studies suggested that heat-shock 70 kDa proteins might be
responsible for chemoresistance in different malignant cell lines,
and increased levels were found in 5-fluorouracil-resistant colon
carcinoma cells suggesting its involvement in colon cancer
chemoresistance (69), whereas
inhibition of heat shock 70 kDa protein helped to overcome
resistance to etoposide and 5-fluorouracil in oral squamous
carcinoma cells (70). Moreover,
TNF- and TRAIL-induced apoptosis could be suppressed by heat-shock
70 kDa in many different cell types (71). We could not be sure which of the
manifold effects of heat-shock proteins are relevant in sarcoma
cell death. Their upregulation might only be a response to TRAIL-
and TRD-induced cell stress. However, heat-shock proteins are
essential to the survival of many cell types and might be one of
the tools in chemoresistance in leiomyosarcoma or epithelioid cell
sarcoma cells.
Microarray analysis revealed a decrease of FADD with
a ≥2-fold change in all three cell lines after combination
treatment. FADD participates in death signalling in the extrinsic
apoptotic pathway and can be recruited by several death receptors,
including TRAIL-receptor. Subsequently this interaction leads to
recruitment of caspase 8 and initiation of apoptosis. The role of
FADD in TRAIL signalling is controversial (73). Some studies showed that the absence
of FADD leads to partial TRAIL-resistance and concluded that FADD
is necessary for TRAIL-induced apoptosis by the death receptors DR4
and DR5 (74,75). Other groups showed that the
induction of apoptosis by TRAIL is independent of FADD in different
cell lines (73,76,77).
Additionally, FADD was described as a negative regulator of the
transcription factor NF-κB (nuclear factor κ-light chain-enhancer
of activated B-cells), which promotes cell survival and tumour
invasiveness of fibrosarcoma cells (78,79).
In our study, we observed consistent downregulation of FADD mRNA in
all cell lines and protein levels in the SK-LMS-1 cells. Repetitive
western blot analyses in A-204 and VA-ES-BJ did not show
significant changes in protein expression. Therefore, a meaningful
interpretion whether the downregulation of FADD plays a role in
activation of apoptosis in the tested soft tissue sarcoma cells is
not possible based on our data.
Expression of the PPM1D gene was downregulated with
a ≥2-fold change in all three sarcoma cell lines. PPM1D, also known
as Wip1, is a member of the nuclear type 2C protein phosphatase
family and is known to be a negative regulator of cell stress
response pathways (80). Previous
studies have shown that PPM1D expression and phosphatase activity
are required for the survival and progression of breast and ovarian
carcinoma cells (81–83). Loss of PPM1D gene function
sensitises mouse embryonic fibroblasts to stress- and DNA
damage-induced apoptosis (84).
PPM1D overexpression has been observed in neuroblastomas,
medulloblastomas, pancreatic adenocarcinomas and gastric carcinomas
(85–89). Since there has been evidence that
PPM1D acts as an oncogene, efforts were made to find selective
inhibitors of PPM1D. As expected, tumour cell lines that
overexpress PPM1D have shown to be more sensitive to PPM1D
inhibition and consecutive apoptosis than cell lines with normal
levels (90). These findings point
to PPM1D as a regulator in tumour cell survival.
The cell cycle gene Wee1 was downregulated in all 3
cell lines after single treatment with TRD as well as combined
treatment with TRD and TRAIL. Though PCR measurements were not
performed to confirm gene expression, microarray analyses and
western blot analyses results were consistent for all cell lines.
The Wee1 protein kinase functions as key regulator of the
G2/M-checkpoint and stabilizes the genome in the S phase (91). Overexpression of Wee1 has
previously been reported in osteosarcoma, glioblastoma, breast
cancer and malignant melanomas (92–95).
Recent studies identified Wee1 as a potential molecular target in
cancer cells and the selective small molecule Wee1-inhibitor
MK-1775 demonstrated promising results in cancer cells with
enhanced levels of Wee1 (96–98).
However, MK-1775 has recently been included in a phase I clinical
trial in patients with advanced solid tumours (95,99).
In a present study, MK-1775 caused significantly apoptotic cell
death in various sarcoma cell lines and patient-derived tumour
explants ex vivo suggesting that Wee1 may represent a new
potential target in the treatment of sarcomas (100).
Combined treatment with TRD and TRAIL led to an
upregulation of the Fyn gene primarily in A-204 and VA-ES-BJ cells.
Fyn belongs to the Src family of kinases and is involved in a
variety of signalling pathways. It is particularly upregulated in
prostate cancer cells and may have a pivotal role in cancer
progression and metastasis (101). Furthermore, high levels of Fyn
activity were correlated with a higher metastatic ability of human
pancreatic carcinoma and murine fibrosarcoma cells (102,103). However, these findings would
indicate an enhanced cancer activity with subsequent disease
aggravation induced by TRAIL and TRD and require further
investigations.
Taken together, all results described above arose
from in vitro tests. To make more concrete conclusions,
further in vivo studies are necessary to specify programmed
cell death following TRD and TRAIL treatment in soft tissue
sarcomas and the results should be validated with primary cultures.
Finally, gene expression and cell viability differed remarkably in
all three analysed sarcoma entities after exposure to TRD and TRAIL
pointing out the pivotal cellular differences among the soft tissue
sarcoma subtypes. Unfortunately, most of the large clinical trials
did not differentiate between histological subtypes because of the
overall rarity of soft tissue sarcomas resulting in generalized
pharmaceutical references and therapy. However, our findings
sustain the approach of individualized therapy and investigation.
Future trials as well as clinical therapy should focus on
histological subtypes and be more indivualized in spite of the
rarity and difficulties.
Acknowledgements
The authors thank Professor W. E.
Schmidt (Department of Medicine I, St. Josef Hospital, Ruhr
University of Bochum) and Professor A. Muegge (Department of
Medicine II, St. Josef Hospital, Ruhr University of Bochum) for
generously supporting our studies. Furthermore, they thank Annegret
Flier, Ilka Werner, Kirsten Mros and Rainer Lebert for technical
assistance. This study was supported by FoRUM Project F544 E-2007,
Ruhr University Bochum, Germany.
References
|
1
|
Hoos A, Lewis JJ and Brennan MF: Soft
tissue sarcoma: prognostic factors and multimodal treatment.
Chirurg. 71:787–794. 2000.(In German).
|
|
2
|
Patrikidou A, Domont J, Cioffi A and Le
Cesne A: Treating soft tissue sarcomas with adjuvant chemotherapy.
Curr Treat Options Oncol. 12:21–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaushal A and Citrin D: The role of
radiation therapy in the management of sarcomas. Surg Clin North
Am. 88:629–646. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O’Brien GC, Cahill RA, Bouchier-Hayes DJ
and Redmond HP: Co-immunotherapy with interleukin-2 and taurolidine
for progressive metastatic melanoma. Ir J Med Sci. 175:10–14.
2006.PubMed/NCBI
|
|
5
|
Solomon LR, Cheesbrough JS, Bhargava R, et
al: Observational study of need for thrombolytic therapy and
incidence of bacteremia using taurolidine-citrate-heparin,
taurolidine-citrate and heparin catheter locks in patients treated
with hemodialysis. Semin Dial. 25:233–238. 2012. View Article : Google Scholar
|
|
6
|
Karavasilis V, Seddon BM, Ashley S,
Al-Muderis O, Fisher C and Judson I: Significant clinical benefit
of first-line palliative chemotherapy in advanced soft-tissue
sarcoma: retrospective analysis and identification of prognostic
factors in 488 patients. Cancer. 112:1585–1591. 2008. View Article : Google Scholar
|
|
7
|
Billingsley KG, Lewis JJ, Leung DH, Casper
ES, Woodruff JM and Brennan MF: Multifactorial analysis of the
survival of patients with distant metastasis arising from primary
extremity sarcoma. Cancer. 85:389–395. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pezzi CM, Pollock RE, Evans HL, et al:
Preoperative chemo-therapy for soft-tissue sarcomas of the
extremities. Ann Surg. 211:476–481. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Donato Di Paola E and Nielsen OS: The
EORTC soft tissue and bone sarcoma group. European Organisation for
Research and Treatment of Cancer. Eur J Cancer. 38(Suppl 4):
S138–S141. 2002.
|
|
10
|
Nedea EA and DeLaney TF: Sarcoma and skin
radiation oncology. Hematol Oncol Clin North Am. 20:401–429. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brodowicz T, Schwameis E, Widder J, et al:
Intensified adjuvant IFADIC chemotherapy for adult soft tissue
sarcoma: a prospective randomized feasibility trial. Sarcoma.
4:151–160. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frustaci S, Gherlinzoni F, De Paoli A, et
al: Adjuvant chemotherapy for adult soft tissue sarcomas of the
extremities and girdles: results of the Italian randomized
cooperative trial. J Clin Oncol. 19:1238–1247. 2001.PubMed/NCBI
|
|
13
|
Bramwell V, Rouesse J, Steward W, et al:
Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma -
reduced local recurrence but no improvement in survival: a study of
the European Organization for Research and Treatment of Cancer Soft
Tissue and Bone Sarcoma Group. J Clin Oncol. 12:1137–1149.
1994.
|
|
14
|
Sarcoma Meta-analysis Collaboration:
Adjuvant chemotherapy for localised resectable soft-tissue sarcoma
of adults: meta-analysis of individual data. Lancet. 350:1647–1654.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hirata T, Yonemori K, Ando M, et al:
Efficacy of taxane regimens in patients with metastatic
angiosarcoma. Eur J Dermatol. 21:539–545. 2011.PubMed/NCBI
|
|
16
|
Penel N, Van Glabbeke M, Marreaud S, Ouali
M, Blay JY and Hohenberger P: Testing new regimens in patients with
advanced soft tissue sarcoma: analysis of publications from the
last 10 years. Ann Oncol. 22:1266–1272. 2011.PubMed/NCBI
|
|
17
|
Mentzel T: Epithelioid sarcoma:
morphologic variants and differential diagnosis. Pathologe.
31:135–141. 2010.PubMed/NCBI
|
|
18
|
Chromik AM, Daigeler A, Bulut D, et al:
Comparative analysis of cell death induction by Taurolidine in
different malignant human cancer cell lines. J Exp Clin Cancer Res.
29:212010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chromik AM, Daigeler A, Hilgert C, et al:
Synergistic effects in apoptosis induction by taurolidine and TRAIL
in HCT-15 colon carcinoma cells. J Invest Surg. 20:339–348. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Daigeler A, Chromik AM, Geisler A, et al:
Synergistic apoptotic effects of taurolidine and TRAIL on squamous
carcinoma cells of the esophagus. Int J Oncol. 32:1205–1220. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Daigeler A, Chromik AM, Haendschke K, et
al: Synergistic effects of sonoporation and taurolidin/TRAIL on
apoptosis in human fibrosarcoma. Ultrasound Med Biol. 36:1893–1906.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yagita H, Takeda K, Hayakawa Y, Smyth MJ
and Okumura K: TRAIL and its receptors as targets for cancer
therapy. Cancer Sci. 95:777–783. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bouralexis S, Findlay DM and Evdokiou A:
Death to the bad guys: targeting cancer via Apo2L/TRAIL. Apoptosis.
10:35–51. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rowinsky EK: Targeted induction of
apoptosis in cancer management: the emerging role of tumor necrosis
factor-related apoptosis-inducing ligand receptor activating
agents. J Clin Oncol. 23:9394–9407. 2005. View Article : Google Scholar
|
|
25
|
Ashkenazi A, Pai RC, Fong S, et al: Safety
and antitumor activity of recombinant soluble Apo2 ligand. J Clin
Invest. 104:155–162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ganten D, Ruckpaul K and Daniel P:
Molekulare Grundlagen der Apoptose. Grundlagen der Molekularen
Medizin. Springer Berlin; Heidelberg: pp. 159–203. 2008
|
|
27
|
Newsom-Davis T, Prieske S and Walczak H:
Is TRAIL the holy grail of cancer therapy? Apoptosis. 14:607–623.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy receptors. Cell Death Differ. 10:66–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Beere HM: Death versus survival:
functional interaction between the apoptotic and stress-inducible
heat shock protein pathways. J Clin Invest. 115:2633–2639. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jacobi CA, Menenakos C and Braumann C:
Taurolidine - a new drug with anti-tumor and anti-angiogenic
effects. Anticancer Drugs. 16:917–921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McCourt M, Wang JH, Sookhai S and Redmond
HP: Taurolidine inhibits tumor cell growth in vitro and in vivo.
Ann Surg Oncol. 7:685–691. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Petrovic L, Schlegel KA, Ries J, et al: In
vitro effect of taurolidine on squamous cell carcinoma in the oral
cavity. Mund Kiefer Gesichtschir. 7:102–107. 2003.(In German).
|
|
33
|
Gallagher KA, Liu ZJ, Xiao M, et al:
Diabetic impairments in NO-mediated endothelial progenitor cell
mobilization and homing are reversed by hyperoxia and SDF-1 alpha.
J Clin Invest. 117:1249–1259. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Calabresi P, Goulette FA and Darnowski JW:
Taurolidine: cytotoxic and mechanistic evaluation of a novel
antineoplastic agent. Cancer Res. 61:6816–6821. 2001.PubMed/NCBI
|
|
35
|
Braumann C, Henke W, Jacobi CA and Dubiel
W: The tumor-suppressive reagent taurolidine is an inhibitor of
protein biosynthesis. Int J Cancer. 112:225–230. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Neary PM, Hallihan P, Wang JH, Pfirrmann
RW, Bouchier-Hayes DJ and Redmond HP: The evolving role of
taurolidine in cancer therapy. Ann Surg Oncol. 17:1135–1143. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Darnowski JW, Goulette FA, Cousens LP,
Chatterjee D and Calabresi P: Mechanistic and antineoplastic
evaluation of taurolidine in the DU145 model of human prostate
cancer. Cancer Chemother Pharmacol. 54:249–258. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stendel R, Biefer HR, Dekany GM, et al:
The antibacterial substance taurolidine exhibits anti-neoplastic
action based on a mixed type of programmed cell death. Autophagy.
5:194–210. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stendel R, Scheurer L,
Stoltenburg-Didinger G, Brock M and Mohler H: Enhancement of
Fas-ligand-mediated programmed cell death by taurolidine.
Anticancer Res. 23:2309–2314. 2003.PubMed/NCBI
|
|
40
|
Daigeler A, Brenzel C, Bulut D, et al:
TRAIL and Taurolidine induce apoptosis and decrease proliferation
in human fibrosarcoma. J Exp Clin Cancer Res. 27:822008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Han Z, Ribbizi I, Pantazis P, Wyche J,
Darnowski J and Calabresi P: The antibacterial drug taurolidine
induces apoptosis by a mitochondrial cytochrome c-dependent
mechanism. Anticancer Res. 22:1959–1964. 2002.PubMed/NCBI
|
|
42
|
Braumann C, Winkler G, Rogalla P,
Menenakos C and Jacobi CA: Prevention of disease progression in a
patient with a gastric cancer-re-recurrence. Outcome after
intravenous treatment with the novel antineoplastic agent
taurolidine Report of a case. World J Surg Oncol. 4:342006.
View Article : Google Scholar
|
|
43
|
Imhof L, Goldinger SM, Baumann K, et al:
The antibacterial substance, taurolidine in the second/third-line
treatment of very advanced stage IV melanoma including brain
metastases: results of a phase 2, open-label study. Melanoma Res.
Nov 3–2010.(Epub ahead of print).
|
|
44
|
Stendel R, Picht T, Schilling A, et al:
Treatment of glioblastoma with intravenous taurolidine. First
clinical experience. Anticancer Res. 24:1143–1147. 2004.PubMed/NCBI
|
|
45
|
Backes C, Keller A, Kuentzer J, et al:
GeneTrail - advanced gene set enrichment analysis. Nucleic Acids
Res. 35:W186–W192. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wiley SR, Schooley K, Smolak PJ, et al:
Identification and characterization of a new member of the TNF
family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tomek S, Koestler W, Horak P, et al:
Trail-induced apoptosis and interaction with cytotoxic agents in
soft tissue sarcoma cell lines. Eur J Cancer. 39:1318–1329. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Clayer M, Bouralexis S, Evdokiou A, Hay S,
Atkins GJ and Findlay DM: Enhanced apoptosis of soft tissue sarcoma
cells with chemotherapy: A potential new approach using TRAIL. J
Orthop Surg (Hong Kong). 9:19–22. 2001.PubMed/NCBI
|
|
50
|
Kondo K, Yamasaki S, Inoue N, et al:
Prospective antitumor effects of the combination of tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL) and cisplatin
against esophageal squamous cell carcinoma. Surg Today. 36:966–974.
2006. View Article : Google Scholar
|
|
51
|
Walters DK, Muff R, Langsam B, Gruber P,
Born W and Fuchs B: Taurolidine: a novel anti-neoplastic agent
induces apoptosis of osteosarcoma cell lines. Invest New Drugs.
25:305–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kluttermann K, Banning U, Kachel M, Krause
C, Korholz D and Mauz-Korholz C: TRAIL-induced cytotoxicity in a
melphalan-resistant rhabdomyosarcoma cell line via activation of
caspase-2. Anticancer Res. 26:351–356. 2006.PubMed/NCBI
|
|
53
|
Komdeur R, Meijer C, Van Zweeden M, et al:
Doxorubicin potentiates TRAIL cytotoxicity and apoptosis and can
overcome TRAIL-resistance in rhabdomyosarcoma cells. Int J Oncol.
25:677–684. 2004.PubMed/NCBI
|
|
54
|
Heikaus S, Matuszek KS, Suschek CV, et al:
Paclitaxel (Taxol)-induced apoptosis in human epithelioid sarcoma
cell lines is enhanced by upregulation of CD95 ligand
(FasL/Apo-1L). J Cancer Res Clin Oncol. 134:689–695. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gheorghescu B, Gherman I, Jovin GH, et al:
Absorption studies in patients with parasitic infestation of the
small intestine, before and after treatment. Med Interne. 14:31–38.
1976.PubMed/NCBI
|
|
56
|
Nedeau AE, Gallagher KA, Liu ZJ and
Velazquez OC: Elevation of hemopexin-like fragment of matrix
metalloproteinase-2 tissue levels inhibits ischemic wound healing
and angiogenesis. J Vasc Surg. 54:1430–1438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nedea ME, Vasilescu F, Gheorghescu B,
Pavelescu E and Runcan V: Determination with thin layer
chromatography of the distribution of lipids in the feces and study
of the incorporation of certain C 14 -labeled 1-fatty acids into
the different lipid fractions. Fiziol Norm Patol. 19:67–73.
1973.(In Romanian).
|
|
58
|
Hollander MC, Poola-Kella S and Fornace
AJ: Gadd34 functional domains involved in growth suppression and
apoptosis. Oncogene. 22:3827–3832. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hollander MC, Zhan Q, Bae I and Fornace AJ
Jr: Mammalian GADD34, an apoptosis- and DNA damage-inducible gene.
J Biol Chem. 272:13731–13737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Adler HT, Chinery R, Wu DY, et al:
Leukemic HRX fusion proteins inhibit GADD34–induced apoptosis and
associate with the GADD34 and hSNF5/INI1 proteins. Mol Cell Biol.
19:7050–7060. 1999.PubMed/NCBI
|
|
61
|
Grishin AV, Azhipa O, Semenov I and Corey
SJ: Interaction between growth arrest-DNA damage protein 34 and Src
kinase Lyn negatively regulates genotoxic apoptosis. Proc Natl Acad
Sci USA. 98:10172–10177. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chromik AM, Hahn SA, Daigeler A, et al:
Gene expression analysis of cell death induction by taurolidine in
different malignant cell lines. BMC Cancer. 10:5952010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Rohde M, Daugaard M, Jensen MH, Helin K,
Nylandsted J and Jaattela M: Members of the heat-shock protein 70
family promote cancer cell growth by distinct mechanisms. Genes
Dev. 19:570–582. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Daugaard M, Rohde M and Jaattela M: The
heat shock protein 70 family: highly homologous proteins with
overlapping and distinct functions. FEBS Lett. 581:3702–3710. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Noguchi T, Takeno S, Shibata T, Uchida Y,
Yokoyama S and Muller W: Expression of heat shock protein 70 in
grossly resected esophageal squamous cell carcinoma. Ann Thorac
Surg. 74:222–226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Huang WJ, Xia LM, Zhu F, et al:
Transcriptional upregulation of HSP70-2 by HIF-1 in cancer cells in
response to hypoxia. Int J Cancer. 124:298–305. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Scieglinska D, Piglowski W, Mazurek A, et
al: The HspA2 protein localizes in nucleoli and centrosomes of heat
shocked cancer cells. J Cell Biochem. 104:2193–2206. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Garg M, Kanojia D, Seth A, et al:
Heat-shock protein 70-2 (HSP70-2) expression in bladder urothelial
carcinoma is associated with tumour progression and promotes
migration and invasion. Eur J Cancer. 46:207–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Grivicich I, Regner A, Zanoni C, et al:
Hsp70 response to 5-fluorouracil treatment in human colon cancer
cell lines. Int J Colorectal Dis. 22:1201–1208. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Park SR, Lee KD, Kim UK, et al:
Pseudomonas aeruginosa exotoxin A reduces chemoresistance of oral
squamous carcinoma cell via inhibition of heat shock proteins 70
(HSP70). Yonsei Med J. 51:708–716. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Beere HM: ‘The stress of dying’: the role
of heat shock proteins in the regulation of apoptosis. J Cell Sci.
117:2641–2651. 2004.
|
|
72
|
Valoti G, Nicoletti MI, Pellegrino A, et
al: Ecteinascidin-743, a new marine natural product with potent
antitumor activity on human ovarian carcinoma xenografts. Clin
Cancer Res. 4:1977–1983. 1998.PubMed/NCBI
|
|
73
|
Zhang L and Fang B: Mechanisms of
resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther.
12:228–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kuang AA, Diehl GE, Zhang J and Winoto A:
FADD is required for DR4- and DR5-mediated apoptosis: lack of
trail-induced apoptosis in FADD-deficient mouse embryonic
fibroblasts. J Biol Chem. 275:25065–25068. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Bodmer JL, Holler N, Reynard S, et al:
TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat
Cell Biol. 2:241–243. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Petak I, Vernes R, Szucs KS, et al: A
caspase-8-independent component in TRAIL/Apo-2L-induced cell death
in human rhabdomyosarcoma cells. Cell Death Differ. 10:729–739.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Piras V, Hayashi K, Tomita M and
Selvarajoo K: Enhancing apoptosis in TRAIL-resistant cancer cells
using fundamental response rules. Sci Rep. 1:1442011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Duckett CS: Apoptosis and NF-kappa B: the
FADD connection. J Clin Invest. 109:579–580. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Park JM, Kim A, Oh JH and Chung AS:
Methylseleninic acid inhibits PMA-stimulated pro-MMP-2 activation
mediated by MT1-MMP expression and further tumor invasion through
suppression of NF-kappaB activation. Carcinogenesis. 28:837–847.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fiscella M, Zhang H, Fan S, et al: Wip1, a
novel human protein phosphatase that is induced in response to
ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci
USA. 94:6048–6053. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lambros MB, Natrajan R, Geyer FC, et al:
PPM1D gene amplification and overexpression in breast cancer: a
qRT-PCR and chromogenic in situ hybridization study. Mod Pathol.
23:1334–1345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Natrajan R, Lambros MB, Rodriguez-Pinilla
SM, et al: Tiling path genomic profiling of grade 3 invasive ductal
breast cancers. Clin Cancer Res. 15:2711–2722. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Tan DS, Lambros MB, Rayter S, et al: PPM1D
is a potential therapeutic target in ovarian clear cell carcinomas.
Clin Cancer Res. 15:2269–2280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Xia Y, Ongusaha P, Lee SW and Liou Y-C:
Loss of Wip1 sensitizes cells to stress- and DNA damage-induced
apoptosis. J Biol Chem. 284:17428–17437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Saito-Ohara F, Imoto I, Inoue J, et al:
PPM1D is a potential target for 17q gain in neuroblastoma. Cancer
Res. 63:1876–1883. 2003.PubMed/NCBI
|
|
86
|
Loukopoulos P, Shibata T, Katoh H, et al:
Genome-wide array-based comparative genomic hybridization analysis
of pancreatic adenocarcinoma: identification of genetic indicators
that predict patient outcome. Cancer Sci. 98:392–400. 2007.
View Article : Google Scholar
|
|
87
|
Fuku T, Semba S, Yutori H and Yokozaki H:
Increased wild-type p53-induced phosphatase 1 (Wip1 or PPM1D)
expression correlated with downregulation of checkpoint kinase 2 in
human gastric carcinoma. Pathol Int. 57:566–571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Castellino RC, De Bortoli M, Lu X, et al:
Medulloblastomas overexpress the p53-inactivating oncogene
WIP1/PPM1D. J Neurooncol. 86:245–256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lu X, Nguyen TA, Moon SH, Darlington Y,
Sommer M and Donehower LA: The type 2C phosphatase Wip1: an
oncogenic regulator of tumor suppressor and DNA damage response
pathways. Cancer Metastasis Rev. 27:123–135. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Rayter S, Elliott R, Travers J, et al: A
chemical inhibitor of PPM1D that selectively kills cells
overexpressing PPM1D. Oncogene. 27:1036–1044. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sorensen CS and Syljuasen RG: Safeguarding
genome integrity: the checkpoint kinases ATR, CHK1 and WEE1
restrain CDK activity during normal DNA replication. Nucleic Acids
Res. 40:477–486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Mir SE, De Witt Hamer PC, Krawczyk PM, et
al: In silico analysis of kinase expression identifies WEE1 as a
gatekeeper against mitotic catastrophe in glioblastoma. Cancer
Cell. 18:244–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Iorns E, Lord CJ, Grigoriadis A, et al:
Integrated functional, gene expression and genomic analysis for the
identification of cancer targets. PloS One. 4:e51202009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Posthuma DeBoer J, Wurdinger T, Graat HC,
et al: WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC
Cancer. 11:1562011.PubMed/NCBI
|
|
95
|
Magnussen GI, Holm R, Emilsen E, Rosnes
AK, Slipicevic A and Florenes VA: High expression of wee1 is
associated with poor disease-free survival in malignant melanoma:
potential for targeted therapy. PloS One. 7:e382542012. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hirai H, Iwasawa Y, Okada M, et al:
Small-molecule inhibition of Wee1 kinase by MK-1775 selectively
sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol
Cancer Ther. 8:2992–3000. 2009. View Article : Google Scholar
|
|
97
|
Rajeshkumar NV, De Oliveira E, Ottenhof N,
et al: MK-1775, a potent Wee1 inhibitor, synergizes with
gemcitabine to achieve tumor regressions, selectively in
p53-deficient pancreatic cancer xenografts. Clin Cancer Res.
17:2799–2806. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bridges KA, Hirai H, Buser CA, et al:
MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes
p53-defective human tumor cells. Clin Cancer Res. 17:5638–5648.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hirai H, Arai T, Okada M, et al: MK-1775,
a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of
various DNA-damaging agents, including 5-fluorouracil. Cancer Biol
Ther. 9:514–522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kreahling JM, Gemmer JY, Reed D, Letson D,
Bui M and Altiok S: MK1775, a selective Wee1 inhibitor, shows
single-agent antitumor activity against sarcoma cells. Mol Cancer
Ther. 11:174–182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Saito YD, Jensen AR, Salgia R and Posadas
EM: Fyn: a novel molecular target in cancer. Cancer. 116:1629–1637.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chen Z-Y, Cai L, Bie P, et al: Roles of
Fyn in pancreatic cancer metastasis. J Gastroenterol Hepatol.
25:293–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Koike K, Kogawa K, Takayama T, et al:
Enhanced expression of type IV collagen-binding protein (p29) in
Fyn-transfected murine fibrosarcoma cells. Jpn J Cancer Res.
93:1090–1099. 2002. View Article : Google Scholar : PubMed/NCBI
|